
Abstract
Multicentric osteolysis with nodulosis and arthropathy (MONA, NAO (OMIM no. 605156)) is an autosomal recessive member of the ‘vanishing bone’ syndromes and is notable for the extent of carpal and tarsal osteolysis and interphalangeal joint erosions, facial dysmorphia, and the presence of fibrocollagenous nodules. This rare disorder has been described previously in Saudi Arabian and Indian families. We now report on the first Turkish family with MONA, further confirming the panethnic nature of this disease. Strikingly, and in addition to the previously noted skeletal and joint features, affected members of this family also had congenital heart defects. Molecular analysis identified a novel MMP2 inactivating mutation that deletes the terminal hemopexin domains and thus confirmed the diagnosis of MONA. On the basis of these findings, we suggest that cardiac defects may also represent a component of this syndrome and thus a physiologically relevant target of MMP-2 activity.
Similar content being viewed by others

Introduction
Multicentric osteolysis–nodulosis–arthropathy (MONA, NAO, Al Aqeel-Sewairi Syndrome, OMIM no. 605156), Torg, and Winchester syndromes represent a group of autosomal recessive multicentric osteolytic disorders of childhood characterized by progressive carpal, tarsal, and interphalangeal involvement.1, 2, 3, 4 The 2001 Nosology of the International Skeletal Dysplasia Society classifies MONA as a variant of Torg syndrome, whereas Winchester syndrome is considered a separate disorder.5 The MONA disease gene was originally localized to 16q12–21, and molecular analysis revealed that all 12 patients affected with MONA from three families lacked MMP-2 gelatinolytic activity resulting from inactivating mutations in the MMP2 gene.6 Since our original report, a number of additional individuals with MONA and MONA-like disorders and MMP2 mutations have been identified, not only revealing the increasing molecular capacity to diagnose the disease,7, 8, 9, 10 but also subtly suggesting that our clinical understanding of the differences between these diseases may still be incomplete. Additional insights into understanding the full spectrum of MMP-2 loss may also come through analysis of Mmp2-deficient mice, wherein the skeletal features of MONA, most notably osteoblastic and osteocytic defects, have now been recently confirmed.11, 12 Similarly, continued identification and characterization of individuals with MMP2 gene mutations may provide unique insights into the physiological role of MMP-2.
We now report the first Turkish family with MONA and describe a novel frameshift mutation deleting the terminal half of the MMP-2 hemopexin domain, a region important in substrate binding and specificity. Intriguingly, the three affected children in this family possessed congenital heart defects ranging from transposition of the great arteries (TGA) to bicuspid aortic valve (BAV) to atrial and ventricular septal defects (ASD and VSD). This finding suggests that the phenotype of MMP-2 deficiency may be broader than originally appreciated.
Material and methods
DNA extraction, PCR, and sequencing
Genomic DNA was extracted from whole blood samples (Puregene DNA isolation kit, Gentra Systems) and the 13 exons, intron/exon boundaries, and flanking 5′-promoter and 3′-untranslated regions of the MMP2 gene were directly sequenced in both directions using the ABI BigDye terminator sequencing kit (Perkin-Elmer) as we have described previously.6 Resulting sequence traces were analyzed using the program Sequencher 4.1 (GeneCodes).
Cell culture, zymography, and RNA isolation
Fibroblast cultures were established from affected individuals using standard skin punch bioposy procedures, as described previously.6 Cultures were grown and maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum until 80% confluent before harvesting.
For zymography, whole blood samples were allowed to clot and centrifuged to isolate serum. Protein concentrations were determined by the Bradford assay. Serum samples were incubated for 10 min with nonreducing Tris-glycine sample buffer (Invitrogen) and electrophoresed and separated (5 μl; 0.2 μg of protein per μl) in 10% gelatin zymogram gels (Novex), which were developed overnight according to the manufacturer's instructions. Gels were stained with Coomassie blue.
RNA was extracted from patient-derived fibroblast cell lines (Qiagen); cDNA was synthesized (Bio-Rad) and directly sequenced in both orientations.
