Introduction
Many individuals take vitamin E supplements under the assumption that they may be good for their health or, if not that, at least can do no harm. The well-documented safety of the micronutrient( Reference Hathcock, Azzi and Blumberg 1 ) started being questioned with the publication of meta-analyses reporting increased mortality in individuals taking high-dose vitamin E supplements( Reference Miller, Pastor-Barriuso and Dalal 2 , Reference Bjelakovic, Nikolova and Gluud 3 ) and interactions with prescription drugs were proposed as one possible explanation for these adverse events( Reference Brigelius-Flohe 4 ). Here, we review the evidence available in the scientific literature for adverse vitamin E–drug interactions and their underlying mechanisms.
Vitamin E
The term vitamin E refers to the eight lipid-soluble substances α-, β-, γ- and δ-tocopherol (T) and -tocotrienol (T3) (Fig. 1), which are exclusively produced by photosynthetic organisms and therefore present at varying concentrations in most plant foods( Reference Frank, Chin and Schrader 5 ). Chemically, T and T3 consist of a chromanol ring attached to a sixteen-carbon saturated alkyl (T), or a threefold unsaturated isoprenoid side chain (T3), respectively. The T side chain has three chiral centres at positions 2, 4’ and 8’, which can be either in the R or S conformation and make possible eight different stereoisomers (RRR, RSR, RRS, RSS, SRR, SSR, SRS and SSS). T3 possess only one chiral centre at position 2, which can be either in the R or S configuration. The biosynthesis of vitamin E in plants is stereo-selective and therefore natural T are exclusively in the RRR configuration, whereas synthetic T consist of an equimolar (all racemic; all rac) mixture of all eight possible stereoisomers. T3 are not synthesised, but extracted from natural sources, and thus present in the diet as stereoisomers with the R conformation at carbon 2 (2R). The prefixes α, β, γ and δ denote the number and positions of methyl groups substituted at the chromanol ring (Fig. 1)( Reference Frank, Chin and Schrader 5 , Reference Kamal-Eldin and Appelqvist 6 ).
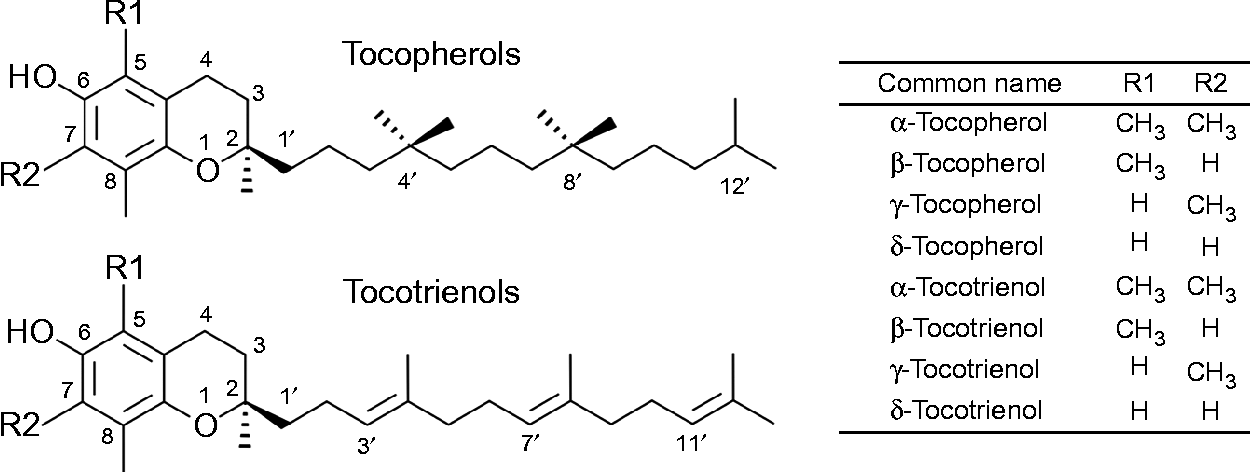
Fig. 1 The chemical structures of tocopherols and tocotrienols.
More than 90 years ago, vitamin E was identified as a dietary factor required for maintaining fertility in female rats by preventing fetal resorption( Reference Evans and Bishop 7 ). The anti-sterility activity of the eight vitamin E congeners differs in rats, with αT being the most active( Reference Leth and Sondergaard 8 ). Furthermore, αT is the major lipid-soluble antioxidant in human plasma( Reference Burton, Joyce and Ingold 9 ) and may (thereby) modulate gene expression and cellular signalling( Reference Azzi, Gysin and Kempna 10 – Reference Frank, de Pascual Teresa and Rimbach 12 ). The biochemical mechanism underlying its essential vitamin function, however, is still unknown.
Although all congeners are ingested with the diet, the organism selectively retains αT and preferentially metabolises and excretes the non-αT congeners( Reference Grebenstein, Schumacher and Graeve 13 ). Therefore, αT is considered the most important form of vitamin E( 14 ) and is the congener typically found in dietary supplements.
In Germany, 28 % of the population regularly take dietary supplements and 11 % frequently take vitamin E( 15 ). Dietary supplement use may be even higher in senior citizens. In a Southern German cohort of individuals aged 65 years or older, 46 % took dietary supplements and 13 % supplemented vitamin E( Reference Schwab, Heier and Schneider 16 ).
In the USA, almost 40 % of the population take over-the-counter vitamin or mineral supplements, of which approximately 37 % contain vitamin E( Reference Balluz, Kieszak and Philen 17 ). These dietary supplements may contain vitamin E in doses as high as 1000 IU per serving; equivalent to 45–60 times the RDA of 18–22 IU/d for adults in the USA( 14 , Reference Wolfram 18 ) or the 16–22 IU/d recommended by the German, Austrian and Swiss nutrition societies( 19 ).
Various meta-analyses questioned the previously established safety (in doses up to 800 IU/d) of vitamin E and suggested an increased risk of gastrointestinal cancer, heart failure, and all-cause mortality in subjects consuming dietary supplements containing vitamin E in high doses (for a review, see Bell & Grochoski( Reference Bell and Grochoski 20 )). Many of the intervention trials included in the meta-analyses studied patients with pre-existing morbidities who took one or more prescription drugs alongside dietary supplements (for reviews, see Bell & Grochoski( Reference Bell and Grochoski 20 ) and Frank & Rimbach( Reference Frank and Rimbach 21 )). In some randomised, controlled human intervention trials, vitamin E was even administered in combination with a drug as part of the study design (for a discussion, see Brigelius-Flohe( Reference Brigelius-Flohe 4 , Reference Brigelius-Flohe 22 ), Bell & Grochoski( Reference Bell and Grochoski 20 ) and Frank & Rimbach( Reference Frank and Rimbach 21 )). Hence, many of these human studies may have suffered from bias due to adverse effects caused by nutrient–drug interactions (for a review, see Frank & Rimbach( Reference Frank and Rimbach 21 )), which questions the validity of the conclusions drawn from the meta-analyses of these trials( Reference Hathcock, Azzi and Blumberg 1 , Reference Bell and Grochoski 20 ).
Nutrient–drug interactions
In order to reach the systemic circulation, orally ingested nutrients and drugs are required to pass a number of physiological barriers. They first need to be liberated from the food matrix or drug formulation, mainly in the stomach and small intestine, before they can pass the second barrier, absorption into gastrointestinal cells. In the endothelial cells of the gastrointestinal tract, dietary factors and drugs may undergo metabolism by phase I and II enzymes, and both the parent compounds and their metabolites may either be excreted back into the intestinal lumen by membrane transporters on the luminal side, or secreted into the mesenteric vein or lymph. Compounds present in the mesenteric blood or lymph ultimately reach the liver via the portal vein. In the liver, the parent compounds and/or metabolites can be further metabolised by hepatic phase I and II enzymes and are either excreted with the bile back into the small intestine or are secreted into the systemic circulation, from where they are distributed through the body. If they are able to pass through the vascular endothelium, they are taken up into peripheral tissues, where they may exert their biological or pharmacological function. Compounds that are not taken up by peripheral tissues are either returned to the liver or delivered to the kidney for renal elimination.
