
Abstract
Interleukin-18 (IL-18) is a key inflammatory molecule suspected of being involved in the etiology of cardiovascular diseases (CVD). Five single nucleotide polymorphisms (SNPs) capturing the common genetic variation of the IL-18 gene (tag SNPs) were genotyped in five European prospective CVD cohorts including 1933 cases and 1938 non-cases as part of the MORGAM Project. Not a single SNP was found associated with CVD. However, a significant (P=0.002) gene–smoking interaction was observed. In smokers, the −105T allele was more frequent in cases than in non-cases (0.29 vs 0.25) and associated with an increased risk of disease (odds ratio (OR)=1.25 (1.07–1.45), P=0.005), whereas the inverse relationship tended to be observed in non-smokers (OR=0.90 (0.78–1.02), P=0.131). The gene–smoking interaction was broadly homogenous across the cohorts and was also observed through haplotype analyses. In conclusion, using the concerted effort of several European prospective CVD cohorts, we are able to show that one IL-18 tag SNP interacts with smoking to modulate the risk of developing CVD.
Similar content being viewed by others

Introduction
Interleukin-18 (IL-18), a pro-inflammatory cytokine, is a source of considerable research interest thanks to its implication in the etiology of several diseases, including immune diseases, type I diabetes and cardiovascular diseases (CVD).1 There is mounting evidence from epidemiological and animal studies that IL-18 plays a key role in CVDs through its contribution to atherosclerosis.2, 3, 4, 5, 6, 7 In particular, increased IL-18 levels were shown to be strong predictors of cardiovascular mortality5, 6 and associated with carotid intima-media thickness.8 More recently, a fresh light has been shed on the relationship between IL-18 and CVD by the finding that IL-18 gene haplotypes are associated with cardiovascular mortality in patients with coronary artery disease, independently of IL-18 levels.7
The purpose of this report was to investigate the association between IL-18 haplotypes derived from five tag IL-18 gene single nucleotide polymorphisms (SNPs)7 and the risk of CVD in the MORGAM Project.9 MORGAM is a collaborative study pooling many European prospective CVD cohorts to identify genetic factors underlying CVD. The pooling of cohorts should provide ample power to detect the moderate genetic effects, gene–gene and gene–environment interactions believed to influence CVD incidence. A case–cohort design10 has been adopted in MORGAM11 to enable the study of multiple end points, including coronary heart disease (CHD) and stroke, while minimizing the genotyping costs. Under such sampling, genotypic information is available only on a random sample of the entire cohort, generally referred to as a subcohort, and for all additional individuals who experienced the events of interest during the follow-up and who were not part of the original subcohort. Case–cohort data analysis requires special statistical methods for handling such non-random data. If the objective is to test the association of a set of covariates (including genotypes) with the disease status, without taking into account the time-to-event information, a standard logistic regression model can be used, provided that the logarithm of the inverse of the probability of being selected in the subcohort is used as an offset.10 If time-to-event information is needed, non-standard methods must be used including pseudolikelihood10, 12, 13 and partial-likelihood score functions.11, 14
In the field of genetic epidemiology, the complexity of a time-to-event case–cohort analysis increases when the objective is to assess the contribution of haplotypes to the risk of disease. Indeed, in the absence of family data, haplotypes cannot generally be deduced from the genotypic information and must be statistically inferred. In ARIC,15 a single imputation strategy16, 17 was applied to investigate haplotypic associations without any previous validation study of an approach that was initially proposed for the analysis of genotypes in random samples of individuals.16, 17 This is why, in MORGAM, before embarking on the study of IL-18 haplotypes using the single imputation strategy, a simulation study was first carried out to investigate the properties of this technique when applied to a case–cohort design.
Materials and methods
Cox's regression analysis under a case–cohort design
Assume that, as in a standard Cox proportional hazard model18 for a classic random prospective design, the hazard at time t for an individual with a covariate vector X is given by λ(t)=λ0(t)exp(βtX), where λ0(t) is an unspecified baseline hazard function and β is a vector of unknown regression parameters to be estimated.
Under a case–cohort design, the set of covariates X is only collected for a random sample of individuals selected at baseline, referred to as the subcohort, and for all subjects with events of interest outside that subcohort. Individuals forming the subcohort can be selected using either equal or unequal probabilities depending on a set of baseline covariates collected on all individuals in the cohort. It is important to emphasize that the subcohort may include both individuals who will develop the event of interest (referred to as ‘cases’) and individuals who would never experience it during the follow-up.
