
Abstract
The inherited prion diseases (IPD) are a group of untreatable neurodegenerative diseases that segregate as autosomal dominant traits. Mutations in the prion protein gene (PRNP) were first found to be causal of IPD in 1989, before the molecular genetic characterisation of any other neurodegenerative disease. Predictive testing for IPD has subsequently been carried out at a single UK clinical and research centre for 22 years. We have analysed the uptake, consequences and factors influencing the decision for predictive testing over this period. In all, 104 predictive tests were done on individuals at 50% risk, compared with 135 positive diagnostic tests. Using genealogies from clinical records, we estimated that 23% of those at 50% risk have completed testing. There was no gender bias, and unsurprisingly, there was a slight excess of normal results because some patients were already partly through the risk period because of their age. An unexpectedly large number of patients developed symptoms shortly after predictive testing, suggesting that undisclosed early symptoms of disease may prompt some patients to come forward for predictive testing. Fifteen per cent of predictive tests were done >10 years after molecular diagnosis in a proband. A strong determinant of the timing of testing in these patients was a second diagnosis in the family. IPD may generate infectious prions that might be transmitted by surgical procedures; however, we found no evidence that public health information influenced decisions about predictive testing.
Similar content being viewed by others

INTRODUCTION
Prion diseases are a group of fatal and transmissible neurodegenerative diseases of humans and animals. Fundamental to the pathogenesis of these disorders is the autocatalytic conversion of the normal cell-surface prion protein (PrPC) to a multimeric conformer (PrPSc) and consequent neuronal toxicity. About 10–15% of prion disease is familial, all of which can be explained by mutation in the coding region of the prion protein gene (PRNP), either as missense mutations leading to amino-acid substitutions, truncating mutations leading to premature stop codons, or alteration in the number of an octapeptide repeat moiety in the N-terminal domain.1, 2, 3
A range of clinical syndromes comprise inherited prion diseases (IPD), including the rapidly progressive myoclonic dementia Creutzfeldt–Jakob disease (CJD), and more slowly progressive ataxic and cognitive syndromes such as Gerstmann–Straussler–Scheinker (GSS). The age of clinical onset in IPD varies according to the mutation in the prion gene. Some mutations towards the C-terminal of the gene (eg, c.598G>A p.E200K) have a typical onset in late middle age. Other mutations (eg, 6-octapeptide repeat insertional mutation, 6-OPRI) can have a strikingly early onset in the third decade of life.4 The normal repeat region of PRNP contains four or five copies of an octapeptide motif. Disease-associated insertional mutations are stable between generations and have more than three extra repeats. An inverse correlation between length and mean age of onset has been reported.5 Different phenotypes can occur in association with the same mutation and among family members. Most of the mutations appear to be fully penetrant; families with the c.598G>A p.E200K, c.532G>A p.D178N and 4-OPRI mutations however have provided examples of elderly unaffected gene carriers who appear to have escaped the disease.2, 5
The NHS National Prion Clinic (NPC) based at the National Hospital for Neurology and Neurosurgery provides a tertiary referral service for patients with or at risk of developing prion disease in the United Kingdom. It is notified of all suspect prion disease patients in the United Kingdom and coordinates its work closely with the National CJD Research and Surveillance Unit in Edinburgh. The NPC, and its predecessor specialist clinic at St Mary’s Hospital, London, has been the only specialist NHS centre for prion disease predictive testing since 1990. The experience of prion disease in the early years informed the subsequent development of predictive testing guidelines for Huntington’s disease (HD) from 1993.6 This long-term experience has allowed a close clinical relationship to develop with large extended pedigrees of 6-OPRI and c.305C>T p.P102L IPD.7, 8
This paper describes the experience of predictive testing for IPD by studying a cohort of families identified in the United Kingdom from 1990 until the end of 2011. While less common than many other genetic disorders, the historically early identification of PRNP mutation, the comprehensive referral patterns and single centre in the United Kingdom and the public health implications of prion disease diagnosis offer an opportunity for distinct insights into the factors that determine the uptake and consequences of predictive testing.
