
Abstract
Tuberous sclerosis complex (TSC) is an autosomal dominant, multisystem disorder, which affects 1 in 6000 people. About half of these patients are affected by mental retardation, which has been associated with TSC2 mutations, epilepsy severity and tuber burden. The bimodal intelligence distribution in TSC populations suggests the existence of subgroups with distinct pathophysiologies, which remain to be identified. Furthermore, it is unknown if heterozygous germline mutations in TSC2 can produce the neurocognitive phenotype of TSC independent of epilepsy and tubers. Genotype–phenotype correlations may help to determine risk profiles and select patients for targeted treatments. A retrospective chart review was performed, including a large cohort of 137 TSC patients who received intelligence assessment and genetic mutation analysis. The distribution of intellectual outcomes was investigated for selected genotypes. Genotype–neurocognitive phenotype correlations were performed and associations between specific germline mutations and intellectual outcomes were compared. Results showed that TSC1 mutations in the tuberin interaction domain were significantly associated with lower intellectual outcomes (P<0.03), which was also the case for TSC2 protein-truncating and hamartin interaction domain mutations (both P<0.05). TSC2 missense mutations and small in-frame deletions were significantly associated with higher IQ/DQs (P<0.05). Effects related to the mutation location within the TSC2 gene were found. These findings suggest that TSC2 protein-truncating mutations and small in-frame mutations are associated with distinctly different intelligence profiles, providing further evidence that different types and locations of TSC germline mutations may be associated with distinct neurocognitive phenotypes.
Similar content being viewed by others

Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant, multisystem disorder caused by heterozygous mutations in the tumor-suppressor genes TSC1 and TSC2.1, 2 Their protein products, hamartin and tuberin respectively, interact to form a protein complex that inhibits signal transduction to the downstream effectors of the mammalian target of rapamycin complex 1 (mTORC1), a serine-threonine kinase with major roles in cell growth signaling. Various regions in the TSC2 C-terminal domain, including the GTPase-activating protein (GAP) domain, appear to be important for normal TSC2 protein function in the mTOR pathway regulation of mTOR.3 Mutations in TSC1 and TSC2 are typically inactivating, resulting in little to no protein activity, leading to upregulation of mTORC1.4, 5 This results in a constitutive growth phenotype with development of hamartomas in various organ systems, including the brain. More than 90% of individuals with TSC show neuroanatomical abnormalities such as tubers, sub-ependymal growths and white matter abnormalities.6 Most patients are affected by epilepsy, often presenting with infantile spasms (IS) as the initial symptom of the disorder.
The prevalence of mental retardation (MR) in TSC is estimated to be between 44 and 70% and has been associated with tuber burden, tuber/brain proportion, early seizure onset, IS, mixed seizure types, TSC2 mutations and poor seizure control.7, 8, 9, 10, 11, 12, 13 A bimodal intellectual quotient (IQ) distribution in the total TSC population has been suggested7 and was recently refined as being observed only in the TSC2 population.10
TSC patients with germline TSC1 and TSC2 mutations have only one fully functional TSC2 allele in all their cells, and this condition could lead to neurocognitive dysfunction through the mechanism of haplo-insufficiency,14, 15, 16 similar to Fragile-X syndrome and Neurofibromatosis type 1.14, 15, 16 However, in TSC there are additional factors which may contribute to cognitive impairment, including loss of heterozygosity, which may contribute to tuber development,17, 18 and effects of early onset and refractory epilepsy. Thus far, no associations have been found between specific TSC mutation types and cognitive outcomes,10, 19 although there are reports on associations with epilepsy and psychiatric features.10, 19, 20, 21, 22 As most of these studies have limited power or do not address all mutation types of interest, more extensive investigations are warranted to determine potential correlations between genotype and neurocognitive phenotype in TSC. Furthermore, as mTOR-inhibitors are now under investigation to prevent or reverse neurocognitive morbidity in TSC, more specific information on genotype–phenotype associations will assist clinicians and caregivers in these important treatment decisions. In this study, we use quantitative intelligence outcomes and genetic mutation results of a large TSC patient cohort to explore the intellectual phenotype and associations with the affected gene and specific gene domains, mutation types and locations.