For a case–cohort of n individuals, a consistent estimator of β, β̃, is available through the maximization of the following pseudolikelihood:

where δi=1{Ti<Ci} is the event indicator, Ti the event time, Ci the censoring time of the ith individual with covariates vector Xi and where R̃(ti) is a risk set containing cases with events at time ti plus all subcohort members at risk at the same time. A case outside the subcohort is considered to be at risk only at the time of his/her failure and not before. The ωj(ti) are weights that are a function of the inverse of the sampling probabilities of being selected in the subcohort.14, 15 In a random prospective cohort, all ωj are equal to 1 and R(ti) only includes the individuals without event by time ti.
Once β̃ and its variance–covariance matrix obtained using the jackniffe method12 are available, the generalized Wald test statistic can be used for testing any specific hypothesis about β.
Application to haplotype association analysis
In the context of association studies based on a case–cohort design, genotypic information is collected only for the individuals in the subcohort and for all additional cases. If single-locus analysis is solely of interest, the genotypes would then form the X covariates in formula (1). When haplotypes are of interest, a simple way to investigate their association with the survival outcome, at the same time dealing with their ambiguity due to multi-heterozygous individuals, would be to apply the single imputation method initially proposed by Zaykin et al17 for standard case–control data. This consists, first, in estimating the haplotypic frequencies derived from the genotypes observed in the subcohort using standard methodology for random samples.19 These frequencies are then used to compute for each genotyped individual (in and out the subcohort) the conditional probabilities of his/her pair of haplotypes given his/her genotypes, under the assumption of Hardy–Weinberg equilibrium (HWE). The unobserved haplotypes are then considered as the X covariates in the pseudolikelihood (formula 1) with the estimated conditional probabilities used now as weights in the regression model βX.
To investigate the statistical properties of this imputation method applied to a case–cohort design, a small simulation study was carried out.
Simulation study
Simulation settings
Data were simulated under a case–cohort design in which five SNPs (A/a, B/b, C/c, D/d and E/e), generating six haplotypes mimicking the IL-18 gene haplotypic structure observed in a previous study,7 were tested for association with survival outcome (Table 1). For each simulated data set, a cohort of 5000 individuals was generated as described in the Supplementary Information to match MORGAM data as closely as possible. Each individual was assigned a sampling probability of being selected in the subcohort, which was randomly drawn from the observed distribution of these probabilities in the MORGAM data. With this sampling, the subcohort was on average composed of 630 subjects, 570 non-cases and 60 cases, whereas the number of cases outside the subcohort was 450 (Table 1). Genotypes at each locus could then be constructed from haplotypes and analyzed using the pseudolikelihood methodology described above.
All simulations were conducted on 1000 replicates for each particular setting.
Evaluation criteria
The performances of the single imputation method for the case–cohort design were investigated in terms of ‘mean bias,’ ‘mean standard error,’ ‘root-mean-square error’ (RMSE), ‘coverage probability,’ which is the probability that the 95% confidence interval of the estimate contains the theoretical value, and the type I error and the power associated with the test for detecting the effect of each single haplotype. Type I error and power of the global haplotypic test of detecting at least one non-zero haplotypic effect were also investigated using the generalized Wald test statistic. In these tests, a significance level of 0.05 was applied.
We also compared this method with standard Cox regression analysis using the full information on all 5000 simulated individuals. For such analysis, a similar single imputation strategy was applied with the exception that the haplotypic frequencies were estimated on the 5000 ‘genotyped’ subjects and not only on the subcohort. This comparison was made to evaluate the loss of power due to the case–cohort design.
MORGAM data analysis
MORGAM study populations
MORGAM is a multinational collaboration of several European cardiovascular cohorts whose description has been published elsewhere.9 A detailed description of MORGAM cohorts is also available online (see Appendix). In the present work, five cohorts were studied, two from Finland (FINRISK, ATBC), one from France and one from Northern Ireland (both from the PRIME Study) and one from Sweden, for a total number of 25 141 subjects with available DNA. All individuals were followed up over a median period of 6.0 years for mortality with specified causes of death and for several cardiovascular phenotypes. In each cohort, subjects were randomly selected to form part of a subcohort according to cohort-specific sampling probabilities depending on age, older subjects having a higher selection probability to ensure that similar age distributions were obtained in cases and subcohort. Globally, the subcohort comprised of 2271 subjects. In this study, we were interested in stroke and CHD events (see detailed event definition online). Subjects with cardiovascular events before the baseline examination were included in the analysis, but the event was not considered as end point event in the analysis. In the presence of multiple events during the follow-up, only the first event was considered. Both fatal and non-fatal events were included. The case–cohort data set was thus composed of 1933 cases (333 cases from the subcohort and 1600 from outside) and 1938 non-cases who were genotyped for the IL-18 gene SNPs.
