


1. Introduction
This journal started at a time when statistical genetics was about to undergo a revolution brought about by the generation of isozyme data with the new technology of electrophoresis. Almost overnight it became possible to score dozens of genetic markers in samples of hundreds of individuals. These new data called into question the predictions about levels of genetic variation expected under theories developed over the previous 50 years. A fierce debate between the ‘neutralists’ and ‘selectionists’ pitted population geneticists against each other, with each side invoking statistical analyses of quantities such as heterozygosity, inbreeding coefficients, linkage disequilibrium and population structure parameters. Publications reporting isozyme data have now almost disappeared, as have papers arguing about the role of selection since there is a realization that both natural selection and random processes have a role in evolution. As the journal celebrates its 50th anniversary another revolution is about to take place – one leading to whole genome sequence data on large numbers of individuals (The 1000 Genomes Consortium, 2010). There does not appear to be any danger of statistical geneticists falling out in the 2010s as they did in the 1960s but surely major shifts in our understanding of evolution will come.
In this discussion we will concentrate on the characterization of allelic associations in the era that has provided whole-genome single nucleotide polymorphism (SNP) datasets, and we will be guided by the experience of our colleagues and ourselves with data collected for genome-wide association studies (GWAS) (Laurie et al., Reference Laurie, Doheny, Mirel, Pugh, Bierut, Bhangale, Boehm, Caporaso, Edenberg, Gabriel, Harris, Hu, Jacobs, Kraft, Landi, Lumley, Manolio, McHugh, Painter, Paschall, Rice, Rice, Zheng and Weir2010). With a million data points per individual we and many other investigators in 2010 are uncovering properties of genomes and populations we could not begin to address in 1960. We have data that give us empirical values where once we had to take limits in mathematical expressions. We have the data but maybe not the statistical tools to exploit them fully.
2. The data
Human geneticists now have array technology that allows the rapid generation of up to 2.5 million SNP genotypes per individual and twice that number will soon be possible. The decisions as to which SNPs of the over 15 million that have been discovered to include on commercial genotyping platforms have been based on uniformity of coverage in terms of physical distances along the genome or in terms of linkage disequilibrium between pairs of markers. In either case, early concerns of bias resulting from the discovery of SNPs in small samples of people of European ancestry, thereby missing variants in other populations, are diminishing with the use of sequencing to discover new variants over many populations in activities such as the 1000-genomes project (http://www.1000genomes.org/).
The amount of SNP data being reported in the literature is substantial. A recent publication on human height reported results from 183 727 individuals with genotype data observed or imputed for 2 834 208 SNPs (Allen et al., Reference Allen2010). An online catalogue of GWAS results (http://wwww.genome.gov/gwastudies) lists over 700 publications and results from over one million study participants.
3. Allelic associations
(i) Hardy–Weinberg testing
The first measure of association considered by population geneticists is that between the two alleles a diploid individual receives at each autosomal locus from its parents. The realization that there should be no such association in random mating populations for neutral genes goes back almost to the rediscovery of Mendel's laws (Hardy, Reference Hardy1908; Weinberg, Reference Weinberg1908). In the biochemical genetic era, examining new data sets for possible departures from Hardy–Weinberg Equilibrium (HWE) was one of the ways in which evidence for the action of natural selection was sought. In the current genome era, Hardy–Weinberg testing is still a frequent activity but the motivation is more of seeking evidence of problems with data. Consistency with the Hardy–Weinberg Law is expected for outcrossing species, so departures raise the possibility of miss-classification of some genotypes. The sheer scale of performing a million tests on a single data set has revealed aspects of the tests that were not previously of concern.
It is well recognized that the classical chi-square goodness of fit test for HWE suffers from spurious significant values when one or more genotype classes have small expected values, and problems follow when the continuous chi-square distribution is used to provide P-values even though the data are discrete. Tests that provide exact P-values are preferred, but it was not until the work of Wigginton et al. (Reference Wigginton, Cutler and Abecasis2005) that the actual nature of exact HWE P-values was widely recognized. If a sample of size n consists of n AA, n Aa, n aa copies of genotypes AA, Aa, aa then an exact test statistic is the multinomial probability of these counts conditional on the allele counts n A=2n AA+n Aa, n a=2n aa+n Aa under the assumption that the HWE hypothesis H 0 is true. Writing this probability as Pr(n Aa|n A,H 0):
where C=(n!n A!n a!)/(2n)!. The P-value for any value of n Aa is this probability for the data plus the probabilities of all sets of genotype counts with the same allele counts and a greater departure from HWE than seen in the data. Under the alternative hypothesis H 1 that HWE does not hold, the probability of the data can be written as:
where ![]() is a function of genotype probabilities in the population and C is chosen to make these probabilities sum to one over all valid values of n Aa. The sum of these quantities for the data and all data sets with a greater departure from HWE gives the power of the test. Note that ψ=2 under HWE. The rejection rule for the exact test specifies those values of n Aa for which the P-value is less than some nominal significance level, such as 0·05. The empirical significance level, the sum of the probabilities under HWE of all n Aa values in the rejection region, however, will always be less than or equal to this nominal value. Rohlfs & Weir (Reference Rohlfs and Weir2008) plotted these empirical significance levels, and corresponding probabilities when HWE does not hold to emphasize the coarseness of the distributions of these statistics. Depending on the allelic counts, the empirical significance levels may be a long way from the nominal values and the power of the test may be quite low.