Materials and methods
Study group
The charts of all 377 patients with a definite diagnosis of TSC who were treated at the Herscot Center for TSC at Massachusetts General Hospital (MGH) were reviewed. TSC patients who had received genetic mutation analysis and neuropsychological assessment at the MGH Psychology Assessment Center were identified. This study was approved by the Institutional Review Board of MGH.
Cognitive assessment
Comprehensive neuropsychological evaluations, including intellectual functioning, were performed by an experienced neuropsychologist (MP). For all patients, the outcome of the most recent full-scale intelligence quotient (IQ) assessment was selected. These were available by one of the following five neuropsychological measures, according to best practice standards: (1) Bayley Scales of Infant Development – 2nd edition (BSID),23 (2) Stanford–Binet Intelligence Scale – 5th edition,24 (3) Wechsler Preschool and Primary Scale of Intelligence – 3rd edition,25 (4) Wechsler Intelligence Scale for Children – 4th edition26 and (5) Wechsler Abbreviated Scale of Intelligence – revised.27 The BSID and Stanford–Binet also provide mental age scores, which are based on a patient's raw score converted to a mental age at which an average child would obtain that score. For the patients who were at the floor of the age-appropriate standardized scores, we calculated developmental quotients (DQs) (mental age/chronological age × 100), where a DQ of 100 would be considered the mean. The presence of MR was recorded for each patient with a score of <70, according to the Diagnostic and Statistical Manual of Mental Disorders, 4th edition.
Clinical data
Clinical data were collected from the patient medical records, including information on gender, history of epilepsy and history of IS. We did not have access to clinical data of two patients, and these missing values were excluded when determining percentages in the results.
Genetic analysis
All patients at MGH followed for TSC are offered genetic testing as part of their comprehensive evaluation. Genetic testing of the TSC1 and TSC2 genes, including detecting of large DNA deletions and rearrangements of the TSC2 gene, was performed at Athena Diagnostics (Worcester, MA, USA) or the MGH Neurogenetic Diagnostic Laboratory (Boston, MA, USA). Pathogenic mutations were confirmed by consultation of two TSC mutation databases (website tsc-project.partners.org, chromium.liacs.nl/LOVD2/TSC). Patients with predicted disease-associated mutations of the TSC1 and TSC2 genes were labeled as such. Patients with no definite findings or only polymorphisms were classified as having no mutation identified (NMI). We examined the possible effects of each of the missense mutations identified in these patients using the Alamut Mutation Interpretation Software. A single mutation (TSC2 A460T) was predicted to have a possible effect on splicing, with a score of −33%. However, as this score was <50%, it was considered unlikely to have an effect on splicing. Patients with two pathogenic mutations were excluded. An individual's specific mutation-type and its exon and nucleotide location within the TSC1 or TSC2 gene were recorded.
To examine the neurocognitive impact of mutations in specific gene domains, functional domains of the TSC1 and TSC2 gene products were selected, including the TSC1 tuberin interaction domain (TID), the TSC2 hamartin interaction domain (HID) and the TSC2 GAP domain.5, 28 Mutations were additionally classified into protein-truncating (PT; nonsense, frame-shift, splice site, large deletions of at least one exon) and non-truncating (missense, small in-frame deletions and insertions) mutations. Protein-truncating mutations were divided into proximal and distal mutations, determined from the middle exon of each gene.
To investigate the effect of gene location of TSC2 missense mutations, these were grouped into three subsets according to the exon (E) location of the affected amino acid: affecting or potentially affecting HID–TID (E1–E22); the GAP domain (E34–E41); and mutations in between these two regions (E23–E33).
Statistical analysis
Statistical analyses were performed using SPSS Version 11.5 (SPSS, Inc., Chicago, IL, USA).
T-tests were used to compare selected genetic mutation domains and types with outcomes on intellectual measures. Owing to relatively small sample size, we restricted analyses in the TSC1 cohort to comparing TID mutations with all remaining mutations and with only proximal PT mutations.
For TSC2 mutations, HID mutations were compared with all other mutations and proximal PT mutations (<E22). Additionally, GAP mutations were compared with other distal mutations, PT mutations were compared with missense mutations and small in-frame deletions, and proximal PT mutations were compared with distal PT mutations. To investigate if, within the TSC2 cohort, PT mutations and missense mutations showed distinctly different intellectual profiles, a two-sample Mann–Whitney test was performed.
