
Abstract
Low molecular weight protein tyrosine phosphatase (LWM-PTP), also known as acid phosphatase, is a highly conserved tyrosine phosphatase in living organisms. However, the function of LWM-PTP homolog has not been reported yet in plants. Here, we revealed a homolog of acid phosphatase, APH, in Arabidopsis plants, is a functional protein tyrosine phosphatase. The aph mutants are hyposensitive to ABA in post-germination growth. We performed an anti-phosphotyrosine antibody-based quantitative phosphoproteomics in wild-type and aph mutant and identified hundreds of putative targets of APH, including multiple splicing factors and other transcriptional regulators. Consistently, RNA-seq analysis revealed that the expression of ABA-highly-responsive genes is suppressed in aph mutants. Thus, APH regulates the ABA-responsive gene expressions by regulating the tyrosine phosphorylation of multiple splicing factors and other post-transcriptional regulators. We also revealed that Tyr383 in RAF9, a member of B2 and B3 RAF kinases that phosphorylate and activate SnRK2s in the ABA signaling pathway, is a direct target site of APH. Phosphorylation of Tyr383 is essential for RAF9 activity. Our results uncovered a crucial function of APH in ABA-induced tyrosine phosphorylation in Arabidopsis.
Similar content being viewed by others


Introduction
Protein phosphorylation is one of the most important post-translational modifications in eukaryotic organisms. Phosphorylation affects the protein conformation, activity, localization, turn-over, or/and protein-protein interaction (Edelman et al., 1987; Zulawski & Schulze, 2015). In eukaryotes, protein phosphorylation mainly occurs on three amino acid residues, Serine, Threonine, and Tyrosine. Tyrosine phosphosite only represents a small portion (1–2%) of total protein phosphorylation sites in animals and plants. In animals, some hormone receptors or signal molecules, like insulin and epidermal growth factor (EGF), are protein tyrosine kinases (PTKs), and the tyrosine phosphorylation regulates cell division, proliferation, metabolism, and oncogenesis. In plants, although only about 1600 tyrosine phosphosites were detected out of more than 40,000 phosphosites (Ahsan et al., 2020; Mergner et al., 2020), increasing evidence showed that tyrosine phosphorylation also has a crucial role in the regulation of plant immunity, growth, and hormone signaling (Kameyama et al., 2000; Kim et al., 2009; Jaillais et al., 2011; Nito et al., 2013; Macho et al., 2015; Li et al., 2018; Luo et al., 2020). Though no genes have been identified in plant genomes encoding canonical protein tyrosine kinase, some uncanonical dual specific protein kinases may mediate the tyrosine phosphorylation in plants (Champion et al., 2004; Macho et al., 2015; Zulawski & Schulze, 2015).
About 150 protein phosphatases have been identified in the Arabidopsis genome (Kerk et al., 2008; Moorhead et al., 2009; Mergner et al., 2020). Twenty of them are protein tyrosine phosphatases (PTPs), including 22 dual specific PTPs, one Classic I PTP PTP1, one Class II low molecular weight PTP (LMW-PTP), and one Class IV Asp-based PTP, according to their sequence silimilty to animal PTPs (Kerk et al., 2008; Moorhead et al., 2009; Mergner et al., 2020). It is known that PTP1 interacts with MPK6 and regulates plant immunity (Kerk et al., 2008; Bartels et al., 2009). Dual-specific PTPs, MKP1 and IBR5, participate in plant immunity and development by regulating the MAP kinase cascade. In animals, LMW-PTP, also known as Acid Phosphatase (APase, or ACP) due to acidic optimum pH range from 5 to 6 (Alho et al., 2013). ACP mediates the dephosphorylation of many tyrosine kinase receptors and other signaling molecules involved in signal transduction, which is considered as a signaling hub of cancer hallmarks (Alho et al., 2013; Faria et al., 2021). In plants, to the best of our knowledge, the function of LMW-PTP has not been reported yet.
In this study, we demonstrated that Arabidopsis ACP homolog, APH (AT3G44620), is a functional protein tyrosine phosphatase and participates in ABA signaling. In aph mutant, but not in wild type, the global tyrosine phosphorylation is strongly increased upon application of ABA. Several splicing factors and post-transcriptional regulators are identified as the putative targets of APH. Consistently, the ABA highly induced genes showed an impaired expression in aph mutant. RAF9, a protein kinase that regulates SnRK2 activation, is also in the list of putative APH target. Mutation of Tyr383 in RAF9 abolishes its kinase activity in vivo. Thus, our results reveal a crucial function of APH in regulating ABA-induced tyrosine phosphorylation in Arabidopsis plants.
Results
Arabidopsis APH is a functional protein tyrosine phosphatase
Our previous study identified APH (AT3G44620), a homolog of acid phosphatase that was co-immunoprecipitated with PYL1 protein (Wang et al., 2018), suggesting a putative function of APH in ABA signaling. APH is encoded by a single gene in human (Hemo sapiens), rat (Rattus norvegicus), Arabidopsis thaliana, maize (Zea mays), and rice (Oryza sativa) genome, and the C-terminal catalytic domain of ACPs are highly conserved in different species (Fig. 1A).

APH is a functional protein tyrosine phosphatase. A Sequence alignment of LMW-PTPs in Rattus norvegicus (NP_067085.1), Hemo sapiens (NP_004291.1), Arabidopsis thaliana (NP_001190010.1), Oryza sativa (XP_015649938.1), and Zea mays (NP_001149867.1). Arrows indicate the conserved Cystine (C) and Aspartic acid (D) residue in the catalytic domain. B The effect of pH on the phosphatase activity of recombinant AtAPH purified from E. coli. C The effect of pH on the phosphatase activity of recombinant human ACP purified from E. coli. D Cys83Ser and Asp231Ala mutations abolish APH phosphatase activity. E The SDS/PAGE of purified SUMO-HIS-AtAPH and SUMO-HIS-HsACP. F The percentage of peptides without (brown) or with (blue) pTyr in the synthetic peptide before and after incubation with recombinant APH. G The rate of peptides without (brown) or with (blue) pSer in the synthetic peptide before and after incubation with recombinant APH
Animal Acid Phosphatase is a ubiquitous lysosomal enzyme with an optimum pH range from 5 to 6. To study the function of APH, we first test the pH optimum of recombinant APH purified from E. coli. The result showed that Arabidopsis APH, unlike its animal homologs, displayed a broader optimum pH range from 5.5 to 7.5, the activity of APH declines at pH 8.0 and 8.5 (Fig. 1B). Mutation of two conserved residues in the catalytic motif, Cys83 to Ser, or the Asp231 to Ala, abolished the phosphatase activity of APH, respectively (Fig. 1D).