is a function of genotype probabilities in the population and C is chosen to make these probabilities sum to one over all valid values of n Aa. The sum of these quantities for the data and all data sets with a greater departure from HWE gives the power of the test. Note that ψ=2 under HWE. The rejection rule for the exact test specifies those values of n Aa for which the P-value is less than some nominal significance level, such as 0·05. The empirical significance level, the sum of the probabilities under HWE of all n Aa values in the rejection region, however, will always be less than or equal to this nominal value. Rohlfs & Weir (Reference Rohlfs and Weir2008) plotted these empirical significance levels, and corresponding probabilities when HWE does not hold to emphasize the coarseness of the distributions of these statistics. Depending on the allelic counts, the empirical significance levels may be a long way from the nominal values and the power of the test may be quite low.
For an experiment on which a million tests for HWE are conducted, a simple way to account for multiple testing is to use the Bonferroni procedure – for an experiment-wise error rate of 5% an individual SNP would be declared significantly out of HWE if it had a P-value less than 5×10−8. This procedure is known to be very conservative and is generally avoided in favour of a Q–Q plot in which the ith of one million ranked P-values is plotted against i/106,i=1,2, …, 106 which are the uniformly distributed expected values if all million SNPs are in HWE. The P-values beyond which observed values start to depart from expected values indicate the ‘significant’ values (Fig. 1). The appeal of this procedure needs to be balanced against the findings of Wigginton et al. (Reference Wigginton, Cutler and Abecasis2005) that the P-values may have a distribution far from uniform. We show an example in Fig. 1, where −log10(p) values are plotted against the values expected if there was HWE. These data are from the Prostate, Lung, Colorectal and Ovarian (PLCO) cancer screening trial (Prorok et al., Reference Prorok, Andriole, Bresalier, Buys, Chia, Crawford, Fogel, Gelmann, Gilbert, Hasson, Hayes, Johnson, Mandel, Oberman, O'Brien, Oken, Rafla, Reding, Rutt, Weissfeld, Yokochi and Gohagan2000) and the figure is from The GENEVA Consortium (2008) report of data on 1651 individuals genotypedon the Illumina HumanHap550v3_B array. The figure shows values for 552 278 SNPs and departures from HWE are occurring for P values in the range of 0·01–0·001 instead of the much lower value of 9·1×10−8 suggested by the Bonferroni correction.

Fig. 1. Q-Q plot of −log10(p) values for tests of HWE in PLCO data (Prorok et al., Reference Prorok, Andriole, Bresalier, Buys, Chia, Crawford, Fogel, Gelmann, Gilbert, Hasson, Hayes, Johnson, Mandel, Oberman, O'Brien, Oken, Rafla, Reding, Rutt, Weissfeld, Yokochi and Gohagan2000). Tests for HWE at 552 278 SNPs are represented. The left panel shows all results, and the right panel shows only those results with P<10−6.
Very large numbers of hypothesis tests that can be conducted with genome data reveal inherent problems with conventional testing theory and basing decisions only on P-values. The use of Bayesian methods as an alternative for HWE inference goes back, implicitly, at least to Altham (Reference Altham1971) and, explicitly, to Pereira & Rogatko (Reference Pereira and Rogatko1984). Wakefield (Reference Wakefield2010) has recently given a spirited account of an approach based on Bayes' factors rather than P-values. He pointed out that the rationale for control of the experiment-wise error rate by the Bonferroni correction is not obvious when it is likely that some of the million HWE hypotheses are false. SNPs in regions under the influence of natural selection, for example, may well depart from HWE while those in linkage disequilibrium with disease susceptibility genes will depart from HWE if testing is confined to affected individuals (Feder, Reference Feder1996; Nielsen et al., Reference Nielsen, Ehm and Weir1998).