Materials and Methods
The family cohort was identified through probands with a molecular diagnosis of IPD in the United Kingdom from 1990 to October 2011 (n=135). This was facilitated by a sample database that has been continuously maintained and records all molecular tests since 1990 at the MRC Prion Unit and predecessor organisations. Clinical files were sought for all these individuals. Seventeen diagnostic tests were excluded because minimal associated genealogical information was found. In all, 14/17 excluded diagnostic tests were from the 1990–1997 period. Pedigrees were constructed using information from clinical notes allowing individuals at risk of IPD to be identified. Only first-degree relatives were included for the purposes of this study (living parents when neither were symptomatic, unaffected siblings and adult offspring). In IPD, there are several examples of a child becoming symptomatic before a parent.9
In counselling for IPD, the Public Health England (formerly the Health Protection Agency) requests that physicians inform families about the potential to transmit prions by invasive surgical or dental procedures. For the purposes of this study, we assume that communication of genetic risk and the public health requirements are universal. Individuals presenting for predictive testing were ascertained in a similar way using the MRC Prion Unit database from 1990 until the end of 2011. The first predictive test for an individual living in the United Kingdom was carried out in September 1991. Eight individuals who had predictive testing and were at 25% risk or less were excluded. Two families, both with a c.598G>A p.E200K mutation, were excluded because feedback of the genetic test results in the proband did not occur. The HD genetic counselling protocol recommends a cooling-off period of at least 1 month between the initial visit and the blood sample being taken. Three individuals fell into this category at the start of the project and were not included in the numbers eligible for predictive testing.
Clinical and molecular genetic research studies at the MRC Prion Unit and NHS NPC have been approved by the local Research Ethics Committee. All mutation nucleotide sequences are available at http://databases.lovd.nl/shared/variants/PRNP. Octapeptide repeat mutations are described in the main text in abbreviated form to assist clarity, please refer to the database for more details.
Statistics were done using the IBM SPSS Statistics package and Excel. We used a permutation/simulation strategy to address questions about whether the observed proportion of abnormal predictive tests was within a range compatible with chance alone. In doing so we assumed a liability curve based on a normal distribution of age at clinical onset, for those mutations in which this was known with confidence, with mean and standard deviations of onsets in years as follows, 6-OPRI 35±7, c.305C>T p.P102L 51±9, c.532G>A p.D178N 49±11 and c.598G>A p.E200K 58±9.2 Estimated prior probabilities of being a mutation carrier were calculated for each individual using their age, and the mean and standard deviation of onset for the specific mutation. This was done with the cumulative NORM.DIST function of Excel. We then simulated mutation discovery in samples of 87 patients based on their prior probabilities of being mutation carriers on 163 780 occasions using random number generation in Excel. Thus, we generated a chance expectation of mutation discovery in the sample that allows for the prior risk for each individual in our sample based on their mutation and age. Estimation of the probability of clinical presentation in the year following testing was calculated by subtraction of the prior probabilities of being a mutation carrier at age at testing from that at age+12 months. Simulation was done in a similar way based on the calculated probabilities of each individual’s presentation with disease in a 12-month period.
RESULTS
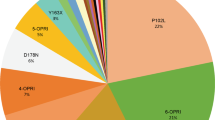
Predictive testing has been carried out on 104 individuals (23% of the 460 eligible at risk individuals) in the United Kingdom over the 22-year period to the end of 2011. Additionally 135 diagnostic tests were carried out (Table 1). Between 1990 and 2011 there has been an increase in uptake of predictive testing (Figure 1). Although this may represent an increasing trend, particularly from around the year 2000 (Pearson’s correlation for annual number of predictive tests 1990–2011 vs duration since 1990: P<0.001), it is also possible that the predictive test number may have plateaued since 2005, with the annual number of tests between 2005 and 2011 fluctuating around an average of 11/year (range 9–14). However, there was a similar trend in abnormal diagnostic tests (Pearson’s correlation for annual number of diagnostic tests 1990–2011 vs duration since 1990: P=0.01), which also may have plateaued between 2005 and 2011 (average 10/year, range 4–17). New diagnosis of IPD might cause an increase in predictive tests as more individuals become aware of their risk status and promptly seek testing. When this was taken into account in regression analysis there was no statistically significant independent linear trend in predictive tests (P=0.25). The IPD mutations most commonly tested were 6-OPRI, c.305C>T p.P102L and c.598G>A p.E200K (Table 1). There were no significant differences in rates of uptake of predictive testing between different mutations.

