
Abstract
Two new types of lentiviral vectors expressing a reporter transgene encoding either firefly luciferase (fLuc) for bioluminescence imaging or the HSV1 thymidine kinase (HSV1-TK) for radiopharmaceutical-based imaging were constructed to monitor human embryonic stem cell (hESC) engraftment and proliferation in live mice after transplantation. The constitutive expression of either transgene did not alter the properties of hESCs in the culture. We next monitored the formation of teratomas in SCID mice to test (1) whether the gene-modified hESCs maintain their developmental pluripotency, and (2) whether sustained reporter gene expression allows noninvasive, whole-body imaging of hESC derivatives in a live mouse model. We observed teratoma formation from both types of gene-modified cells as well as wild-type hESCs 2-4 months after inoculation. Using an optical imaging system, bioluminescence from the fLuc-transduced hESCs was easily detected in mice bearing teratomas long before palpable tumors could be detected. To develop a noninvasive imaging method more readily translatable to the clinic, we also utilized HSV1-TK and its specific substrate, 1-(2′-deoxy-2′-fluoro-β-D-arabinofuranosyl)-5-[125I]iodouracil ([125I]FIAU), as a reporter/probe pair. After systemic administration, [125I]FIAU is phosphorylated only by the transgene-encoded HSV1-TK enzyme and retained within transduced (and transplanted) cells, allowing sensitive and quantitative imaging by single-photon emission computed tomography. Noninvasive imaging methods such as these may enable us to monitor the presence and distribution of transplanted human stem cells repetitively within live recipients over a long term through the expression of a reporter gene.
Similar content being viewed by others

Introduction
Several methods for transient labeling of transplanted cells with contrast-generating reagents have been developed for optical, nuclear [positron emission tomography (PET) and single-photon emission computed tomography (SPECT)], and magnetic resonance (MR) imaging. However, it has been difficult to label human stem cells stably, which divide extensively in vivo and generate an array of differentiated cells after transplantation. We investigated techniques to track the presence and proliferation of transplanted human stem cells in vivo based on transgenic reporter expression. We have earlier shown stable gene transduction of human hematopoietic stem cells (HSCs) by lentiviral vectors expressing the green fluorescent protein (GFP) reporter gene under the control of either a ubiquitous or tissue-specific promoter 1, 2. Although GFP fluorescence can be easily detected using ex vivo analysis months after engraftment, a high background signal (in the visible light spectrum) prevents efficient GFP detection in vivo particularly from deep tissues and organs 1, 2, 3, 4, 5. Consequently, postmortem histology remains necessary when using a GFP-based fluorescent reporter 3, 4, 5.
hESCs are established cell lines that proliferate in culture in an unlimited fashion, while retaining their pluripotency to differentiate into essentially any cell types 6. The pluripotency can be tested using in vitro assays and in vivo using severe combined immune deficient (SCID) mouse models. Several months after being injected intramuscularly or into another site in a SCID mouse, hESCs slowly form a benign tumor (teratoma) comprising various cell types derived from all of the three embryonic germ layers 6. To monitor hESC engraftment and proliferation in live animal models, we constructed two new types of lentiviral vectors co-expressing GFP and an additional transgene encoding an in vivo imaging reporter. In the first reporter vector, the gene encoding the firefly luciferase (fLuc) is used for bioluminescence imaging. To develop a noninvasive imaging method more readily translatable to the clinic, we also constructed a second vector in which the HSV1 thymidine kinase (HSV1-TK) is used as a reporter gene for radiopharmaceutical-based, planar γ or SPECT imaging. For this purpose, the HSV1-TK and its specific substrate, such as 1-(2′-deoxy-2′-fluoro-β-D-arabinofuranosyl)-5-[125I]iodouracil ([125I]FIAU), serve as a reporter gene/reporter probe pair 7, 8, 9. FIAU, which is inert to uninfected mammalian cells, can be synthesized in radiolabeled form with a variety of isotopes that enable imaging by PET (iodine-124) or SPECT (iodine-123, or in small animals, iodine-125). After systemic administration at a non-pharmacological dose, [125I]FIAU is phosphorylated by HSV1-TK encoded by the HSV1-tk transgene, but not by the mammalian nuclear TK genes. Only phosphorylated [125I]FIAU is retained within transduced (and transplanted) cells, whereas the unphosphorylated substrate freely crosses the cell plasma membrane and is cleared much more quickly from the extracellular fluid. Although this method has been successfully used for in vivo imaging studies of human tumor cells 7, 8, 9, it has not been applied to monitor transplanted human stem cells in a live recipient until recently. We report here that we can easily detect teratomas by SPECT in live mice months after transplantation of hESCs labeled by the HSV1-tk-GFP fusion reporter gene, or by a standard bioluminescence imaging system for hESCs labeled with the fLuc reporter gene after injection of a fLuc substrate D-luciferin.
We will discuss the difference and applications of these types of noninvasive imaging methods that enable us to monitor repetitively the presence and biological activity of transplanted human and mouse stem cells through the expression of reporter genes within live organisms at the whole-body level.