Wakefield (Reference Wakefield2010) made use of Dirichlet prior distributions on genotype frequencies and he saw the need for a decision rule that depends on sample size and on allele frequencies. The Bayes' factor is the probability of the observed genotypic data under HWE divided by the probability under the alternative hypothesis and this does depend on both sample size and allele frequencies. Using the Bayes' factor as a test statistic gives a procedure by which the type I and type II errors (false rejections of H 0 and false failures to reject H 0) decrease to zero with increasing sample size. To pick a threshold for rejection of HWE using Bayes' factors it is necessary to specify the prior odds of H 0, and the ratio of costs of type II to type I errors. The costs of avoiding both types of error will vary with the context: if HWE tests are being used to detect genotyping errors there may be little cost in retaining SNPs that do depart from HWE (type II error) or in discarding SNPs that do not (type I error). Type I errors would be of concern, however, if SNPs that were truly associated with a disease were discarded because of departures from HWE.
The multiple-testing issues surrounding HWE testing in the genome era apply more generally of course. Wakefield (Reference Wakefield2009) looked at case-control association testing where the costs of both type I and type II errors can be significant. A false rejection of the null hypothesis of no association of an SNP with a disease may waste resources in following up this SNP in a replication study, whereas a failure to detect a real association may delay the location of causal variants. Wakefield invoked a Bayesian decision theory approach by specifying the costs of false non-discovery C FND and false discovery C FD and setting R=C FND/C FD. He would flag an SNP as significant if the posterior odds on the null hypothesis drop below the ratio R: an association is called noteworthy if the Bayes' factor times the prior odds are less than R. There are three elements to the decision problem: the ratio of the probabilities of the data under null and alternative, the prior odds on the null hypothesis and the ratio of costs. The use of Bayes' factors could also be applied to tests of linkage disequilibrium, population structure and so forth.
(ii) Estimation of inbreeding and relatedness
Many applications of statistical-genetic theory rest on knowledge of the relatedness of pairs of individuals in a study sample. Two individuals are related when their alleles are associated because of descent from common ancestral alleles. A single individual is inbred when the two alleles it receives at a locus have descended from a single common allele. Inbreeding and relatedness here refer to allelic associations (identity by descent) brought about by past events, unlike the within-population associations (identity in state) that result in departures from HWE. A recent application concerns the search for ‘missing heritability’ (Manolio et al., Reference Manolio, Collins, Cox, Goldstein, Hindorff, Hunter, McCarthy, Ramos, Cardon, Chakravarti, Chio, Guttmacher, Kong, Kruglyak, Mardis, Rotimi, Slatkin, Valle, Whittemore, Boehnke, Clark, Eichler, Gibson, Haines, Mackay, McCarroll and Visscher2009). When evidence is sought for associations between single SNPs and a trait of interest the SNPs that pass genome-wide threshold values for statistical significance account for only a small fraction of the genetic variation for the trait found in pedigree-based studies (Manolio et al., Reference Manolio, Collins, Cox, Goldstein, Hindorff, Hunter, McCarthy, Ramos, Cardon, Chakravarti, Chio, Guttmacher, Kong, Kruglyak, Mardis, Rotimi, Slatkin, Valle, Whittemore, Boehnke, Clark, Eichler, Gibson, Haines, Mackay, McCarroll and Visscher2009). For human height, for example, analyses of data on twin pairs suggests a heritability of 80% (Visscher et al., Reference Visscher, Macgregor, Benyamin, Zhu, Gordon, Medland, Hill, Hottenga, Willemsen, Boomsma, Liu, Deng, Montgomery and Martin2007), whereas 180 highly associated SNPs account for only 10% (Allen et al., Reference Allen2010). There have been several explanations for the discrepancy, including the possibility of epigenetic effects or that the observed SNPs are not causal but are in linkage disequilibrium with the causal variants. Yang et al. (Reference Yang, Benyamin, McEvoy, Gordon, Henders, Nyholt, Madden, Heath, Martin, Montgomery, Goddard and Visscher2010) used 294 831 observed SNPs in 3925 individuals of European descent to estimate the actual inbreeding of individuals and the actual relatedness of pairs of individuals and then estimated the heritability of height with a method that rests on the relationships
Here ![]() X is the actual inbreeding coefficient for individual X,
X is the actual inbreeding coefficient for individual X, ![]() is the actual coancestry coefficient for individuals X and Y, σA2 is the additive component of genetic variance for the trait and σE2 is the non-genetic component of trait variance. Dominance and epistatic components of variance are ignored. The ‘actual’ inbreeding and coancestry values reflect Mendelian sampling and linkage as opposed to the expected values that follow from pedigree information (Hill & Weir, Reference Hill and Weir2010). Yang et al. were able to account for 45% of the variance in height and they concluded (Yang et al., Reference Yang, Benyamin, McEvoy, Gordon, Henders, Nyholt, Madden, Heath, Martin, Montgomery, Goddard and Visscher2010, p. 565). ‘Thus, most of the heritability is not missing but has not previously been detected because the individual effects are too small to pass stringent significance tests.’