Genotyping
Five tag SNPs were genotyped in this study, which captured the haplotypic diversity of the regulatory, coding and flanking intronic regions of the IL-18 gene: G-887T (rs1946519), C-105T (rs360717), S35S (A/C) (rs549908), A+183G (rs5744292) and T+533C (rs4937100). Genotyping was performed using the TaqMan 5′-nuclease detection assay on a 7000 Sequence Detection System (Applied Biosystems). The genotyping success rate was at least 98% for all SNPs and we observed less than 0.85% genotyping errors based on the blinded duplicate comparison. All information for genotyping (PCR primers, probes, conditions of amplification and hybridization) can be found on the GeneCanvas website (http://genecanvas.ecgene.net).
Statistical analysis
Deviation from HWE was tested in each subcohort by a standard χ2 with 1 degree of freedom (d.f.). SNPs were first tested for association with the case/non-case status without taking into account the censoring/event time information using a logistic regression model including the inverse of the sampling probabilities as an offset. Single-locus and haplotypic analyses with THESIAS software20 were carried out. As a second step, time-to-event survival analysis was conducted using the pseudolikelihood methodology described above. Time from the baseline examination was used in the analysis. For single-locus analyses, the ‘coxph’ R language function, with an offset as defined above, was used.21 For haplotype analysis, haplotypic frequencies were first estimated in the subcohort and then used for computing individual conditional probabilities employing the ‘haplo.em’ R function. These probabilities were then incorporated as weights in the regression model of the ‘coxph’ R function.
Analyses were first carried out in each population separately, adjusting for age at baseline, gender and smoking status when appropriate. The Mantel–Haenszel statistic was used for testing the homogeneity of the results across cohorts and subsequently for providing combined odds ratio (OR) and HR estimates in the whole study.
Results
Results of the simulations
Different patterns of association including various values of HR (1.0, 1.3, 1.5 and 1.8) associated with different common or rare haplotypes were investigated. As all these resulted in similar conclusions, we present only the results when one common haplotype with a frequency of 0.25 was associated with an HR of 1.3 by comparison with the most frequent haplotype (Table 1).
The six haplotypes were always correctly inferred from the genotypes, and the bias associated with haplotypic frequency estimates was very small (data not shown). Similarly, the bias associated with HR estimates was relatively small and increased as the corresponding haplotypic frequency decreased, but no bias was significantly different from 0. These properties were observed both for the pseudolikelihood method on case–cohort data and for the partial likelihood on the whole cohort. However, the SE and RMSE of the HR estimates in the case–cohort analysis were higher than those of the whole cohort, leading to a loss of power in the case–cohort analysis compared with the full-cohort analysis. For example, for the settings considered in Table 1, the power to detect the effect of the ‘at risk’ haplotype decreased from 87 to 52% when the analysis was restricted to the case–cohort design. Similarly, the power of the test to detect a global haplotypic effect (ie a χ2 test with 5 d.f.) was 83% using the genotypic information on the full cohort, whereas it decreased to 37% using the case–cohort data.
Under the null hypothesis of no haplotypic effect, the type I error of the global haplotypic test was always in agreement with the nominal 0.05 level, for both methods. As expected, testing all single haplotypes without correcting for multiple testing would lead to the erroneous identification of an effect in about 22% of the replicates simulated under the null hypothesis (data not shown). Correcting for the number of estimated haplotypic effects (ie five in these simulations) led to a type I error rate of 5.8%, which was consistent with the expected 5% level.
MORGAM data
A brief description of the populations used for this analysis is given in Table 2. Three cohorts out of five, ATBC, PRIME/France and PRIME/N.Ireland, included only men. In the ATBC cohort, the prevalence of smokers was very high (77.7%) because smoking a few years earlier was one of the inclusion criteria. In the other cohorts, the percentage of smokers was much lower (about 25%). Of the 1933 cases of CVD, 1275 experienced a CHD event, 557 a stroke event and 101 experienced both. The first event was fatal for 401 CHD cases (31%) and 73 stroke cases (13%).