With the exception of parenteral nutrition, nutrients and other dietary factors are generally ingested orally. A drug, however, may also be administered by injection, inhalation or other routes, which may reduce the number of barriers it has to pass before reaching the systemic circulation.
The term pharmacokinetics (‘what the body does to the drug’) is used to describe the extent (concentration) to which a drug or its active metabolite is present in the systemic circulation and how long it stays there. The term pharmacodynamics (‘what the drug does to the body’), on the other hand, designates the biological activity of a drug, for example, the lowering of blood lipids, inhibition of platelet aggregation and so forth( Reference Benet, Mitchell and Horning 23 ).
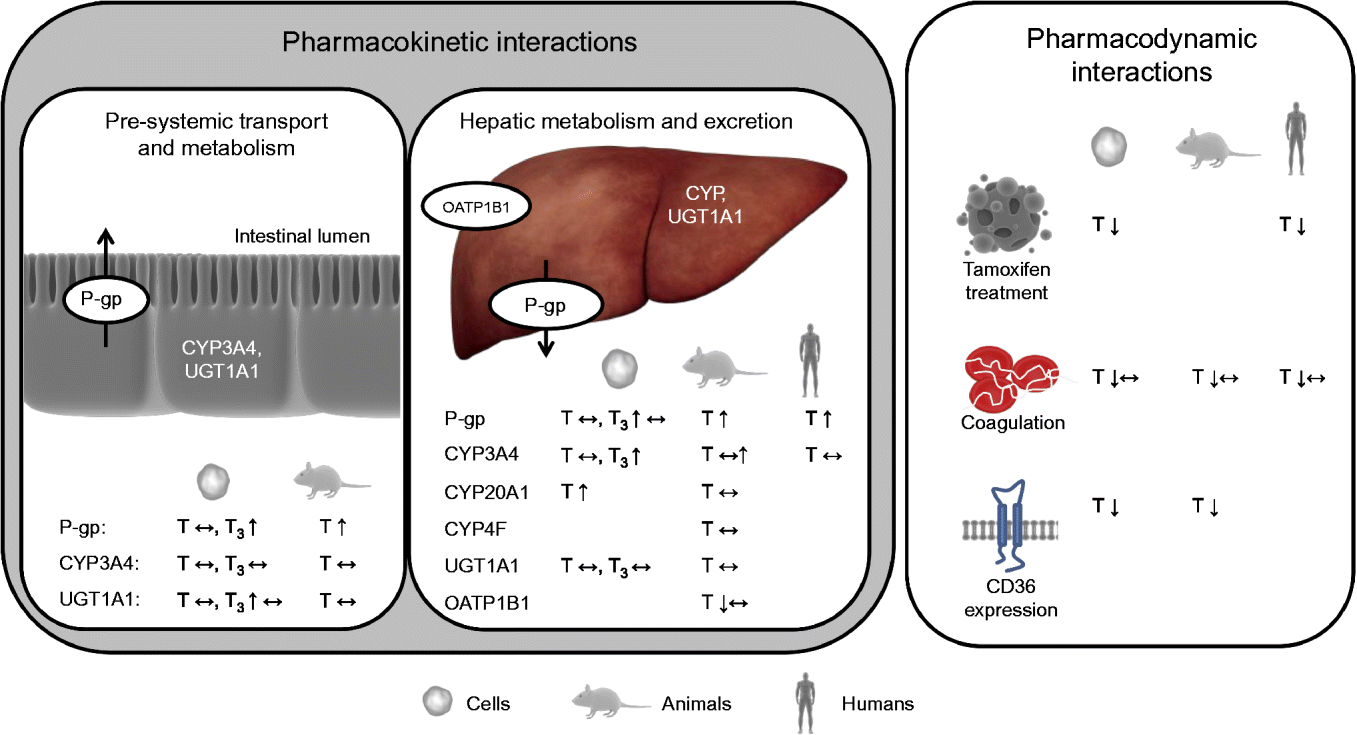
Interactions between a nutrient and a drug may occur in either of two ways: a nutrient may alter the pharmacokinetics and/or pharmacodynamics of a drug (nutrient–drug interaction) or a drug may alter the concentration and/or biological activity of a nutrient in the organism (drug–nutrient interaction)( Reference Chan 24 ). Drug–nutrient interactions, such as impaired absorption of vitamin E when administered together with drugs that reduce fat-absorption, are not within the scope of the present review. Nutrient–drug interactions can be roughly categorised, based on the underlying mechanism and nature of the interaction, into: (1) drug inactivation occurring ex vivo (in the drug formulation and outside of the body); (2) interactions affecting pre-systemic transport (uptake) and metabolism; (3) interactions affecting systemic metabolism and elimination of the drug; and (4) interactions directly altering drug activity (pharmacodynamics). However, overlap between these hypothetical classes may, and is likely to, occur. Interactions of the second and third kind alter the pharmacokinetics and interactions of the fourth type the pharmacodynamics of drugs (Fig. 2).

Fig. 2 Effects of vitamin E observed in cell-culture, animal and/or human trials on targets potentially altering the pharmacokinetics and pharmacodynamics of drugs. P-gp, P-glycoprotein; CYP, cytochrome P450 mixed-function mono-oxygenase; UGT, UDP-glucuronosyltransferase; T, tocopherol; ↔ , no change; T3, tocotrienol; ↑ , increase; OATP, organic anion transporting polypeptide; ↓ , decrease. For detailed information on the experiments and outcomes, see Tables 1 and 2. (A colour version of this figure can be found online at www.journals.cambridge.org/nrr).
Table 1 Effects of vitamin E on mRNA or protein expression or activity of transporters and enzymes potentially affecting the pharmacokinetics of drugs in cell-culture, animal and human experiments
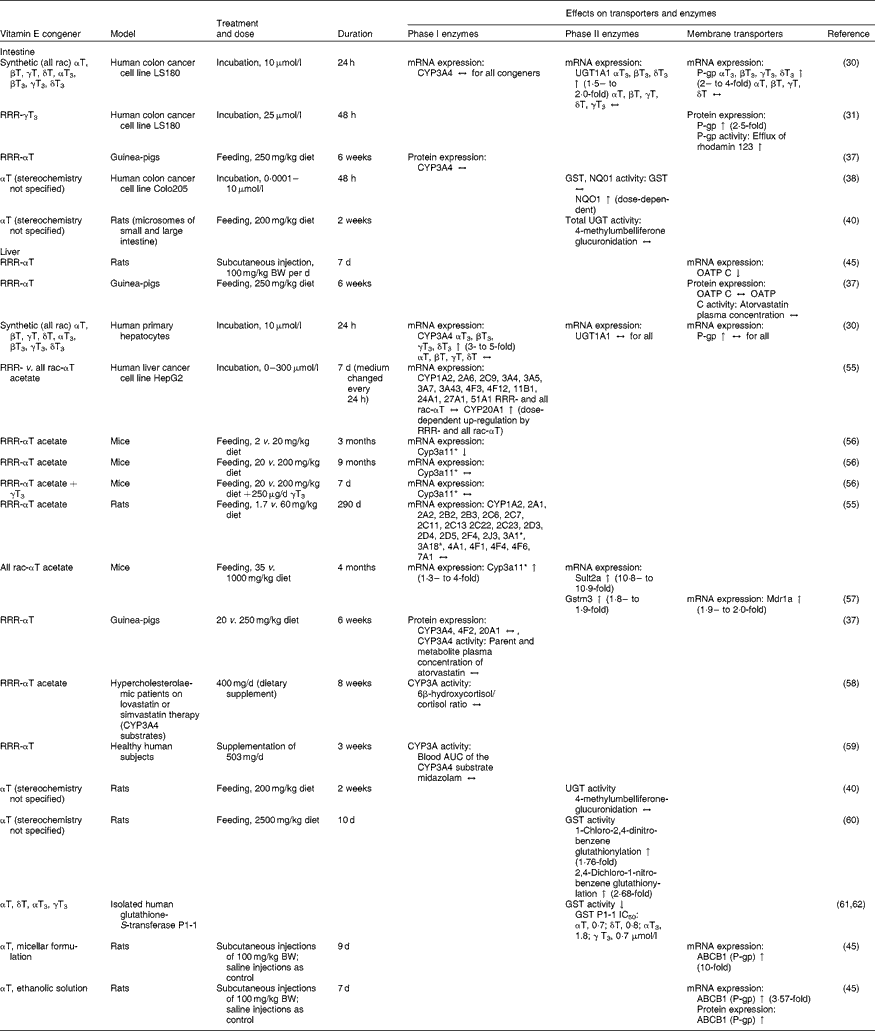
All rac, all racemic; T, tocopherol; T3, tocotrienol; CYP, cytochrome P450 mixed-function mono-oxygenase; ↔ , no change; UGT, UDP-glucuronosyltransferase; ↑ increase; P-gp, P-glycoprotein; GST, glutathione-S-transferase; NQO1, NAD(P)H:quinone oxidoreductase; BW, body weight; OATP, organic anion transporting polypeptide; ↓ , decrease; Sult2a, sulfotransferase 2A; Mdr1a, multidrug resistance protein 1a; Gstm3, glutathione S-transferase M3; IC50, 50% inhibition; ABCB1, ATP-binding cassette transporter B1.
