
Abstract
Outbreak of COVID-19 by SARS-CoV-2 infection caused severe acute respiratory syndrome that has been declared a public health emergency of international concern. To control infections, there is urgent need to develop an effective therapeutic strategy. COVID-19 viral spike glycoprotein and proteases play major role in viral entry and mediating virus replication and spread and thus can serve as potential antiviral drug target. Being highly specific, efficacious and safe, peptides hold their place in therapeutics. In present study, molecular docking of 21 pharmacologically active non ribosomal peptides (NRPs) from marine microbes with SARS-CoV-2 spike glycoprotein and papain such as protease was done. Dactinomycin, Tyrocidine A and Gramicidin S showed highest binding interaction with target proteins. The binding affinity of Dactinomycin and Gramicidin S docked with SARS-CoV-2 spike glycoprotein was − 12.4 kcal/mol and − 11.4 kcal/mol, respectively. This suggested their potential to destabilize SARS spike protein binding with human host ACE2 receptor and thus hindering viral entry to the cells. Binding affinity of Tyrocidine A and Gramicidin S with SARS-CoV-2 papain-like protease was − 13.1 kcal/mol and − 11.4 kcal/mol, respectively which might be inhibited COVID-19 by acting on the protease. Gramicidin S showed same binding affinity for both target proteins and thus expected to be most potent. Based on the binding energy score, it was suggested that these pharmacologically active NRPs are potential molecules to be tested against SARS-CoV-2 and used to develop effective antiviral drugs.
Similar content being viewed by others


Introduction
Recent outbreak of corona global pandemic has posed serious threat to humans across the world. The disease first commenced in Wuhan, China in December 2019 (Kim et al. 2020) that later spreaded worldwide with 30,675,675 confirmed cases and approximately 3,992,680 deaths till date (WHO report). COVID-19 is an infectious disease caused by novel β-coronavirus having novel enveloped RNA and phylogenetic similarity to SARS-CoV. It was also named as coronavirus 2 (SARS-CoV-2) (Owis et al. 2020). The affected person/patient manifests the symptoms of fever, fatigue, dry cough, muscle ache, difficulty in breathing, shortness of breath and in some cases pneumonia. In serious conditions, it causes severe acute respiratory syndrome resulting in death due to massive alveolar damage and respiratory failure (Shah et al. 2020; Owis et al. 2020). Symptoms and severity of this disease vary from patient to patient. Elderly people, children (below 6 years), immunocompromised patients and patients with past medical history of comorbidities viz. asthma, diabetes and cardiovascular diseases are reportedly more vulnerable to this disease (Shah et al. 2020).
Reports are available for certain drugs, i.e., oseltamivir, lopinavir, ritonavir, remdesivir, favipiravir, ribavirin, chloroquine and hydroxychloroquine to have potency against COVID-19 (Narkhede et al. 2020), but these drugs were not effective in patients with comorbidities and immunocompromised state (Peele et al. 2020). Therefore, researchers have been continuously working to find novel effective drugs to combat infection. Inhibiting attachment of viral particles to host cells is one of the effective strategy to prevent viral infection. COVID-19 is a viral disease caused by Coronaviruses that belong to Coronaviridae family, the largest family of RNA viruses having long RNA strands as genome (Chen et al. 2020; Peele et al. 2020). Spike (S), Membrane (M), Envelope (E) and nucleocapsid (N) are major structural proteins of coronaviruses. These proteins promote replication, transmission, host cell interaction of the virus and they can act as major virulent factors (Skariyachan et al. 2020). S-glycoprotein is a fusion viral protein comprising two subunits S1 and S2. It facilitates the entry of viral particles into the host cell after binding with the host angiotensin-converting enzyme 2 (ACE-2) receptor and also determines the host range and cellular tropism. This highly conserved S-glycoprotein of SARS-CoV-2 essential for its entry in human cells to start the infection process and thus one of the major drug target to neutralize the virus (Hiremath et al. 2021). Similarly, the main protease (Mpro), the papain-like protease (PLpro) and RNA-dependent RNA polymerase (RdRp) played crucial role in coronavirus replication and targeting one or multiple of these proteins may lead to potential treatment for COVID-19 (Hosseini et al. 2021). PLpro is reportedly involved in viral polyprotein processing and contributes to biogenesis of virus replication complex (Lindner et al. 2005). It also participate in stripping ubiquitin and ISG15 from host-cell proteins to aid evasion of coronaviruses from host’s innate immune response. Drugs targeting the PLpro might have an advantage in inhibiting viral replication and inhibiting dysregulation of signaling cascades in infected cells that might lead to cell death of surrounding uninfected cells (Báez-Santos et al. 2015). Therefore, inhibition of PLpro activity can prevent virus replication and destroy its role in host immune response evasion, making it a promising target for developing new antiviral drugs.