DHPLC screening
Genomic DNA was isolated from 100 unrelated control subjects of Turkish descent and screened for the presence of the identified mutation, using denaturing high-performance liquid chromatography (DHPLC) (Transgenomic). All samples with evidence of heteroduplex formation were then directly sequenced as described above.
Results
Clinical description
Patient 1 (IV-2)
The proband was a 6-year-old Turkish boy who was the second child of healthy parents who were first cousins (Figure 1). The patient was well until 6 months of age, but then was noted to have swelling and tenderness on the fourth finger of his left hand. This progressed with time to include swelling and tenderness on all fingers of his other hand and feet. The patient's weight, height, and head circumference were 17 kg (10th centile), 120 cm (50th centile), and 49 cm (3rd to 10th centile), respectively. He had minor craniofacial anomalies that consisted of narrow forehead with prominent metopic suture, mild hypertelorism, antimongoloid axis, strabismus, bulbous nose, large ears, micrognathia, and gingival hypertrophy (Figure 2a and b). He also had fusiform painful swellings on all fingers and camptodactyly of the fourth finger of the left hand and the fifth finger of the right hand (Figure 2c). Fixed hyperextension of the halluces and palmar and plantar subcutaneous nodules, erythematous lesions on the arms, and mild body hirsutism, especially on the knees, were noted (Figure 2d and e). Hypospadias was also noted. Cardiac auscultation revealed the presence of a mild systolic ejection murmur. Psychomotor development was appropriate for age (IQ: 95), but he had also been diagnosed with attention deficit hyperactivity disorder. Opthalmological examination revealed strabismus and hypermetrophia but without evidence of corneal clouding.

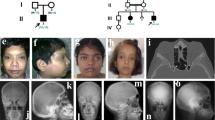
Pedigree of the Turkish family. Arrow indicates proband (patient 1, IV-2).

Patient 1 (IV-2) at age 6 years. (a) Note the mild hypertelorism and antimongoloid axis, strabismus, bulbous nose, large ears, small chin, and (b) gingival hypertrophy. (c) Fusiform painful swellings on all fingers and camptodactyly of the fourth finger of the left hand and the fifth finger of the right hand. (d and e) Fixed hyperextension of the halluces; palmar and plantar subcutaneous nodules are indicated by arrows.
Radiological examination revealed marked generalized osteopenia (BMD Z-score –2.3 SD), a narrowed pelvis, minimal tibial curvature, wide metacarpal bones with irregular contours, especially in the fourth and the fifth fingers bilaterally, and symmetric osteolytic areas in the metatarsal bones and first phalanges of the halluces (Figure 3a and b). A small left kidney was noted on ultrasonography. Consistent with his heart murmur, a BAV was detected by echocardiography (ECHO). Cytogenetic analysis was performed and a normal karyotype was identified (46, XY).

Radiographs of proband (IV-2) and affected sibling. (a) Wide metacarpal bones with irregular contours especially in the fourth and the fifth fingers bilaterally. (b) Symmetric osteolytic areas in the metatarsal bones and first phalanges of the halluces. (c) Radiographs of deceased older sister (IV-1) of patient 1. Note the width of the metacarpal bones and phalanges with camptodactyly of the second and third finger of the left hand and the second finger of the right hand.
The patient was started on a trial of prednisolone therapy (2 mg/kg) and, after 6 months of treatment, the number of painful swellings and nodules decreased and camptodactyly of the fingers improved. At the following yearly examination, his improvements were noted and steroid treatment was not restarted at that time.
Interestingly, his elder sister (IV-1), whom the parents described as also having painful swellings on all fingers, had died due to TGA when she was 6 years old. Examination of his older sister's photographs and hand radiographs revealed the presence of similar craniofacial features and osteopenia and metacarpal and phalangeal osteolysis, respectively, which strongly suggested the same underlying bone disease (Figure 3c).