Results
Characterization of lentiviral vectors co-expressing GFP and an in vivo reporter gene
To deliver vectors encoding reporter genes stably and efficiently to a variety of cell types, including those refractory to plasmid transfection, we used a virus-mediated gene transfer and expression system based on lentiviral vectors that we had developed earlier 1. The central purine track (cPPT) element was added to this lentiviral vector system to increase vector integration efficiency (Figure 1). A ubiquitous promoter from the human housekeeping gene EF1α was used to drive the expression of two types of imaging reporter genes. In the first vector, cEF.tk-GFP, the cDNA encoding the HSV1-TK-GFP fusion protein 9 was inserted as the sole transgene (Figure 1A). The bi-functional HSV1-TK-GFP fusion protein allows us to monitor transduced cells by both optical imaging (of GFP signal) and PET or SPECT-CT (of HSV1-TK using radiolabeled probes such as [125I]FIAU) from a single reporter protein 9. Similarly, we made several lentiviral vectors in which the fLuc encoding sequence was fused in frame with that of the GFP gene, without or with a 10-amino-acid spacer. We found that in all cases GFP intensity was severely reduced. After repeated failure, we tried a second strategy in which the fLuc and GFP genes were expressed from the same transcript, but as two separate proteins. Specifically, the fLuc was placed as the first gene to allow maximal expression, whereas the GFP was expressed as the second gene through an internal ribosomal entry site (IRES) element (Figure 1B).

A diagram of lentiviral vectors used in this study, in the form after viral integration. (A) cEF.tk-GFP; (B) cEF.Luc-IRES.GFP. EF1α: (human) elongation factor 1α promoter; HSV1-tk: Herpes simplex virus 1 (HSV1) thymidine kinase gene (encoding the HSV1-TK enzyme); fLuc: firefly luciferase gene; GFP: enhanced green fluorescent protein gene; LTR: long-terminal repeat of lentiviral DNA (The one used here is mutated and lacking viral promoter and enhancer activities.); cPPT: central purine track (of viral) element that enhances viral integration; IRES: internal ribosomal entry site sequence (to express two proteins from one mRNA). See Materials and Methods for more details.
Characterization of the tk-GFP lentiviral vector and transduced cell lines
We first tested the cEF.tk-GFP lentiviral vector in several human cell lines, including 293T and TF1 (hematopoietic progenitor) cells. The viral titer of this new vector is similarly high as the cEF.GFP parental vector (∼10 × 106 transducing units [TU]/ml). After one round of transduction, > 50% of transduced cells stably expressed GFP. We observed a moderate reduction (∼2-3 fold) of GFP fluorescence as expected, as compared with the cells transduced by cEF.GFP expressing non-fused GFP in 293T cells. Similar results were seen with transduced TF1 hematopoietic progenitor cells (data not shown). The cEF.tk-GFP lentiviral vector offers long-lasting transgene expression: ≥ 4 months in continuous culture of both human cell types.
We next transduced hESCs with the cEF.GFP lentiviral vector. Transduced (GFP+) cells were expanded by passage on top of mouse feeder cells. As evident in Figure 2A, undifferentiated hESC colonies expressed GFP. We further analyzed the whole cell population by staining for cell surface markers, such as SSEA-3, which is expressed preferentially in undifferentiated hESCs but not in feeder cells or differentiated human cells (Figure 2B). Approximately 50% of cells expressed both GFP and SSEA-3. The GFP expression in transduced hESCs remained stable during the following cell expansion and characterization (lasting for 1-2 months). We next examined the biological functionality of the HSV1-TK enzyme in cEF.tk-GFP-transduced cells (Figure 2C). One assay is to use HSV1-TK-specific prodrugs, such as FIAU, that are cytotoxic to the HSV1-TK-positive cells during cell proliferation 9. We measured the sensitivity of cEF.GFP-transduced hESCs in comparison with control (untransduced) hESCs to the exposure of FIAU at various concentrations for 5 days followed by a colony-forming assay of undifferentiated hESCs. As evident in Figure 2C, FIAU selectively blocked the growth of cEF.tk-GFP-transduced hESCs. We conclude that the tk-GFP fusion reporter protein made by the cEF.tk-GFP lentiviral vector is bi-functional as intended.

Human ESCs transduced with the tk-GFP reporter. After transduction by the cEF.tk-GFP lentiviral vector, hESCs were expanded by passage on top of mouse feeder cells. Many undifferentiated hESC colonies expressed GFP (A). We further analyzed the whole cell population by staining for cell surface marker SSEA-3, which is expressed only in undifferentiated hESCs but not on feeder cells or differentiated human cells (B). The percentage of cells in each quadrant (measuring SSEA-3 and GFP expression) is denoted. Approximately 50% of cells expressed both GFP and SSEA-3. After expansion, tk-GFP-transduced or control hESCs were tested for their sensitivity to the cell killing by FIAU, a selective prodrug for HSV1-TK (C). Human ESCs were cultured under a feeder-free condition and treated with various concentrations of FIAU for 4 days. The harvested cells were then plated under the optimal culture condition (with feeders) to numerate colony-forming (undifferentiated) hESCs. Numbers of hESC colonies were plotted as a function of the FIAU concentration and normal by those when FIAU was absent (100%), for both tk-GFP or control hESC groups. A representative experiment is shown with mean and SD (n = 3). (D) The expanded tk-GFP or control hESCs were also injected into SCID mice to allow teratoma formation. The excised teratoma cells were also subjected to RT-PCR analysis for the expression of lineage markers, as evidence of hESC pluripotency. Specific primers for PAX6 (ectoderm), CD34 (mesoderm) and AFP (endoderm) as well as β-actin (a normalization control) were used to detect expression of these genes in either undifferentiated hESCs (hESC) or teratoma cells either from the control (Cont) hESCs or from the tk-GFP transduced hESCs. The normalized levels (mean and SD, n = 4) of gene expression are plotted using a log scale. As expected, the expression levels of lineage markers such as PAX6, CD34 or AFP are very low or undetectable (*) after 40 cycles of PCR. The expression levels of these three marker genes are not significantly different between the two types of teratomas from control or tk-GFP-transduced hESCs.