is the actual coancestry coefficient for individuals X and Y, σA2 is the additive component of genetic variance for the trait and σE2 is the non-genetic component of trait variance. Dominance and epistatic components of variance are ignored. The ‘actual’ inbreeding and coancestry values reflect Mendelian sampling and linkage as opposed to the expected values that follow from pedigree information (Hill & Weir, Reference Hill and Weir2010). Yang et al. were able to account for 45% of the variance in height and they concluded (Yang et al., Reference Yang, Benyamin, McEvoy, Gordon, Henders, Nyholt, Madden, Heath, Martin, Montgomery, Goddard and Visscher2010, p. 565). ‘Thus, most of the heritability is not missing but has not previously been detected because the individual effects are too small to pass stringent significance tests.’
In the past, values for inbreeding and coancestry have been inferred from pedigree information. Yu et al. (Reference Yu, Pressoir, Briggs, Bi, Yamasaki, Doebley, McMullen, Gaut, Nielsen, Holland, Kresovich and Buckler2006) were among the first to suggest that more appropriate values may be obtained from genetic marker information. They were concerned with situations where pedigree records may not be accurate or where artificial selection for crop species altered the relationships at selected loci. The results of Yang et al. (Reference Yang, Benyamin, McEvoy, Gordon, Henders, Nyholt, Madden, Heath, Martin, Montgomery, Goddard and Visscher2010) go much of the way to accounting for missing heritability but there are additional complexities surrounding the estimation of inbreeding and relatedness that can be addressed with genomic data.
(a) Estimation methods
For non-inbred individuals, inbreeding and relatedness parameters can be estimated by ad hoc methods of moments or by maximum likelihood. Either procedure requires large numbers of genetic markers to provide reliable estimates. Yang et al. (Reference Yang, Benyamin, McEvoy, Gordon, Henders, Nyholt, Madden, Heath, Martin, Montgomery, Goddard and Visscher2010) phrased estimators in terms of indicator variables defined for locus j and individual X as x j=2, 1, 0 for genotypes AA, Aa, aa. If the frequencies of A, a at locus j are p j, q j in the population to which the individuals of interest belong then for one individual E(x j)=2p j, Var(x j)=2p jq j(1+F X) and for two individuals X,Y with indicator variables x j and y j, Cov(x j,y j)=4p jq jθ XY. Means and variances here refer to averages over all evolutionary histories that have led to the current pair of genotypes at this locus for these two individuals. The estimators used by Yang et al. (Reference Yang, Benyamin, McEvoy, Gordon, Henders, Nyholt, Madden, Heath, Martin, Montgomery, Goddard and Visscher2010) build on these values and are

These estimators have smaller variance than those implicit in the work of Price et al. (Reference Price, Patterson, Plenge, Weinblatt, Shadick and Reich2006) for the EIGENSTRAT package for population structure. The variance may be further reduced by taking the ratios of the sums over loci of the numerators and denominators instead of averaging the ratios.
There may well be interest in a more detailed description of the relatedness of two individuals, with the three k-coefficients of Thompson (Reference Thompson1975) serving to distinguish, say, parent-offspring from full-siblings even though both pairs have coancestries of 0·25. The k i are the probabilities that two non-inbred relatives share i=0, 1, 2 pairs of alleles identical by descent from a recent common ancestor, and these are summarized by the coancestry θ=k 2/2+k 1/4. Moment estimates of the k i were given by Purcell et al. (Reference Purcell, Neale, Todd-Brown, Thomas, Ferreira, Bender, Maller, Sklar, de Bakker, Daly and Sham2007) in their very useful computer package PLINK. For a pair of individuals, they equated the numbers N i of loci for which two individuals share i pairs of alleles identical-in-state to the expected numbers for these categories expressed in terms of the identity-by-descent probabilities k i and solved these equations for the k i. At locus j with alleles A, a and allele frequencies p j, q j, the first two states and their probabilities are

Ignoring finite-sampling and other corrections to ensure valid estimates (Purcell et al., Reference Purcell, Neale, Todd-Brown, Thomas, Ferreira, Bender, Maller, Sklar, de Bakker, Daly and Sham2007) this provides

which lead to
Moment estimators are not unique and care is needed to ensure that they provide valid estimates. In general, maximum likelihood estimates are preferred although the computational burden can be substantial. If loci j can be regarded as being independent then the likelihood is the product over loci of the probabilities Pr(G j) of the observed genotypes, to estimate the inbreeding coefficient, or over pairs of genotypes to estimate the k i's and hence θ. The correlations that are observed to exist among SNPs, especially those within a few megabases of each other, may not affect the bias of the resulting estimates although they will increase the variance. It would be appropriate to limit the SNPs used in relationship estimation to those not in strong linkage disequilibrium with each other. If F represents the probability the individual in question has two ibd alleles at locus j,

From Bayes' theorem then

This suggests an iterative scheme: assign an initial value to F, and then average the updated values over loci. If G j is the genotype at locus j, the updated value F′ is
This value is then substituted into the right-hand side and the process continues until convergence.