Patient 2 (IV-6)
A 4-year-old girl, the second cousin of patient 1 (IV-2), was the third child of healthy parents who were also first cousins (Figure 1). She had two healthy elder sisters (IV-4, IV-5). The patient was well until 3 months of age. She then developed progressive painful swellings and tenderness on the proximal parts of her hands and feet. Her weight, height, and head circumference were 13 kg (<3rd centile), 96 cm (<3rd centile), and 48 cm (2nd centile), respectively. She had minor facial anomalies, including narrow forehead, bilateral mild proptosis and hypertelorism, thick lips, full cheeks, micrognathia, bulbous nose, and gingival hypertrophy with high narrow palate and large ears (Figure 4a and b). All fingers had painful fusiform swellings with hyperextension of the metacarpophalangeal joints, flexion contractures of the interphalangeal joints, and palmar and plantar subcutaneous nodules (Figure 4d and f). Severe flexion contractures were found on the left elbow and knees. Deformed feet with foreshortening of all toes with hyperextension of the fifth toe of the left foot and second toe of the right foot were present (Figure 4c). The patient also had single palmer creases bilaterally and hyperpigmented lesions (12 × 5 mm) on the left foot. Opthalmological examination revealed the presence of corneal opacities. Serum chemistry, including a thyroid hormone profile, was normal. Psychomotor development was compatible with her chronological age.

Patient 2 (IV-6) at age 4 years. (a) Note the bilateral mild proptosis and hypertelorism, thick lips, full cheeks, micrognathia, bulbous nose, large ears, and (b) gingival hypertrophy with high narrow palate. (c) Deformed feet with foreshortening of all toes with hyperextension of the fifth toe of the left foot and second toe of the right foot were present. (d) All fingers had painful fusiform swellings with hyperextension of the metacarpophalangeal joints and flexion contractures of the interphalangeal joints. (e) Radiographs showing carpal and metacarpal osteolysis associated with severe interphalangeal joint destruction and erosions. (f) Palmar and plantar subcutaneous nodules (indicted by arrows). (g) Radiographs showing tarsal osteolysis associated with severe interphalangeal joint destruction and erosions. (h) Note the broad medial ends of the clavicles indicated by arrowheads.
Skeletal X-ray survey revealed carpal, tarsal, and metacarpal osteolysis that was associated with severe interphalangeal joint destruction and erosions (Figure 4e and g). In addition, broad medial ends of both clavicles were also noted (Figure 4h). A marked generalized osteopenia was also present, which on DEXA exam revealed a BMD Z-score of −3.2 SD. Finally, an ECHO revealed the presence of both atrial and ventricular septal defects (ASD and VSD).
A karyotype was performed and no obvious chromosomal abnormalities were identified (46, XX).
The same dose of prednisolone (2 mg/kg) was given to patient 2 for 6 months followed by a month-long treatment of methotrexate (15 mg/m2/week). However, both therapies were deemed unsuccessful, as her hand and feet deformities increased with time and she was unable to walk without difficulty. At the following yearly examination, we began a second round of prednisolone therapy (2 mg/kg). During the third month of treatment, the number of painful nodules and contractures mildly decreased and she started to walk more comfortably.
Molecular analysis
DNA sequence and biochemical analysis
Given the clinical similarities of these children with previously described MONA-affected patients, we analyzed the MMP2 gene sequence and MMP-2 biochemical activity. Genomic DNA was directly sequenced in both affected and six unaffected family members. In addition, fibroblast cell lines of the affected patients were established, mRNA was extracted, and cDNA was generated and directly sequenced. Both the affected children were homozygous for a novel single nucleotide deletion, 1732delA, in exon 11 (Figure 5a) in both genomic DNA and cDNA samples. This is predicted to result in the loss of the second half of the hemopexin domain. Parents were heterozygous for the deletion, whereas siblings were homozygous for the wild-type allele. The mutation was not present in 200 chromosomes from 100 control individuals of Turkish descent (data not shown).

Frameshift mutation results in the loss of MMP-2 activity. (a) Chromatogram showing the 1732delA frameshift mutation as homozygous in the affected and heterozygous in parents of Patient. (b) Gelatin zymogram shows complete loss of MMP-2 activity in affected serum, 50% activity in that from parents, and 100% activity in noncarrier sibling and population-control serum samples. (c) Structural model of the MMP-2 mutation was based on the coordinates of the wild-type protein (PDB ID 1CK7). Yellow represents the amino portion of the molecule that is expressed before the deletion, whereas the purple C-terminal 83 amino acids are deleted in the truncation. The orange ball represents the zinc ion required for activity of the metalloprotease. Rendering was done using the program O v.12 (courtesy of Dr Alwyn Jones, University of Uppsala, Uppsala, Sweden).