Our data indicate that expression of the tk-GFP reporter by our lentiviral vector did not retard the growth of undifferentiated hESCs. Similarly, the tk-GFP-transduced hESCs formed teratomas at a normal rate using the assay we have described earlier 10, as did the hESCs expressing HSV1-tk or GFP alone 11, 12. We analyzed teratoma cells derived from tk-GFP-transduced hESCs for the expression of specific markers of the three embryonic germ layers, as an evidence of pluripotency of the transduced hESCs. Whereas the expression of PAX6 (ectoderm), CD34 (mesoderm), and AFP (endoderm) is very low or undetectable in undifferentiated (Undiff) hESCs, it was vastly elevated in teratoma cells generated from either the control (cont) or tk-GFP-transduced hESCs (Figure 2D).
Characterization of the fLuc lentiviral vector and transduced human cell lines
Similarly, the new fLuc vector (cEF.Luc-IRES.GFP, Figure 1B) was tested in 293T human cells, together with the parental vector cEF.X-IRES.GFP (Figure 3A). Although the GFP intensity in the bi-cistronic vector was weaker than that from the tk-GFP vector, the intensity sufficed to allow cell sorting based on GFP signal. Sorted GFP 293T cells were cultured and expanded for > 3 months, as for other lentivirally transduced cells. The fLuc activity of the transduced 293T cells was measured, after a limiting cell dilution, both in vitro (in 96-well plates after overnight culture) and in vivo (imaging 1 day after injection into SCID mice) using the Xenogen IVIS 200 imaging system (Figures 3A and 3B, respectively). Figure 3B represents an example of composite images showing density maps of three mice 2 weeks after injection of 293T cells transduced with the fLuc vector. We estimated that ∼500 cells can be readily detected in vitro and 10 000 cells can be detected in SCID mice 1 day after intramuscular injection.

In vitro and in vivo imaging of the luciferase gene (fLuc)-labeled human cells. The human 293T cell line was transduced by either the lentiviral cEF.luc-IRES.GFP or the cEF.X-IRES.GFP control vector, and transduced (GFP+) cells were selected. (A) Transduced or control human cells were plated in 96-well plates (0.34 cm2) at different cell doses. D-Luciferin was added the next day into the culture medium. The fLuc activity was measured by the Xenogen IVIS 200 system. The average signal (n = 2) from each well was recorded as photon counts per minute. Note that ≤ 500 fLuc cells per well (or ≤ 1 800 cells/cm2) could be easily detected in vitro. (B) In vivo imaging of three mice in each group harboring tumors derived from fLuc-transduced 293T cells. Measured in photons/second/cm2/steradian, signal densities were calculated and are illustrated with a color bar in gradient and over-laid on the black and white image of mice. (C) Mice harboring teratomas derived from parental hESCs (no. 1), hESCs expressing the tk-GFP reporter (no. 2) or the fLuc reporter (no. 3). Note that both thighs of no. 3 mouse were injected with hESCs transduced with the fLuc lentiviral vector 75 days before imaging. Injected fLuc-transduced cells on the right thigh of no. 3 mouse (T2, although no sign of a palpable tumor) were detected as in the left thigh (T1) 20 min after D-luciferin systematic administration. The region of interest (ROI) and sum (total photons/s) were identified and calculated by the commercial (Xenogen IVIS) software for both T1 and T2 tumors. See Figure 4 for corresponding longitudinal data.
hESCs were transduced by the same cEF.Luc-IRES.GFP vector, using a method identical to that described in Figure 2 for cEF.tk-GFP. GFP transduced hESCs were sorted and expanded. After confirming the fLuc activity in vitro, hESCs transduced by the cEF.Luc-IRES.GFP were injected intramuscularly in SCID mice: 2-5 million cells per site in one or both thighs. The implanted hESCs, which form palpable tumors (teratomas) typically after 2-4 months 10, can be monitored virtually daily after systemic administration of fLuc substrate D-luciferin. Figure 3C represents an example of composite images showing density maps of three mice with hESC-derived tumors (teratomas). A mouse (no. 3) inoculated with the fLuc-transduced hESCs showed optical signals (co-localized with tumor) 75 days after hESC injection, and only after D-luciferin administration. In contrast, two mice that were inoculated with either parental vector (no. 1) or tk-GFP-transduced hESCs (no. 2) and bore a small but palpable tumor did not show signals above background after the same D-luciferin administration. Using Xenogen's IVIS software, the region of interest (ROI) and sum (total photons/second in the ROI) were identified and calculated for both detected tumors (T1 on the left and T2 on the right thigh) of no. 3 mouse. We noticed that the density as well as sum of optical signals reached the plateau at 20 min after D-luciferin administration (Figure 4), and then declined after 30 – 40 min (data not shown).

Longitudinal data for the three tumors over a period of 122 days after inoculation of hESCs labeled with the fLuc vector. Data were compiled from the mouse with two tumors (T1, red and T2, blue) in Figure 4 and an additional mouse with tumor T3 (gray). Animals were imaged at either 5 or 20 min after injection of D-luciferin at various days after inoculation. The total signal (photons/second) from the tumor ROI was plotted using a log scale as a function of time. p: palpable tumor detected at the site of inoculation. A dashed horizontal line (blue) denotes the threshold of an optical signal level (2 × 107 photons/s) when a palpable tumor was first detected.