* Homologous to human CYP3A4.
Table 2 Effects of vitamin E on the activity (pharmacodynamics) of drugs in cell culture, animal and human experiments

T, tocopherol; ↓ , decrease; all rac, all racemic; PMA, phorbol 12-myristate 13-acetate; ↔ , no change; PKC, protein kinase C; BW, body weight; PIVKA-II, protein induced by vitamin K absence factor II; ↑ , increase; p-ERK, phosphorylated extracellular signal-regulated kinase.
Nutrient–drug interactions, that is, a decrease or increase in the concentration of the active drug and/or altered pharmacodynamics, may increase the risk for and rate of adverse effects. In the present review, we summarise and discuss the evidence for potentially adverse interactions of vitamin E with drugs (nutrient–drug interactions) according to these four categories.
Potential interactions of vitamin E with drugs
Ex vivo inactivation of drugs
The first class of nutrient–drug interactions comprises chemical reactions (for example, complexation, hydrolysis, precipitation, oxidation) between a nutrient and a drug, which occur within the galenic formulation and mainly outside of the body( Reference Chan 24 ). T and T3 are potent antioxidants in homogeneous non-aqueous solutions and liposomal formulations( Reference Kamal-Eldin and Appelqvist 6 ) and could thus protect easily oxidisable drugs with reduction potentials higher than those of the respective vitamin E congener. For drugs with reduction potentials lower than those of vitamin E, the nutrient could act as oxidising agent and, at least theoretically, oxidatively damage the drug molecule( Reference Buettner 25 ). T and their radical forms (tocopheroxyl radicals) are capable of reducing transition metal ions to a lower valence state (for example, reduction of Fe3+ to Fe2+), which could favour pro-oxidant reactions catalysed by these metals( Reference Kamal-Eldin and Appelqvist 6 ). Hence, drug formulations with considerable amounts of free transition metals could theoretically be at risk of increased Fenton or Fenton-like oxidation reactions. To date, however, no vitamin E–drug interactions of this kind have been reported in the scientific literature.
Vitamin E and pharmacokinetics
Pre-systemic transport and metabolism
The second class of interactions discussed considers interactions that alter the absorption of a drug and mostly occur before the drug appears in the systemic circulation. Such interactions may be facilitated by modulation of the activity of membrane transporters or enzymes involved in xenobiotic metabolism or drug binding( Reference Chan 24 ) and occur on the level of the pre-systemic transport and metabolism.
Vitamin E and pre-systemic drug transport
Epithelial efflux transporters, such as the multidrug resistance proteins (for example, P-glycoprotein (P-gp), also known as ATP-binding cassette transporter B1 (ABCB1) and multidrug resistance protein 1 (MDR1))( Reference Roninson, Chin and Choi 26 ), may limit the intestinal absorption of drugs. Intestinal P-gp is expressed on the apical surface of enterocytes( Reference Thiebaut, Tsuruo and Hamada 27 ) and limits the bioavailability and ultimately the activity of its substrates by increasing their efflux back into the intestinal lumen( Reference Mayer, Wagenaar and Beijnen 28 ). A prominent example for a P-gp-mediated drug interaction is the herbal remedy St John's wort, which induces P-gp and thereby reduces the plasma concentrations of the orally administered heart medication digoxin( Reference Dürr, Stieger and Kullak-Ublick 29 ).
All four T3, but none of the T, induced P-gp mRNA expression via activation of the transcription factor pregnane X receptor (PXR; also known as steroid and xenobiotic sensing nuclear receptor) in the human colorectal adenocarcinoma cell line LS180 (incubated with 10 μmol/l of the respective T or T3 for 24 h)( Reference Zhou, Tabb and Sadatrafiei 30 ). Consistent with the effects on mRNA, P-gp protein expression and export of the P-gp substrate rhodamine 123 were increased in LS180 cells incubated with γT3 (25 μmol/l; 48 h)( Reference Abuznait, Qosa and O'Connell 31 ). No studies on effects of any of the remaining vitamin E congeners on intestinal P-gp protein expression or activity have been reported in the literature.
These in vitro studies suggest that, albeit at very high concentrations, T3 may potentially enhance the transfer activity of P-gp in intestinal cells. However, the scientific literature does not contain reports from animal or human trials suggesting that intestinal P-gp-mediated interactions of vitamin E with drugs may actually occur. Thus, controlled in vivo trials are now warranted to elucidate the biological relevance of this potential mechanism.
Vitamin E and pre-systemic drug metabolism
The intestine is responsible for the uptake of nutrients, but also serves as a barrier against ingested xenobiotics( Reference Kaminsky and Zhang 32 ). Intestinal phase I enzymes, such as cytochrome P450 mixed-function mono-oxygenases (CYP), and phase II enzymes, such as UDP-glucuronosyltransferases (UGT), glutathione-S-transferases (GST) or NAD(P)H:quinone oxidoreductase (NQO1), to name only a few, play a key role in the first-pass metabolism of drugs, which determines their and their metabolites’ plasma concentrations. CYP3A4 is a phase I enzyme involved in the metabolism of approximately 50 % of all prescription drugs( Reference Guengerich 33 ) and the most abundant CYP in the intestine( Reference Benet and Cummins 34 ). Inhibition of CYP3A4, for example, by drinking a glass of grapefruit juice, reduces the oral availability of the sedative and CYP3A4 substrate( Reference Paine, Shen and Kunze 35 ) midazolam in humans( Reference Kupferschmidt, Ha and Ziegler 36 ).
In LS180 colon cells, neither T nor T3 (10 μmol/l; 24 h) altered the mRNA expression of CYP3A4( Reference Zhou, Tabb and Sadatrafiei 30 ). In agreement with these in vitro findings, the intestinal protein expression of CYP3A4 did not differ between guinea-pigs fed RRR-αT at normal dietary (20 mg/kg diet) or pharmacological doses (250 mg/kg diet) for 6 weeks( Reference Podszun, Grebenstein and Hofmann 37 ).
During phase I metabolism, reactive quinones and epoxides may be generated. These reactive metabolites are inactivated by phase II enzymes, including NQO1, which reduces quinones to less reactive hydroquinones, and GST, which conjugates electrophiles with glutathione. In the colon cancer cell line Colo205, αT dose-dependently (0·0001–10 μmol/l; 48 h) increased NQO1 activity, whereas GST activity remained unchanged( Reference Wang and Higuchi 38 ).