To determine if the 1732delA mutation resulted in the loss of MMP-2 activity, serum from the affected and unaffected family members was then analyzed by zymography. Serum from both the affected individuals was notable for almost complete absence of enzymatic activity. As would be predicted for heterozygous carriers of the mutation, parents had approximately 50% activity compared with noncarrier siblings and population controls (Figure 5b).
Structural analysis
The X-ray crystallographic-derived structure of the pro-MMP-2 (Protein Data Bank ID-1ck7) molecule contains a prodomain, catalytic domain, and fibronectin and hemopexin domains.13 The truncation mutation identified in these children mapped onto the amino-acid residue N577 of the expressed enzyme (Figure 5c). On the basis of this structure,13 the majority of the third and entire fourth hemopexin domain would be predicted to be deleted, as well as one of the calcium-binding ligands contributed by the carbonyl oxygen on D618 (truncated region is highlighted in purple). The hemopexin domain consists of approximately 200 amino acids forming a four-bladed propeller around a central calcium ion. Each ‘propeller’ contains an α-helix and four short antiparallel β-sheets. In the wild-type structure, a disulfide bond between C440 and C641 stabilizes the entire domain. This disulfide bond is lacking in the truncated mutant protein.
Discussion
Multicentric osteolysis, nodulosis, and arthropathy (OMIM no. 605156) is a rare osteolytic syndrome, which was originally described by Al-Aqeel et al3 and Al-Mayouf et al4 in a number of Saudi Arabian families. This condition is primarily characterized by the development of severe osteoporosis and flexion contractures of interphalangeal joints, with fusiform swelling and tenderness of the hands and feet, palmar and plantar subcutaneous nodules in the first years of life, and distinctive facies.3, 4 A number of additional nonosseous features, including hirsutism, have also been described.3 Given the rarity of the syndrome, it is often difficult to prove causation of the novel nonosseous features unless confirmed in additional families.
In this report, we describe progressive distal arthropathy, palmar and plantar subcutaneous nodules, facial changes, osteolysis of bones of hands and feet, and hirsutism in a 6-year-old boy (IV-2) and a 4-year-old girl (IV-6) who were second cousins. The 4-year-old girl (IV-6) demonstrated a more severe phenotype than that of her male cousin (IV-2). Both developed symptoms in the first year of life. Patient 1 (IV-2) had fusiform painful swellings on all fingers and camptodactyly of the fourth finger of the left hand and the fifth finger of the right hand, whereas patient 2 (IV-6) had painful fusiform swellings with hyperextension of the metacarpophalangeal joints in all fingers and flexion contractures of the interphalangeal joints. Both children had similar facial features, which were notable for proptosis, hypertelorism, bulbous nose, thick lower lip, gum hypertrophy, and large and low-set ears. Radiological evaluation revealed a generalized osteopenia, fusiform swelling of fingers with flexion of interphalangeal joints, and osteolysis of carpal and tarsal bones.
The patients were started on a trial of prednisolone therapy and, after 6 months, the number of painful swellings and nodules decreased and camptodactyly of the fingers improved in patient 1. This therapeutic intervention was, however, unsuccessful in patient 2, who had a more severe phenotype. We restarted prednisolone therapy and reexamined her after 1 year.
In addition, and possibly related to the syndrome, patient 1 (IV-2) had attention deficit hyperactivity disorder (ADHD), whereas patient 2 (IV-6) had corneal clouding. Corneal clouding and gum hypertrophy has not been reported previously in the MONA literature (Table 1).