For two individuals with genotype pair G j at locus j, there are three unobserved identity-by-descent states D i, i=0, 1, 2 that have probabilities k i:
and an iterative scheme similar to that for the inbreeding coefficient was described by Choi et al. (Reference Choi, Wijsman and Weir2009). Since Pr(D i|G j)=Pr(G j|D i)k i/Pr(G j) from Bayes' theorem, initial values k i0 assigned to the k i's can be updated to k ij's at locus j:
and these values averaged over loci to provide new estimates. This pair of operations is repeated until the likelihood changes by less than a specified amount. Estimates given by this procedure for the PLCO data referred to above are similar to those shown in Fig. 2 which were produced by the moment method in PLINK. Estimates are shown only for those pairs of individuals with a coancestry coefficient greater than 1/32, which accounts for the angling of points away from the line k 0+k 1=1 near k 0=1. There is a parent– offspring pair at k 0=0, k 1=1, several pairs of full sibs centred on k 0=0·25, k 1=0·50, pairs of half sibs centred on k 0=k 1=0·5 and various pairs of less-related individuals on the line k 0+k 1=1. Unrelated pairs of individuals, not shown in the figure, would have k 0=1, k 1=0.
(b) Variation in actual relatedness
It has long been recognized that there is variation in actual inbreeding and relatedness about the values predicted from pedigrees and indeed there is variation about expected values for the estimates shown in Fig. 2. Half siblings, for example, are expected to share one pair of alleles by descent from their common parent with probability k 1=0·5. At any one SNP, however, half siblings either have one pair of identical alleles or they do not: the actual identity coefficient ![]() 1 has values 0 or 1. Over the genome this quantity has a mean of k 1=0·5 and a variance of k 1(1−k 1)=0·25. The variance over a chromosome with m SNPs of the actual proportion of SNPs with one pair of identical alleles is the average over all pairs of SNPs j, j′ of the covariances of actual identities: ∑j,j′ Cov(
1 has values 0 or 1. Over the genome this quantity has a mean of k 1=0·5 and a variance of k 1(1−k 1)=0·25. The variance over a chromosome with m SNPs of the actual proportion of SNPs with one pair of identical alleles is the average over all pairs of SNPs j, j′ of the covariances of actual identities: ∑j,j′ Cov(![]() 1j,
1j, ![]() 1j′)/m 2. In their prediction of the variances and covariances of the
1j′)/m 2. In their prediction of the variances and covariances of the ![]() i's for any degree of relatedness, Hill & Weir (Reference Hill and Weir2010) recognized that the only wayhalf siblings can have
i's for any degree of relatedness, Hill & Weir (Reference Hill and Weir2010) recognized that the only wayhalf siblings can have ![]() 1=1 at loci j, j′ is for them each to receive the same recombinant or the same non-recombinant haplotype from their common parent. This provides
1=1 at loci j, j′ is for them each to receive the same recombinant or the same non-recombinant haplotype from their common parent. This provides
where c jj′ is the recombination fraction between loci j and j′. Although this simplifies to (1−2c jj′)2/4 it helps later generalizations to write the covariance as
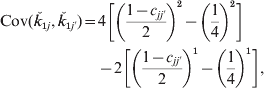
which is a special case of the expression ∑na n [b n−(1/4)n] with b=(1−c)/2. Here there are two values of n and a 2=4, a 1=−2.

Fig. 2. Estimates of relationship coefficients k 0, k 1 for participants in the PLCO study (Prorok et al., Reference Prorok, Andriole, Bresalier, Buys, Chia, Crawford, Fogel, Gelmann, Gilbert, Hasson, Hayes, Johnson, Mandel, Oberman, O'Brien, Oken, Rafla, Reding, Rutt, Weissfeld, Yokochi and Gohagan2000). Only estimates for pairs of individuals where k 0+k 1/2⩽15/16 are shown. The orange bars centred on k 0=0·25, k 1=0·5 (full sibs), k 0=k 1=0·5 (half sibs) and k 0=0·75, k 1=0·25 (first cousins) are two predicted standard deviations in length each side of the centre points.