For ease of presentation, most of the results presented are those obtained in the whole study, after having checked the homogeneity of the associations across cohorts.
Single-locus analysis
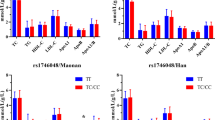
In all the subcohorts, genotypic distributions were compatible with HWE. Allele frequencies did not differ between subcohorts (P-values from 0.42 to 0.89 for the five SNPs), and none of the polymorphisms were significantly associated with disease status (Table 3). We further investigated whether SNPs could potentially interact with the studied covariates (age, sex, smoking) to modulate the risk of disease. Three SNPs were found differentially associated with disease according to smoking (Supplementary Table 1). In smokers, the −105T allele was significantly more frequent in cases than in controls (0.29 vs 0.25) and associated with an increased OR for disease of 1.25 (95% confidence interval 1.07–1.45) (P=0.005), whereas no association was observed in non-smokers (OR=0.90 (0.78–1.03), P=0.131). The test for homogeneity of these two ORs was highly significant (P=0.002). The association found in smokers was consistently observed in the five cohorts (Figure 1) and was even stronger after exclusion of the ATBC cohort in which all non-smokers are former smokers (OR=1.43 (1.06–1.92), P=0.017). Similar associations were observed for two other SNPs, G-887T and S35S(A/C), for which the minor allele was associated with an increased OR in smokers only (Supplementary Table 1).

Association between the C-105T polymorphism and CVD risk in non-smokers (squares) and smokers (diamonds) by cohort. *Population-adjusted.
Time-to-event pseudolikelihood analyses yielded similar observations (Supplementary Table 1).
Haplotypic analysis
Owing to strong linkage disequilibrium, the five IL-18 SNPs defined six major haplotypes with frequencies higher than 1% (Supplementary Table 2). The haplotypic structures were broadly homogeneous across the five cohorts even though the Finnish cohorts were more alike than the three others. As the frequency of the TCAAC haplotype was very low, about 2% in non-Finnish cohorts and ∼0.5% in Finnish populations, its effect on the risk of disease was not estimated.
No difference in haplotypic frequency was observed between cases and controls in any of the five cohorts (Supplementary Table 3) or in the whole MORGAM cohort (χ2=1.051 with 4 d.f.; P=0.902). However, consistent with single-locus analyses, IL-18 haplotypes were differentially associated with the disease according to smoking habit. Although no association with the disease status was observed in non-smokers (χ2=3.801 with 4 d.f., P=0.433), IL-18 haplotypes were significantly associated with the disease in smokers (χ2=10.358 with 4 d.f., P=0.035). In smokers, the TTCAT haplotype was more frequent in cases than in controls (0.29 vs 0.25) and was associated with a population-adjusted OR of 1.27 (1.06–1.52) (P=0.009) in comparison with the most frequent GCAAC haplotype (Figure 2). The corresponding OR in non-smokers was 0.87 (0.78–0.97) (P=0.015), the test of homogeneity between these two ORs being highly significant (P<10−3). The effect observed for the TTCAT haplotype in smokers was homogeneous across the five cohorts (Figure 3) and was maintained even after excluding the ATBC cohort (OR=1.55 (1.14–2.11), P=0.005).

Association (HRs and ORs) between IL-18 haplotypes and CVD risk in non-smokers (squares) and smokers (diamonds). ‘a’ indicates Mantel–Haenszel test for homogeneity between smokers and non-smokers. Polymorphisms are ordered according to their position in the genomic sequence.

Association between TTCAT haplotype and CVD risk in non-smokers (squares) and smokers (diamonds) by cohort. *Population-adjusted.
In a time-to-event analysis, the population-adjusted HR associated with the TTCAT haplotype observed in smokers (1.20 (0.99–1.45), P=0.062) was significantly different (P=0.022) from that observed in non-smokers (0.89 (0.74–1.05), P=0.174) (Figure 2).
As the TTCAT haplotype is the only one associated with the disease and the only one carrying the −105T allele, this haplotype analysis confirmed the differential effect of the C-105T SNP on disease risk according to smoking habit and suggests that the effect of the S35S(C) and the −887T alleles observed in single-locus analyses could be due to their LD with the −105T allele.