The number of predictive (positive and negative results) and diagnostic tests (only positive results) in the United Kingdom since the discovery of causal mutations for prion disease in 1989.
From the 104 predictive tests, 42 were abnormal (62 normal, no indeterminate findings). Although this proportion is marginally statistically significant compared with an expectation of 50% abnormal (P=0.03, binomial test), this analysis does not take into account the fact that individuals present for predictive testing having lived through part of the risk period. For four of the most common mutations (6-OPRI, c.305C>T p.P102L, c.532G>A c.532G>A p.D178N, c.598G>A p.E200K), we modelled the true prior probability of being a mutation carrier based on the known liability curves for these mutations. We estimated the proportion of the risk period survived by each individual using mean age of onset and standard deviation from the published literature (see Methods). In all, 35 mutations were found in 87 individuals in this subset of mutation types, whereas 30 would be most likely (95% confidence interval 23–37, based on 163 780 permutations).
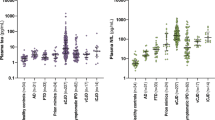
Age at predictive testing varied from 18 to 90, most commonly in the fourth decade of life (Figure 2). The mean age for predictive testing across the mutations is 39.8 years (SD=14.6). The age distribution was distinct between different mutations (P=0.01, one-way ANOVA), driven by a much older age at testing for the c.598G>A p.E200K mutation vs the 6-OPRI mutation (P<0.001, Tukey post hoc test), consistent with the differences in age of clinical onset in these mutations. Predictive testing was similarly frequent in men and women (Table 2, P=0.18).

Requests for predictive testing vary by age and mutation type. Much of this variation can be explained by the expected age at clinical onset of each mutation.
We went on to consider other factors that might determine the timing of predictive testing. Sixty per cent of individuals were tested within 2 years of the proband’s molecular diagnosis (Figure 3). Of those who came forward for predictive testing >10 years after becoming aware of their risk, 8/16 had a further relative who had either predictive or diagnostic testing in the previous 2 years. Of these 16 individuals, 3 were younger than age 18 when they became aware of their risk and were not eligible for predictive testing at that time according to current UK practice. The timing in relation to the decision to have children was not addressed in this study.

Duration between diagnosis in a proband and referral for predictive testing. Many of the late referrals can be explained by proximity to a second diagnosis in the family. Parentheses indicate number of individuals who were under the age of 18 when they were made aware of risk (U=unknown).
We also considered whether social factors or comprehension might influence testing, perhaps because of better access to health care. Higher proportions who were parents, a predominance of individuals living as a couple, and an overrepresentation of those in the higher socio-economic brackets has been observed in predictive testing for other neurodegenerative conditions.10, 11, 12 From 42 individuals who came forward for predictive testing and had an abnormal test result, 20 are enrolled in an observational cohort study National Prion Monitoring Cohort (NPMC) of prion disease that involves longitudinal neuropsychological assessments. The mean IQ for these individuals was 96.6 (SD=11.8), suggesting no strong association between intelligence and predictive testing.
We further considered the possibility that individuals were privately concerned about disease onset because of mild or transient symptoms, and presented for predictive testing to resolve diagnostic issues. From 42 individuals with a positive gene result, 10 became symptomatic in the study period, notably 7/10 became symptomatic within 12 months of predictive testing, more than would be expected by chance (P<0.0001, permutation testing see Methods). These data suggest that proximity to clinical onset is strongly associated with requests for predictive testing.
The proportion of abnormal predictive tests for the 6-OPRI mutation was low (7 patients from 25 tests, P=0.02 vs 50% expectation), particularly so when three individuals who became symptomatic within 12 months were excluded (4 patients, P=0.002 vs 50% expectation). This finding might be explained by the presence of a premorbid personality disorder in some gene mutation carriers that influences the decision to present for predictive testing; however, permutation testing did not show that the proportion of positive results in 6-OPRI were different from chance expectation when taking into account the liability curve for this mutation (P=0.12).