We used two synthetic peptides, EGpSMDTTTETASLLLGMNDSR from SIZ1, and 375DKSHNRpYSGV from RAF9, as the substrate to verify the specificity of APH enzymatic activity on phosphotyrosine (pTyr) or phosphoserine/phosphothreonine (pSer/pThr), respectively. As the result, incubation with APH strongly dephosphorylated the pTyr in the RAF9 phosphopeptide (Fig. 1F), but could not affect the abundance of pSer in the SIZ1 phosphopeptide (Fig. 1G). Thus, APH is a functional protein tyrosine phosphatase with an broad optimal pH from 5.5 to 7.5.
aph mutants are insensitive to ABA in post-germination growth
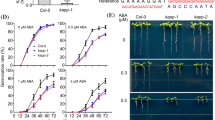
To study the function of APH, we obtained 3 T-DNA insertion lines aph-1 (Salk_120974), aph-2 (Salk_083590) and aph-3 (Salk_101423) from ABRC (Fig. 2A). The aph-3 is a null allele, in which the T-DNA is inserted within the second exon, and no expression of APH was detected in aph-3 (Fig. 2B). In aph-1, the T-DNA insertion is located in the promoter region of APH (Fig. 2A). The RNA sequencing result showed that the T-DNA fragment and the coding region of APH formed a fused transcript in aph-1, which may result in a truncated or lose-function form of APH (Fig. S1). In aph-2, the T-DNA insertion is also located in the promoter region of APH. Phenotypic analysis revealed that all the three aph mutant lines were insensitive to ABA in the post-germination growth compared to the wild type (Col-0) (Fig. 2C and D, Fig. S2A-C). No difference in the germination rate or water loss was observed between wild type and aph mutants (Fig. 2E and F). In addition, overexpression of APH does not affect the ABA response in APH-OE lines (Fig. S2D). Application of ABA only very slightly (fold change = 1.6, p < 0.05) induced the expression of APH at later stage.

aph mutants are insensitive to ABA in the post-germination growth. A APH gene model and T-DNA insertion sites. B qRT-PCR analysis of the APH expression in the aph-1, aph-2, and aph-3 T-DNA insertion lines. C The percentage of seedlings showing green cotyledons after 7 days of germination and growth on 1/2 Murashige and Skoog (MS) medium containing 1 μM ABA. Error bars, SD (n = 3). D Photographs of seedlings after 10 days of germination and growth on 1/2 MS medium containing 1 μM ABA. E The germination rate of seeds after 3 days of germination and growth on 1/2 MS medium containing 1 μM ABA. Error bars, SD (n = 3). F Water loss rate of the 4-week-old wild type and aph mutants. Error bars, SD (n = 3). Asterisks indicate significant differences between wild type and aph mutants (Student’s t-test; *p < 0.05, **p < 0.01, ***p < 0.001)
Identifying the APH-regulated tyrosine phosphorylation by phosphoproteomics
To dissect the function of APH in ABA signaling, we performed a quantitative phosphoproteomics to compare the APH-mediated tyrosine dephosphorylation in the presence or absence of ABA in the wild type and aph-1 mutant, the mutant showed the strongest ABA phenotype. As shown in Fig. 3A, phosphoproteomics were conducted with the total protein extracted from wild type and aph-1 mutant with or without ABA treatment. The tryptic peptides were then subjected to affinity purification using anti-phosphotyrosine (pTyr) antibody 4G10 and subsequently quantified by LC-MS/MS. We detected 16 and 29 pTyr phosphopeptides in the wild-type seedlings without or with ABA-treatment, respectively (Table S1). We also identified 36 and 203 phosphopeptides containing pTyr from the aph-1 without or with ABA-treatment, respectively. A total of 298 pTyr sites were identified by proteome-scale pTyr enrichment and LC-MS/MS analysis (Table S1). Compared to a total of 6492 phosphosites identified in this phosphoproteomic analysis, 298 phosphopeptides (4.6%) contain at least one phosphorylated tyrosine residue (Fig. 3B). Compared to previous works showing 0.4% (Al-Momani et al., 2018), and 1.8% (Wang et al., 2018) of pTyr in total phosphosites, the 4G10 affinity purification demonstrated a 2.5 to 12-fold enrichment of pTyr peptides.

Quantitative phosphoproteomics analysis of global tyrosine phosphorylation in wild type and aph-1 mutant. A Overview of the experimental workflow. B The rate of phosphotyrosine (pY), phosphoserine (pS), and phosphothreonine (pT) in the phosphosites identified in this study. C The number of pY phosphopeptides identified in wild type and aph-1 mutant without or with ABA treatment. Error bars, SD (n = 3). D The tyrosine phosphorylation in some splicing factors and post-transcriptional regulators in the wild type and aph-1 mutant without or with ABA treatment. The phosphosites identified in these phosphopeptides are highlighted in red. Error bars, SD (n = 3). Asterisks indicate significant differences between wild type and aph-1 mutant (Student’s t-test; *** p < 0.001)
We then performed a quantitative comparison of the pTyr phosphorylation in the wild type and aph-1 mutant with or without ABA treatment, using the pTyr sites identified in at least twice out of three biological replicates (Fig. S3A). Eighty pTyr sites corresponding to 76 pTyr proteins were significantly induced in aph-1 mutant after ABA treatment, compared to the very slight increment of pTyr induced by ABA in the wild-type seedlings (Fig. 3C and S3A). These results suggest that APH may be a negative regulator of ABA-dependent tyrosine phosphorylation, given that dysfunction of APH resulted in an accumulation of pTyr in aph-1 mutant under ABA treatment.