The phase II enzyme UGT1A conjugates lipophilic substances with UDP-glucuronic acid. UGT1A1 is the quantitatively most important UGT in the small intestine in humans and its activity in intestinal microsomes is higher than in hepatic microsomes. Hence, intestinal glucuronidation contributes significantly to the first-pass metabolism and low bioavailability of UGT1A1 substrates( Reference Fisher, Vandenbranden and Findlay 39 ). In LS180 cells, UGT1A1 mRNA increased at most 2-fold upon incubation with αT3, βT3 or δT3 (10 μmol/l; 24 h), while neither γT3 nor any of the T altered UGT1A1 mRNA( Reference Zhou, Tabb and Sadatrafiei 30 ). Such small increases in UGT1A1 mRNA, however, are unlikely to have a significant impact on UGT1A1 protein expression and/or activity. In agreement with this notion, the mean total UGT activity in the small and large intestine of rats fed 200 mg αT/kg diet for 2 weeks was unchanged relative to control animals( Reference Van der Logt, Roelofs and van Lieshout 40 ).
Thus, there is no evidence from in vivo studies to suggest that T or T3 may alter the intestinal metabolism of drugs even though modest effects on NQO1 and UGT1A1 activity have been observed in cultured colonic adenocarcinoma cells.
Interactions of vitamin E with hepatic drug metabolism and excretion
Hepatic interactions of nutrients with drugs occur predominantly on the level of the uptake of drugs into the liver and/or their conversion by hepatic phase I and II enzymes.
Vitamin E and hepatic uptake of drugs
Transport proteins in the plasma membrane are involved in the uptake of many drugs into hepatocytes and strongly determine their overall hepatic clearance, regardless of other subsequent reactions( Reference Shitara, Sato and Sugiyama 41 ). In humans, the organic anion transporting polypeptide (OATP C, also known as solute carrier organic anion transporter family member 1B1 (OATP1B1) or OATP2) is a liver-specific transporter facilitating the uptake of various endogenous (for example, 17β-glucuronosyl oestradiol)( Reference König, Cui and Nies 42 ) and xenobiotic substances (for example, the drug pravastatin)( Reference Hsiang, Zhu and Wang 43 , Reference Hagenbuch and Meier 44 ) from the blood into the liver.
In rats, daily subcutaneous injections with 100 mg RRR-αT/kg body weight for 1 week, a mode of administration that was chosen to load the liver with extremely high concentrations of αT, reduced hepatic OATP C mRNA compared with control( Reference Traber, Labut and Leonard 45 , Reference Farley, Leonard and Labut 46 ). In humans, subcutaneous injection of αT, which has been used to treat retinopathy (retrolental fibroplasia) in prematurely born infants( Reference Bougle, Boutroy and Heng 47 ), as well as intramuscular( Reference Guggenheim, Ringel and Silverman 48 ) and intravenous injections of vitamin E( Reference Brion, Bell and Raghuveer 49 ) are rare modes of administration and thus of minor relevance in the context of vitamin E–drug interactions. Also, changes in mRNA expression often do not mirror changes in protein concentration and/or activity. In a model that more closely reflects the co-ingestion of nutrients and drugs observed in human subjects, feeding guinea-pigs for 6 weeks with either 20 or 250 mg RRR-αT/kg diet in the presence or absence of the OATP C substrate atorvastatin (300 mg/kg diet) did not alter hepatic OATP C protein expression, atorvastatin plasma concentrations (reflecting OATP C activity) or the lipid-lowering activity of the drug( Reference Podszun, Grebenstein and Hofmann 37 ).
In summary, these studies suggest that subcutaneous and potentially also intramuscular and intravenous injections with high doses of αT may carry a limited risk of altering OATP C-mediated hepatic drug uptake, but there is currently no in vivo evidence for the biological importance of this potential mechanism of vitamin E–drug interactions. Also the oral ingestion of high doses of αT does not appear to affect OATP C-mediated drug transport and efficacy in vivo. None of the other vitamin E congeners has been studied for impact on the hepatic uptake of drugs yet.
Vitamin E and hepatic phase I metabolism
During phase I metabolism in the liver, cytochrome P450 enzymes (CYP) and other phase I enzymes oxidise, reduce or hydrolyse xenobiotics (such as drugs) and thereby activate them for the subsequent phase II conjugation reactions. All vitamin E congeners undergo phase I metabolism and are ω-hydroxylated at the terminal methyl group in the side chain by a CYP, in humans probably CYP4F2( Reference Sontag and Parker 50 , Reference Bardowell, Duan and Manor 51 ). Another member of the cytochrome family, CYP3A4, is involved in the metabolism of many drugs and can either contribute to the formation of the active agent from a parent drug or to the generation of metabolites destined for elimination( Reference Guengerich 33 ). Since the metabolism of all vitamin E congeners and of many drugs is catalysed by the same enzyme family, it is feasible that interactions on the level of hepatic metabolism may occur.
The impact of vitamin E on CYP3A4 expression and activity has been extensively studied in numerous in vitro and in vivo models. The expression of CYP3A4 is controlled by the nuclear receptor PXR( Reference Lehmann, McKee and Watson 52 ). In a reporter gene assay with HepG2 cells, T3 at 10 and 50 μmol/l activated PXR (5- and 10-fold relative to untreated control cells), while T at these concentrations were much less potent activators (maximum 2-fold induction relative to control)( Reference Zhou, Tabb and Sadatrafiei 30 , Reference Landes, Pfluger and Kluth 53 ). We, on the other hand, did not observe an induction of PXR at all in a HEK293 reporter gene assay with concentrations of up to 200 μmol/l RRR- or all rac-αT( Reference Podszun, Grebenstein and Spruss 54 ). In primary human hepatocytes, 10 μmol/l T3 induced CYP3A4 mRNA 3- to 5-fold, whereas T were ineffective( Reference Zhou, Tabb and Sadatrafiei 30 ). Incubation of HepG2 cells with increasing concentrations (0–300 μmol/l) of either RRR- or all rac-αT for 7 d dose-dependently increased mRNA expression of the orphan CYP20A1 only, but not of any of the other thirteen CYP (including CYP3A4) expressed in these cells( Reference Hundhausen, Frank and Rimbach 55 ).
A number of in vivo studies investigated the effects of high-dose supplementation as well as vitamin E deficiency on the expression of hepatic CYP, especially CYP3A4. In mice, feeding of a vitamin E-deficient diet (2 mg RRR-αT/kg diet) for 3 months reduced, whereas RRR-αT supplementation up to 9 months (200 mg/kg diet) did not alter hepatic mRNA expression of Cyp3a11 (the murine homologue of the human CYP3A4) compared with control (20 mg/kg diet)( Reference Kluth, Landes and Pfluger 56 ). In a similar study, feeding rats a diet deficient or adequate in vitamin E ( < 2 or 60 mg RRR-αT) for up to 9 months did not change the mRNA of any of the thirty-three hepatic CYP expressed (including CYP3A4)( Reference Hundhausen, Frank and Rimbach 55 ). On the other hand, feeding of high doses of all rac-αT acetate (1000 mg/kg diet) for 4 months induced the hepatic mRNA expression of Cyp3a11 approximately 1·3- to 4-fold (depending on the analytical method used) compared with control mice on a standard diet (35 mg/kg diet)( Reference Mustacich, Gohil and Bruno 57 ). However, 1000 mg/kg diet of synthetic all rac-αT acetate is a supraphysiological dose, which corresponds to about 7–10 g/d for an average human, and might explain the differences between the latter and the aforementioned mouse and rat studies. Although T3 have a higher potential to induce CYP3A4 in reporter gene assays as well as in primary hepatocytes, hepatic mRNA expression of the murine homologue Cyp3a11 remained unchanged after oral administration of mice with γT3 (250 μg/d for 7 d)( Reference Kluth, Landes and Pfluger 56 ).