Compounds from different natural sources need to be evaluated for development of therapeutics against their biological targets. NRPs are secondary bioactive metabolites synthesized by a multi-modular enzyme complex known as non-ribosomal peptide synthetases (NRPSs) found only in bacteria, fungi and cyanobacteria. NRPs exhibit broad spectrum of biological activities ranging from antimicrobial to anticancer (Agrawal et al. 2017). Studies revealed that several NRPs from marine microbes also possessed promising antiviral effects (Agrawal et al. 2017; Vinothkumar and Parameswaran 2013). More than 50% of the drug in clinical use till date either belong to NRPs or mixed polyketide–NRP family of natural products. Moreover, screening of large number of NRPs with antiviral activity against novel coronavirus will be a challenge in very short period. Docking is fast and cost effective approach to select or prioritize potential drug-like molecules for experimental studies. Therefore, the objective of the current study was to find out which NRP can potentially inhibit these viral targets and thus molecular docking of NRPs of marine microbes with S-glycoprotein and PLpro of SARS-CoV-2 was done.
Materials and methods
Receptor preparation
Based on the literature survey, the proteins, which played a major role in the pathogenicity of SARS-CoV-2 were selected as molecular targets. Pre-fusion spike glycoprotein with single receptor binding domain (PDB: 6VSB; electron microscopy, resolution 3.46 Å) was selected as it plays role in viral entry inside the host. This makes it a key target for developing therapy against novel coronavirus (Wrapp et al. 2020). PLpro (PDB: 6W9C; X-ray crystal structure, resolution 2.7 Å) (Osipiuk et al. 2020) is an attractive target as it is known to play essential role in cleavage and maturation of viral polyproteins, assembly of the replicase–transcriptase complex and disruption of host responses (Osipiuk et al. 2021). The three-dimensional crystal structures of spike glycoprotein and papain-like protease from SARS-CoV-2 were downloaded from Research Collaboratory Structural Bioinformatics-Protein Data Bank (RCSB-PDB) (Berman et al. 2000) using PDB IDs of 6VSB and 6W9C, respectively. Models were cleaned by excluding water molecules and heteroatoms.
Ligands preparation
Twenty-one NRPs from marine microbes were selected for virtual screening (Agrawal et al. 2017; Khalifa et al. 2019). Ligands library was prepared manually by downloading NRPs in structure data file (SDF) format from Pubchem database. The drug likeliness of all the ligands was assessed by the Lipinski’s rule of five viz. molecular weight < 500 Da, having up to 5 H-bond donors and less than 10 H-bond acceptors and logP (octanol–water partition coefficient) should not be more than 5. The potential SARS-CoV-2 inhibitor hydroxychloroquine was used as positive control for comparative studies (Alazmi & Motwalli 2020).