We further characterized the biological functions of the ABA-induced pTyr proteins in aph-1 mutant. Fisher’s exact test (p < 0.05) was performed to select the biological function of gene ontology (GO) significantly enriched in ABA-induced pTyr proteins in aph-1 mutant. Only one GO term, mRNA metabolic process (GO: 0016071), is 8.76-fold enriched in the 76 pTyr proteins. The pTyr of some splicing factors like SR45, SCR25, SCL30A, and RSZ22a were only detected in the ABA-treated aph-1, but not in control or ABA-treated wild type (Fig. 3D, Table S1). The protein-protein interaction data obtained from the String database also showed that SR45, SCL30A, RSZ22a, and RRP5 are in the cluster of mRNA splicing (Fig. S3B), which is consistent with the result of GO analysis. SR45 is one of the spliceosome components and is involved in pre-mRNA splicing and RNA metabolism. sr45–1 mutant confers hypersensitivity to glucose and ABA during early seedling growth (Carvalho et al., 2010). PRP4 KINASE A (PRP4KA) is a spliceosome-associated kinase involved in alternative splicing (Kanno et al., 2018). A tyrosine phosphosite pTyr634 in PRP4KA was only detected in ABA-treated aph-1seedlings, but not in untreated aph-1, nor control and ABA-treated wild type (Fig. 3D, Table S1). The tyrosine phosphorylation of PAT1, a component of the mRNA decapping machinery, also showed a similar aph-1 dependent pattern to pTyr167 in RSZ22a (Fig. 3D, Table S1). Thus, these results imply that the APH-mediated tyrosine phosphorylation may play an essential role in transcriptional response to ABA.
Arabidopsis APH regulates transcription of ABA-responsive genes
We then performed a transcriptome-sequencing to further explore the role of APH in the regulation of ABA-responsive gene expression. A shown in Fig. 4A and Table S3, a major proportion of ABA highly induced genes showed a reduced expression level in aph-1 mutant. For example, 124 genes showed at least 5-fold induction by applying ABA in the wild type (Fig. 4B, rightmost panel, and Table S3). Forty-nine (39.5%) out of these 124 genes showed a significant reduced expression (fold change < 0.5, p < 0.05) in aph-1 compared to wild type in the control condition (Fig. 4B, rightmost panel). None of the 124 genes showed increased expression in aph-1 in the control condition (Fig. 4B, rightmost panel). Thus, most genes that showed reduced expression in aph-1 are ABA highly induced genes (Fig. 4C).

Transcriptome analysis of ABA-responsive genes expression in wild type and aph-1 mutant. A Heat map showing the expression levels of ABA-responsive genes in wild type and aph-1 seedlings. B A major proportion of ABA-induced genes in wild type showed a reduced expression in aph-1 mutant. DEGs with 5- to 1-fold (p < 0.05) are classified according to their expression in response to ABA in Col-0 wild type. Blue, DEGs showed more than two-fold reduction in aph1 mutant, compared to that in the wild type (FC < 0.5, p < 0.05), Red, DEGs showed more than two-fold increment in aph-1 mutant, compared to that in the wild type (FC > 2, p < 0.05). C The ABA-responsive patterns of DEGs showed reduced expression (FC < 0.5, p < 0.05) in aph-1 mutant compared to that in the wild type. D GO analysis of the DEGs showed reduced expression in the untreated aph-1 mutant, compared to that in the untreated wild-type seedlings. E Expression of the ABA-inducible marker genes in wild type and aph-1 seedlings upon 6 h of ABA treatment. Error bars, SEM (n = 3 biological replicates). Asterisks indicate significant differences between wild type and aph-1 mutant (Student’s t-test; * p < 0.05, ** p < 0.01, *** p < 0.001)
GO analysis revealed that these genes with reduced expression in aph-1, are mainly involved in water deprivation, ABA, lipid, abiotic stress, cold, osmotic stress, and aging and developmental processes (Fig. 4D and Table S3). Quantitative RT-PCR analysis of several ABA-inducible genes, like RAB18, KIN1, LEA7, COR15A, and HAI2, showed an impaired induction in aph-1 (Fig. 4E). Thus, alteration of ABA-responsive genes in aph-1 supported the function of APH-mediated tyrosine phosphorylation in transcription regulation of ABA response.
Tyrosine phosphorylation mediates RAF9 activation
Recent studies revealed that B2, B3, and B4 subgroup Raf-like protein kinases directly phosphorylate SnRK2s and are required for ABA- and osmotic-induced SnRK2 activation (Saruhashi et al., 2015; Katsuta et al., 2020; Soma et al., 2020; Takahashi et al., 2020; Lin et al., 2021). One phosphopeptide in RAF9, DKpSHQINRpYpSGVK, contains a pTyr site Y383 and two phosphoserine residues pSer377 and pSer384, showed an ABA-induced accumulation in aph-1 mutant (Fig. 5A and B). To evaluate the role of Tyrosine and Serine phosphorylation in RAF9, we used a transient activation assay in Arabidopsis mesophyll cell protoplasts. The LUCIFERASE (LUC) reporter gene driven by the ABA-responsive RD29B promoter was used as a reporter of ABA response (Wang et al., 2018; Lin et al., 2021). In the wild type, ABA-induced the expression of RD29B-LUC in the mesophyll cell protoplasts, while ABA-induced RD29B-LUC expression was abolished entirely in the protoplasts of OK100-nonu, a mutant lacking nine out of 12 B2 and B3 RAFs (Fig. 5C). Co-expression of wild-type RAF9 partially rescued the ABA-induced expression of RD29B-LUC in the protoplasts of OK100-nonu. Similarly, co-expression of the RAF9S377A and RAF9S384A, the non-phosphorylatable mutations of Ser377 and Ser384 residues, could also partially rescue the ABA-induced expression of RD29B-LUC in the protoplasts of OK100-nonu mutant (Fig. 5C). Interestingly, co-expression of RAF9Y383F, a non-phosphorylatable mutation form of RAF9, cannot rescue the ABA-induced expression of RD29B-LUC in the protoplasts of OK100-nonu mutant (Fig. 5C). As the tyrosine residue is also existing in RAF3 and RAF11, two other B2 and B3 RAFs, we also introduced non-phosphorylatable mutation Tyr (Y) to Pho (F) in RAF3 and RAF11 and tested the activity of RAF3Y525F and RAF11Y425F mutant forms in the protoplasts of OK100-nonu (Fig. 5D). The result showed that the phosphorylation of Tyr525 in RAF3 and Tyr425 in RAF11 is not essential for their activity. Thus, the Tyr383 is a unique phosphosite that is essential for RAF9 function in vivo. Additionally, the phosphorylation of Ser377 and Ser384 is also required for the full activation of RAF9 in vivo.