Most striking, however, was that 3 children in this consanguineous family all shared cardiac defects. Patient 1 (IV-2) had a BAV and his older sister, who on the basis of history, facial features, and radiological examination we believe to also have shared the same MMP-2 mutation, died from complications associated with TGA. Patient 2 (IV-6) had both an ASD and VSD. We are aware of only one other reported child with MONA who has been described as having ‘heart disease’;4 however, no information was provided on the nature of the defect. Given the preponderance of congenital heart defects clustering within this single family, and despite the apparently discordant anatomic features – TGA, BAV, ASD, and VSD – we nonetheless suggest that this cardiac association may represent a previously unrecognized component of MONA. At this time, however, the less likely possibilities that this family may also have had an inherited form of BAV or a secondary genetic defect associated with cardiac defects cannot be ruled out.
As has been recently reviewed,14 the increasing recognition of single gene defects underlying congenital heart disease is consistent with our hypothesis that the range of cardiac features in this family may be related to a single gene defect. For example, mutations in a number of cooperative transcription factors, including TBX5,15 NKX2-5,16 and GATA4,17 have now been shown to result in cardiac septation defects during early development. Of these, mutations in TBX5 have also been associated with outflow tract defects, including TGA.18 In addition to transcription factors, mutations in signaling molecules also result in cardiac defects. In particular, mutations in Notch1, which also plays an important role in osteoblast differentiation,19 have been associated with multiple defects in valvulogenesis, including the development of BAV.20 Finally, mutations in genes along a single signaling pathway can also result in cardiac defects. Mutations in the ras-mitogen-activated protein-kinase signaling pathway have been demonstrated to underlie Noonan's syndrome, an autosomal dominant cardio-facio-cutaneous syndrome, associated with septation defects and pulmonary valve defects (reviewed by Gelb and Tartaglia21).
Although an increasing literature is establishing an association between the MMPs and cardiac function, most have focused on their possible role in coronary and carotid atherosclerosis (reviewed by Abilleira et al22). Despite the recent demonstration that MMP-2-deficient mice display features of MONA,11, 12 no reports have described cardiac defects in these mice. If cardiac defects do occur in MMP-2-deficient mice, early lethality may result and possibly explain our finding of lower-than-expected numbers of surviving Mmp2−/− mice obtained in heterozygous crosses (Mosig RA and Martignetti JA, personal communication, 2008). Two studies, on the basis of MMP-2 inhibition, also support our hypothesis for a role of MMP-2 in cardiac formation. First is a study examining developing chick hearts following targeted inhibition of MMP-2. In these studies, at specific early developmental time points, either whole chick embryos were exposed to MMP-2 neutralizing antibodies or these antibodies were directly microinjected in the chick heart fields. Beating cardiac tissues developed, but were differentially associated with defects in ventral tissue fusion and directional defects in looping.23 Second, inhibition of MMP-2 also blocked the epithelial–mesenchymal transition, migration, and invasion of explanted mouse atrioventricular canal cells involved in the endocardial cushion formation necessary for proper heart development.24 Although the defects in chick and mouse do not prove the causality of the defects identified in our patients, they are nonetheless supportive for a role of MMP-2 in heart organogenesis. On the basis of these accumulated findings, and until additional MMP-2-deficient patients with cardiac defects are identified or MMP-2-deficient mice are shown to possess similar developmental defects, we suggest that newly identified patients be carefully screened for cardiac disease.