When not specifically mentioned, all mutation subgroups were compared with the remaining cohort of the respective affected gene. All reported P-values used two-tailed tests of significance with α set at 0.05.
Results
Patient characteristics
Of all 377 patients with a definite diagnosis of TSC, 164 (44%) had received IQ/DQ assessment. Genetic testing had been performed on 137 (85%) of these patients, including 66 men and 71 women, with a mean age of 17 years old (range 3–57) and 4 patients under the age of 5. Of this study group of 137 patients, 35 (26%) patients were affected by a pathogenic TSC1 mutation, 81 (59%) patients by a pathogenic TSC2 mutation and in 21 (15%) patients no mutation could be identified (NMI). The distribution of the pathogenic mutations within the TSC1 and TSC2 genes is shown in Figure 1.

TSC1 and TSC2 gene exon map, depicting mutation types of patients with and without MR. CaMD, calmodulin-binding domain; CCD, coil–coil domain; ERM, ezrin–radixin–moesin; GAP, GTPase-activating protein; LZD, leucine zipper domain; TAD, transcription-activating domain; TMD, transmembrane domain.
Intellectual profiles of TSC1, TSC2 and NMI cohorts
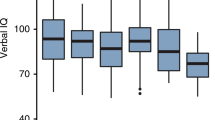
Of the total study group, the mean IQ/DQ was 71.1 (range 7–135). In 36 (22%) patients, conversion to DQ was performed. The prevalence of MR was 23% for the TSC1 population, 57% for TSC2 patients and 29% for the NMI cohort (Figure 1, Table 1). Intelligence scores for the total study group and according to mutation subtype are illustrated in Figure 2a and confirm the bimodal appearance of the IQ/DQ distribution of the total and TSC2 cohort (Figures 2a and b). The mean IQ/DQ of the TSC1 and TSC2 mutation subgroups was 83 and 64, respectively, where male and female patients showed identical mean IQ/DQs (Figure 3a). The mean IQ/DQ of the total NMI subgroup was 79, with the 13 men showing a mean IQ/DQ of 77, and the 8 women a mean of 84. Of note is that the NMI subgroup consisted of 13 men and 8 women while the TSC1 and TSC2 cohorts each had a slight preponderance of women.

Histograms depicting intelligence distributions of selected TSC mutation groups. (a) Intelligence outcomes of the total TSC cohort, including the TSC1 (dark gray), TSC2 (shaded gray) and NMI (light gray) cohorts. (b) Intelligence outcomes of TSC2 mutation subgroups with missense mutations and small in-frame deletions (dark gray), proximal protein-truncating mutations (shaded gray) and distal protein-truncating mutations (light gray).

Boxplots depicting intelligence outcomes of selected TSC mutation cohorts. (a) Intelligence outcomes of TSC1, TSC2 and NMI mutation cohort, including the mean intelligence, SD and outliers. (b) Intelligence distributions for TSC2 subgroups with small in-frame deletions, proximal protein-truncating mutations, distal protein-truncating mutations. Small in-frame mutations included missense mutations and deletions <1 exon.
Genotype–phenotype analyses TSC1 mutations
In the TSC1 cohort, the 11 (33%) patients with a mutation in the TID showed a significantly lower mean IQ/DQ of 66 (P<0.03) compared with 88 in the remaining TSC1 cohort (for epilepsy characteristics, see Table 1). Compared with patients with proximal PT mutations (<E15) not affecting the TID domain who showed a higher mean IQ/DQ of 84, the IQ/DQs of patients with TID mutations remained lower, although not significantly (P<0.12). Of the four TSC1 missense mutations, one patient had an IQ of 37, which lowered the mean of the other three related patients who had IQ/DQs between 72 and 103. Of interest are the relatively high IQ/DQs associated with the three splice site mutations affecting the proximal TID, contrary to the more distal splice site mutation in I14, which was associated with an IQ/DQ of 21. Excluding this latter splice site mutation, no TSC1 patients with a mutation distal of the TID were affected by MR or IS, although most had a positive history of epilepsy (Figure 1 and Table 1).