We also carried out longitudinal studies by imaging a group of mice that had been injected with fLuc-transduced hESCs, before and after palpable tumors were evident (Figure 4). Three representative tumors (T1, T2, and T3) with various tumor sizes were scanned twice at 5 and 20 min, respectively, after D-luciferin administration. The total signal (photons/s) in ROI was plotted as a function of time (days after hESC injection). For the mouse bearing a tumor (T3), imaging scans were carried out periodically from day 1 to day 122. Although variable growth rates occurred, we observed a rapid cell growth from 42 to 56 days after hESC inoculation. For mice with tumor T1 and T3, the formation of a palpable tumor was evident at day 84 and 98, respectively. However, the presence of fLuc-labeled hESC cells (or derived tumors) can be detected long before the detection of palpable tumors (T1 and T3), or for a small tumor like T2, which was never palpable. An estimated threshold for detecting a palpable tumor was determined (∼2 × 107 photons/s), which is more than four orders of magnitudes higher than the detection limit of the current optical technology (≤ 103 photons/s).
Planar γ and SPECT-CT imaging of tumors derived from hESCs
Similarly, we used clinically translatable in vivo imaging technologies, such as SPECT to detect hESCs after transplantation into SCID mice. We used a dedicated small-animal SPECT/CT device to obtain high-resolution imaging, while concurrently providing anatomic details (by CT). Figure 5 shows the ability to use the tk-GFP vector for radiopharmaceutical-based imaging (Figures 5A and 5B). The longitudinal imaging shown in Figure 5C demonstrates the typical time course for growth of a teratoma derived from hESCs.

Noninvasive and whole-body SPECT imaging of human ESCs in vivo. Representative planar γ (A) and SPECT-CT (B) imaging of tumors derived from hESC cells transduced with the cEF.tk-GFP vector. This animal was scanned at 87 days after tumor inoculation (when palpable tumor was detected). In all, 671 μCi of [125I]FIAU was injected through the tail vein, and the animal was scanned at 24 h after radiopharmaceutical injection. The tumor is shown by the yellow arrows in (A) and (B). Tumor radiopharmaceutical uptake: 0.646 ± 0.007 nCi/cm3. (C) Longitudinal SPECT-CT imaging of another SCID-beige mouse harboring a teratoma from hESCs transduced with the cEF.tk-GFP vector. The same mouse was imaged at 61, 81, and 95 days after inoculation. The animal was treated with ∼1 mCi of [125I]FIAU at each time point and imaged at 24 h after injection. Radiopharmaceutical uptake: 61 days (0.018 ± 0.001 nCi/cm3), 81 days (0.066 ± 0.001 nCi/cm3), and 95 days (0.098 ± 0.001 nCi/cm3).
Discussion
hESCs that can divide indefinitely in culture, while retaining their pluripotency to form tissues derived from all of the embryonic germ layers, provide unprecedented opportunities to develop new cell therapies and basic research models for human biology and disease. To realize the full potential of hESCs, however, understanding their disposition, i.e., their location, viability, and ability to differentiate into the tissue of interest, once administered in vivo, is critical (see two recent reviews, 13, 14). An ideal way to follow the disposition of hESCs is to use molecular-genetic imaging, which is a highly sensitive, noninvasive technique that employs a variety of modalities, including those that can be translated to human studies. We describe the use of molecular-genetic imaging, an “indirect” reporter imaging technique, to study the expansion and differentiation of hESCs into teratomas – their inevitable fate when undifferentiated hESCs are transplanted to immune-deficient mice.
As an initial step, we labeled undifferentiated hESCs by either the fLuc or the HSV1-tk reporter gene with lentiviral vectors that deliver transgenes efficiently and more stably. These two reporters have been widely used in cancer cell in vivo imaging, but few studies on human stem cells have been published 15, 16, 17. We also found that the expression of the two reporters had no detrimental effects on the growth of hESCs. Under the control of the EF1α housekeeping gene promoter, fLuc reporter expression can be readily detected even 4 months after hESC inoculation. (The animal had to be terminated at this point because of a large tumor size.) Similarly, transplanted hESCs labeled by the HSV1-tk reporter (as a GFP fusion protein) can be detected by SPECT 95 days after inoculation. Therefore, use of molecular-genetic imaging techniques is a viable approach to imaging hESCs and other types of human stem cells that divide extensively in vivo and require long-term monitoring to assess their biological activities. During the course of this study, however, we also observed partial silencing of the GFP transgene signal in the majority of teratoma cells when being analyzed postmortem. This is not unique to tk-GFP or fLuc reporter as we also observed similar silencing of transgene expression when the GFP reporter gene was used, after extensive cultures 18 or after teratoma formation lasting for several months (data not shown). The transgene silencing was observed even when a housekeeping gene promoter such as the EF1α promoter was used 18. Although we could always select GFP+ cells in vitro, gene silencing poses a problem for in vivo experiments, such as the in vivo imaging studies described in this paper. Progressive silencing of the transgene not only reduces the sensitivity of our detection methods, but also causes a heterogeneity problem as we can no longer assume all the engrafted cells will be imaged in the same way. We are currently testing several approaches to reduce the silencing. One way is to drive the reporter gene by a promoter from other housekeeping genes, such as that of Ubiquitin C (UbC), which is slightly weaker but also less dependent on cell proliferation than the EF1α promoter (18; ZY and LC, unpublished data). An improved transgene expression system will likely help us in conducting long-term in vivo imaging of other cell types, in addition to teratoma formation. This sensitive method of detecting teratoma formation derived from undifferentiated hESCs may also help us to estimate the levels of residual undifferentiated hESCs (as contaminants) after they are induced to differentiate into a mature or committed cell type for clinical applications. Similarly, this method may help us to measure whether there is an increased level of teratoma formation or other forms of tumorigenesis from induced pluripotent stem (iPS) cells, which are reprogrammed from human and mouse somatic cells by ectopic expression of a set of transcription factors delivered by retroviral or lentiviral vectors 19, 20, 21, 22.