Tyr383 phosphorylation regulates RAF9 activity. A The relative abundance of RAF9 phosphopeptide in the wild type and aph-1 mutant without or with ABA treatment. The tyrosine phosphosite in the phosphopeptide are highlighted in red. Error bars, SD (n = 3). B Sequence alignment of the peptides in Arabidopsis B2 and B3 RAFs and PpARK/PpCTR1 from Physcomitrella patens. The phosphosites showed in (A) are highlighted in yellow. The conserved pTyr in RAF3 and RAF11 are highlighted in grey. C Activation of the reporter gene by wild type and the non-phosphorylatable mutants of Ser377, Tyr383, and Ser384 in RAF9 in the transient assay in the protoplasts of OK100-nonu. Error bars, SEM (n = 3 individual transfections). Fold change (FC) and p-value (Student’s t-test) are indicated. D Activation of the reporter gene by RAF3WT, RAF3Y525F, RAF9WT, RAF9Y383F, RAF11WT, and RAF11Y425F, in the transient assay in the protoplasts of OK100-nonu. Error bars, SEM (n = 3 individual transfections). Asterisk indicates significant differences between wild-type and mutated RAFs (Student’s t-test; * p < 0.05). E A proposed model of APH-mediated tyrosine phosphorylation in the ABA signaling
Discussion
Although tyrosine phosphorylation only represents a small proportion in total protein phosphorylation, emerging evidence supports a vital role of tyrosine phosphorylation in diverse processes, including immunity, growth/development, and abiotic stress responses in plants (Champion et al., 2004; de la Fuente van Bentem & Hirt, 2009; Macho et al., 2015; Zulawski & Schulze, 2015; Liu et al., 2018). It has been known that tyrosine phosphorylation may have some potential roles in ABA signaling. Application of phenylarsine oxide, an inhibitor of PTP, abolishes ABA-induced stomatal closure (MacRobbie, 2002), and regulates ABA-mediated post-germination development (Reyes et al., 2006). Several dual-specific PTPases, like IBR5 (Monroe-Augustus et al., 2003), PHS1 (Quettier et al., 2006), and AtDsPTP1 (Liu et al., 2015), are known to be involved in ABA signaling. Here we identified APH, a highly conserved tyrosine-specific protein phosphatase, functioning in ABA signaling (Fig. 5E). We demonstrate that APH represses the ABA-mediated protein tyrosine phosphorylation (Fig. 3). Several proteins involved in different mRNA processing steps, including transcription, mRNA splicing, and mRNA decay, undergo ABA-induced tyrosine phosphorylation in aph-1 mutant. Consistent with this observation, the basal expression of ABA-highly-induced genes is repressed in aph-1 mutant. APH may mediate ABA-responsive gense expression by dephosphorylating effectors in both transcriptional and post-transcriptional processes. Among putative targets of APH, SR45 has been known to be involved in cotyledon greening (Carvalho et al., 2010). Thus, we reason that the ABA-insensitivity phenotype of aph mutants in post-germination growth may be attributed at least in part to the dysfunction of SR45 (Fig. 2).
In addition to transcriptional and post-transcriptional regulation, APH may directly control the function of RAF9, a member of B2 and B3 RAF kinases. Recent studies suggested B2 and B3 subgroup RAFs are essential for ABA- and osmotic stress-mediated SnRK2 activation in Arabidopsis and moss Physcomitrella patens (Saruhashi et al., 2015; Katsuta et al., 2020; Soma et al., 2020; Takahashi et al., 2020; Lin et al., 2021). The B2 and B3 have a basal level of activity, which is not induced by ABA (Takahashi et al., 2020; Lin et al., 2021). Such a basal-level activation of RAFs is sufficient to phosphorylate and activate SnRK2s, upon release from PP2C inhibition in the presence of ABA (Lin et al., 2021). In this study, we identified one pTyr site in RAF9, Tyr383, as a putative target site of APH (Fig. 5). The non-phosphorylatable Y383F mutation completely abolished RAF9 activity in the transient assay in the protoplasts. Our results imply that unidentified protein tyrosine kinase(s) may function as an activator to regulate RAF9 activity through phosphorylating RAF9 Tyr383 in ABA signaling. The unknown tyrosine kinase(s) and APH reciprocally coordinate the phosphorylation and function of RAF9. In the future, it would be of great interest to identify the protein kinase(s) that phosphorylate Tyr383 and to dissect how ABA regulates the enzyme activity of APH and unidentified kinase(s). Besides the RAF9 and some proteins involved in transcriptional and post-transcriptional regulation, we also identified dozens of proteins whose tyrosine phosphorylation are up-regulated in aph-1 (Table S1). These putative APH targets could be a valuable resource for further study of the function of tyrosine phosphorylation in plants.
Although we revealed a crucial role of APH in ABA signaling, how ABA regulates APH activity/function is unknown. The APH was originally co-immunoprecipitated with PYL1 protein. However, we failed to detect the physical interaction between APH and ABA receptor PYR1/PYL/RCAR proteins. In addition, ABA likely does not regulate the APH function through transcriptional regulation, as the expression of APH is only very slightly induced by ABA (Fig. S2D). The activity of animal LWM-PTP is regulated by tyrosine phosphorylation and redox regulation. It would be of interest to verify whether Arabidopsis APH also undergoes such post-translational modifications upon ABA signaling. It is also worth noting that the APH over-expression lines did not show any observable ABA phenotype (Fig. S2). That is likely because the APH activity is tightly controlled at the post-translational level, or some feedback machinery enhanced these kinases mediating ABA-responsive tyrosine phosphorylation. We also noticed T-DNA insertion in aph-1 resulted in a fused transcript of the T-DNA fragment and APH CDS (Fig. S1). Interestly, aph-1 showed a stronger phenotype than the “knock-out” mutant aph-3, the mechanism is worth to study in the future.
