
Abstract
Background:
This phase 1 trial utilising a Bayesian continual reassessment method evaluated bortezomib and sunitinib to determine the maximum tolerated dose (MTD), dose-limiting toxicities (DLT), and recommended doses of the combination.
Methods:
Patients with advanced solid organ malignancies were enrolled and received bortezomib weekly with sunitinib daily for 4 weeks, every 6 weeks. Initial doses were sunitinib 25 mg and bortezomib 1 mg m−2. Cohort size and dose level estimation was performed utilising the Escalation with Overdose Control (EWOC) adaptive method. Seven dose levels were evaluated; initially, sunitinib was increased to a goal dose of 50 mg with fixed bortezomib, then bortezomib was increased. Efficacy assessment occurred after each cycle using RECIST criteria.
Results:
Thirty patients were evaluable. During sunitinib escalation, DLTs of grade 4 thrombocytopenia (14%) and neutropenia (6%) at sunitinib 50 mg and bortezomib 1.3 mg m−2 were seen. Subsequent experience showed tolerability and activity for sunitinib 37.5 mg and bortezomib 1.9 mg m−2. Common grade 3/4 toxicities were neutropenia, thrombocytopenia, hypertension, and diarrhoea. The recommended doses for further study are bortezomib 1.9 mg m−2 and sunitinib 37.5 mg. Four partial responses were seen. Stable disease >6 months was noted in an additional six patients.
Conclusion:
Bortezomib and sunitinib are well tolerated and have anticancer activity, particularly in thyroid cancer. A phase 2 study of this combination in thyroid cancer patients is planned.
Similar content being viewed by others

Main
The development of receptor- and protein complex-targeted anticancer agents has improved disease outcomes with favourable tolerability profiles. While monotherapy with these drugs has had success in cancers with historically poor outcomes (e.g., gastrointestinal stromal tumours and renal cell carcinoma), single agent responses in other solid tumour malignancies have been limited (Barrios et al, 2010; Gallagher et al, 2010). Combinations with conventional DNA-damaging agents have also generally failed to show improvement over cytotoxics alone (Socinski et al, 2010). Alternative approaches inhibiting multiple interacting and interdependent pathways, vertically and horizontally, with two or more molecularly targeted therapies holds greater potential for improved outcomes.
Sunitinib is an oral, small-molecule inhibitor of multiple receptors (platelet-derived growth factor, vascular endothelial growth factor, and others). In addition to known activity in RCC and GIST, sunitinib is efficacious in refractory differentiated and medullary thyroid cancers. Bortezomib is a first-in-class reversible inhibitor of the 26S proteasome, which leads to apoptosis through multiple mechanisms. It is approved by the US Food and Drug Administration (FDA) for the treatment of multiple myeloma and relapsed mantle cell lymphoma. To date, single agent response rates in heavily pretreated patients with solid tumour malignancies have been low; however, combination strategies with conventional chemotherapy and molecularly targeted agents have activity and are tolerable (Fanucchi et al, 2006; Dees et al, 2008; Dudek et al, 2009).
Based on complementary antiangiogenic activity (Williams et al, 2003; Ebos et al, 2007) and an expected synergy between the two compounds (Wright 2010), we conducted a phase 1 evaluation of sunitinib and bortezomib in patients with refractory solid tumours (NCT00720148). The primary objective was to establish the safety and determine the maximum tolerated doses (MTD) of bortezomib and sunitinib in combination. Secondary objectives were to define toxicities and obtain preliminary information on the anticancer activity of the regimen.
Methods
Patients with confirmed solid organ malignancies refractory to standard therapy, or for whom no standard therapy existed, and measurable disease by RECIST criteria (version 1.0) were eligible. Other eligibility criteria included age ⩾18 years; Eastern Cooperative Group (ECOG) performance status ⩽2; adequate bone marrow reserve, renal and hepatic function. Patients were excluded if they had prior radiation to >30% of bone marrow volume; ejection fraction of ⩽45%; uncontrolled cardiovascular disease; peripheral neuropathy of ⩾grade 2 by NCI Common Toxicity Criteria for Adverse Events (CTCAE), version 4; and/or recent (within 30 days) history of venous thromboembolism. The study was approved by the Emory University Institutional Review Board and was conducted in accordance with the guidelines from the Helsinki declaration of 1975. All participants gave written, informed consent.