Another way to illustrate this gene–smoking interaction is to look at the OR for disease associated with smoking according to the C-105T genotypes (Supplementary data). In the whole MORGAM cohort, smoking was associated with a population-adjusted OR of 1.53 (1.31–1.80) (P=2 × 10−7). This effect was stronger in carriers of the −105T allele (OR=1.93 (1.53–2.44), P=3.5 × 10−8) than in homozygous carriers of the −105C allele (OR=1.25 (1.00–1.55), P=0.05), these two ORs being significantly different (P=0.003).
Similar analyses were performed treating CHD and stroke events separately, as well as fatal and non-fatal events, and excluding cases with CVD at baseline. All analyses consistently identified a C-105T–smoking interaction (data not shown).
Discussion
This work reports the results of an association analysis of tag IL-18 gene SNPs with CVD in five European cohorts, contributing to the MORGAM Project, as this gene has been seen as a good candidate for CVD.2, 3, 4, 5, 6, 7
The allele-frequency distribution of the five SNPs studied and the resulting haplotypic structures did not differ significantly across the five cohorts. The −105T allele, and therefore the unique haplotype carrying this allele, was found to be associated with an increased risk of CVD in smokers, whereas an inverse relationship tended to be observed in non-smokers. The C-105T polymorphism and four other SNPs were genotyped in MORGAM because they were found to well reflect IL-18 gene variability.7 The C-105T SNP is nearly in complete association with the rs187238 that has been found associated with IL-18 transcription activity,22, 23 type I diabetes and atopic asthma even if conflicting results have been reported (see Thompson and Humphries1 for a review). One possible explanation for these discrepancies could be that the effect of this SNP is modulated by an unmeasured factor the prevalence of which varied across the different studies investigating the role of IL-18 SNPs. Supporting this hypothesis, we demonstrate that the C-105T SNP (and therefore any SNP in complete association with it) might interact with smoking to influence the risk of CVD. The consistency of this finding across the five cohorts reduces the likelihood that this is a chance finding. However, even if nicotine has been found to be associated with IL-18 levels24, 25 and interactions between smoking and inflammatory gene SNPs have already been observed in the context of atherosclerosis,26 genetic results of this significance do not often replicate, given the small prior that an individual SNP, no matter how good a candidate gene it resides in, has to show a gene–smoking interaction effect on CVD. Further work in large sample studies are therefore required to confirm the result observed in this report.
The MORGAM Project has adopted a case–cohort design to investigate genetic associations with CVD, a design that needs recourse to non-standard methods to deal with time-to-event analysis. To cope with the additional complexity of haplotypic inference, we decided upon a single imputation technique coupled with a pseudolikelihood approach for testing the association of haplotypes with a survival outcome in a case–cohort setting. The validity of this approach was checked by simulation. We incorporated our method into an R procedure that can be used routinely and that also enables a posteriori power calculations to be made easily. For example, in a case–cohort data set of ∼1800 individuals with equal proportions of cases and non-cases such as in our subgroup of smokers, the power to detect an HR of 1.3 associated with a haplotype with a frequency of 0.28 was 70%. More complex statistical methods for the case–cohort design exist27, 28, 29 and their application to haplotypic association analysis would warrant further investigation.
In conclusion, we have demonstrated that the MORGAM Project is a powerful tool for investigating the association of candidate genes with CVD and to detect, if any, their interaction with environmental factors. In particular, we report here that one IL-18 tag SNP was associated with CVD and that its effect could be modulated by cigarette smoking. Six other European cohorts with DNA available are in the process of joining the MORGAM Project, which will make it even more powerful.
References
Thompson S, Humphries S : Interleukin-18 genetics and inflammatory disease susceptibility. Genes Immun 2007; 8: 91–99.
de Nooijer R, von der Thusen JH, Verkleij CJ et al: Overexpression of IL-18 decreases intimal collagen content and promotes a vulnerable plaque phenotype in apolipoprotein-E-deficient mice. Arterioscler Thromb Vasc Biol 2004; 24: 2313–2319.
Mallat Z, Corbaz A, Scoazec A et al: Expression of interleukin-18 in human atherosclerotic plaques and relation to plaque instability. Circulation 2001; 104: 1598–1603.