Although, as mentioned earlier, changes in mRNA may not necessarily result in similar changes in protein expression and activity, none of the above studies investigated the effects of vitamin E on CYP protein expression and activity. In an attempt to close this gap, we fed guinea-pigs with 20 or 250 mg RRR-αT/kg diet for 6 weeks and studied the metabolism of atorvastatin, a drug that is metabolised by CYP3A4, and its lipid-lowering efficacy. The hepatic protein expression of CYP3A4 and CYP20A1 and the serum concentrations of the CYP3A4 products of atorvastatin, para- and ortho-hydroxy-atorvastatin were similar in both groups. In agreement with this, the cholesterol-lowering activity of atorvastatin was unaltered by high-dose αT feeding( Reference Podszun, Grebenstein and Hofmann 37 ). This is in agreement with studies in human subjects addressing the effects of αT on CYP3A4-mediated drug metabolism. Patients receiving either lovastatin or simvastatin (both CYP3A4 substrates) were supplemented with 400 mg/d RRR-α-tocopheryl acetate for 8 weeks and CYP3A4 activity was determined by quantification of urinary cortisol and its CYP metabolite 6β-hydroxycortisol. In these patients, vitamin E supplementation neither altered CYP3A4 activity (urinary 6β-hydroxycortisol), nor the lipid-lowering activity of the statins( Reference Leonard, Joss and Mustacich 58 ). In another human intervention trial, healthy volunteers were intravenously injected with 1 mg of the CYP3A4 substrate midazolam and its area under the plasma concentration–time curve (AUC) was determined before and after a 3-week intervention with 750 IU (503 mg) RRR-αT/d orally or placebo (six subjects per group). By using intravenous injection of the test compound, the investigators were able to circumvent intestinal phase I metabolism and to investigate the effects of vitamin E on hepatic phase I metabolism only. The plasma AUC for midazolam and its metabolite hydroxymidazolam did not differ between the placebo and vitamin E groups( Reference Clarke, Burnett and Wu 59 ).
In summary, vitamin E–drug interactions due to altered phase I metabolism have not been reported and appear unlikely based on the available evidence. Even at high oral doses, neither T nor T3 appear to significantly alter CYP activity in animals or humans.
Vitamin E and hepatic phase II metabolism
Phase II of xenobiotic metabolism, which takes place to a significant extent in the liver, comprises the conjugation of xenobiotics or their phase I metabolites with polar groups, such as glucuronic acid, sulfate, glutathione or amino acids, to enable rapid biliary and urinary elimination. The inhibition of phase II enzymes, such as the UGT and GST, results in increased blood and tissue concentrations of their substrates. The induction of phase II enzymes, on the other hand, may increase the clearance and thereby reduce the activity of drugs.
None of the eight vitamin E congeners (incubation with 10 μmol/l for 24 h) altered UGT1A1 mRNA in primary human hepatocytes( Reference Zhou, Tabb and Sadatrafiei 30 ) and feeding rats 200 mg αT/kg diet for 2 weeks had no effect on hepatic UGT activity( Reference Van der Logt, Roelofs and van Lieshout 40 ). A small increase (1·76– to 2·68-fold, depending on assay) in hepatic GST activity was observed in rats fed very high doses of αT (2500 mg/kg diet) for 10 d( Reference Manson, Ball and Barrett 60 ). This dose, however, would be equal to more than 17 g/d of αT for a 70 kg human. Similarly, feeding mice with 1000 mg/kg diet of all rac-αT acetate for 4 months led to a small increase (maximum 1·9-fold) in the hepatic mRNA expression of the murine Gstm3 compared with mice on a standard diet (35 mg all rac-αT acetate/kg diet)( Reference Mustacich, Gohil and Bruno 57 ). In disagreement with the above-described effects in vivo, studies with isolated human GST P1-1 suggest that the concentrations at which T and T3 inhibit GST activity by 50 % (IC50) are: αT, 0·7; δT, 0·8( Reference Van Haaften, Evelo and Penders 61 ); αT3, 1·8; and γT3, 0·7 μmol/l( Reference van Haaften, Haenen and Evelo 62 ). However, direct interactions of vitamin E with enzymes in artificial in vitro systems may differ significantly from cellular systems and the situation in vivo, where vitamin E resides in membranes and may thus not have direct access to these soluble cytosolic proteins.
The mRNA expression of another phase II enzyme, sulfotransferase 2A, was significantly induced (11-fold) in the aforementioned mouse study, where animals were fed 1000 compared with 35 mg/kg diet all rac-αT acetate for 4 months( Reference Mustacich, Gohil and Bruno 57 ).
Overall, there is no evidence to support the notion that T or T3 ingested at normal dietary or supplemental doses may alter the activity of hepatic phase II metabolism in the living organism.
Vitamin E and hepatic drug export
Some researchers consider the export of xenobiotics or their phase I and II metabolites from cells, facilitated by the activity of membrane transporters such as the multidrug resistance proteins (for example, P-gp) and other ATP-binding cassette transporters, as phase III of xenobiotic metabolism. In the liver, hepatocytes lining the biliary canalicular surface express P-gp( Reference Thiebaut, Tsuruo and Hamada 27 ) and secrete substrates, including vitamin E and its metabolites( Reference Mustacich, Shields and Horton 63 ), into the bile.
In primary human hepatocytes, P-gp mRNA expression was not affected by incubation with any of the T or T3 (10 μmol/l; 24 h)( Reference Zhou, Tabb and Sadatrafiei 30 ). In mice fed very high doses of all rac-αT acetate (1000 mg/kg diet compared with 35 mg/kg diet) for 4 months, P-gp mRNA expression increased 1·9-fold in the liver( Reference Mustacich, Gohil and Bruno 57 ). The same authors, in another in vivo experiment, investigated the effects of daily subcutaneous injections of two different formulations (micellised vitamin E (EmcellE) and vitamin E dissolved in 20 % ethanol and 1 % benzyl alcohol (VitalE)) of high-dose RRR-αT (100 mg/kg body weight for up to 9 d) in rats. Both formulations increased P-gp mRNA (VitalE, 3·57-fold; EmcellE, about 10-fold), while P-gp protein expression was only reported for VitalE injections, which significantly increased it 3-fold( Reference Traber, Labut and Leonard 45 ).
Thus, two animal studies report that αT may induce hepatic P-gp expression when injected intraperitoneally or subcutaneously in high doses, which are unusual routes of administration and as such of limited relevance in a nutritional context. Overall, there are no publications to suggest that the consumption of T or T3 at conventional dietary or supplemental doses may induce the expression of hepatic efflux transporters or cause hepatic membrane-transporter-mediated drug interactions in vivo. However, it cannot presently be excluded that vitamin E taken at extremely high doses may affect the expression and perhaps the activity of hepatic drug exporters.
Vitamin E and pharmacodynamics
The effects of vitamin E on pharmacodynamics can be mediated indirectly by altering drug pharmacokinetics, as described in the previous sections, or more directly by targeting the molecular mechanisms underlying drug activity. The latter may be achieved by direct interactions with the drug's molecular targets or by actions on additional (independent) targets, such as signalling molecules, which alter the activity of a drug.
Vitamin E and blood coagulation
The best-known vitamin E–drug interactions are those with drugs that affect platelet aggregation and blood coagulation, which are presently the only interactions listed by the US Food and Drug Administration (FDA) in their consumer health information( 64 ). Blood coagulation is the process during which blood is (locally) converted from a liquid to a gel and is the body's answer to injury ensuring that ruptured blood vessels are rapidly sealed off to minimise blood loss.