In this study, we used anti-pTyr antibody 4G10-based affinity purification to enrich the phosphopeptides containing tyrosine phosphosites. This method allows us to compare the tyrosine phosphorylation in wild type and aph-1 (Fig. 3B). However, the specificity and efficiency of this workflow need to be further improved. One of the reasons might be the very low concentration of pTyr in plants. Compared to about 90 typical tyrosine protein kinases in human kinome, no canonical tyrosine protein kinase was identified in Arabidopsis, even though Arabidopsis kinome contains more than 1100 protein kinases (Zulawski et al., 2014). In the future, more efficient and specific strategies, like Src homology 2 (SH2)-domain-derived pY superbinder (Bian et al., 2016), are expected to be helpful for in-depth analysis of tyrosine phosphorylation in plants (Ahsan et al., 2020).
Materials and methods
Protein expression, purification, and phosphatase assay
cDNA fragment encoding a truncated APH (aa 57–262) was cloned into pSUMO vector (LifeSensors). The resulting plasmids were transformed into E. coli BL21 (DE3) cells. The recombinant proteins were expressed and purified using standard protocols. The phosphatase activity was measured using the colorimetric substrate pNPP (Sigma-Aldrich). Reactions were performed in a buffer containing 150 mM NaCl, 2 mM DTT, 1 mM EDTA, 50 mM Tris-Cl/HEPES-NaOH, and the indicated pH from 5.5 to 8.5. Reactions were initiated by the addition of pNPP to a final concentration of 5 mM and terminated by adding 2 M NaOH. The hydrolysis of pNPP was measured by the absorbance reads at 405 nm (A405). The primers used for vector construction and site-directed mutagenesis was listed in Table S4.
For peptide dephosphorylation assay, 500 pmol of synthetic RAF9 peptide DKSHNRpYSGVK and SIZ1 peptide EGpSMDTTTETASLLLGMNDSR (GenScript) were incubated with 100 pmol recombinant in 100 μL buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 2 mM DTT, 1 mM EDTA) at 37 °C for 2 h. The reactions were quenched by acidifying with 10 μL 10% TFA. After desalted with Sep-Pak C18 tips, the peptides were dissolved in 20 μL 0.2% FA and subjected into LC-MS/MS analysis.
Phenotyping of germination and post-germination growth
Seeds were surface-sterilized for 10 min in 70% ethyl alcohol, and then rinsed four times in sterile-deionized water. For germination assays, sterilized seeds were grown on medium containing 1/2 MS nutrients, pH 5.7, with or without the indicated concentration of ABA and kept at 4 °C for 3 days. Radicle emergence and green cotyledons were analyzed after placing the plates at 23 °C under a 16 h light/8 h dark photoperiod. Photographs of seedlings were taken at indicated times after transfer to light.
Water loss measurement
To measure water loss, detached rosette leaves of 4-week-old plants were placed in weighing dishes and left on the laboratory bench with light. Fresh weight was monitored at the indicated times. Water loss was expressed as a percentage of initial fresh weight.
Protein extraction and digestion
Protein extraction and digestion were performed as previously described (Hsu et al., 2018; Wang et al., 2018). Plants were lysed in lysis buffer (6 M guanidine hydrochloride in 100 mM Tris-HCl, pH 8.5), with 10 mM NaF, EDTA-free protease, and phosphatase inhibitor cocktails (Sigma-Aldrich). Protein lysate was precipitated using the methanol-chloroform precipitation method. Precipitated protein pellets were suspended in digestion buffer (12 mM sodium deoxycholate and 12 mM sodium lauroyl sarcosinate in 100 mM Tris-Cl, pH 8.5) and then were 5-fold diluted. The protein amount was quantified using the BCA assay (ThermoFisher). One mg of protein was then digested with Lys-C and trypsin overnight. The detergents were separated from digested peptides by acidifying using 10% TFA and then centrifuged at 16,000 g for 20 min. The digests were then desalted using a 100 mg SEP-PAK C18 cartridge (Waters, Milford, MA).
4G10 antibody affinity enrichment
The anti-phosphotyrosine antibody, clone 4G10 (Merck Millipore) was used to enrich the peptides containing phosphotyrosines. About 200 μg tryptic peptides were incubated with 20 μg 4G10 antibody coated on 20 μg Dynabeads (ThermoFisher) at 4 °C overnight, in incubating buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA) with phosphatase inhibitor cocktail 3 (Sigma-Aldrich). After washing with washing buffer (50 mM Tris-Cl, pH 7.5, 150 mM NaCl, 0.5 mM EDTA) for 5 times. The bound peptides were eluted with 150 μL of 0.1% TFA, then desalted using a C18 beads StageTip. The Tyr phosphopeptides were dried using a SpeedVac.
LC-MS/MS analysis
The phosphopeptides were dissolved in 5 μL of 0.25% formic acid (FA) and injected into an Easy-nLC 1000 (Thermo Fisher Scientific). Peptides were separated on a 45 cm in-house packed column (360 μm OD × 75 μm ID) containing C18 resin (2.2 μm, 100 Å, Michrom Bioresources). The mobile phase buffer consisted of 0.1% FA in ultra-pure water (Buffer A) with an eluting buffer of 0.1% FA in 80% ACN (Buffer B) run over a linear 90 min gradient of 6%–30% buffer B at flow rate of 250 nL/min. The Easy-nLC 1000 was coupled online with a Orbitrap Fusion mass spectrometer (ThermoFisher). The mass spectrometer was operated in the data-dependent mode in which a full-scan MS (from m/z 350–1500 with the resolution of 60,000 at m/z 400) was followed by top 10 higher-energy collision dissociation (HCD) MS/MS scans of the most abundant ions with the dynamic exclusion for 60 s and exclusion list of 500. The normalized collision energy applied for HCD was 40% for 10 ms activation time.