Nishihira K, Imamura T, Hatakeyama K et al: Expression of interleukin-18 in coronary plaque obtained by atherectomy from patients with stable and unstable angina. Thromb Res 2007; 16: 16.
Blankenberg S, Luc G, Ducimetiere P et al: Interleukin-18 and the risk of coronary heart disease in European men: the Prospective Epidemiological Study of Myocardial Infarction (PRIME). Circulation 2003; 108: 2453–2459.
Blankenberg S, Tiret L, Bickel C et al: Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation 2002; 106: 24–30.
Tiret L, Godefroy T, Lubos E et al: Genetic analysis of the interleukin-18 system highlights the role of the interleukin-18 gene in cardiovascular disease. Circulation 2005; 112: 643–650.
Yamagami H, Kitagawa K, Hoshi T et al: Associations of serum IL-18 levels with carotid intima-media thickness. Arterioscler Thromb Vasc Biol 2005; 25: 1458–1462.
Evans A, Salomaa V, Kulathinal S et al: MORGAM (an international pooling of cardiovascular cohorts). Int J Epidemiol 2005; 34: 21–27.
Prentice RL : A case–cohort design for epidemiologic cohort studies and disease prevention trials. Biometrika 1986; 73: 1–11.
Kulathinal S, Karvanen J, Saarela O, Kuulasmaa K, Project FT : Case–cohort design in practice – experiences from the MORGAM Project. Epidemiol Perspect Innov 2007; 4: 15.
Barlow WE, Ichikawa L, Rosner D, Izumi S : Analysis of case–cohort designs. J Clin Epidemiol 1999; 52: 1165–1172.
Kim S, De Gruttola V : Strategies for cohort sampling under the Cox proportional hazards model, application to an AIDS clinical trial. Lifetime Data Anal 1999; 5: 149–172.
Chen K, Lo S-H : Case–cohort and case–control analysis with Cox's model. Biometrika 1999; 86: 755–764.
Lee CR, North KE, Bray MS et al: Genetic variation in soluble epoxide hydrolase (EPHX2) and risk of coronary heart disease: The Atherosclerosis Risk in Communities (ARIC) study. Hum Mol Genet 2006; 15: 1640–1649.
Chiano MN, Clayton DG : Fine genetic mapping using haplotype analysis and the missing data problem. Ann Hum Genet 1998; 62: 55–60.
Zaykin DV, Westfall PH, Young SS, Karnoub MA, Wagner MJ, Ehm MG : Testing association of statistically inferred haplotypes with discrete and continuous traits in samples of unrelated individuals. Hum Hered 2002; 53: 79–91.
Cox DR : Regression models and life tables. J R Stat Soc B 1972; 34: 187–220.
Excoffier L, Slatkin M : Maximum-likelihood estimation of molecular haplotype frequencies in a diploid population. Mol Biol Evol 1995; 12: 921–927.
Tregouet DA, Garelle V : A new JAVA interface implementation of THESIAS: testing haplotype effects in association studies. Bioinformatics 2007; 23: 1038–1039.
Therneau TM, Li H : Computing the Cox model for case cohort designs. Lifetime Data Anal 1999; 5: 99–112.
Liang XH, Cheung W, Heng CK, Wang DY : Reduced transcriptional activity in individuals with IL-18 gene variants detected from functional but not association study. Biochem Biophys Res Commun 2005; 338: 736–741.
Giedraitis V, He B, Huang WX, Hillert J : Cloning and mutation analysis of the human IL-18 promoter: a possible role of polymorphisms in expression regulation. J Neuroimmunol 2001; 112: 146–152.
Takahashi HK, Iwagaki H, Hamano R, Yoshino T, Tanaka N, Nishibori M : alpha7 Nicotinic acetylcholine receptor stimulation inhibits lipopolysaccharide-induced interleukin-18 and -12 production in monocytes. J Pharmacol Sci 2006; 102: 143–146.
McKay A, Komai-Koma M, MacLeod KJ et al: Interleukin-18 levels in induced sputum are reduced in asthmatic and normal smokers. Clin Exp Allergy 2004; 34: 904–910.
Jerrard-Dunne P, Sitzer M, Risley P, Buehler A, von Kegler S, Markus HS : Inflammatory gene load is associated with enhanced inflammation and early carotid atherosclerosis in smokers. Stroke 2004; 35: 2438–2443.