Molecular docking of ligands with proteins
PyRx v0.8 (https://pyrx.sourceforge.io/downloads) software was used for virtual screening and molecular docking. It utilizes AutoDock Vina (Trott and Olson 2010) program for molecular docking between protein and ligands. Screened ligands were energy minimized by applying Universal Force Field (UFF) and converted into.pdbqt by OpenBabel (O'Boyle et al. 2011). All docking studies were performed as blind docking (i.e., grid box large enough to cover whole protein structure to encounter any possible protein–ligand interaction) to screen against ligand library using exhaustiveness value of 10 so to increase the probability of detecting global minimum of scoring function inbuilt in AutoDock Vina. Molecules binding with high affinity were analyzed using Discovery Studio Visualizer 4.0 and PyMOL.
Results and discussion
Rapid spread of novel coronavirus across the world necessitated the search of potential therapeutics to control this fatal infection. There is an urgent need to identify effective drugs with lower side effects to fight against SARS-CoV-2. Scientists across the world are investigating for cure/drug against COVID-19. Computer-aided virtual screening is cost effective and it takes relatively less time to reach the drug to the market. In silico studies suggested mechanism of binding of lead molecules to the targets of SARS-CoV-2 (Skariyachan et al. 2020). As S-glycoproteins plays important role in viral entry and PLpro enzyme is essentially required for processing of viral polyproteins to facilitate the viral spread thus evaluation of potential drug molecule from natural sources (marine microbes). S-glycoproteins and proteases is considered the effective therapeutic strategy to control virus entry and its spread in humans.
Natural products are considered the most important resource for drug research and development especially for infectious diseases (Alrasheid et al. 2021). Natural products of microbial origin reported to be unique with respect to their chemical diversity and 53% of FDA-approved natural product based drugs, including antiviral agent are of microbial origin, (Sayed et al. 2020). Therefore, molecular docking study of 21 potential NRPs from marine microbes was studied. All these molecules were docked against the target protein and ranked based on their dock score.
Screening and docking results
Molecular docking studies showed strong interaction of 21 potential ligands with SARS-CoV-2 S-glycoprotein and papain-like protease in different modes of ligand–protein interactions with particular binding affinity (docking score) is summarized in Table 1. Docked model with least binding energy and highest binding affinity indicated the most stable binding for ligand with target protein. Ligands with appreciable binding affinities were selected. Visualization of structures with high dock score in PyMOL and Discovery Studio 4.0 revealed specific amino acids involved in ligand–protein binding. The S-glycoprotein (PDB IDs 6VSB) and papain-like protease (PDB IDs 6W9C) from SARS-CoV-2 contained three chains A, B and C that might participate in interaction with ligand.
Hydroxychloroquine was used as control and its interaction with target proteins (S-glycoprotein and PLpro) was studied by docking. Binding affinity for SARS-CoV-2 S-glycoprotein-Hydroxychloroquine complex was estimated to be − 6.0 kcal/mol. Hydroxychloroquine interacted with S-glycoprotein through H-bonds with Gln954 of A-chain and through alkyl and π-alkyl interactions with B-chain. Van der Waal interactions were observed between ligand and Gln1010 (of A-chain) and Arg765 (of B-chain) of S-protein (Fig. 1A). Blind docking studies of viral spike protein with hydroxychloroquine revealed considerable binding through hydrogen bonding between only one amino acid (Ile472) of the viral spike protein with binding affinities of − 6.0 kcal/mole (Nimgampalle et al. 2020). Binding affinity for SARS-CoV-2 S-glycoprotein–hydroxychloroquine complex was − 5.2 kcal/mol and Ser730, Thr778, Pro863, Thr866, Asp867, Glu868 and His1058 were reported to be interacting residues (Skariyachan et al. 2020). The docking of Hydroxychloroquine with SARS-CoV-2 PLpro showed significant interactions in the central pocket near chains A and C with binding affinity of − 6.3 kcal/mol. The major interaction of the ligand was through H-bonds with Gly160 of chain A and Gln269 residue of chain C of protease (Fig. 1B). Binding affinity for SARS-CoV-2 S-glycoprotein–hydroxychloroquine complex was − 5.0 kcal/mol with Tyr213, Glu214, Lys217, Tyr305, Tyr310 and Thr313 identified as interacting residues and Thr313 possessed hydrogen bond with Hydroxychloroquine was reported (Skariyachan et al. 2020).