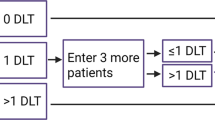
All patients received sunitinib (Sutent; Pfizer Labs; New York, NY, USA) and bortezomib (Velcade; Millennium Pharmaceuticals, Inc.; Cambridge, MA, USA) orally daily and intravenously weekly, respectively, for 4 weeks, followed by a 2-week rest. The study was conducted in two phases. Initially, the dose of bortezomib was fixed at 1 mg m−2 and sunitinib escalated from 25 to 37.5 to 50 mg (Table 1). Once maximum tolerated dose (MTD) was determined for the first phase, the bortezomib dose was escalated with fixed sunitinib. Dose selection was carried out utilising the flexible Bayesian method of Escalation with Overdose Control (EWOC) (Tighiouart and Rogatko 2010), a fully adaptive, real time dose-finding method that uses individual patient experiences to select subsequent doses, allowing for more rapid dose escalation while minimising the number of patients who are underdosed.
Adverse events were characterised using the NCI CTCAE version 4.0, and patients were evaluated weekly. Dose limiting toxicity (DLT) was defined as grade 4 neutropenia, anaemia, or thrombocytopenia; grade 4 fatigue, or a two-point decline in ECOG performance status unrelated to underlying malignancy; grade 3 or 4 gastrointestinal toxicity despite the use of maximal medical intervention; or any other clinically significant toxicity of grade ⩾3 attributed to one or both agents during the first cycle.
Treatment cycles were initiated if the ANC was ⩾1000 per mm3, the platelet count ⩾100 000 per mm3. Patients experiencing grade 4 haematologic toxicity had both treatments held for up to 2 weeks, until recovery of ANC ⩾1000 per mm3 and platelet count ⩾100 000 per mm3. For grade 3 haematologic toxicities prior to beginning a treatment cycle, treatment was held up to 2 weeks until resolution to ⩽grade 2. For uncontrolled hypertension due to sunitinib, maximum doses of up to three antihypertensive agents were used prior to dose reduction or discontinuation. Bortezomib was dose-reduced by one level for neuropathy. Patients were deemed evaluable for DLT if they completed ⩾75% of the first cycle.
Tumour assessments were done at baseline and the end of each cycle, and responses were classified using RECIST criteria. The initial dose level was sunitinib 25 mg and bortezomib 1.0 mg m−2. Dose escalation was conducted in two stages; initially bortezomib was fixed whereas sunitinib was escalated to its MTD. Specifically, each patient received 1.0 mg m−2 bortezomib and the level of sunitinib determined by EWOC so that, on the basis of all available data, the probability that it exceeded the MTD was equal to a prespecified value α. In the first stage, we started at α=0.2 and increased α in increments of 0.05 until α=0.4, this value being a compromise between the therapeutic aspect of the agent and side effects. Sixteen patients were enrolled in the first stage of the trial. In the second stage, sunitinib was fixed at the newly established MTD and bortezomib escalated from 1.0 mg m−2. During this stage, the parameter α was increased, after each successive patient, in increments of 0.05 from an initial value of 0.4 to a terminal value of 0.5. At the end of the second stage of the trial, an MTD was estimated using data from the second stage as the median of the posterior distribution of the MTD of bortezomib given that sunitinib was =37.5 mg.
Results
Thirty-seven patients consented, of whom 31 received at least one dose of the study drug and 30 were evaluable for the primary endpoint. Demographic data are presented in Table 2. The seven inevaluable patients had rapidly progressive disease and did not complete cycle 1. Because enrolment occurred real time as patients were referred, the majority of experience was with the sunitinib 50 mg dose level and bortezomib 1 or 1.3 mg m−2. Cycle 1 adverse event data is summarised in Table 3. DLTs were seen at dose level four (sunitinib 50 mg and bortezomib 1.3 mg m−2) and were grade 4 thrombocytopenia (16%) and neutropenia (4%). Following cycle 1, the most common treatment-emergent adverse events were thrombocytopenia, diarrhoea, mucositis, and fatigue. Two patients developed varicella zoster infections at dose level three, prompting the institution of acyclovir prophylaxis in subsequent subjects. One patient at dose level three who developed grade 3 hypertension in cycle 1 (subsequently controlled on lisinopril), went on to develop grade 2 proteinuria (3239 mg over 24 h), which spontaneously resolved.