Teratoma formation remains the only in vivo assay for pluripotency of hESCs and iPS cells. For early-passage hESCs that retain a normal karyotype and grow slowly in culture (doubling time is about 36 h), it normally takes months to form a palpable tumor. Noninvasive imaging of fLuc-labeled hESCs provides a sensitive and simple means to monitor the presence of engrafted hESCs. We noticed that the fLuc activity dropped initially but increased eventually above the initial level (Figure 4). The levels of fLuc activity roughly correlate with the tumor size: palpable tumors displayed a greater fLuc activity (> 2 × 107 photons/s), but non-palpable tumors can also be detected even when their fLuc activities are as low as 103 photons/s (Figure 4). Our in vivo imaging data are consistent with the notion that the majority of injected hESCs died out after injection, and a teratoma is formed after cell proliferation from a small fraction of the injected hESCs 23. It is currently unclear whether tumor formation is initiated from a subpopulation of distinct hESCs, or whether all hESCs are capable of tumor formation but only a few randomly surviving hESCs eventually form tumors in vivo. Sensitive and noninvasive techniques such as the fLuc bioluminescence reporter system will help us answer this important question 23. A possible approach is to fractionate and monitor a subpopulation of hESCs that is enriched for teratoma-forming cells. Alternatively, one could inject more hESCs and harvest surviving hESCs at nadir (but still detectable using the fLuc bioluminescence reporter system) from animals, and then analyze these survived hESCs. Further investigations are necessary with increasing numbers of recipient animals and monitoring time points.
With the ability to track a small number of cells in vivo, the fLuc bioluminescence reporter system may help to monitor teratoma formation and provide an experimental model for human teratomas and teratocarcinomas. It has also been proposed that an hESC-derived teratoma system may provide an experimental platform for studying the growth and invasiveness of human tumor cells within a microenvironment supplied by an hESC-derived teratoma 24, 25. For example, teratoma may provide human cell-derived endothelium (24, 25, and our published data), which may in turn promote angiogenesis for human tumor cells inoculated into a teratoma. Noninvasive imaging using a ubiquitous promoter as described here or a cell-specific promoter may help to monitor the location as well as functions of the cells derived from hESCs.
A major goal of this project was to compare the strengths and weaknesses of the available modalities (fLuc-based bioluminescence and HSV1-tk-based SPECT) to visualize the disposition of hESCs in vivo and over time. The fLuc bioluminescence reporter system is easy to use and is very sensitive. It allowed us to detect inoculated hESCs daily and tumor formation long before a palpable tumor could be detected (Figure 4). It also offers a dynamic range (at least four orders of magnitudes) of detection. This optical system will likely be useful in basic research for studies of hESC differentiation and other functions in vivo, when fLuc is controlled by a specific promoter 15, 16, 17. However, it is not clinically translatable as a high dose of D-luciferin substrate must be administered and because the light output produced has limited penetration through tissues of a large animal. In addition, fLuc bioluminescence imaging is primarily a 2D in vivo assay (although tomographic systems are under development) and reveals little spatial information. As expected, SPECT imaging of hESCs labeled with the HSV1-tk reporter (here as the GFP fusion protein) is less sensitive, i.e., the signal-to-noise ratio is significantly lower. However, there are several ways in which we can improve the sensitivity of this radiopharmaceutical-based technique. First, we could switch to iodine-124 and use PET, as PET is at least an order of magnitude more sensitive than SPECT. From a biological perspective, we could use a more efficient version of the HSV1-tk reporter gene, such as the HSV1-sr39tk, which uses acycloguanosine substrates 26. That reporter represents an improvement over the wild-type version used herein because of the use of substrates that interact less with the mammalian TK than do analogs of FIAU. Other strategies to improve the sensitivity of the radiopharmaceutical-based technique would involve the use of alternative genes, i.e., other than HSV1-tk altogether 26, 27, 28. Such reporter genes could be designed to provide a signal amplification step to further increase the sensitivity 26, 27, 28.
In terms of extension of this work, we will test the engraftment of lineage-specific progeny derived from hESCs. So far, the differentiated progeny from hESCs have been detected only by using a fLuc optical bioluminescence imaging system similar to that described here 15, 16, 17, although both optical bioluminescence and HSV1-tk-based nuclear imaging have been reported with mouse ESC derivatives 29, 30. In the near future, a radiopharmaceutical-based technique for hESCs is more likely to be used in clinical translation 27, 28. This report provides a foundation for further improvements of in vivo detection of transplanted hESCs, and for other forms of cell therapy, by using various imaging techniques, including those that are clinically translatable.
Materials and Methods
Lentiviral vectors containing imaging reporter genes
We have made several lentiviral vectors expressing the HSV1-tk gene as a reporter. The most studied and useful among these is cEF.tk-GFP, which is based on the EF.GFP lentiviral vector backbone with the EF1a housekeeping gene promoter 1. The improved version, cEF.GFP (vs. EF.GFP), contains the central purine track (PPT, 200 bp) element, which was added to increase vector-integration efficiency. The chimeric gene encoding the TK-GFP fusion protein was from the pCMV.TK-GFP plasmid 9. Briefly, the 337-amino-acid TK enzyme is fused to the N-terminus of the GFP reporter (238 amino acids), encoded by a single transgene 9.