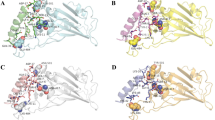
Docking of Hydroxychloroquine with S-glycoprotein (A) and papain-like protease (B) of SARS-CoV-2. Best binding mode in the pocket of protein and binding interaction of ligands with amino acids for Hydroxychloroquine
Out of 21 screened ligands, compounds having binding affinity higher than that of hydroxychloroquine were selected. Dactinomycin and Gramicidin S showed highest binding affinities, i.e., − 12.4 kcal/mol and − 11.4 kcal/mol, respectively, for SARS-CoV-2 S-glycoprotein. Tyrocidine A and Gramicidin S showed highest binding affinities with − 13.1 kcal/mol and − 11.4 kcal/mol, respectively, for SARS-CoV-2 PLpro (Table 1). Gramicidin S showed same binding affinity for both target proteins. Selected NRPs were docked with target proteins and docked structures are shown in Figs. 1 and 2.

Docking of NRPs with S-glycoprotein of SARS-CoV-2. Best binding mode in the pocket of protein and binding interaction of ligands with amino acids for Dactinomycin (A) and Gramicidin S (B)
SARS-CoV-2 S-glycoprotein–Dactinomycin complex
The docking of SARS-CoV-2 S-glycoprotein–Dactinomycin complex showed significant binding affinity of − 12.4 kcal/mol. This ligand interacted through H-bonds with Thr207,302, Leu303 residue of chain B and Arg765 of C-chain. Dactinomycin also formed π-cation interactions Arg765 of C-chain (Fig. 2A).
SARS-CoV-2 S-glycoprotein–Gramicidin S complex
Binding affinity of docked SARS-CoV-2 S-glycoprotein–Gramicidin S complex was estimated to be − 11.4 kcal/mol. Significant binding was observed with chain A and B. Gramicidin S formed H-bond with Asp950 of chain A and Thr761 of chain B. π-anion and π-sigma interactions were formed with Glu309 of chain A and Thr761,768 residues of chain B. Gramicidin S formed Van der Waal bonds with Gln341 of chain A (Fig. 2B).
SARS-CoV-2 papain-like protease–Tyrocidine A complex
Docking of Tyrocidine A with SARS-CoV-2 protease showed its binding in pocket of chains B and C with binding affinity of − 13.1 kcal/mol. The ligand exhibited H-bonding with Lys105 of chain C. Tyrocidine A established π-sigma and π-alkyl interactions with Leu162 and Met208 of chain. It also showed π-cation interactions with Lys157, Asp164 and Glu167 residues of B-chain (Fig. 3A).

Docking of NRPs with papain-like protease of SARS-CoV-2. Best binding mode in the pocket of protein and binding interaction of ligands with amino acids for Tyrocidine A (A) and Gramicidin S (B)
SARS-CoV-2 papain-like protease–Gramicidin S complex
Docking of Gramicidin S with SARS-CoV-2 PLpro showed binding affinity of − 11.4 kcal/mol. Ligand formed four hydrogen bonds with Lys157, Glu161 residues of B-chain and Lys105, Asp108 residues of C-chain of SARS-CoV-2 protease. Lys157, Asp164, Glu167 residues of chain B of protease formed π-cation and anion interactions with ligand. Alkyl and π-alkyl interactions were observed with Met208 and Pro247 residues of B-chain and ligand (Fig. 3B).