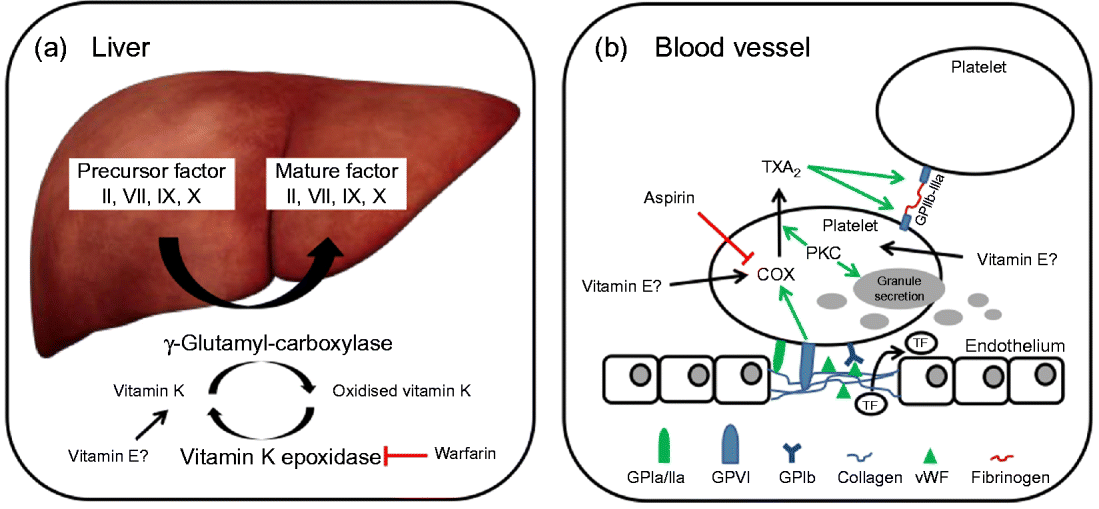
In brief, coagulation is initiated when collagen, which is normally buried underneath the epithelium, is exposed and platelets adhere to it via collagen-specific surface receptors (glycoprotein Ia/IIa). Platelet adhesion and the release of signalling molecules (including tissue factor and von Willebrand factor) from both the injured endothelium and the platelets activate further platelets and the intrinsic and extrinsic coagulation pathways. These coagulation cascades include a number of enzymes that are synthesised as precursors in the liver, where they undergo vitamin K-dependent carboxylation by γ-glutamyl-carboxylase, and are secreted as mature (carboxylated) proteins into the bloodstream (Fig. 3). The adhesion and aggregation of platelets at the site of injury result in the formation of a patch that is stabilised by the deposition and maturation of the fibrous protein fibrin (also known as factor Ia). Fibrin is formed by polymerisation of its precursor fibrinogen through the activity of the enzyme thrombin, which itself is activated by cleavage of prothrombin (factor II). Activated platelets secrete granules containing ADP, serotonin, platelet-activating factor, von Willebrand factor, platelet factor 4 and thromboxane A2 (TXA2) that induce platelet aggregation through cross-linkage of activated glycoprotein IIb-IIa and fibrinogen. TXA2 is synthesised from arachidonic acid through the cyclo-oxygenase (COX) and hydroperoxidase catalytic activities of PG synthase, via the intermediate PGH2. The COX reaction is the rate-limiting step in the synthesis of prostaglandins and thromboxanes and is inhibited by drugs, such as acetylsalicylic acid, which thereby reduce platelet aggregation.
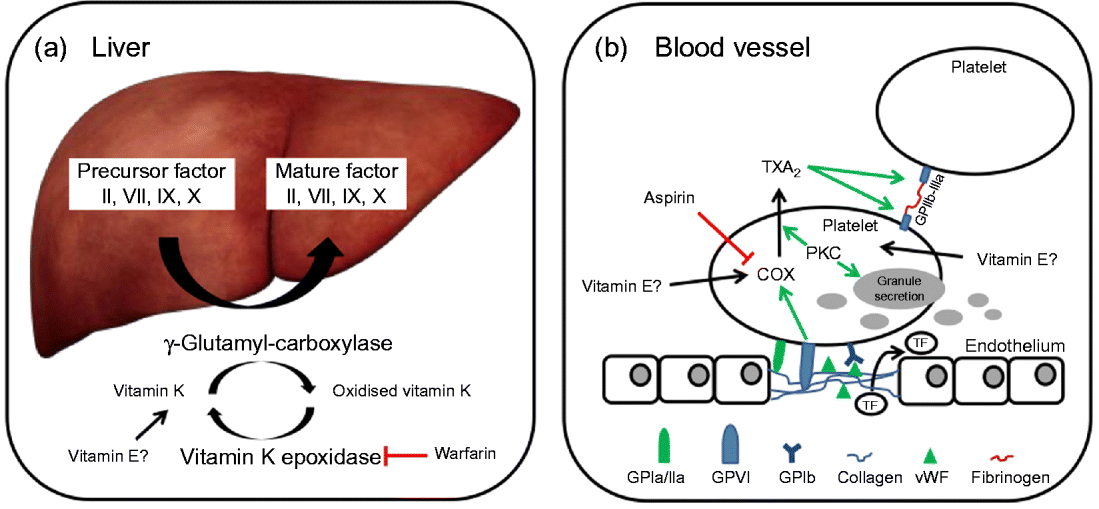
Fig. 3 (a) The precursors of mature factor II, VII, IX and X are carboxylated by γ-glutamyl-carboxylase to their mature forms. The cofactor (vitamin K) for this reaction is oxidised and then regenerated by vitamin K epoxidase. The inhibition of vitamin K epoxidase by, for example, warfarin decreases the recycling of oxidised vitamin K and results in a shortage of the cofactor for the carboxylation reaction. (b) Upon endothelial injury, platelets bind to sub-endothelial collagen (via glycoprotein Ia/IIa (GPIa/IIa) and von Willebrand factor (vWF)). Tissue factor (TF) is also released to activate the extrinsic pathway. Interaction between GPVI and collagen induces cyclo-oxygenase (COX) and triggers the formation of thromboxane A2 (TXA2), which cross-links platelets via GPIIb-IIIa and fibrinogen. Protein kinase C (PKC) activates, among other things, TXA2 formation and granule secretion. (A colour version of this figure can be found online at www.journals.cambridge.org/nrr).
Platelet activation is a complex regulatory pathway with multiple agonists, including ADP, TXA2, collagen, adrenaline and many more. Ex vivo platelet aggregation can be activated by either endogenous agonist or phorbol myristate actetate (PMA), a protein kinase C (PKC) activator. PKC is a family of enzymes catalysing the phosphorylation and thus controlling the activity of many regulatory proteins. PKC activation stimulates, among other things, platelet granule secretion, TXA2 synthesis and thereby the aggregation of platelets( Reference Harper and Poole 65 ).