As we observed that several versions of fusion proteins between fLuc and GFP severely diminished GFP fluorescence, we decided to make a bi-cistronic vector coupling fLuc expression and GFP by an internal ribosomal entry site (IRES) sequence. The fLuc coding sequence (1.7 kb for 551 amino acids in a BsmI to XbaI DNA fragment) was excised from pGL3-basic plasmid (Promega, Madison, WI) and inserted into the lentiviral vector cEF.GFP3, which is derived from cEF.GFP and contains an IRES element (600 bp) upstream from the GFP sequence. The resulting lentiviral vector cEF.Luc-IRES.GFP (also called L3GFP) expresses both fLuc and GFP from the same transcript, although GFP intensity is weaker than that obtained by cEF.GFP or cEF.GFP3.
Production of lentiviral vectors
All recombinant lentiviruses were produced by transient transfection of 293T cells, using the CMV ΔR8.91 and pMD.G helper plasmids 1, 2, 31. After overnight incubation, the culture medium was replaced with Dulbecco's modified Eagle's medium (DMEM) containing 1% fetal bovine serum (FBS). The medium containing secreted viral particles was collected daily and filtered through a 0.45-μm filter (Corning Life Sciences, www.corning.com). Higher titers of virus were obtained using Amicon Ultra-15 centrifugal filter devices (Millipore, Billerica, MA) as described earlier 1, 2, 31. Recombinant lentiviral viruses were confirmed as replication-incompetent as described earlier 1. This is based on the lack of production of HIV-1 p24 viral proteins in stably transduced cells or their culture supernatants 1, 31, using the HIV-1 p24 ELISA kit from RETROtek (Buffalo, NY).
Cell culture
Human 293T cells were cultured in DMEM (Invitrogen, www.invitrogen.com) containing 10% FBS. The NIH-approved hESC cell lines H1 (WA01) and H9 (WA09) were used in this study. They were obtained from WiCell Research Institute, Inc. (www.wicell.org) at passage 22 and 23, respectively. Both H1 and H9 hES cells were propagated on top of mitotically inactive primary mouse embryonic fibroblast (pMEF) feeder cells or human feeder cells as described 10. A human CD34 hematopoietic progenitor cell line TF1 (ATCC CRL-2003) was cultured as described earlier 1, 2, 18. Briefly, TF1 cells were cultured in RPMI-1640 medium supplemented with 10% FBS and 1 ng/ml GM-CSF (Peprotech, www.peprotech.com).
Lentiviral transduction
293T and TF1 cells were transduced as described earlier 1, 2, 18. For transduction of hESCs cultured under a feeder-free condition, undifferentiated cell clumps were passaged onto Matrigel (BD Biosciences, www.bdbiosciences.com) and cultured in pMEF-conditioned medium (pMEF-CM) for 1-2 days 18, 31. At the time of transduction, a mixture of 50 μl of concentrated (30×) lentiviral supernatant and 4 μg/ml of polybrene in pMEF-CM was applied to hESCs for 8 h (MOI = 30-100). Transduced cells were then cultured with daily replacement of pMEF-CM. At 3 days after transduction, the cells were replated onto fresh pMEF feeders 10, 31.
Flow cytometric analysis of transgene expression
To analyze regulated transgene expression, target cells were first transduced and sorted for GFP cells by either FACSvantage or FACSAria flow cytometry (BD Biosciences, www.bdbiosciences.com).
HSV1-tk activity in transduced cells
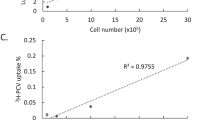
FIAU, ganciclovir (GCV) and penciclovir (PCV) were purchased from Moravek Biochemicals (www.moravek.com). [8-3H]PCV (22 Ci/mmol, 1 mCi/ml, purchased from Moravek Biochemicals) was also used to monitor specific uptake by the tk-GFP-transduced cells. [8-3H]PCV at a non-cytotoxic concentration of 0.02, 0.1 or 0.5 μM was added into the transduced or parental 293T cells (5 × 104 cells per well). After 1 h of incubation, the medium containing radioactive probes was replaced by PCV-free fresh medium. After an additional 2 h of incubation, adherent cells were washed and harvested, and the cell-associated tritium-labeled probes were counted by a scintillation counter.
Tumor formation in mice
The pluripotency of hESCs can be estimated by testing their ability to form teratomas in immuno-deficient mouse models. The following procedure, based on earlier reports 10, was approved by the Animal Care and Use Committee at Johns Hopkins University and has been used since 2002. When 2 × 106 293T cells or transduced derivatives were used, they formed a palpable tumor within 2 weeks of inoculation into SCID mice. For teratoma formation that typically takes 2-4 months, we found that C.B-17 SCID-Beige mice (Taconic, www.taconic.com, catalog no. CBSCBG-MM) are more efficient in supporting teratoma formation than NOD/SCID and NOD/SCID/β2-microglobulin−/− mice, as reported elsewhere 32. Cells from one 6-well plate (2-5 × 106 hESCs) per injection were used for teratoma formation. Cells were washed and resuspended in 250-300 μl of HBSS for intramuscular injection. Histological analysis of the resulting teratomas was performed by H&E staining of fixed sections to identify cells derived from each of the three embryonic germ layers 10.