If there are many loci on a chromosome, adding variances and covariances over pairs of loci is equivalent to integrating over all pairs of positions on the chromosome. Assuming Haldane's mapping function (Haldane, Reference Haldane1919) relating recombination fraction c to map positions x, y: (1−c)=(1+e −2|x−y|)/2, Hill & Weir (Reference Hill and Weir2010) found it convenient to define the function ɸn(l) for a chromosome of length l map units:

This let them write the variance of ![]() 1 on a chromosome of length l for half sibs as
1 on a chromosome of length l for half sibs as
There is an immediate extension to descendants of half sibs. For each additional generation (strictly, each meiosis) separating the descendants, two-locus haplotypes remain intact and the expectation of the product of two ![]() 1j's is reduced by a factor of (1−c)/2. For separation by g generations/meioses (g=2 for half sibs, g=3 for half-uncle nephew, g=4 for half-cousins etc):
1j's is reduced by a factor of (1−c)/2. For separation by g generations/meioses (g=2 for half sibs, g=3 for half-uncle nephew, g=4 for half-cousins etc):
It is straightforward to average over chromosomes with different map lengths. Hill & Weir (Reference Hill and Weir2010) gave similar expressions for an individual with a lineal descendant and for pairs of individuals descending from full sibs. The variance of ![]() 1 for first cousins, for example, is
1 for first cousins, for example, is
Because ![]() 1 for first cousins refers to identity for pairs of alleles carried on gametes from full sibs, this variance also applies to the actual inbreeding coefficient for an offspring of full sibs. The variances for other degrees of inbreeding follow from the variances for appropriate pairs of related parents.
1 for first cousins refers to identity for pairs of alleles carried on gametes from full sibs, this variance also applies to the actual inbreeding coefficient for an offspring of full sibs. The variances for other degrees of inbreeding follow from the variances for appropriate pairs of related parents.
Estimation of the inbreeding coefficient for an individual or the coancestry coefficient for a pair of individuals requires many thousands of SNPs and was not possible in the pre-genome era. Even with substantial data, however, the estimates will reflect the inherent variation of actual identity along the genome. Subject to computational resources, maximum likelihood estimation is preferred over the method of moments but both methods are affected by the need to use sample allele frequencies rather than population values and this can be an issue for structured populations when the target allele frequencies for a specific individual or pair of individuals are not the same as the frequencies in the study population to which the specific individual(s) belong (Anderson & Weir, Reference Anderson and Weir2007). There is a further complication in estimating the relatedness of a pair of individuals when they are inbred as then there are nine measures of identity by descent instead of three (Jacquard, Reference Jacquard1970; Weir et al., Reference Weir, Anderson and Hepler2006).
(iii) Population structure
Not only are genomic data revealing the structure of allelic associations along the genome within individuals but also they are revealing information about the structure of populations. The F-statistics of Wright (Reference Wright1951) can be regarded as reflecting the history of populations in the same way that coancestry coefficients reflect the pedigrees of individuals. The F-statistics describe the associations of alleles within and between populations. Early treatments of the variances of these quantities have now been augmented by empirical studies. Weir et al. (Reference Weir, Cardon, Anderson, Nielsen and Hill2005) presented plots of F ST estimated from all the SNPs in five Mb windows along the genome and these plots showed substantial variation. A similar plot is shown in Fig. 3 for chromosome 19 andthree of the HapMap III populations, Han Chinese from Beijing (CHB), Japanese from Tokyo (JPT) and Yoruba from Nigeria (YRI). The full data set has information from 11 populations and all those populations were used for the population-specific values in Fig. 3. The values are for 100-SNP windows of 11 906 SNPs. For each population i, suppose p ij is the minor allele frequency for SNP j and n ij the number of individuals typed at that SNP in that population. The average SNP j frequency over populations is ![]() j=∑in ijp ij/∑in ij. Then, the analogue of F ST for population i is
j=∑in ijp ij/∑in ij. Then, the analogue of F ST for population i is
where n ij*=n ij−n ij2/∑in ij. As Weir & Hill (Reference Weir and Hill2002) pointed out, βi is the value of F ST for population i relative to the relationship between pairs of alleles among all pairs of populations in the study. The plots in Fig. 3 show great similarity between the CHB and JPT values with differences from the YRI values, but great variation along the chromosome.

Fig. 3. Population-specific values of F ST (Weir & Hill, Reference Weir and Hill2002) for CHB, JPT and YRI samples in the HapMap III data for markers on chromosome 19. Each value is based on a window of 100 SNPs, and there is a 20 SNP overlap between adjacent windows.