Proteomics data search
The raw files were searched directly against the Arabidopsis thaliana database (TAIR10 with 35,386 entries) with no redundant entries using MaxQuant software (version 1.5.4.1) with reporter ion MS2 type. Peptide precursor mass tolerance was set at 20 ppm, and MS/MS tolerance was set at 20 ppm. Search criteria included a static carbamidomethylation of cysteines (+ 57.0214 Da) and variable modifications of (1) oxidation (+ 15.9949 Da) on methionine residues, (2) acetylation (+ 42.011 Da) at N-terminus of protein, and (3) phosphorylation (+ 79.996 Da) on serine, threonine or tyrosine residues were searched. The search was performed with full tryptic digestion and allowed a maximum of two missed cleavages on the peptides analyzed from the sequence database. The false discovery rates of proteins, peptides and phosphosites were set at 1% FDR. The phosphorylation sites induced by ABA treatment in Col-0 and aph-1 mutant plants were selected using Perseus software (version 1.6.2.1). The intensities of phosphorylation sites were log2 transformed, and the quantifiable phosphorylation sites were selected from the identification in at least two replicates in Col-0 and aph-1 mutant plants. For hierarchical clustering, the intensities of the pTyr phosphorylation sites were first z-scored and clustered using Euclidean as a distance measure for row clustering. The number of clusters was set at 250, with a maximum of 10 iterations and 1 restart. The protein annotation search was performed using the PANTHER database, and enrichment of cellular component was performed using Fisher’s exact test with a cut-off of p-value < 0.05. Protein network analysis was performed in the String database (version 11.0) with medium confident score (> = 0.4) as the cut-off, and protein-protein interaction networks were visualized using Cytoscape (version 3.8.2).
RNA sequencing and data analysis
Total RNA was isolated from two-week-old seedlings of Col-0 and aph-1 mutant, with and without ABA treatment, using RNeasy Plant Mini Kit (Qiagen). Total RNA (1 μg) was used for library preparation with NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England BioLabs, E7765) following the manufacturer’s instructions. Prepared libraries were assessed for fragment size using NGS High-Sensitivity kit on a Fragment Analyzer (AATI), and for quantity using Qubit 2.0 fluorometer (Thermo Fisher Scientific) and KAPA Library Quantification Kit (Kapa, KK4824). All libraries were sequenced in paired-end 150 bases protocol (PE150) on an Illumina Nova sequencer.
The paired-end reads were cleaned by Trimimomatic (Bolger et al., 2014) (version 0.36). After trimming the adapter sequence, removing low quality bases, and filtering short reads, clear read pairs were retained for further analysis. The Arabidopsis thaliana reference genome sequence was downloaded from TAIR10. Clean reads were mapped to the genome sequence by HISAT (Kim et al., 2015) with default parameters. Number of reads that were mapped to each gene was calculated with the htseq-count script in HTSeq (Anders et al., 2015). EdgeR (Robinson et al., 2010) was used to identify genes that were differentially expressed. Genes with at least three-fold change in expression with an FDR < 0.05 were considered differentially expressed genes (DEGs).
Analysis of gene expression by qRT-PCR
Total RNA was extracted from wild type and aph-1 seedlings treated with 50 mM ABA for 6 h, and isolated using the RNeasy Plant Mini Kit (QIAGEN) according to the manufacturer’s instructions. For real-time PCR assays, reactions were set up with iQ SYBR Green Supermix (Bio-Rad). A CFX96 Touch Real-Time PCR Detection System (Bio-Rad) was used to detect amplification levels. Quantification was performed using three independent biological replicates.
Protoplast isolation and transactivation assay
Protoplast isolation and transactivation assays were performed as previously described (Wang et al., 2018). Briefly, protoplasts were isolated from leaves of 4-week-old plants grown under a short photoperiod (10 h light at 23 °C/14 h dark at 20 °C). The RD29B-LUC (7 μg of plasmid per transfection) and ZmUBQ-GUS (1 μg per transfection), wild type and mutated RAFs (3 μg of plasmid per transfection) were used as an ABA-responsive reporter gene, internal control, and effectors, respectively. After transfection, protoplasts were incubated for 5 h under light in washing and incubation solution (0.5 M mannitol, 20 mM KCl, 4 mM MES, pH 5.7) with or without 5 μM ABA. The primers used form site-direct mutagenesis are listed in Table S4.
Availability of data and materials
The RNA sequencing data were deposited to the GEO database with the dataset identifier GSE186664. The phosphoproteomic data were deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD029650. The materials used in this study will be available for research upon request.
Abbreviations
- LWM-PTP:
-
Low molecular weight protein tyrosine phosphatase.