Vitamin E and platelet adhesion, activation and aggregation
Platelets obtained from healthy volunteers and pre-incubated with RRR-αT for 5 min (0·016–16·22 μmol/l) exhibited a dose-dependent reduction in the metabolism of arachidonic acid to TXA2 and PGD2. The authors suggested that this may be due to an inhibition of COX activity in αT-treated platelets( Reference Ali, Gudbranson and McDonald 66 ). Incubation of platelets from healthy human subjects with supraphysiological concentrations (500 μmol/l) of RRR-αT or RRR-α-tocopheryl acetate, but not all rac-αT, inhibited arachidonic acid- or PMA-induced platelet aggregation and PKC-mediated protein phosphorylation( Reference Freedman, Farhat and Loscalzo 67 , Reference Freedman and Keaney 68 ). Although 500 μmol/l αT is a supranutritional dose, the authors observed that the cellular content of αT in the incubated platelets was comparable with that of platelets isolated from human subjects receiving 800 mg αT/d for 14 d( Reference Freedman, Farhat and Loscalzo 67 ). In the latter study, platelets isolated from human subjects supplemented for 14 d with 267, 533 or 800 mg αT/d, PMA-induced platelet aggregation was dose-dependently reduced, arachidonic acid-induced aggregation was reduced in the 800 mg/d group only, and ADP-induced aggregation was not altered by αT( Reference Freedman, Farhat and Loscalzo 67 ). PKC-mediated phosphorylation was determined in two subjects and was inhibited after 2 weeks’ supplementation with 800 mg RRR αT/d compared with baseline( Reference Freedman, Farhat and Loscalzo 67 ). In another study where healthy volunteers were given placebo, 100 mg all rac-αT acetate, or mixed T (20 mg RRR-αT, 100 mg RRR-γT and 40 mg RRR-δT) daily for 8 weeks, only mixed T significantly reduced ADP-induced, but not PMA-induced platelet aggregation, although phosphorylation of PKC was significantly inhibited by both vitamin E interventions( Reference Liu, Wallmon and Olsson-Mortlock 69 ). In yet another study, platelet adhesion to collagen was dose-dependently reduced in twelve healthy subjects supplemented with increasing doses of all rac-αT acetate (400, 800 and 1200 mg/d for 2 weeks, each; 6 weeks supplementation in total) compared with eleven healthy control subjects. Platelet aggregation (induced with collagen, adrenaline or ADP) was only modestly decreased in these subjects and only collagen-induced platelet aggregation was significantly reduced (P< 0·05) in women, but not men( Reference Steiner 70 ). The same study also analysed the combination of all rac-αT with the analgesic, antipyretic and anti-inflammatory drug acetylsalicylic acid (aspirin)( Reference Steiner 70 ), which itself inhibits the COX-catalysed formation of TXA2 from arachidonic acid( Reference Vane and Botting 71 ). The combined oral administration of all rac-αT acetate in doses up to 1200 mg/d with 300 mg aspirin every other day had no effect on collagen-, adrenaline- or ADP-induced platelet aggregation or platelet adhesion to collagen compared with vitamin E or acetylsalicylic acid alone( Reference Steiner 70 ). In a subset of the Alpha-Tocopherol Beta-Carotene Cancer Prevention Study, male smokers aged 50–59 years who received a combination of 100–3200 mg acetylsalicylic acid and 50 mg all rac-αT acetate/d and were followed for 5–7 years showed a significant increase in oral ‘bleeding on probing’ (gingival bleeding) compared with subjects receiving acetylsalicylic acid alone( Reference Liede, Haukka and Saxen 72 ). Accordingly, in patients who previously suffered from an ischaemic event, the combination of acetylsalicylic acid (325 mg) and all rac-αT acetate (400 mg/d) for up to 2 years (n 52), compared with acetylsalicylic acid alone (n 48), was more effective in the prevention of transient ischemic attacks, suggesting a synergistic activity of αT with aspirin( Reference Steiner, Glantz and Lekos 73 ).
The mechanism for the reduction in ex vivo platelet aggregation observed in the above studies still remains to be fully elucidated. The inhibition of PKC by αT may be an important factor, since platelet aggregation induced by the PKC activator PMA was reduced by vitamin E in most studies concomitantly with a reduction in PKC activity( Reference Freedman, Farhat and Loscalzo 67 – Reference Liu, Wallmon and Olsson-Mortlock 69 ). Interactions with vitamin K-dependent coagulation cascades, however, may be part of the explanation, as discussed in the next section.
Vitamin E, vitamin K and the extrinsic and intrinsic coagulation pathways
As mentioned earlier, the coagulation factors II, VII, IX and X undergo post-translational carboxylation by γ-glutamyl-carboxylase (Fig. 3), an enzyme that requires vitamin K as a cofactor, in the liver. Vitamin K is oxidised in the carboxylation reaction and requires subsequent reduction by vitamin K epoxide reductase in order to take part in another cycle of carboxylation( Reference Suttie 74 ). Some anticoagulants, such as warfarin, act by reducing the vitamin K epoxide reductase-catalysed recycling of oxidised vitamin K, thereby limiting the carboxylation of the precursors of factors II (prothrombin), VII, IX and X to their mature forms (Fig. 3)( Reference Hirsh, Dalen and Anderson 75 ).
In rats with decreased concentrations of reduced vitamin K (induced by intraperitoneal injections of warfarin), intramuscular injections of RRR-αT acetate (1000 mg/kg body weight/d for 7 d) reduced mature prothrombin coagulant activity compared with animals treated with warfarin only, but not the amount of the prothrombin precursor produced in the liver( Reference Corrigan and Ulfers 76 ). Since αT influences the formation of the mature prothrombin rather than the production of the precursor, this might be caused by a vitamin E-induced aggravation of vitamin K deficiency. Prothrombin activity in warfarin-treated patients supplemented with vitamin E (800 or 1200 IU vitamin E (congener not specified)/d, n 13; or 100 or 400 IU all rac αT-acetate/d, n 6) for 4 weeks did not differ significantly from that of placebo-treated patients( Reference Corrigan and Ulfers 76 , Reference Kim and White 77 ). Although these trials are limited due to their small sample size, they suggest that doses up to 1200 IU vitamin E have no clinical effects in patients on warfarin therapy. Nevertheless, vitamin E may interfere with vitamin K metabolism to some extent. Human subjects supplemented with 1000 IU RRR-αT/d (12 weeks) showed an increase in PIVKA-II (protein induced by vitamin K absence factor II) plasma concentrations( Reference Booth, Golly and Sacheck 78 ), which is a marker of subclinical vitamin K deficiency( Reference Widdershoven, Van Munster and De Abreu 79 ). Interactions between the metabolism of vitamins E and K are feasible, since both appear to share the same metabolic fate and may be ω-hydroxylated by the same enzyme(s) (inter alia CYP4F2)( Reference Sontag and Parker 50 , Reference Bardowell, Duan and Manor 51 , Reference Edson, Prasad and Unadkat 80 ). However, vitamin K ω-hydroxylation was neither affected in cells incubated with, nor in animals fed high doses of, vitamin E( Reference Farley, Leonard and Labut 46 , Reference Farley, Leonard and Taylor 81 ). Thus, the mechanism of vitamin E-induced changes in vitamin K status remains unclear and warrants further investigation.
In summary, reduced blood coagulation and increased bleeding have been observed in some cases in animals and human subjects treated with aspirin or warfarin simultaneously with high doses of vitamin E (mainly αT). Such a synergistic activity may be desirable, for example in patients at risk for CVD or stroke, or undesirable, as in the case of increased (gastrointestinal) bleeding, a known side effect of aspirin( Reference Shiotani, Kamada and Haruma 82 ). Vitamin K status and coagulation activity should be monitored in patients supplementing vitamin E while on warfarin therapy (or taking other medication interfering with vitamin K-dependent activation of coagulation factors) to detect and counteract changes in haemostasis.
Interactions of vitamin E of an unknown kind that alter drug efficacy
Vitamin E may impair tamoxifen drug activity in breast cancer patients
Oestrogen-responsive cancer may be treated with the selective oestrogen receptor modulator tamoxifen( Reference Fisher, Redmond and Legault-Poisson 83 ), but most patients eventually develop resistance to the drug( Reference Shou, Massarweh and Osborne 84 ). Tamoxifen rapidly increases intracellular concentrations of the second messenger Ca2+ ( Reference Chang, Huang and Wang 85 ). Cell-culture studies suggest that vitamin E may alter the activity of tamoxifen. In oestrogen-responsive mammary carcinoma cells (MCF-7, T47D), incubation with tamoxifen in the presence of αT (10 μmol/l; 24 h) inhibited the tamoxifen-induced rise in intracellular Ca2+ and reduction in cell proliferation( Reference Peralta, Viegas and Louis 86 ). In a small prospective study with seven breast cancer patients on tamoxifen therapy, supplementation with 400 mg/d αT acetate for 30 d reduced tamoxifen blood concentrations in five patients and increased markers of tamoxifen resistance in six of the seven patients. In four of these patients, tamoxifen blood concentrations dropped to below the minimum therapeutic concentration( Reference Peralta, Brewer and Louis 87 ).