Genotype–phenotype analyses TSC2 mutations
Within the TSC2 cohort, the patients with mutations in the HID (n=18, 21%) showed a significantly lower mean IQ/DQ of 51 (P<0.05) (for epilepsy characteristics, see Table 1). The patients with a proximal PT mutation (<E22, excluding HID mutations) showed a similar mean IQ/DQ of 49. Distal PT mutations showed a significantly higher mean IQ/DQ of 69 (P<0.04) compared with proximal PT mutations (Figures 2b and 3b). When PT mutations were compared with small in-frame deletions and missense mutations combined, the latter group had a significantly higher mean IQ/DQ of 76 (P<0.05), which was only slightly higher with an IQ/DQ of 78 when only missense mutation were included in the analysis (P<0.04) (Figures 2b and 3b). The Mann–Whitney test confirmed significantly different intellectual profiles for PT and missense mutations. Mutations in the GAP-domain were associated with a mean IQ/DQ of 72, which was higher than remaining mutations with a mean IQ/DQ of 63, but not significantly so (P<0.38). Although the GAP-related IQ/DQ profile was higher than the mean IQ/DQ of all PTs (P<0.12), it was only slightly higher than distal PTs (Table 1). When missense GAP mutations were compared with the remaining GAP mutations, the mean IQ/DQs were similar (71 vs 68).
Grouping all TSC2 missense mutations according to their position on the gene (see Materials and Methods) revealed mean IQ/DQs that were relatively lower for proximal and distal missense mutations, whereas missense mutations in the middle of the TSC2 gene were associated with a relatively normal cognitive phenotype, excluding one outlier (Figure 4).

Boxplot depicting intelligence outcomes for all patients with TSC2 missense mutations, specified per location of the mutation on the TSC2 gene. The outlier in the middle group represents a female patient with a neurological history of refractory partial complex seizures and epilepsy surgery; genetic analysis revealed a P1497R mutation.
Discussion
The data provided by our large study cohort and quantitative intelligence outcomes are the first to indicate significant relationships between specific mutation types and intellectual outcomes in patients with TSC. We confirm the more severe neurocognitive phenotype of the total TSC2 population, and within this cohort, found subgroups showing significantly different intellectual profiles associated with specific genotypes. TSC2 patients with proximal PT mutations and HID mutations showed very similar, significantly lower mean IQ/DQs compared with patients with small, in-frame deletions or missense mutations, confirming case reports finding a milder phenotype in patients with missense mutations.20, 22, 29, 30, 31 This apparent phenotypic dichotomy corresponds with the reported bimodal IQ/DQ distribution in the TSC2 population and we showed that this bimodal appearance can, at least partly, be explained by the effects of different mutation subtypes. Furthermore, these findings correlate well with functional considerations of the effects of different mutations on the TSC1–TSC2 protein complex. Two pathophysiological mechanisms have been reported in TSC, where truncating TSC1 and TSC2 mutations undergo mRNA nonsense-mediated decay, and what little aberrant truncated protein is produced is likely rapidly cleared from cells in the cytosol with no functional protein production. In contrast, missense and other small in-frame mutations may produce an intact, albeit dysfunctional, protein that remains present in the cell with variable remaining function.32, 33 In addition to this ‘all-or-nothing’ theory, we found that PT mutations occurring in the latter half of TSC2 were associated with significantly higher intellectual outcomes than PT mutations in the first half of the protein, suggesting that mutations in TSC2 which leave the HID intact may result in production of some functional protein. This suggests a third pathophysiological mechanism, applying to distal TSC2 truncating mutations that leave the HID intact and result in appropriate formation of the hamartin–tuberin complex, but perhaps disrupt functions exerted by domains in the distal part of TSC2, such as GAP-expression, transcription and binding of kinases.22, 34 There was some suggestion that mutations in the GAP domain were associated with a relatively better neurocognitive profile, although this did not reach significance, perhaps because the relatively small sample size limited the power of this observation as we only investigated mutations directly in this domain.
Investigating the location of mutations more in detail, we found compelling clinical support for previous observations that TSC2 missense mutations that do not affect the hamartin–tuberin interaction or the GAP domain produce a milder phenotype, possibly by retaining some GAP activity.31, 35 The single severely affected patient in the ‘milder’ missense group possibly reflects a remote splice site mutation,36 a severe ‘second hit’ in the other allele, secondary effect of seizures or another pathophysiologic phenomenon in this complex syndrome.