Although some of the variations along the chromosome in Fig. 3 reflects Mendelian sampling, some of it will reflect the effects of natural selection (e.g. Akey et al., Reference Akey, Zhang, Khang, Jin and Shriver2002). There has been some success with using F ST variation for detecting selection, but there is the difficulty of the high variances of single-SNP estimates predicted by the following argument. By assuming allele frequencies were approximately normally distributed across populations, Weir & Hill (Reference Weir and Hill2002) were able to find a maximum likelihood estimate of the population-average value of F ST. For a locus with sample allele frequencies ![]() iu for the uth of m alleles in the ith of r sampled populations, and averages
iu for the uth of m alleles in the ith of r sampled populations, and averages ![]() u of the
u of the ![]() iu's over populations,
iu's over populations,
This estimate divided by the true value has a chi-square distribution with (m−1)(r−1) degrees of freedom. For SNPs, m=2 and the df are 1 or 2 when data from two or three populations are used. In either case the distribution peaks at F ST=0 and has a very long tail to the right. The variances are substantial and it may be difficult to conclude significantly different values at different loci. The degrees of freedom are summed over the loci when multiple loci are used for estimation of F ST, the chi-square distribution tends to normality and the variances are reduced. The possibility of declaring significant differences is offset by these differences now referring to regions larger than a single SNP.
(a) Principal component (PC) analysis
An alternative approach to characterizing population structure is to reduce the high dimensionality of the number of SNPs to a small number of PCs. These refer to the matrix with dimensions equal to the total number of individuals in the study and with elements being multiples of estimates of one plus the inbreeding coefficient of the individuals on the diagonal and the coancestry coefficients of pairs of individuals off the diagonal. When individuals are plotted in two dimensions for pairs of the first few PCs they tend to cluster in populations (Novembre et al., Reference Novembre, Johnson, Bryc, Kutalik, Boyko, Auton, Indap, King, Bergmann, Nelson, Stephens and Bustamante2008) in ways that often bear striking resemblances to geographic maps of population locations. A novel finding of such analyses is that chromosomal regions with low recombination, such as polymorphic inversions, are revealed in samples from the same population. Tian et al. (Reference Tian, Plenge, Ransom, Lee, Villoslada, Selmi, Klareskog, Pulver, Qi, Gregersen and Seldin2008) reported the clustering into three groups of a sample of European-ancestry individuals corresponding to the genotypes of a cluster of highly correlated SNPs in chromosomal region 8p23, a region that contains a polymorphic inversion. Laurie et al. (Reference Laurie, Doheny, Mirel, Pugh, Bierut, Bhangale, Boehm, Caporaso, Edenberg, Gabriel, Harris, Hu, Jacobs, Kraft, Landi, Lumley, Manolio, McHugh, Painter, Paschall, Rice, Rice, Zheng and Weir2010) report a process of searching systematically for such genomic features by looking for regions where SNPs are highly correlated with one of the first few PCs. An illustration of their approach is shown in Fig. 4, using data from PLCO and the companion EAGLE (Environment and Genetics in Lung Cancer Etiology) study (Landi et al. Reference Landi, Consonni, Rotunno, Bergen, Goldstein, Lubin, Goldin, Alavanja, Morgan, Subar, Linnoila, Previdi, Corno, Rubagotti, Marinelli, Albetti, Colombi, Tucker, Wacholder, Pesatori, Caporaso and Bertazzi2008).

Fig. 4. Correlations of SNPs on chromosomes 2 and 8 with the first and third PCs for the PLCO and EAGLE data (Prorok et al., Reference Prorok, Andriole, Bresalier, Buys, Chia, Crawford, Fogel, Gelmann, Gilbert, Hasson, Hayes, Johnson, Mandel, Oberman, O'Brien, Oken, Rafla, Reding, Rutt, Weissfeld, Yokochi and Gohagan2000; Landi et al., Reference Landi, Consonni, Rotunno, Bergen, Goldstein, Lubin, Goldin, Alavanja, Morgan, Subar, Linnoila, Previdi, Corno, Rubagotti, Marinelli, Albetti, Colombi, Tucker, Wacholder, Pesatori, Caporaso and Bertazzi2008). Set A is when all SNPs were used to calculate the PCs, set B was when the SNPs in the LCT gene or the chromosome 8 inversion were omitted before calculating the principal components. The vertical dashed red lines mark LCT on chromosome 2 and the 8p23 inversion on chromosome 8.
PC analysis was performed with unrelated PLCO and EAGLE study subjects. PC3 showed a remarkable separation of both studies into three clusters. This distinct clustering by a PC that accounts for only 0·06% of the variance suggests the strong influence of one polymorphism. To investigate this possibility, the correlation between each PC and the genotypic scores of each SNP was computed (The GENEVA Consortium, 2008). This was done ignoring study and also for each study separately in order to find SNPs that influence the separation within each study group (rather than between the studies). The results for both studies are similar to one another and to the overall correlation (ignoring study), so attention is restricted now to the overall correlation results.