In summary, the limited evidence from cell-culture and human intervention trials suggests that it might be undesirable for women on tamoxifen therapy to consume dietary supplements containing high doses of αT, as they may impair drug efficacy. However, before valid conclusions can be drawn, sufficiently powered randomised clinical trials investigating the interactions of αT with tamoxifen pharmacokinetics and pharmacodynamics are required.
High-dose vitamin E supplementation may alter the pharmacokinetics of cyclosporine A
Cyclosporine A is an immunosuppressant administered to prevent graft rejection in organ transplant recipients. One of the side effects of cyclosporine A is an increased generation of reactive oxygen species, which was suggested to be involved in cyclosporine A-induced nephrotoxicity( Reference Baliga, Ueda and Walker 88 ). To investigate if supplementation with antioxidants may be beneficial in cyclosporine A-treated renal transplant recipients, ten patients were supplemented with an antioxidant cocktail (800 IU vitamin E (αT), 1000 mg vitamin C and 6 mg β-carotene per d) or placebo for 6 months in a cross-over study with a 6-month washout period( Reference Blackhall, Fassett and Sharman 89 ). All cyclosporine A trials reported in this paragraph used αT in the dietary supplements (ML Blackhall, RG Fassett, JE Sharman, DP Geraghty and JS Coombes, personal communication). The antioxidant cocktail did not alter any of the markers of oxidative stress, but decreased cyclosporine A concentrations in blood by 24 %( Reference Blackhall, Fassett and Sharman 89 ). In a comparable trial, kidney transplant recipients on cyclosporine A therapy were supplemented for 3 months with placebo (n 28) or 1000 mg vitamin C plus 300 mg vitamin E (αT; n 25) per d and reduced cyclosporine A blood concentrations were reported for the vitamin-supplemented group( Reference de Vries, Oterdoom and Gans 90 ). Similar observations were made retrospectively in heart transplant patients; compared with baseline, cyclosporine A blood concentrations decreased by 30 % when patients were supplemented with 1000 mg vitamin C and 800 IU vitamin E (αT) per d( Reference Lake, Aaronson and Gorman 91 ). However, in all studies, vitamin E was administered in combination with vitamin C, which makes it impossible to discern the contribution of each individual vitamin.
The effect of vitamin E alone on cyclosporine A pharmacokinetics was studied earlier in healthy volunteers before and after supplementation with 800 IU vitamin E (αT) per d for 6 weeks. Vitamin E supplementation had no impact on the glomerular filtration rate or peak plasma concentration, but reduced the AUC of a single oral dose of cyclosporine A (5 mg) by 21 % compared with baseline( Reference Barany, Stenvinkel and Ottosson-Seeberger 92 ).
The mechanism underlying the interaction of vitamin E with cyclosporine A is currently unknown. In the trials discussed above, vitamin E and cyclosporine A were administered separately, which eliminates the possibility of a direct interaction. Cyclosporine A is a substrate for intestinal P-gp and CYP3A4 as well as hepatic CYP3A4 and CYP3A5( Reference Hebert 93 ), which makes pre-systemic interactions on the level of drug absorption and intestinal and systemic interactions possible. Induction of intestinal and hepatic metabolism would both decrease the plasma concentration and AUC of cyclosporine A. However, as discussed above, orally administered αT does not induce the expression of CYP or membrane transporters( Reference Podszun, Grebenstein and Hofmann 37 , Reference Hundhausen, Frank and Rimbach 55 , Reference Kluth, Landes and Pfluger 56 , Reference Leonard, Joss and Mustacich 58 ), rendering this mode of interaction rather unlikely.
Since vitamin E supplementation at daily doses of ≥ 300 mg affected the pharmacokinetics of cyclosporine A in four independent human trials, patients treated with this drug should abstain from the consumption of dietary supplements containing such high doses of vitamin E or at least have the blood concentration and efficacy of the drug tested regularly by their physician.
α-Tocopherol supplementation reverses the overexpression of the fatty acid translocase CD36
The potential interaction of vitamin E with ritonavir described below differs from the cases discussed above in that it relates to a potential adverse interaction in the case of vitamin E deficiency rather than under vitamin E supplementation. Ritonavir is a drug that specifically inhibits the HIV 1 protease and thereby HIV replication( Reference Vacca and Condra 94 ) and is used for the treatment of HIV-infected patients. Adverse effects observed upon long-term treatment with ritonavir include hyperlipidaemia and accelerated atherosclerosis, which may in part be caused by an overexpression of the scavenger receptor CD36 protein in macrophages( Reference Dressman, Kincer and Matveev 95 ).
In THP1 monocytes, ritonavir-induced CD36 (over-) expression was reversed by incubation with RRR-αT (50 μmol/l; 24 h)( Reference Munteanu, Zingg and Ricciarelli 96 ). Further evidence to support a role of vitamin E in the expression of CD36 came from two studies in rats fed vitamin E-deficient (1·7 mg αT acetate; < 1 mg total T/kg diet, respectively) compared with vitamin E-sufficient (60 mg αT acetate; 12 mg RRR-αT and 24 mg RRR-γT/kg, respectively) diets for 6 and up to 9 months, respectively, in which an up-regulation of hepatic CD36 mRNA was observed in the deficient animals from the third month onwards( Reference Barella, Muller and Schlachter 97 , Reference Gaedicke, Zhang and Schmelzer 98 ). In guinea-pigs fed a high-fat diet (21 % fat by weight), CD36 protein expression in the liver was almost twice that observed in control animals on a 5 % fat diet. Inclusion of high doses of RRR-αT (250 mg/kg diet) in the high-fat diet reversed the increase in hepatic CD36 expression back to the expression level observed in animals fed the 5 % fat control diet( Reference Podszun, Grebenstein and Spruss 54 ).
Taken together, these data suggest that vitamin E deficiency might increase CD36 expression and could thus potentially aggravate ritonavir-induced hyperlipidaemia. Therefore, a sufficient intake (and perhaps even low-dose supplementation) of vitamin E may be beneficial in ritonavir-treated patients. However, there are presently no in vivo data available to corroborate that such an interaction may be biologically relevant in humans and further studies addressing the potential benefit as well as the safety of a co-administration of ritonavir and vitamin E are necessary before valid recommendations can be given.
Conclusions
There is no convincing evidence from animal models or controlled human intervention trials to suggest that T–drug interactions may be caused by an induction of xenobiotic metabolism or trans-membrane transport. Corresponding in vivo studies for T3 are presently not available and are therefore warranted. Based on the documented safety of αT in randomised clinical trials( Reference Meydani, Meydani and Rall 99 , Reference Meydani, Lipman and Han 100 ), the FDA has set its tolerable upper intake level for adults at 1000 mg/d( Reference Hathcock, Azzi and Blumberg 1 ). There is little evidence to suggest potent health benefits from the intake of such high doses in healthy individuals; specific subgroups of the population (for example, type 2 diabetics with a haptoglobin 2–2 genotype), however, may benefit from αT supplementation at doses below 300 mg/d( Reference Frank and Rimbach 21 , Reference Milman, Blum and Shapira 101 – Reference Blum, Vardi and Brown 103 ). The dietary intake of T and T3 and the consumption of medium-dose supplements ( < 300 mg/d) appear safe and unlikely to cause adverse drug interactions and are in accordance with the tolerable upper intake levels for adults set by the European Food Safety Authority (300 mg/d)( 104 ). Consumption of high-dose vitamin E supplements may lead to interactions with the drugs aspirin, warfarin, tamoxifen and cyclosporine A and may alter their activities. Patients treated with these drugs should therefore consult their physician and have drug efficacy tested if they wish to consume vitamin E supplements. For the majority of drugs, however, interactions with vitamin E, even at high doses, are unlikely and have not been observed.
Acknowledgements
There are no conflicts of interest.