The median number of cycles delivered was 3 (range 1–12), for a median time on study of 18 weeks (range 6–72). Four patients achieved partial response by RECIST criteria, two at dose level three (medullary thyroid cancer and squamous cell cancer of the nasopharynx), and one each at dose levels four (Hurthle cell thyroid) and seven (papillary thyroid cancer). Stable disease lasting >6 months was noted in an additional six subjects, specifically in patients with papillary (two) and medullary (one) thyroid cancers, pancreatic neuroendocrine tumour, melanoma, and pleomorphic sarcoma. Taken together, the clinical benefit rate was 30%. At the conclusion of dose escalation and after considering the overall toxicity profile, the recommended phase 2 doses of the combination using EWOC were sunitinib 37.5 mg PO daily and bortezomib 1.9 mg m−2 IV weekly, each given 4 weeks of 6.
Discussion
Anticancer drug development has evolved to include combinations of targeted agents without traditional cytotoxic partners. Although multitargeted receptor tyrosine kinase inhibitors such as sunitinib have advanced therapy in the single agent setting, the goal of further tumour burden reduction and clinically meaningful prolongation of disease control is likely going to require multiple agents to achieve. The combination of bortezomib and sunitinib is a rational one, as proteasome inhibition impairs cycle progression and proliferation, activates apoptosis, and inhibits angiogenesis and metastasis (Boccadoro et al, 2005). Additionally, bortezomib-induced inhibition of the NFκB pathway is augmented in the presence of sunitinib, suggesting at least additive if not synergistic activity in combination (Sorolla et al, 2012). Furthermore, sunitinib sensitises cancer cells to bortezomib-induced apoptosis (Yeramian et al, 2012). These preclinical data, along with favourable toxicity profiles, support the combination evaluated.
The addition of bortezomib to sunitinib was well tolerated and demonstrated meaningful anticancer activity in a number of solid tumours. We observed intolerable haematologic toxicity with sunitinib 50 mg and bortezomib 1.3 mg m−2, leading to escalation of bortezomib at a lower dose of sunitinib to gain further insight into the relative contribution of each agent to haematologic toxicity. The adaptive Bayesian design allowed accrual to both the known safest and most effective dose during the trial, as well as informed decision-making for subsequent evaluation. When compared with different up-and-down schemes, including ‘3+3’ designs, EWOC assigns fewer patients to either subtherapeutic or toxic dose levels, treats more patients at optimal dose levels and estimates the MTD with smaller average bias and mean squared error (Babb et al, 1998).
We preferentially enrolled patients with thyroid cancers, due to the previously reported activity of sunitinib (Carr et al, 2010). Both the radiographic and biochemical activity noted in thyroid cancer in our trial compares favourably to prior data with single agent sunitinib. The sunitinib dose selected here is slightly lower than prior experiences; however, improved long-term tolerability in combination with bortezomib formed the basis of the recommendation. Subsequent evaluation of the combination in thyroid cancer is warranted.
Change history
05 March 2013
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
Babb J, Rogatko A, Zacks S (1998) ‘Cancer phase I clinical trials: efficient dose escalation with overdose control’. Stat Med 17 (10): 1103–1120
Barrios CH, Liu MC, Lee SC, Vanlemmens L, Ferrero JM, Tabei T, Pivot X, Iwata H, Aogi K, Lugo-Quintana R, Harbeck N, Brickman MJ, Zhang K, Kern KA, Martin M (2010) ‘Phase III randomized trial of sunitinib versus capecitabine in patients with previously treated HER2-negative advanced breast cancer’. Breast Cancer Res Treat 121 (1): 121–131
Boccadoro M, Morgan G, Cavenagh J (2005) ‘Preclinical evaluation of the proteasome inhibitor bortezomib in cancer therapy’. Cancer Cell Int 5 (1): 18
Carr LL, Mankoff DA, Goulart BH, Eaton KD, Capell PT, Kell EM, Bauman JE, Martins RG (2010) ‘Phase II study of daily sunitinib in FDG-PET-positive, iodine-refractory differentiated thyroid cancer and metastatic medullary carcinoma of the thyroid with functional imaging correlation’. Clin Cancer Res 16 (21): 5260–5268