Molecular docking studies presented in this communication suggested 3 candidate NRPs viz. Dactinomycin, Tyrocidine A and Gramicidin S as potential drugs with highest binding affinities for SARS-CoV-2 proteins. S-glycoprotein plays an important role in attachment of coronavirus on host cell surface via ACE-2 receptors. Binding studies of Dactinomycin and Gramicidin S with S-glycoprotein resulted in disruption of interaction of S-glycoprotein and ACE-2 resulting in loss of connection between viral S-glycoprotein and host’s ACE-2 receptor. Tyrocidine A and Gramicidin S showed high binding affinity for PLpro of SARS-CoV-2 thereby further suggesting that these peptides might be used for inhibiting key viral proteases which are required for viral replication in host indicated NRPs could be validated as drugs to treat COVID-19 infection. These NRPs drugs reportedly exhibited potent effects in different therapeutic clinical conditions (Khalifa et al. 2019; Fang et al. 2016). Dactinomycin, a RNA synthesis inhibitor, has already been approved for treatment of various types of cancer and for inhibition of growth of feline enteric coronaviruses. It acts synergistically with sirolimus (immunosuppressive drug) for treatment of human coronaviruses (Zhou et al. 2020). Tyrocidine A and Gramicidin S are pharmacologically active NRPs drugs being used in clinics because of their potent antibacterial effects (Agrawal et al. 2017; Fang et al. 2016).
Conclusion
COVID-19 is a pandemic that has become a challenge for the global scientific community. To overcome life threatening SARS-CoV-2 infection, there is continuous search of new drugs with antiviral effects. Molecular docking has become a promising tool for drug discovery and development. NRPs are promising therapeutic antiviral candidates that might be used against SARS-CoV-2 strain with minimal side effects. Molecular docking of 21 potential NRPs from marine microbes revealed different binding interaction of with target protein. Dactinomycin, Tyrocidine A and Gramicidin S showed favorable interactions with highest affinity with SARS-CoV-2 S-glycoprotein and protease. Molecular docking studies indicated that these peptides could be used to block SARS-CoV-2 S-glycoprotein and ACE-2 interaction and thus could be used as protease inhibitors. Furthermore, in-vitro and in-vivo studies of these NRPs are required before using these as drug to combat COVID-19 infection.
References
Agrawal S, Acharya D, Adholeya A, Barrow CJ, Deshmukh SK (2017) Nonribosomal peptides from marine microbes and their antimicrobial and anticancer potential. Front Pharmacol 8:828
Alazmi M, Motwalli O (2020) Molecular basis for drug repurposing to study the interface of the S protein in SARS-CoV-2 and human ACE2 through docking, characterization, and molecular dynamics for natural drug candidates. J Mol Model 26:338
Alrasheid AA, Babiker MY, Awad TA (2021) Evaluation of certain medicinal plants compounds as new potential inhibitors of novel corona virus (COVID-19) using molecular docking analysis. Silico Pharmacol 9:10
Báez-Santos YM, John SE, Mesecar AD (2015) The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res 115:21–38
Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE (2000) The protein data bank. Nucleic Acids Res 28:235–242
Chen B, Tian EK, He B, Tian L, Han R, Wang S, Xiang Q, Zhang S, El Arnaout T, Cheng W (2020) Overview of lethal human coronaviruses. Signal Transduct Target Ther 5:89
Fang WY, Dahiya R, Qin HL, Mourya R, Maharaj S (2016) Natural proline-rich cyclopolypeptides from marine organisms: chemistry, synthetic methodologies and biological status. Mar Drugs 14:194
Hiremath S, Kumar HDV, Nandan M, Mantesh M, Shankarappa KS, Venkataravanappa V, Basha CRJ, Reddy CNL (2021) In silico docking analysis revealed the potential of phytochemicals present in Phyllanthus amarus and Andrographis paniculata, used in Ayurveda medicine in inhibiting SARS-CoV-2. 3 Biotech 11:44