Although the TSC1 cohort was relatively smaller, the findings that mutations in the TID were associated with a more severe cognitive phenotype, even when compared with only proximal PT mutations, confirms the importance of unimpaired binding of hamartin and tuberin through their interaction domains. The three patients with splice site mutations in the TID without cognitive impairment were noteworthy and future studies should focus on protein studies of splice site mutations to learn more about their effect. The relatively low mean IQ/DQ of the five TSC1 missense mutations conflicts with previous observations of a relatively mild phenotype,33, 37 possibly due to the fact that all of these missense mutations occurred in the N-terminal region, which is essential for TSC1 function.32, 38 Of note is that, excluding splice site mutations, none of the patients with more distal mutations were affected by MR.
Previous studies have reported a more severe cognitive phenotype in men compared with women with TSC, using dichotomous outcomes such as ‘MR’.19, 39, 40 However, the nearly identical IQ/DQs in men and women in our large TSC1 and TSC2 cohorts are more consistent with previous data on the prevalence of autism, ADHD and other neuropsychiatric disorders in the TSC population,41, 42 suggesting that genetic effects override gender effects.
The neurocognitive phenotype of patients with TSC is highly variable, because of several effects. Apart from the effect of the genetic mutations, the effect of epilepsy comorbidity, neurosurgery and other anti-epileptic treatments may influence cognitive development in patients with TSC and thus complicate genotype–phenotype associations. Our explorations confirm that cognition and epilepsy are interrelated in TSC, showing greater frequencies of epilepsy and IS in mutation subgroups with a higher prevalence of MR and low mean IQ/DQs, which may contribute to the large ranges of IQ/DQ per mutation subgroup. In addition, it is still unclear if second hits are absolutely necessary for the formation of tubers,17, 18 which are also associated with intellectual outcomes.9, 43
A drawback for this type of study is the use of multiple cognitive measures, which is inherent to the inclusion of different age groups. We accounted for this by using both dichotomous and quantitative outcomes of cognitive functioning, MR and IE, in order to validate and strengthen our findings. As the patients were assessed at different ages, it is unclear if cognitive development is sufficiently stable in patients with TSC to perform such a study. Although thus far there has been little investigation on cognitive development in children and adults with TSC, we recently found that the mean IQ/DQ of a large TSC cohort remained stable over time, albeit showing variability,44 confirming a previous study in infants with TSC.45 As this study group represents only TSC patients who were referred for neuropsychological assessment, this may represent a bias toward more severely affected patients. For this study, we selected intelligence as the primary outcome because these quantitative data provide more precise and powerful information, although this reduced the size of the study cohort. We limited our statistical analysis to intelligence outcomes per mutation type, as similar investigations on epilepsy parameters are ongoing in a larger sample. Of importance is that some subjects in categories associated with a more severe neurocognitive phenotype had excellent intelligence outcomes. This, together with the described missense mutation finding, limits the use of our findings as prognostic indicators and should remind clinicians to be very cautious in attempting phenotype predictions. Future genotype–phenotype correlations in larger cohorts should expand on our findings and include seizure variables, psychiatric burden and the neuroanatomical endophenotype of TSC. Functional analysis on the biochemical effects of specific missense mutations, small in-frame deletions and splice site mutations may identify more genotype–phenotype correlations.
References
van Slegtenhorst M, de Hoogt R, Hermans C et al: Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 1997; 277: 805–808.
European Chromosome 16 Tuberous Sclerosis, Consortium: Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 1993; 75: 1305–1315.
Li Y, Inoki K, Guan KL : Biochemical and functional characterizations of small GTPase Rheb and TSC2 GAP activity. Mol Cell Biol 2004; 24: 7965–7975.
Garami A, Zwartkruis FJ, Nobukuni T et al: Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell 2003; 11: 1457–1466.
van Slegtenhorst M, Nellist M, Nagelkerken B et al: Interaction between hamartin and tuberin, the TSC1 and TSC2 gene products. Hum Mol Genet 1998; 7: 1053–1057.
Ridler K, Bullmore ET, De Vries PJ et al: Widespread anatomical abnormalities of grey and white matter structure in tuberous sclerosis. Psychol Med 2001; 31: 1437–1446.
Winterkorn EB, Pulsifer MB, Thiele EA : Cognitive prognosis of patients with tuberous sclerosis complex. Neurology 2007; 68: 62–64.