The inactivating mutation identified in this family is also of interest. The majority of previously reported MMP2 mutations have been postulated to directly affect enzymatic function through disruption of either the catalytic site itself or activation mechanism through mutations in the cysteine-switch domain.6, 7, 8, 9, 10 In general, the hemopexin domain is responsible for dimerization, activation, inhibition, and other functions for numerous proteins across many species.25 Specifically, the MMP-2 hemopexin domain is believed to be critical to a number of higher-order interactions. The two deleted hemopexin domains are believed critical for MMP-2 activation and inhibition, as they are responsible for interaction of several molecules, most notably TIMP-2.13 The activation mechanism of MMP-2 requires binding of the hemopexin domain by TIMP-2 tethered to MTI-MMP, which coordinates the cleavage of the MMP-2 propeptide by a second MTI-MMP molecule.26 Previous studies using site-directed mutagenesis demonstrated that the third and fourth hemopexin repeats are largely responsible for this interaction.26 Specifically, hemopexin domain 3 interacts with the C-terminal ‘tail’ of TIMP-2, and hemopexin domain 4 interacts with the GH loop of TIMP-2. A number of other functions have also been assigned to this region and include the following: the C-terminal PEX domain's inhibitory interaction with integrin αvβ3 on the surface of angiogenic blood vessels;26 requirement of the hemopexin domain for cleavage of collagen type 1, the largest constituent of bone ECM;27, 28 and cleavage of both monocyte chemoattractant protein-3 and stromal-derived factor-1 into antagonists.29, 30
Taken together, these findings in a Turkish family with MONA not only highlight a potential cardiac involvement to the syndrome, but also highlight the importance of the terminal portion of the MMP-2 domain. In turn, the clinical findings may prove helpful in providing better prospective patient care and in understanding the physiologic role of MMP-2 in human development. The molecular findings may clarify which substrates and MMP-2 activities are critical in bone, joint, and possibly cardiac biology, and in the development of this syndrome.
References
Hardegger F, Simpson LA, Segmueller G : The syndrome of idiopathic osteolysis. classification, review, and case report. J Bone Joint Surg Br 1985; 67: 88–93.
Petit P, Fryns JP : Distal osteolysis, short stature, mental retardation, and characteristic facial appearance: delineation of an autosomal recessive subtype of essential osteolysis. Am J Med Genet 1986; 25: 537–541.
Al Aqeel A, Al Sewairi W, Edress B, Gorlin RJ, Desnick RJ, Martignetti JA : Inherited multicentric osteolysis with arthritis: a variant resembling torg syndrome in a saudi family. Am J Med Genet 2000; 93: 11–18.
Al-Mayouf SM, Majeed M, Hugosson C, Bahabri S : New form of idiopathic osteolysis: nodulosis, arthropathy and osteolysis (NAO) syndrome. Am J Med Genet 2000; 93: 5–10.
Hall CM : International nosology and classification of constitutional disorders of bone (2001). Am J Med Genet 2002; 113: 65–77.
Martignetti JA, Aqeel AA, Sewairi WA et al: Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat Genet 2001; 28: 261–265.
Phadke SR, Ramirez M, Difeo A, Martignetti JA, Girisha KM : Torg-winchester syndrome: lack of efficacy of pamidronate therapy. Clin Dysmorphol 2007; 16: 95–100.
Rouzier C, Vanatka R, Bannwarth S et al: A novel homozygous MMP2 mutation in a family with winchester syndrome. Clin Genet 2006; 69: 271–276.
Zankl A, Pachman L, Poznanski A et al: Torg syndrome is caused by inactivating mutations in MMP2 and is allelic to NAO and winchester syndrome. J Bone Miner Res 2007; 22: 329–333.
Zankl A, Bonafe L, Calcaterra V, Di Rocco M, Superti-Furga A : Winchester syndrome caused by a homozygous mutation affecting the active site of matrix metalloproteinase 2. Clin Genet 2005; 67: 261–266.
Inoue K, Mikuni-Takagaki Y, Oikawa K et al: A crucial role for MMP-2 in osteocytic canalicular formation and bone metabolism. J Biol Chem 2006; 281: 33814–33824.
Mosig RA, Dowling O, Difeo A et al: Loss of MMP-2 disrupts skeletal and craniofacial development and results in decreased bone mineralization, joint erosion and defects in osteoblast and osteoclast growth. Hum Mol Genet 2007; 16: 1113–1123.
Morgunova E, Tuuttila A, Bergmann U et al: Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science 1999; 284: 1667–1670.
Bruneau BG : The developmental genetics of congenital heart disease. Nature 2008; 451: 943–948.
Basson CT, Huang T, Lin RC et al: Different TBX5 interactions in heart and limb defined by holt-oram syndrome mutations. Proc Natl Acad Sci USA 1999; 96: 2919–2924.
Schott JJ, Benson DW, Basson CT et al: Congenital heart disease caused by mutations in the transcription factor NKX2-5. Science 1998; 281: 108–111.