Scheike TH, Martinussen T : Maximum likelihood estimation for Cox's regression model under case–cohort sampling. Scand J Stat 2004; 31: 283–293.
Zeng D, Lin DY, Avery CL, North KE, Bray MS : Efficient semiparametric estimation of haplotype–disease associations in case–cohort and nested case–control studies. Biostatistics 2006; 7: 486–502.
Kulathinal S, Arjas E : Bayesian inference from case–cohort data with multiple end-points. Scand J Stat 2006; 33: 25–36.
Acknowledgements
The research was supported by the MORGAM Project grant under the Biomed 2 Programme, by the GenomEUtwin Project grant under the Programme ‘Quality of Life and Management of the Living Resources’ of fifth Framework Programme (no. QLG2-CT-2002-01254) and by the European Community's Seventh Framework Programme (FP7/2007-2013), ENGAGE project, grant agreement HEALTH-F4-2007-201413.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary information
Appendix
Appendix
MORGAM Website: http://www.ktl.fi/publications/morgam/index.html
Sites and key personnel of contributing MORGAM centers:
Finland
FINRISK, National Public Health Institute, Helsinki: V Salomaa (principal investigator), A Juolevi, E Vartiainen, P Jousilahti; ATBC, National Public Health Institute, Helsinki: J Virtamo (principal investigator), H Kilpeläinen, M Virtanen; MORGAM Data Centre, National Public Health Institute, Helsinki: K Kuulasmaa (head), Z Cepaitis, A Haukijärvi, B Joseph, J Karvanen, S Kulathinal, M Niemelä, O Saarela; MORGAM Central Laboratory, National Public Health Institute, Helsinki: L Peltonen (responsíble person), M Perola, K Silander, M Alanne, P Laiho, K Kristiansson, K Ahonen.
France
National Coordinating Centre, National Institute of Health and Medical Research (U258), Paris: P Ducimetière (national coordinator), A Bingham; PRIME/Strasbourg, Department of Epidemiology and Public Health, Louis Pasteur University, Faculty of Medicine, Strasbourg: D Arveiler (principal investigator), B Haas, A Wagner; PRIME/Toulouse, Department of Epidemiology, Toulouse University School of Medicine, Toulouse: J Ferrières (principal investigator), J-B Ruidavets, V Bongard, D Deckers, C Saulet, S Barrere; PRIME/Lille, Department of Epidemiology and Public Health, Pasteur Institute of Lille: P Amouyel (principal investigator), M Montaye, B Lemaire, S Beauchant, D Cottel, C Graux, N Marecaux, C Steclebout, S Szeremeta; MORGAM Laboratory, INSERM U525, Paris: F Cambien (responsible person), L Tiret, V Nicaud.
Sweden
Northern Sweden, Umeå University Hospital, Department of Medicine, Umeå: B Stegmayr (principal investigator), K Asplund (former principal investigator), S Nasic, G Rönnberg, Å Johansson, V Lundberg, E Jägare-Westerberg, T Messner.
United Kingdom
PRIME/Belfast, Queen's University Belfast, Belfast, Northern Ireland: A Evans (principal investigator), J Yarnell, E Gardner; MORGAM Coordinating Centre, Queen's University Belfast, Belfast, Northern Ireland: A Evans (MORGAM coordinator), S Cashman, F Kee.
MORGAM Management Group: A Evans (chair), S Blankenberg (Mainz, Germany), F Cambien, M Ferrario (Varese, Italy), K Kuulasmaa, L Peltonen, M Perola, V Salomaa, D Shields (Dublin, Ireland), B Stegmayr, H Tunstall-Pedoe (Dundee, Scotland), K Asplund (honorary consultant, Stockholm Sweden).
Rights and permissions
About this article
Cite this article
Grisoni, ML., Proust, C., Alanne, M. et al. Haplotypic analysis of tag SNPs of the interleukin-18 gene in relation to cardiovascular disease events: the MORGAM Project. Eur J Hum Genet 16, 1512–1520 (2008). https://doi.org/10.1038/ejhg.2008.127
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ejhg.2008.127
Keywords
This article is cited by
-
The role of the inflammasome in cardiovascular diseases
Journal of Molecular Medicine (2014)
-
Lack of association between polymorphisms of the IL18R1 and IL18RAP genes and cardiovascular risk: the MORGAM Project
BMC Medical Genetics (2009)