Goodman M, Lamm SH, Engel A, Shepherd CW, Houser OW, Gomez MR : Cortical tuber count: a biomarker indicating neurologic severity of tuberous sclerosis complex. J Child Neurol 1997; 12: 85–90.
Jansen FE, Vincken KL, Algra A et al: Cognitive impairment in tuberous sclerosis complex is a multifactorial condition. Neurology 2008; 70: 916–923.
Jansen FE, Braams O, Vincken KL et al: Overlapping neurologic and cognitive phenotypes in patients with TSC1 or TSC2 mutations. Neurology 2008; 70: 908–915.
Shepherd CW, Houser OW, Gomez MR : MR findings in tuberous sclerosis complex and correlation with seizure development and mental impairment. Am J Neuroradiol 1995; 16: 149.
O’Callaghan FJ, Harris T, Joinson C et al: The relation of infantile spasms, tubers, and intelligence in tuberous sclerosis complex. Arch Dis Child 2004; 89: 530–533.
Takanashi J, Sugita K, Fujii K, Niimi H : MR evaluation of tuberous sclerosis: increased sensitivity with fluid-attenuated inversion recovery and relation to severity of seizures and mental retardation. Am J Neuroradiol 1995; 16: 1923.
Cui Y, Costa RM, Murphy GG et al: Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 2008; 135: 549–560.
Narayanan U, Nalavadi V, Nakamoto M et al: S6K1 phosphorylates and regulates fragile X mental retardation protein (FMRP) with the neuronal protein synthesis-dependent mammalian target of rapamycin (mTOR) signaling cascade. J Biol Chem 2008; 283: 18478–18482.
Sharma A, Hoeffer CA, Takayasu Y et al: Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci 2010; 30: 694–702.
Crino PB, Aronica E, Baltuch G, Nathanson KL : Biallelic TSC gene inactivation in tuberous sclerosis complex. Neurology 2010; 74: 1716–1723.
Qin W, Chan JA, Vinters HV et al: Analysis of TSC cortical tubers by deep sequencing of TSC1, TSC2 and KRAS demonstrates that small second-hit mutations in these genes are rare events. Brain Pathol 2010; 20: 1096–1105.
Sancak O, Nellist M, Goedbloed M et al: Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype – phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet 2005; 13: 731–741.
Jansen AC, Sancak O, D’Agostino MD et al: Unusually mild tuberous sclerosis phenotype is associated with TSC2 R905Q mutation. Ann Neurol 2006; 60: 528–539.
Numis AL, Major P, Montenegro MA, Muzykewicz DA, Pulsifer MB, Thiele EA : Identification of risk factors for autism spectrum disorders in tuberous sclerosis complex. Neurology 2011; 76: 981–986.
Mayer K, Goedbloed M, van Zijl K, Nellist M, Rott HD : Characterisation of a novel TSC2 missense mutation in the GAP related domain associated with minimal clinical manifestations of tuberous sclerosis. J Med Genet 2004; 41: e64.
Bayley N : Bayley Scales of Infant Development – Second Edition (BSID-II). San Antonio, TX: Psychological corporation, 1993.
Roid GH : Stanford-Binet Intelligence Scales, 5th edn Itasca, IL: Riverside Publishing, 2003.
Wechsler D : Wechsler Preschool and Primary Scale of Intelligence – Revised. San Antonio, TX: Psychological Corporation, 1989.
Wechsler D : Wechsler Intelligence Scale for Children, 4th edn San Antonio, TX: PsychCorp, 2003.
Wechsler D : Wechsler Abbreviated Scale of Intelligence (WASI). San Antonio, TX: Harcourt Assessments, 1999.
Nellist M, van Slegtenhorst MA, Goedbloed M, van den Ouweland AM, Halley DJ, van der Sluijs P : Characterization of the cytosolic tuberin-hamartin complex. Tuberin is a cytosolic chaperone for hamartin. J Biol Chem 1999; 274: 35647–35652.
Khare L, Strizheva GD, Bailey JN et al: A novel missense mutation in the GTPase activating protein homology region of TSC2 in two large families with tuberous sclerosis complex. J Med Genet 2001; 38: 347–349.