Hosseini M, Chen W, Xiao D, Wang C (2021) Computational molecular docking and virtual screening revealed promising SARS-CoV-2 drugs. Precis Clin Med 4(1):1–16
Khalifa SA, Elias N, Farag MA, Chen L, Saeed A, Hegazy ME, Moustafa MS, El-Wahed A, Al-Mousawi SM, Musharraf SG, Chang FR (2019) Marine natural products: a source of novel anticancer drugs. Mar Drugs 17:491
Kim J, Zhang J, Cha Y, Kolitz S, Funt J, Chong RE, Barrett S, Kusko R, Zeskind B, Kaufman H (2020) Advanced bioinformatics rapidly identifies existing therapeutics for patients with coronavirus disease-2019 (COVID-19). J Transl Med 18:257
Lindner HA, Fotouhi-Ardakani N, Lytvyn V, Lachance P, Sulea T, Ménard R (2005) The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J Virol 79:15199–15208
Narkhede RR, Cheke RS, Ambhore JP, Shinde SD (2020) The molecular docking study of potential drug candidates showing anti-COVID-19 activity by exploring of therapeutic targets of SARS-CoV-2. EJMO 4:185–195
Nimgampalle M, Devanathan V, Saxena A (2020) Screening of Chloroquine, Hydroxychloroquine and its derivatives for their binding affinity to multiple SARS-CoV-2 protein drug targets. J Biomol Struct Dyn 1–13. https://doi.org/10.1080/07391102.2020.1782265
O’Boyle NM, Banck M, James CA, Morley C, Vandermeersch T, Hutchison GR (2011) Open babel: an open chemical toolbox. J Cheminform 3:33
Osipiuk J, Azizi SA, Dvorkin S, Endres M, Jedrzejczak R, Jones KA, Kang S, Kathayat RS, Kim Y, Lisnyak VG, Maki SL, Nicolaescu V, Taylor CA, Tesar C, Zhang Y-A, Zhou Z, Randall G, Michalska K, Snyder SA, Dickinson BC, Joachimiak A (2021) Structure of papain-like protease from SARS-CoV-2 and its complexes with non-covalent inhibitors. Nat Commun 12:743
Osipiuk J, Jedrzejczak R, Tesar C, Endres M, Stols L, Babnigg G, Kim Y, Michalska K, Joachimiak A (2020) The crystal structure of papain-like protease of SARS CoV-2. RSCB PDB. https://doi.org/10.2210/pdb6W9C/pdb
Owis AI, El-Hawary MS, El Amir D, Aly OM, Abdelmohsen UR, Kamel MS (2020) Molecular docking reveals the potential of Salvadora persica flavonoids to inhibit COVID-19 virus main protease. RSC Adv 10:19570–19575
Peele KA, Chandrasai P, Srihansa T, Krupanidhi S, Sai AV, Babu DJ, Indira M, Reddy AR, Venkateswarulu TC (2020) Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: a computational study. Inform Med Unlocked 19:100345
Sayed AM, Alhadrami HA, El-Gendy AO, Shamikh YI, Belbahri L, Hassan HM, Abdelmohsen UR, Rateb ME (2020) Microbial natural products as potential inhibitors of SARS-CoV-2 main protease (Mpro). Microorganisms 8:970
Shah B, Modi P, Sagar SR (2020) In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci 252:117652
Skariyachan S, Gopal D, Chakrabarti S, Kempanna P, Uttarkar A, Muddebihalkar AG, Niranjan V (2020) Structural and molecular basis of the interaction mechanism of selected drugs towards multiple targets of SARS-CoV-2 by molecular docking and dynamic simulation studies-deciphering the scope of repurposed drugs. Comput Biol Med 126:104054
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem 31:455–461
Vinothkumar S, Parameswaran PS (2013) Recent advances in marine drug research. Biotechnol Adv 31:1826–1845
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367(6483):1260–1263
Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F (2020) Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov 6:14
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors Poonam Bansal, Raman Kumar, Jasbir Singh and Suman Dhanda declare that they have no conflict of interest.
Ethical approval
No human or animal samples were included in study, so no ethical clearance was required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bansal, P., Kumar, R., Singh, J. et al. In silico molecular docking of SARS-CoV-2 surface proteins with microbial non-ribosomal peptides: identification of potential drugs. J Proteins Proteom 12, 177–184 (2021). https://doi.org/10.1007/s42485-021-00072-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42485-021-00072-z