Finally, we considered some of the consequences of living with the risk of IPD. The NPC is aware of two individuals (50% at risk) who were aware of IPD risk and committed suicide. They had not come forward for predictive testing and it is unknown whether they had early symptoms at the time. No asymptomatic individuals who had requested predictive testing have attempted suicide as far as the NPC is aware. A recent study of behavioural and psychiatric symptoms in participants in the MRC Prion Unit’s NPMC study included 24 individuals at risk of IPD (14 mutation carriers and 10 untested first-degree relatives). Current depressive symptoms were reported in at least one assessment by six of these individuals (25%). Although this is a small group from which to draw conclusions, this is above the expected prevalence in the general UK population,13 is similar to that reported in studies of presymptomatic HD,14 and it is certainly our clinical experience that the knowledge of being at risk can have a significant and persistent psychological impact in some individuals.
DISCUSSION
We have analysed the uptake, consequences and factors influencing the decision for predictive testing over a 22-year period at a single national centre. Overall, the uptake of predictive testing in IPD is high (23%) when compared with typical uptake of predictive testing for HD which ranges from 3 to 24% in similar studies.11, 15, 16, 17, 18, 19, 20, 21, 22 The uptake of diagnostic and predictive testing for IPD (Figure 1) increased over the first 16 years of testing, but the data suggest that this may then have reached a plateau, with levels of testing in the subsequent 6 years fluctuating around an average. In comparison, in HD, a decline in the uptake of testing has been observed.22, 23 There are several factors that may have influenced these trends.
First in HD a suggested explanation for driving trends is expectation about experimental therapies.22 In IPD, increasing uptake is associated with increased ascertainment and diagnosis of symptomatic patients and an increase in clinical trial-like research at the MRC Prion Unit since the PRION-1 trial (2001–2007), which involved biannual information days. Twenty individuals who have had predictive testing are enrolled in the on-going NPMC study. The opportunity to contribute to efforts towards the development of treatments is a motivating factor for some patients. A significant difference with other inherited neurodegenerative diseases is that patients with IPD may generate infectious prions that potentially can be transmitted by invasive medical procedures. The NPC therefore delivers a public health message to the family when a proband is diagnosed. We found no evidence that this information influenced decisions for predictive testing; however, it is conceivable that this message was a prompt to encourage sharing of genetic information in families.
Second, initially in HD there was a relatively high uptake of predictive testing which then declined after 4 years and has fluctuated at a lower level since. When a test first becomes available there will be a backlog of people who are asymptomatic who wish to be tested whereas after this group has been tested there will be a fixed, and lower, number of asymptomatic patients accrued each year. In the case of prion diseases, the situation is more complex as there are multiple, phenotypes and mutations which were discovered over a period of years. This would have the effect of offsetting the backlog effect at least for many years. If correct, then one would predict a gradual lowering of the number of asymptomatic patients coming forward now that all of the common mutations are known.
Several factors may influence the proportion of abnormal predictive tests. Excess normal results have also been observed in HD studies of individuals at 50% risk.22, 24 Similarly in the current study the proportion of patients with normal results was greater than half (62 vs 42). However, this analysis is flawed as it assumes that the population studied is the same throughout the entire lifetime of that group. In fact, the number of people who become symptomatic with age increases and these individuals who would have tested positive in the asymptomatic period are removed from the at-risk population so that the chances of getting a positive presymptomatic test decrease. We have overcome this problem by correcting for the risk period. Furthermore, the calculation is probably confounded by the fact some of those tested near to the likely onset of symptoms are thought by the clinician to be symptomatic a situation that certainly occurs in prion disease. After considering these issues we have shown that the IPD results are no different from chance expectation.
The majority of patients asked for predictive testing within 2 years of diagnosis in the proband. In HD, it has been observed that 32.4% of candidates presented for testing within 1 year of becoming aware of their risk.25 Half of those who wait more than 10 years before predictive testing for IPD had a second relative who had recently presented for predictive or diagnostic testing. These data suggest that the major drive for predictive testing is personal exposure to the disease, prompting individuals to think further about their own risk. In studies of other inherited conditions, anxiety reduction, family planning and to inform their children are common reasons for genetic testing.26, 27, 28 Although we were not able to assess objectively these aspects, where this information has been recorded in patient notes, similar reasons were documented.