- EGF:
-
Epidermal growth factor
- PTKs:
-
Protein tyrosine kinases (PTKs)
- PTPs:
-
Protein tyrosine phosphatases
- ACP:
-
Acid Phosphatase
- APH:
-
Arabidopsis ACP homolog
- SIZ1:
-
SAP and Miz
- GO:
-
Gene ontology
- SR45:
-
ARGININE/SERINE-RICH 45
- SCL30A:
-
SC35-LIKE SPLICING FACTOR 30A
- RSZ22A:
-
RS-CONTAINING ZINC FINGER PROTEIN 22A
- PRP4KA:
-
PRP4 KINASE A
- LUC:
-
LUCIFERASE
- RD29B:
-
RESPONSIVE TO DESICCATION 29B
- RAB18:
-
RESPONSIVE TO ABA 18
- LEA7:
-
LATE EMBRYOGENESIS ABUNDANT 7
- COR15A:
-
Cold-regulated 15A
- HAI2:
-
HIGHLY ABA-INDUCED PP2C GENE 2
- OK:
-
Osmotic stress activated protein kinase
- IBR5:
-
INDOLE-3-BUTYRIC ACID RESPONSE 5
- PHS1:
-
PROPYZAMIDE-HYPERSENSITIVE 1
- SnRK2:
-
Sucrose nonfermenting 1 (SNF1)-related protein kinase 2 s
- PYR1/PYL/RCAR:
-
PYRABACTIN RESISTANCE1/PYR1-LIKE/REGULATORY COMPONENTS OF ABA RECEPTORS
- SH2:
-
Src homology 2
- BCA:
-
Bicinchoninic acid
- EDTA:
-
Ethylene Diamine Tetraacetic Acid
- TFA:
-
Trifluoroacetic acid
- FA:
-
Formic acid
- ACN:
-
Acetonitrile
- MS:
-
Mass Spectrometry
- HCD:
-
Higher-energy collision dissociation
- FDR:
-
False discovery rate
- DEGs:
-
Differentially expressed genes
References
Ahsan N, Wilson RS, Rao RSP, Salvato F, Sabila M, Ullah H et al (2020) Mass spectrometry-based identification of phospho-Tyr in plant proteomics. J Proteome Res 19(2):561–71. https://doi.org/10.1021/acs.jproteome.9b00550
Alho I, Costa L, Bicho M, Coelho C (2013) The role of low-molecular-weight protein tyrosine phosphatase (LMW-PTP ACP1) in oncogenesis. Tumour Biol 34(4):1979–89. https://doi.org/10.1007/s13277-013-0784-1
Al-Momani S, Qi D, Ren Z, Jones AR (2018) Comparative qualitative phosphoproteomics analysis identifies shared phosphorylation motifs and associated biological processes in evolutionary divergent plants. J Proteom 181:152–9. https://doi.org/10.1016/j.jprot.2018.04.011
Anders S, Pyl PT, Huber W (2015) HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31(2):166–9. https://doi.org/10.1093/bioinformatics/btu638
Bartels S, Anderson JC, González Besteiro MA, Carreri A, Hirt H, Buchala A et al (2009) MAP kinase phosphatase1 and protein tyrosine phosphatase1 are repressors of salicylic acid synthesis and SNC1-mediated responses in Arabidopsis. Plant Cell 21(9):2884–97. https://doi.org/10.1105/tpc.109.067678
Bian Y, Li L, Dong M, Liu X, Kaneko T, Cheng K et al (2016) Ultra-deep tyrosine phosphoproteomics enabled by a phosphotyrosine superbinder. Nat Chem Biol 12(11):959–66. https://doi.org/10.1038/nchembio.2178
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–20. https://doi.org/10.1093/bioinformatics/btu170
Carvalho RF, Carvalho SD, Duque P (2010) The plant-specific SR45 protein negatively regulates glucose and ABA signaling during early seedling development in Arabidopsis. Plant Physiol 154(2):772–83. https://doi.org/10.1104/pp.110.155523
Champion A, Kreis M, Mockaitis K, Picaud A, Henry Y (2004) Arabidopsis kinome: after the casting. Funct Integr Genomics 4(3):163–87. https://doi.org/10.1007/s10142-003-0096-4
de la Fuente van Bentem S, Hirt H (2009) Protein tyrosine phosphorylation in plants: more abundant than expected? Trends Plant Sci 14(2):71–6. https://doi.org/10.1016/j.tplants.2008.11.003
Edelman AM, Blumenthal DK, Krebs EG (1987) Protein serine/threonine kinases. Ann Rev Biochem 56(1):567–613. https://doi.org/10.1146/annurev.bi.56.070187.003031
Faria AVS, Fonseca EMB, Cordeiro HG, Clerici SP, Ferreira-Halder CV (2021) Low molecular weight protein tyrosine phosphatase as signaling hub of cancer hallmarks. Cell Mol Life Sci 78(4):1263–73. https://doi.org/10.1007/s00018-020-03657-x
Hsu C-C, Zhu Y, Arrington JV, Paez JS, Wang P, Zhu P et al (2018) Universal plant phosphoproteomics workflow and its application to tomato signaling in response to cold stress*. Mol Cellu Proteom 17(10):2068–80. https://doi.org/10.1074/mcp.TIR118.000702
Jaillais Y, Hothorn M, Belkhadir Y, Dabi T, Nimchuk ZL, Meyerowitz EM et al (2011) Tyrosine phosphorylation controls brassinosteroid receptor activation by triggering membrane release of its kinase inhibitor. Genes Dev 25(3):232–7. https://doi.org/10.1101/gad.2001911
Kameyama K, Kishi Y, Yoshimura M, Kanzawa N, Sameshima M, Tsuchiya T (2000) Tyrosine phosphorylation in plant bending. Nature 407(6800):37. https://doi.org/10.1038/35024149
Kanno T, Venhuizen P, Wen T-N, Lin W-D, Chiou P, Kalyna M et al (2018) PRP4KA, a putative spliceosomal protein kinase, is important for alternative splicing and development in Arabidopsis thaliana. Genetics 210(4):1267–85. https://doi.org/10.1534/genetics.118.301515
Katsuta S, Masuda G, Bak H, Shinozawa A, Kamiyama Y, Umezawa T et al (2020) Arabidopsis Raf-like kinases act as positive regulators of subclass III SnRK2 in osmostress signaling. Plant J 103(2):634–44. https://doi.org/10.1111/tpj.14756
Kerk D, Templeton G, Moorhead GBG (2008) Evolutionary radiation pattern of novel protein phosphatases revealed by analysis of protein data from the completely sequenced genomes of humans, green algae, and higher plants. Plant Physiol 146(2):351–67. https://doi.org/10.1104/pp.107.111393
Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12(4):357–60. https://doi.org/10.1038/nmeth.3317
Kim T-W, Guan S, Sun Y, Deng Z, Tang W, Shang J-X et al (2009) Brassinosteroid signal transduction from cell-surface receptor kinases to nuclear transcription factors. Nat Cell Biol 11(10):1254–60. https://doi.org/10.1038/ncb1970
Li B, Tunc-Ozdemir M, Urano D, Jia H, Werth EG, Mowrey DD et al (2018) Tyrosine phosphorylation switching of a G protein. J Biol Chem 293(13):4752–66. https://doi.org/10.1074/jbc.RA117.000163
Lin Z, Li Y, Wang Y, Liu X, Ma L, Zhang Z et al (2021) Initiation and amplification of SnRK2 activation in abscisic acid signaling. Nat Commun 12(1):2456. https://doi.org/10.1038/s41467-021-22812-x