In vitro and in vivo luciferase assays
D-luciferin (potassium salt), purchased from Xenogen Corporation (www.xenogen.com), was dissolved in sterile PBS to make a stock solution of 15 mg/ml. The Xenogen IVIS and IVIS 200 systems were used for both in vitro imaging of cells in 96-well plates and in vivo imaging of live mice. For in vitro imaging, cells were plated at various densities in 96-well plates. The wells were aspirated before addition of 100 μl of a 150-μg/ml solution of D-luciferin. Plates were incubated at 37 °C in 5% CO2 for 5 min before imaging. Optical signals reached a plateau at 5-10 min after addition of D-luciferin substrate and were then recorded as a sum using a 1-min exposure. Photon counts per minute from each duplicate well were recorded and plotted as a function of cell number.
For imaging in vivo, each mouse received 150 mg of D-luciferin per kg of body weight. Mice were anesthetized using isoflurane gas (2% in oxygen at 0.6 l/min flow rate) throughout imaging, and images were collected at indicated times (normally 5 or 20 min) after D-luciferin injection (monitored up to 40 min after the injection). Composite images obtained were comprised of black and white digital photos with an overlay of images reflecting bioluminescent intensity. The density map, measured as photons/second/cm2/steradian (p/s/cm2/sr), were created using the Xenogen software and represented as a color gradient centered at the maximal spot. (The number is also denoted.) A sum in the ROI was also automatically selected and calculated by the software.
[125I]FIAU preparation
We used a modification of an earlier-described procedure 9. Briefly, 1-(2′-deoxy-2′-fluoro-β-D-arabinofuranoside)-uracil (FAU, 300 μg, 1.22 mmol, Moravek Biochemicals, www.moravek.com) was dissolved in 170 μl of 2 M HNO3. To this solution, 1.5 mCi of [125I]NaI (MP Biomedicals, Cost Mesa, CA) was added and the contents heated at 130 °C for 45 min. The reaction was quenched with 150 μl of HPLC mobile phase (20:79.9:0.1% MeCN:H2O:triethylamine). The resulting [125I]FIAU was purified by reverse-phase HPLC through two passages over a Phenomenex Luna C18 semiprep column (10 μm, 4.6 × 250 mm, Phenomenex, Torrance, CA), by using the above-mentioned isocratic mobile phase at a flow rate of 2 ml/min. The product was concentrated under reduced pressure and formulated in 0.9% physiological saline before sterile filtration through a 0.22-μm syringe filter. Formulations were kept at 1 mCi/ml to minimize the injection volume. The final radiochemical yield was 50%, the radiochemical purity was > 99%, and the specific radioactivity was > 2 000 Ci/mmol.
In vivo SPECT-CT
Mice were injected with 200-250 μCi of [125I]FIAU through the tail vein and imaged at various time points thereafter. Mice were anesthetized through s.c. administration of a combination of ketamine (72 mg/kg), xylazine (6 mg/kg) and acepromazine (6 mg/kg) before imaging. Planar gamma scintigraphy was performed over 10 min using a dedicated small-animal SPECT/CT device (Gamma Medica X-SPECT, Northridge, CA). The X-SPECT has a γ-ray detector head with dimensions of 20.5 cm × 15 cm × 9 cm and a 120 mm × 125 mm field of view (FOV). The high-resolution parallel hole collimator used in this study has the following specifications: 1.22 mm hole diameter, 0.20 mm septa thickness, 25.4 mm bore-hole length. The detector material or scintillator crystal is composed of NaI[Tl], which has a pixel size of 2 mm × 2 mm × 6 mm. Mice were placed in a prone position on the parallel-hole collimator. The static acquisition protocol of the LumaGEM™ software provided with the X-SPECT was used. High-resolution scans of each mouse were obtained. SPECT-CT was performed using the above-mentioned γ-ray detector and the CT detector, sequentially, without removing the mouse from the gantry.
References
Cui Y, Golob J, Kelleher E, et al. Targeting transgene expression to antigen-presenting cells derived from lentivirus-transduced engrafting human hematopoietic stem/progenitor cells. Blood 2002; 99:399–408.
Yu X, Zhan X, D'Costa J, et al. Lentiviral vectors with two independent internal promoters transfer high-level expression of multiple transgenes to human hematopoietic stem-progenitor cells. Mol Ther 2003; 7:827–838.
Yang X, Atalar E, Li D, et al. MRI permits in vivo monitoring catheter-based vascular gene transfer. Circulation 2001; 104:1588–1590.
Chen HH, Zhan X, Kumar A, et al. Detection of dual gene expression in arteries using an optical imaging method. J Biomed Opt 2004; 9:1223–1229.
Du X, Qiu B, Zhan X, et al. Intravascular MR/radiofrequency-enhanced vascular gene transduction/expression: feasibility study in pigs. Radiology 2005; 236:939–944.
Thomson JA, Itskovitz-Eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science 1998; 282:1145–1147.
Tjuvajev JG, Stockhammer G, Desai R, et al. Imaging the expression of transfected genes in vivo. Cancer Res 1995; 55:6126–6132.
Tjuvajev JG, Avril N, Oku T, et al. Imaging herpes virus thymidine kinase gene transfer and expression by positron emission tomography. Cancer Res 1998; 58:4333–4341.
Jacobs A, Dubrovin M, Hewett J, et al. Functional coexpression of HSV-1 thymidine kinase and green fluorescent protein: implications for noninvasive imaging of transgene expression. Neoplasia 1999; 1:154–161.
Dravid G, Hammond H, Cheng L . Culture of human embryonic stem cells on human and mouse feeder cells. Methods Mol Biol 2006; 331:91–104.