Although suicide rates are increased in patients with HD, this has not been found in presymptomatic patients having predictive testing.11, 29, 30 The decline of adverse events over time is a common occurrence in predictive testing programs.31, 32 Our protocol is always to involve local Clinical Geneticists in predictive testing, and to date we are not aware of any suicides or attempts in patients who have had predictive testing. Nevertheless, psychiatric morbidity is prevalent, particularly mild-moderate mood disorder, emphasising the need for long-term follow-up and support.
We noted a low proportion of positive predictive tests with the 6-OPRI IPD mutation; however, this was not statistically significant and further studies are required. Clinical assessment of patients from the large British kindred affected by 6-OPRI led to the observation that patients carrying the mutation were often reported to have abnormal, antisocial personality traits from a very young age, predating the onset of the classical neurological or cognitive symptoms of the disease by many years.4, 33 Interestingly, this has also been reported in a small number of patients with different OPRI mutations.34, 35 Whether this effect is due to the mutation per se or to environmental factors remains unclear. We have made efforts to address this intriguing issue with prospective assessment of at-risk individuals using personality assessment and neuropsychological tools, but have so far been unable to engage sufficient numbers of these individuals with this research to allow meaningful conclusions to be drawn.
References
Owen F, Poulter M, Lofthouse R et al: Insertion in prion protein gene in familial Creutzfeldt-Jakob disease. Lancet 1989; 1: 51–52.
Mead S : Prion disease genetics. Eur J Hum Genet 2006; 14: 273–281.
Hsiao K, Baker HF, Crow TJ et al: Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature 1989; 338: 342–345.
Collinge J, Brown J, Hardy J et al: Inherited prion disease with 144 base pair gene insertion: II: Clinical and pathological features. Brain 1992; 115: 687–710.
Kaski DN, Pennington C, Beck J et al: Inherited prion disease with 4-octapeptide repeat insertion: disease requires the interaction of multiple genetic risk factors. Brain 2011; 134: 1829–1838.
International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Huntington’s Chorea. Guidelines for the molecular genetics predictive test in Huntington’s disease. Neurology 1994; 44: 1533–1536.
Mead S, Poulter M, Beck J et al: Inherited prion disease with six octapeptide repeat insertional mutation—molecular analysis of phenotypic heterogeneity. Brain 2006; 129: 2297–2317.
Webb TEF, Poulter M, Beck J et al: Phenotypic heterogeneity and genetic modification of P102L inherited prion disease in an international series. Brain 2008; 131: 2632–2646.
Beck JA, Poulter M, Campbell TA et al: PRNP allelic series from 19 years of prion protein gene sequencing at the MRC Prion Unit. Hum Mutat 2010; 31: E1551–E1563.
Binedell J, Soldan JR, Harper PS : Predictive testing for Huntington’s disease: I. Predictors of uptake in South Wales. Clin Genet 1998; 54: 477–488.
Goizet C, Lesca G, Durr A : Presymptomatic testing in Huntington's disease and autosomal dominant cerebellar ataxias. Neurology 2002; 59: 1330–1336.
Rodrigues CS, de Oliveira VZ, Camargo G et al: Presymptomatic testing for neurogenetic diseases in Brazil: assessing who seeks and who follows through with testing. J Genet Couns 2012; 21: 101–112.
Jenkins R, Lewis G, Bebbington P et al: The National Psychiatric Morbidity surveys of Great Britain—initial findings from the household survey. Psychol Med 1997; 27: 775–789.
Julien CL, Thompson JC, Wild S et al: Psychiatric disorders in preclinical Huntington’s disease. J Neurol Neurosurg Psychiatry 2007; 78: 939–943.
Presymptomatic testing for Huntington’s disease: a world wide survey. The World Federation of Neurology Research Group on Huntington’s Disease. J Med Genet 1993; 30: 1020–1022.
Laccone F, Engel U, Holinski-Feder E et al: DNA analysis of Huntington’s disease: five years of experience in Germany, Austria, and Switzerland. Neurology 1999; 53: 801–806.
Maat-Kievit A, Vegter-van der Vlis M, Zoeteweij M et al: Paradox of a better test for Huntington’s disease. J Neurol Neurosurg Psychiatry 2000; 69: 579–583.
Harper PS, Lim C, Craufurd D : Ten years of presymptomatic testing for Huntington’s disease: the experience of the UK Huntington’s Disease Prediction Consortium. J Med Genet 2000; 37: 567–571.