Garg V, Kathiriya IS, Barnes R et al: GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature 2003; 424: 443–447.
McElhinney DB, Geiger E, Blinder J, Benson DW, Goldmuntz E : NKX2.5 mutations in patients with congenital heart disease. J Am Coll Cardiol 2003; 42: 1650–1655.
Engin F, Yao Z, Yang T et al: Dimorphic effects of notch signaling in bone homeostasis. Nat Med 2008; 14: 299–305.
Garg V, Muth AN, Ransom JF et al: Mutations in NOTCH1 cause aortic valve disease. Nature 2005; 437: 270–274.
Gelb BD, Tartaglia M : Noonan syndrome and related disorders: dysregulated RAS-mitogen activated protein kinase signal transduction. Hum Mol Genet 2006; 15 (Spec No 2): R220–R226.
Abilleira S, Bevan S, Markus HS : The role of genetic variants of matrix metalloproteinases in coronary and carotid atherosclerosis. J Med Genet 2006; 43: 897–901.
Linask KK, Han M, Cai DH, Brauer PR, Maisastry SM : Cardiac morphogenesis: matrix metalloproteinase coordination of cellular mechanisms underlying heart tube formation and directionality of looping. Dev Dyn 2005; 233: 739–753.
Enciso JM, Gratzinger D, Camenisch TD, Canosa S, Pinter E, Madri JA : Elevated glucose inhibits VEGF-A-mediated endocardial cushion formation: modulation by PECAM-1 and MMP-2. J Cell Biol 2003; 160: 605–615.
Piccard H, Van den Steen PE, Opdenakker G : Hemopexin domains as multifunctional liganding modules in matrix metalloproteinases and other proteins. J Leukoc Biol 2007; 81: 870–892.
Overall CM, King AE, Sam DK et al: Identification of the tissue inhibitor of metalloproteinases-2 (TIMP-2) binding site on the hemopexin carboxyl domain of human gelatinase A by site-directed mutagenesis. the hierarchical role in binding TIMP-2 of the unique cationic clusters of hemopexin modules III and IV. J Biol Chem 1999; 274: 4421–4429.
Patterson ML, Atkinson SJ, Knauper V, Murphy G : Specific collagenolysis by gelatinase A, MMP-2, is determined by the hemopexin domain and not the fibronectin-like domain. FEBS Lett 2001; 503: 158–162.
Tam EM, Moore TR, Butler GS, Overall CM : Characterization of the distinct collagen binding, helicase and cleavage mechanisms of matrix metalloproteinase 2 and 14 (gelatinase A and MT1-MMP): The differential roles of the MMP hemopexin c domains and the MMP-2 fibronectin type II modules in collagen triple helicase activities. J Biol Chem 2004; 279: 43336–43344.
McQuibban GA, Butler GS, Gong JH et al: Matrix metalloproteinase activity inactivates the CXC chemokine stromal cell-derived factor-1. J Biol Chem 2001; 276: 43503–43508.
McQuibban GA, Gong JH, Tam EM, McCulloch CA, Clark-Lewis I, Overall CM : Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3. Science 2000; 289: 1202–1206.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tuysuz, B., Mosig, R., Altun, G. et al. A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur J Hum Genet 17, 565–572 (2009). https://doi.org/10.1038/ejhg.2008.204
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.204
Keywords
This article is cited by
-
Clinical, radiographic and molecular characterization of two unrelated families with multicentric osteolysis, nodulosis, and arthropathy
BMC Musculoskeletal Disorders (2023)
-
Identification and development of the novel 7-genes diagnostic signature by integrating multi cohorts based on osteoarthritis
Hereditas (2022)
-
Identification of fibrinogen as a natural inhibitor of MMP-2
Scientific Reports (2019)
-
Bisphosphonates in multicentric osteolysis, nodulosis and arthropathy (MONA) spectrum disorder – an alternative therapeutic approach
Scientific Reports (2016)
-
Functional characterisation of a novel mutation affecting the catalytic domain of MMP2 in siblings with multicentric osteolysis, nodulosis and arthropathy
Journal of Human Genetics (2014)