Schuldiner M, Itskovitz-Eldor J, Benvenisty N . Selective ablation of human embryonic stem cells expressing a “suicide” gene. Stem Cells 2003; 21:257–265.
Gropp M, Itsykson P, Singer O, et al. Stable genetic modification of human embryonic stem cells by lentiviral vectors. Mol Ther 2003; 7:281–287.
van der Bogt KE, Swijnenburg RJ, Cao F, Wu JC . Molecular imaging of human embryonic stem cells: keeping an eye on differentiation, tumorigenicity and immunogenicity. Cell Cycle 2006; 5:2748–2752.
Swijnenburg RJ, van der Bogt KE, Sheikh AY, et al. Clinical hurdles for the transplantation of cardiomyocytes derived from human embryonic stem cells: role of molecular imaging. Curr Opin Biotechnol 2007; 18:38–45.
Barberi T, Bradbury M, Dincer Z, et al. Derivation of engraftable skeletal myoblasts from human embryonic stem cells. Nat Med 2007; 13:642–648.
Bradbury MS, Tomishima M, Panagiotakos G, et al. Optical bioluminescence imaging of human ES cell progeny in the rodent CNS. J Neurochem 2007; 102:2029–2039.
Li Z, Suzuki Y, Huang M, et al. Comparison of reporter gene and iron particle labeling for tracking fate of human embryonic stem cells and differentiated endothelial cells in living subjects. Stem Cells 2008; 26:864–873.
Zhou BY, Ye Z, Gao PZ, et al. Inducible and reversible transgene expression in human stem cells after efficient and stable gene transfer. Stem Cells 2007; 25:779–789.
Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007; 131: 861–872.
Yu J, Vodyanik MA, Smuga-Otto K, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007; 318:1917–1920.
Park IH, Zhao R, West JA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature 2008; 451:141–146.
Mali P, Ye Z, Hammond H, et al. Improved efficiency and pace of generating induced pluripotent stem cells from human adult and fetal fibroblasts. Stem Cells 2008; 26:1998–2005.
Blum B, Benvenisty N . Clonal analysis of human embryonic stem cell differentiation into teratomas. Stem Cells 2007; 25:1924–1930.
Tzukerman M, Rosenberg T, Ravel Y, et al. An experimental platform for studying growth and invasiveness of tumor cells within teratomas derived from human embryonic stem cells. Proc Natl Acad Sci USA 2003; 100:13507–13512.
Tzukerman M, Rosenberg T, Reiter I, et al. The influence of a human embryonic stem cell-derived microenvironment on targeting of human solid tumor xenografts. Cancer Res 2006; 66:3792–3801.
Iyer M, Barrio JR, Namavari M, et al. 8-[18F] Fluoropenciclovir: an improved reporter probe for imaging HSV1-tk reporter gene expression in vivo using PET. J Nucl Med 2001; 42:96–105.
Blasberg RG, Tjuvajev JG . Molecular-genetic imaging: current and future perspectives. J Clin Invest 2003; 111:1620–1629.
Massoud TF, Gambhir SS . Integrating noninvasive molecular imaging into molecular medicine: an evolving paradigm. Trends Mol Med 2007; 13:183–191.
Cao F, Drukker M, Lin S, et al. Molecular imaging of embryonic stem cell misbehavior and suicide gene ablation. Cloning Stem Cells 2007; 9:107–117.
Cao F, Lin S, Xie X, et al. In vivo visualization of embryonic stem cell survival, proliferation, and migration after cardiac delivery. Circulation 2006; 113:1005–1014.
Ye Z, Yu X, Cheng L . Lentiviral gene transduction of mouse and human stem cells. Methods Mol Biol 2008; 430:243–253.
Tian X, Woll PS, Morris JK, et al. Hematopoietic engraftment of human embryonic stem cell-derived cells is regulated by recipient innate immunity. Stem Cells 2006; 24:1370–1380.
Acknowledgements
We thank Drs Vladimir Ponomarev and Juri Gelovani (Johns Hopkins University) for providing the pCMV.TK-GFP plasmid. We thank Ms Lu Qin, Drs Hyam Levitsky and Richard Wahl (Johns Hopkins University) for their helpful discussions. This work was funded in part by the Stem Cell Research Foundation (S2005-026), Johns Hopkins Institute for Cell Engineering and NIH grants R01 HL073781, U24 CA92871 and P50 CA103175. L Cheng is a recipient of an International Collaboration Award (#30428010) from the National Natural Sciences Foundation of China.
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Pomper, M., Hammond, H., Yu, X. et al. Serial imaging of human embryonic stem-cell engraftment and teratoma formation in live mouse models. Cell Res 19, 370–379 (2009). https://doi.org/10.1038/cr.2008.329
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cr.2008.329
Keywords
This article is cited by
-
Bright ferritin for long-term MR imaging of human embryonic stem cells
Stem Cell Research & Therapy (2023)
-
Current Status and Progress in Stem Cell Therapy for Intracerebral Hemorrhage
Translational Stroke Research (2023)
-
A Human-derived Dual MRI/PET Reporter Gene System with High Translational Potential for Cell Tracking
Molecular Imaging and Biology (2022)
-
CRYβB2 enhances tumorigenesis through upregulation of nucleolin in triple negative breast cancer
Oncogene (2021)
-
124I-PET Assessment of Human Sodium Iodide Symporter Reporter Gene Activity for Highly Sensitive In Vivo Monitoring of Teratoma Formation in Mice
Molecular Imaging and Biology (2015)