Dees EC, O’Neil BH, Lindley CM, Collichio F, Carey LA, Collins J, Riordan WJ, Ivanova A, Esseltine D, Orlowski RZ (2008) ‘A phase I and pharmacologic study of the combination of bortezomib and pegylated liposomal doxorubicin in patients with refractory solid tumors’. Cancer Chemother Pharmacol 63 (1): 99–107
Dudek AZ, Lesniewski-Kmak K, Shehadeh NJ, Pandey ON, Franklin M, Kratzke RA, Greeno EW, Kumar P (2009) ‘Phase I study of bortezomib and cetuximab in patients with solid tumours expressing epidermal growth factor receptor’. Br J Cancer 100 (9): 1379–1384
Ebos JM, Lee CR, Christensen JG, Mutsaers AJ, Kerbel RS (2007) ‘Multiple circulating proangiogenic factors induced by sunitinib malate are tumor-independent and correlate with antitumor efficacy’. Proc Natl Acad Sci USA 104 (43): 17069–17074
Fanucchi MP, Fossella FV, Belt R, Natale R, Fidias P, Carbone DP, Govindan R, Raez LE, Robert F, Ribeiro M, Akerley W, Kelly K, Limentani SA, Crawford J, Reimers HJ, Axelrod R, Kashala O, Sheng S, Schiller JH (2006) ‘Randomized phase II study of bortezomib alone and bortezomib in combination with docetaxel in previously treated advanced non-small-cell lung cancer’. J Clin Oncol 24 (31): 5025–5033
Gallagher DJ, Milowsky MI, Gerst SR, Ishill N, Riches J, Regazzi A, Boyle MG, Trout A, Flaherty AM, Bajorin DF (2010) ‘Phase II study of sunitinib in patients with metastatic urothelial cancer’. J Clin Oncol 28 (8): 1373–1379
Socinski MA, Scappaticci FA, Samant M, Kolb MM, Kozloff MF (2010) ‘Safety and efficacy of combining sunitinib with bevacizumab+paclitaxel/carboplatin in non-small cell lung cancer’. J Thorac Oncol 5 (3): 354–360
Sorolla A, Yeramian A, Valls J, Dolcet X, Bergadà L, Llombart-Cussac A, Martí RM, Matias-Guiu X (2012) 'Blockade of NFkappaB activity by Sunitinib increases cell death in Bortezomib-treated endometrial carcinoma cells'. Mol Oncol 6 (5): 530–541
Tighiouart M, Rogatko A (2010) 'Dose finding with escalation with overdose control (EWOC) in cancer clinical trials'. Stat Sci 25: 217–226
Williams S, Pettaway C, Song R, Papandreou C, Logothetis C, McConkey DJ (2003) ‘Differential effects of the proteasome inhibitor bortezomib on apoptosis and angiogenesis in human prostate tumor xenografts’. Mol Cancer Ther 2 (9): 835–843
Wright JJ (2010) ‘Combination therapy of bortezomib with novel targeted agents: an emerging treatment strategy’. Clin Cancer Res 16 (16): 4094–4104
Yeramian A, Sorolla A, Velasco A, Santacana M, Dolcet X, Valls J, Abal L, Moreno S, Egido R, Casanova JM, Puig S, Vilella R, Llombart-Cussac A, Matias-Guiu X, Martí RM (2012) 'Inhibition of activated receptor tyrosine kinases by Sunitinib induces growth arrest and sensitizes melanoma cells to Bortezomib by blocking Akt pathway'. Int J Cancer 130 (4): 967–978
Acknowledgements
We thank Pfizer for their global support of the trial and Millennium Pharmaceuticals for the provision of bortezomib.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Disclaimer
The authors take full responsibility for the scope, direction, and content of the manuscript and have approved the submitted manuscript.
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article
Cite this article
Harvey, R., Owonikoko, T., Lewis, C. et al. A phase 1 Bayesian dose selection study of bortezomib and sunitinib in patients with refractory solid tumor malignancies. Br J Cancer 108, 762–765 (2013). https://doi.org/10.1038/bjc.2012.604
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2012.604
Keywords
This article is cited by
-
Is more better? An analysis of toxicity and response outcomes from dose-finding clinical trials in cancer
BMC Cancer (2021)
-
The Treatment of Advanced Thyroid Cancer in the Age of Novel Targeted Therapies
Drugs (2017)
-
Treatment of advanced thyroid cancer: role of molecularly targeted therapies
Targeted Oncology (2015)
-
Discovery of novel covalent proteasome inhibitors through a combination of pharmacophore screening, covalent docking, and molecular dynamics simulations
Journal of Molecular Modeling (2014)