Creighton S, Almqvist EW, MacGregor D et al: Predictive, pre-natal and diagnostic genetic testing for Huntington's disease: the experience in Canada from 1987 to 2000. Clin Genet 2003; 63: 462–475.
Tassicker RJ, Marshall PK, Liebeck TA et al: Predictive and pre-natal testing for Huntington Disease in Australia: results and challenges encountered during a 10-year period (1994–2003). Clin Genet 2006; 70: 480–489.
Panas M, Karadima G, Vassos E et al: Huntington’s disease in Greece: the experience of 14 years. Clin Genet 2011; 80: 586–590.
Morrison PJ, Harding-Lester S, Bradley A : Uptake of Huntington disease predictive testing in a complete population. Clin Genet 2011; 80: 281–286.
Bernhardt C, Schwan AM, Kraus P, Epplen JT, Kunstmann E : Decreasing uptake of predictive testing for Huntington’s disease in a German centre: 12 years’ experience (1993–2004). Eur J Hum Genet 2009; 17: 295–300.
Tyler A, Morris M, Lazarou L et al: Presymptomatic testing for Huntington's disease in Wales 1987-90. Br J Psychiatry 1992; 161: 481–488.
Trembath MK, Tassicker RJ, Collins VR et al: Fifteen years of experience in predictive testing for Huntington disease at a single testing center in Victoria, Australia. Genet Med 2006; 8: 673–680.
Molinuevo JL, Pintor L, Peri JM et al: Emotional reactions to predictive testing in Alzheimer’s disease and other inherited dementias. Am J Alzheimers Dis Other Demen 2005; 20: 233–238.
Williamson J, LaRusse S : Genetics and genetic counseling: recommendations for Alzheimer’s disease, frontotemporal dementia, and Creutzfeldt-Jakob disease. Curr Neurol Neurosci Rep 2004; 4: 351–357.
Dufrasne S, Roy M, Galvez M, Rosenblatt DS : Experience over fifteen years with a protocol for predictive testing for Huntington disease. Mol Genet Metab 2011; 102: 494–504.
Almqvist EW, Brinkman RR, Wiggins S, Hayden MR : Psychological consequences and predictors of adverse events in the first 5 years after predictive testing for Huntington's disease. Clin Genet 2003; 64: 300–309.
Gargiulo M, Lejeune S, Tanguy ML et al: Long-term outcome of presymptomatic testing in Huntington disease. Eur J Hum Genet 2009; 17: 165–171.
Shaw C, Abrams K, Marteau TM : Psychological impact of predicting individuals’ risks of illness: a systematic review. Soc Sci Med 1999; 49: 1571–1598.
Broadstock M, Michie S, Marteau T : Psychological consequences of predictive genetic testing: a systematic review. Eur J Hum Genet 2000; 8: 731–738.
Alner K, Hyare H, Mead S et al: Distinct neuropsychological profiles correspond to distribution of cortical thinning in inherited prion disease caused by insertional mutation. J Neurol Neurosurg Psychiatry 2012; 83: 109–114.
Cochran EJ, Bennett DA, Cervenakova L et al: Familial Creutzfeldt-Jakob disease with a five-repeat octapeptide insert mutation. Neurology 1996; 47: 727–733.
Goldfarb LG, Brown P, McCombie WR et al: Transmissible familial Creutzfeldt-Jakob disease associated with five, seven, and eight extra octapeptide coding repeats in the PRNP gene. Proc Natl Acad Sci USA 1991; 88: 10926–10930.
Acknowledgements
We would like to thank Ray Young who assisted with figure design. The work was funded by The Department of Health and University College London (UCL). Some of this work is supported by UCL hospitals NHS Trust Biomedical Research Centre and the UK Medical Research Council. We would like to thank the National CJD Surveillance Unit in Edinburgh which refers patients to the NPC, therefore helping to make this research possible. We are also grateful to patients and relatives who have participated in this research.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Owen, J., Beck, J., Campbell, T. et al. Predictive testing for inherited prion disease: report of 22 years experience. Eur J Hum Genet 22, 1351–1356 (2014). https://doi.org/10.1038/ejhg.2014.42
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2014.42