Liu J, Liu B, Chen S, Gong BQ, Chen L, Zhou Q et al (2018) A tyrosine phosphorylation cycle regulates fungal activation of a plant receptor Ser/Thr kinase. Cell Host Microbe 23(2):241-253.e246. https://doi.org/10.1016/j.chom.2017.12.005
Liu R, Liu Y, Ye N, Zhu G, Chen M, Jia L et al (2015) AtDsPTP1 acts as a negative regulator in osmotic stress signalling during Arabidopsis seed germination and seedling establishment. J Exp Bot 66(5):1339–53. https://doi.org/10.1093/jxb/eru484
Luo X, Wu W, Liang Y, Xu N, Wang Z, Zou H et al (2020) Tyrosine phosphorylation of the lectin receptor-like kinase LORE regulates plant immunity. EMBO J 39(4):e102856. https://doi.org/10.15252/embj.2019102856
Macho AP, Lozano-Durán R, Zipfel C (2015) Importance of tyrosine phosphorylation in receptor kinase complexes. Trends Plant Sci 20(5):269–72. https://doi.org/10.1016/j.tplants.2015.02.005
MacRobbie EAC (2002) Evidence for a role for protein tyrosine phosphatase in the control of ion release from the guard cell vacuole in stomatal closure. Proceed Natl Acad Sci 99(18):11963–8. https://doi.org/10.1073/pnas.172360399
Mergner J, Frejno M, List M, Papacek M, Chen X, Chaudhary A et al (2020) Mass-spectrometry-based draft of the Arabidopsis proteome. Nature 579(7799):409–14. https://doi.org/10.1038/s41586-020-2094-2
Monroe-Augustus M, Zolman BK, Bartel B (2003) IBR5, a dual-specificity phosphatase-like protein modulating auxin and abscisic acid responsiveness in arabidopsis. Plant Cell Online 15(12):2979–91. https://doi.org/10.1105/tpc.017046
Moorhead GB, De Wever V, Templeton G, Kerk D (2009) Evolution of protein phosphatases in plants and animals. Biochem J 417(2):401–9. https://doi.org/10.1042/BJ20081986
Nito K, Wong CC, Yates JR 3rd, Chory J (2013) Tyrosine phosphorylation regulates the activity of phytochrome photoreceptors. Cell Rep 3(6):1970–9. https://doi.org/10.1016/j.celrep.2013.05.006
Quettier A-L, Bertrand C, Habricot Y, Miginiac E, Agnes C, Jeannette E et al (2006) The phs1–3 mutation in a putative dual-specificity protein tyrosine phosphatase gene provokes hypersensitive responses to abscisic acid in Arabidopsis thaliana. Plant J 47(5):711–9. https://doi.org/10.1111/j.1365-313X.2006.02823.x
Reyes D, Rodríguez D, Nicolás G, Nicolás C (2006) Evidence of a role for tyrosine dephosphorylation in the control of postgermination arrest of development by abscisic acid in Arabidopsis thaliana L. Planta 223(2):381–5. https://doi.org/10.1007/s00425-005-0135-6
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26(1):139–40. https://doi.org/10.1093/bioinformatics/btp616
Saruhashi M, Kumar Ghosh T, Arai K, Ishizaki Y, Hagiwara K, Komatsu K et al (2015) Plant Raf-like kinase integrates abscisic acid and hyperosmotic stress signaling upstream of SNF1-related protein kinase2. Proceed Natl Acad Sci 112(46):E6388-96. https://doi.org/10.1073/pnas.1511238112
Soma F, Takahashi F, Suzuki T, Shinozaki K, Yamaguchi-Shinozaki K (2020) Plant Raf-like kinases regulate the mRNA population upstream of ABA-unresponsive SnRK2 kinases under drought stress. Nature Commun 11(1):1373. https://doi.org/10.1038/s41467-020-15239-3
Takahashi Y, Zhang J, Hsu P-K, Ceciliato PHO, Zhang L, Dubeaux G et al (2020) MAP3Kinase-dependent SnRK2-kinase activation is required for abscisic acid signal transduction and rapid osmotic stress response. Nature Commun 11(1):12. https://doi.org/10.1038/s41467-019-13875-y
Wang P, Zhao Y, Li Z, Hsu C-C, Liu X, Fu L et al (2018) Reciprocal regulation of the TOR kinase and ABA receptor balances plant growth and stress response. Molecular Cell 69(1):100-112.e106. https://doi.org/10.1016/j.molcel.2017.12.002
Zulawski M, Schulze G, Braginets R, Hartmann S, Schulze WX (2014) The Arabidopsis Kinome: phylogeny and evolutionary insights into functional diversification. BMC Genomics 15(1):548. https://doi.org/10.1186/1471-2164-15-548
Zulawski M, Schulze WX (2015) The plant kinome. Methods Mol Biol 1306:1–23. https://doi.org/10.1007/978-1-4939-2648-0_1
Acknowledgements
We thank Dr. Zhizhong Gong of China Agriculture University for helpful discussion.
Funding
This work was supported by the National Natural Science Foundation of China, Grant 31771358 (to PW), and National Insitutes of Health, Grant 3RF1AG064250 (to WAT).
Author information
Authors and Affiliations
Contributions
PW designed the research; YD, YW, YM, XL, RL, and PW performed the research; YD, SX, RL, C-PS, WAT, and PW contributed towards data analysis; YD, XZ, WAT, and PW wrote the paper. The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
C-PS and PW are members of the editorial board but were not involved in the journal’s review, or any decisions, related to this submission.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Fig. S1.
The fused expression of a T-DNA fragment and AT3G44620 exons in aph-1 mutant. Fig. S2. Overexpression of APH does not affect ABA sensitivity in APH-OE lines. Fig. S3. Quantitative comparison of tyrosine phosphorylation in wild type and aph-1.
Additional file 2: Table S1.
The tyrosine phosphosites identified in wild type and aph-1 mutant, without or with ABA treatment.
Additional file 3: Table S2.
The DEG in wild type and aph-1 mutant, with or without ABA treatment.
Additional file 4: Table S3.
The GO analysis of the down-regulated genes in aph-1 mutant.
Additional file 5: Table S4.
Sequences of primers used in this study.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Du, Y., Xie, S., Wang, Y. et al. Low molecular weight protein phosphatase APH mediates tyrosine dephosphorylation and ABA response in Arabidopsis.. Stress Biology 2, 23 (2022). https://doi.org/10.1007/s44154-022-00041-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44154-022-00041-6