The correlation between each SNP and each of the first three PCs revealed two distinct clusters of SNPs with high correlations. PC1 is highly correlated with SNPs on chromosome 2 in a region containing the LCT gene, which is a well-known marker of the north–south European cline (Bersaglieri, Reference Bersaglieri, Sabeti, Patterson, Vanderploeg, Schaffner, Drake, Rhodes, Reich and Hirschhorn2004). PC3 is highly correlated with a cluster on chromosome 8. A previously documented inversion in 8p23 most likely accounts for this cluster of SNPs, which are in strong linkage disequilibrium. The genotypes of the most highly correlated SNP in this region (rs2409798) largely define the three clusters of samples separated by PC3. These highly localized features underlying some PCs may limit their usefulness in detecting and controlling for population structure. In fact, they may even be counterproductive when used as covariates in association testing for traits affected by SNPs in those chromosomal regions. Therefore, SNPs in the two regions were removed (to make SNP set B) and the PCs recalculated and compared with the full set of autosomal SNPs (set A). Figure 4 shows the effects of removing the SNP clusters on chromosomes 2 and 8 in calculating the PCs for set B. The very prominent cluster of SNPs having high correlation with PC3 in set A is no longer evident, as expected. However, the cluster of chromosome 2 SNPs in the LCT region is evident in both sets A and B, even though those SNPs were not used in the calculation of PCs for set B, contrary to naïve expectation. The same result is obtained when all SNPs on chromosome 2 are removed from the PC calculation. It seems likely that the LCT region is correlated with multiple SNPs on other chromosomes that all contribute to the north–south European cline.
4. Discussion
Allelic associations are quantities of primary interest to population geneticists. They can be regarded as being purely descriptive, as in measures of departure from HWE, or they may be interpreted as indicators of chromosomal proximity when they refer to the relationship between genetic markers and disease genes (Weir, Reference Weir2008). Although there is still an issue of making inferences about evolutionary mechanisms on the basis of statistics calculated from data collected at a single time point, there is no doubt that the genome era has provided a wealth of data for estimating association measures and for demonstrating the variation of associations along the genome within individuals, among individuals and among populations.
This review has shown the impact of dense sets of SNP markers on associations at single loci and averaged over chromosomes. One of the striking observations is that any measure of association varies greatly over the genome. If the association of interest was that between a genetic marker and an individual's disease status then a small number of genomic regions with significant associations would offer hope of developing a small number of targeted therapies, although the report of Allen et al. (Reference Allen2010) of 180 significant associations with height suggests that complex traits are affected by many genes. If the association under study refers to departures from HWE or to population structure, however, then variation over the genome might not be expected as the whole genome has been subjected to the same set of population size and mating structure parameters. Differences might well reflect differential effects of natural selection, as is thought likely for the LCT gene on chromosome 2 (Bersaglieri et al., Reference Bersaglieri, Sabeti, Patterson, Vanderploeg, Schaffner, Drake, Rhodes, Reich and Hirschhorn2004), but this review has used the example of relationship measures to point out that Mendelian or genetic sampling and linkage can lead to substantial variation in measures of association. Genome-era data are making very concrete these theoretical predictions.
This review has not addressed the substantial current activity in association mapping. Whereas departures from HWE are measured by the association between pairs of alleles at single locus, association mapping seeks evidence for an association between an observed marker allele and an allele at an unobserved trait gene. There is population genetic theory and substantial empirical evidence that such associations will decrease with distance on a chromosome between the two genes. Current SNP-based association studies are therefore indirect, in the sense they seek markers that are associated (in linkage disequilibrium with) with trait genes. The expectation of whole-genome DNA sequence studies (The 1000 Genomes Consortium, 2010) is that the causal variants themselves will be observed and the associations will be direct.
The change in the scale of genetic data over the past 50 years has been dramatic and has led to new understanding of genomic structure and evolutionary processes. We can expect no less over the next 50 years.
This work was supported in part by NIH grants R01 GM075091 and U01 HG 004446. Neil Caporaso and Maria Teresa Landi, National Cancer Institute, consented to inclusion of Figs 1, 2 and 4 that display results from their study of lung cancer (supported by NIH GEI: HG-06-033-NCI-01 and the Intramural Research Program of National Institutes of Health, National Cancer Institute, Division of Cancer Epidemiology and Genetics). Genotyping for the lung cancer study was performed at Johns Hopkins University Centerfor Inherited Disease Research, with support from the NIH GEI (U01HG004438) and the NIH contract ‘High throughput genotyping for studying the genetic contributions to human disease’ (HHSN268200782096C). Data were cleaned by the GENEVA Coordinating Center that receives support from U01 HG 004446. Assistance with data cleaning was provided by the National Center for Biotechnology Information with support from the Intramural Research Program of the NIH, National Library of Medicine. Caitlin McHugh, University of Washington, plotted the figures. Helpful comments were made by Bill Hill.