O’Connor SE, Kwiatkowski DJ, Roberts PS, Wollmann RL, Huttenlocher PR : A family with seizures and minor features of tuberous sclerosis and a novel TSC2 mutation. Neurology 2003; 61: 409–412.
Wentink M, Nellist M, Hoogeveen-Westerveld M et al: Functional characterization of the TSC2 c. 3598C>T (p.R1200W) missense mutation that co-segregates with tuberous sclerosis complex in mildly affected kindreds. Clin Genet 2011; e-pub ahead of print 17 February 2011; doi:10.1111/j.1399-0004.2011.01648, PMID: 21332470.
Hoogeveen-Westerveld M, Wentink M, van den Heuvel D et al: Functional assessment of variants in the TSC1 and TSC2 genes identified in individuals with tuberous sclerosis complex. Hum Mutat 2011; 32: 424–435.
Nellist M, van den Heuvel D, Schluep D et al: Missense mutations to the TSC1 gene cause tuberous sclerosis complex. Eur J Hum Genet 2009; 17: 319–328.
Mayer K, Goedbloed M, van Zijl K, Nellist M, Rott HD : Characterisation of a novel TSC2 missense mutation in the GAP related domain associated with minimal clinical manifestations of tuberous sclerosis. J Med Genet 2004; 41: e64.
Nellist M, Verhaaf B, Goedbloed MA, Reuser AJ, van den Ouweland AM, Halley DJ : TSC2 missense mutations inhibit tuberin phosphorylation and prevent formation of the tuberin-hamartin complex. Hum Mol Genet 2001; 10: 2889–2898.
Mayer K, Ballhausen W, Leistner W, Rott H : Three novel types of splicing aberrations in the tuberous sclerosis TSC2 gene caused by mutations apart from splice consensus sequences. Biochim Biophys Acta 2000; 1502: 495–507.
Mozaffari M, Hoogeveen-Westerveld M, Kwiatkowski D et al: Identification of a region required for TSC1 stability by functional analysis of TSC1 missense mutations found in individuals with tuberous sclerosis complex. BMC Med Genet 2009; 10: 88.
Hoogeveen-Westerveld M, Exalto C, Maat-Kievit A, van den Ouweland A, Halley D, Nellist M : Analysis of TSC1 truncations defines regions involved in TSC1 stability, aggregation and interaction. Biochim Biophys Acta 2010; 1802: 774–781.
Au KS, Williams AT, Roach ES et al: Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med 2007; 9: 88–100.
Sancak O, Nellist M, Goedbloed M et al: Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype – phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet 2005; 13: 731–741.
Numis AL, Muzykewicz DA, Pulsifer MB, Thiele EA : Clinical and genetic risk factors for autism spectrum disorders in Tuberous Sclerosis Complex. Neurology 2011; 11: 981–987.
Lewis JC, Thomas HV, Murphy KC, Sampson JR : Genotype and psychological phenotype in tuberous sclerosis. J Med Genet 2004; 41: 203–207.
Kassiri J, Snyder TJ, Bhargava R, Wheatley BM, Sinclair DB : Cortical tubers, cognition, and epilepsy in tuberous sclerosis. Pediatr Neurol 2011; 44: 328–332.
van Eeghen AM, Chu-Shore CJ, Pulsifer MB, Camposano SE, Thiele EA : Cognitive and adaptive development of patients with tuberous sclerosis complex: A retrospective, longitudinal investigation. Epilepsy Behav 2011; e-pub ahead of print 16 November 2011. PMID: 22099526.
Humphrey A, Williams J, Pinto E, Bolton PF : A prospective longitudinal study of early cognitive development in tuberous sclerosis – a clinic based study. Eur Child Adolesc Psychiatry 2004; 13: 159–165.
Acknowledgements
We are very grateful for the helpful suggestions regarding the selection of missense mutation location by Mark Nellist, PhD, and for the graphing assistance by Peter Pulsifer, PhD, and Anna Larson. Statistical analysis was performed with assistance of Brian Healy of the Biostatistics Center of our hospital. This work was supported by the Herscot Center for TSC and NIH/NINDS 1P01NS24279.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
van Eeghen, A., Black, M., Pulsifer, M. et al. Genotype and cognitive phenotype of patients with tuberous sclerosis complex. Eur J Hum Genet 20, 510–515 (2012). https://doi.org/10.1038/ejhg.2011.241
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2011.241
