
Abstract
The World Health Organization has declared the rapidly spreading coronavirus to be a global pandemic. The FDA is yet to approve a vaccine for human novel coronavirus. Here, we developed a peptide-based vaccine and used high-throughput screening by molecular dynamics simulation to identify T-cell- and β-cell-recognized epitopes for producing specific antibodies against SARS-nCoV-2. We construct ~ 12 P′ antigenic epitope peptides to develop a more effective vaccine and identify specific antibodies. These epitope peptides selectively presented the best antigen presentation scores for both human pMHC class I and II alleles to develop a strong binding affinity. All antigens identified of SARS-nCoV-2 different proteins by each attached specific ~ 1–7 L linker adaptor were used to construct a broad single peripheral peptide vaccine. It is expected to be highly antigenic with a minimum allergic effect. As a result of these exciting outcomes, expressing a vaccine using the intimated peptide was highly promising and positive to be highly proposed as epitope-based peptide vaccine of specific antibody against SARS-nCoV-2 by initiating T cells and β-cells. An in vitro study for the proposed peptide-based vaccine is mostly recommended. Further clinical trials are required to check the efficacy of this vaccine.
Similar content being viewed by others


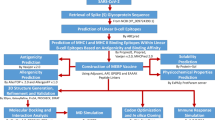
Introduction
Novel coronaviruses are large enveloped single-stranded RNA viruses (size ranging from ~ 26 to ~ 32 kb in length) in the coronaviridae family that cause the common cold, influenza-like illness and more serious acute respiratory illnesses, including pneumonia, exacerbations of underlying lung disease, croup and bronchiolitis [1–3]. The subgroups of coronavirus families are alpha (α), beta (β), gamma (γ) and delta (δ) coronaviruses. In particular, this virus was reported to be a member of the β group of coronaviruses. The novel virus was Wuhan coronavirus or 2019 novel coronavirus (2019-nCoV) by the Chinese [4–7]. The International Committee on Taxonomy of Viruses (ICTV) named the virus SARS-CoV-2 and the disease COVID-19 [8–10]. Globally, to date, there has been 1 death every ~ 1166 people under ~ 65 years old compared to ~ 1 death every ~ 358 people in the general population. In addition, 89% of the time, the person who died had one or more underlying medical conditions. A total of ~ 8,256,725 coronavirus cases in approximately ~ 3.4% of reported COVID-19 cases have died according to the COVID-19 outbreak [4–7] as of June 17, 2020, 02:16 GMT. By comparison, seasonal flu generally kills far fewer than 1% of those infected [9], estimated by simply looking at the value of current total deaths plus current total recovered and pairing it with a case total in the past that has the same value, i.e., ~ 445,959/(445,959 + 4,306,426) = 9% CFR (crude fatality ratio) worldwide [11].
Since the 1930s, when the coronavirus family of viruses was first identified, until now commercially, there have been no successful vaccines or antiviral drugs that have been able to prevent or treat infections for COVID-19, and past and current vaccine development efforts against this disease might be of high value for the development of an effective vaccine for COVID-19 [12–14]. According to the WHO, ~ 41 candidate vaccines are being developed for COVID-19 from different countries as of March 13, 2020 (WHO 15 Mar 2020). Many research teams worldwide have been working on monoclonal and polyclonal antibodies against novel coronaviruses produced in vitro using tissue culture techniques, but few of them have entered into trials. Monoclonal antibodies are generally very rarely used in the treatment of infectious diseases. As polyclonal antibodies are composed of a mixture that represents the natural immune response to an antigen, they are prone to a higher risk since they are not all successful. Second, drugs and small molecules can calm the immune system, but patients become seriously ill when their immune system overreacts and starts causing collateral damage to the body. The US Food and Drug Administration today dated June 4, 2020, announced the following actions taken in its ongoing response effort to the COVID-19 pandemic. Dexmedetomidine hydrochloride in ~ 0.9% sodium chloride injection (ANDA-209307) [15] and hydroxychloroquine and chloroquine to treat COVID-19 [16], but side effects such as irregular heartbeats, dizziness or fainting, require medical attention immediately. Another drug use of remdesivir in treatment of COVID-19 is having current drug with possible mechanism of action and chemistry of remdesivir against viral infection [12–14].
The inhaled virus SARS-nCoV-2 likely binds to epithelial cells in the nasal cavity and starts replicating [17]. Human angiotensin-converting enzyme-2 (hACE2) is the main receptor for cell entry in both SARS-CoV and SARS-nCoV-2 outbreaks [18, 19]. The spike glycoprotein (S) on the surface of coronaviruses is essential for virus entry through binding to the hACE2 receptor and for viral fusion with the host cell [20]. Thus, one could target this interaction site between hACE2 and SARS-nCoV-2 spike protein with antibodies or small drug molecules [21]. In vitro data with SARS-nCoV-2 indicate that ciliated cells are primary cells infected in the conducting airways [22]. At this time, the disease COVID-19 is clinically manifest. Overexpression of hACE2 enhanced the disease severity in a physiologically relevant model for investigating hACE2 as a therapeutic target for antiviral intervention against the COVID-19 pandemic. Without a vaccine, we should not think of herd immunity as a light at the end of the tunnel. A vaccine is the only lifetime way to move directly from susceptibility to immunity, bypassing the pain from becoming infected and possibly dying. Moreover, the aim of this study is to focus on identifying selected fragmented (protein subunit) genetic code of antigenic epitope peptides from each novel coronavirus protein domain such as spike protein (S), membrane glycoprotein (M), envelop protein (E) and nucleocapsid protein (N) of the virus and use that as our vaccine. When the vaccine is injected into the body, muscle cells naturally “amplify” it by producing copies of each antigenic epitope peptide from each protein domain, which the immune system detects as a threat. This trains the body’s immune system to defend against novel coronaviruses to produce specific antibodies, such as IgG, IgA, IgM and IgM, by recognizing all antigenic peptides. Finally, only a solution is vaccine that produces antibodies by our own body’s β-cells to fight off infections by novel coronavirus other than any side effects.
Materials and methods
Creation of the clinical dataset
SARS-nCoV-2-infected patient complete gene sequences and metadata for all COVID-19 were obtained from different databases in the NCBI Virus SARS-nCoV-2 Data Hub, GenBank SARS-nCoV-2 sequences, Broad Terra Cloud and Geo COVID Datasets. All nucleotide sequences were translated using the Biopython [23] package. There is a well set of ~ 281 complete genome sequences of SARS-nCoV-2, each one comprising approximately ~ 30,000 nucleotides in length using CLC Bio [24]. We selected mapped to the complete genome of the SARS-nCoV-2 Wuhan-Hu-1 isolate (GenBank accession number: MN908947.3). The protein sequences of ~ 2666 coronaviruses were collected from the 2019 Novel Coronavirus Resource (2019nCoVR) Database of China National Genomics Data Center NGDC on January 29, 2020 [25]. The corresponding data file contains information about the length of each sequence, geographical location, isolation source, collection date of the sample, etc. Clinical information was obtained from UCSF COVID-19 Clinical Data, Vivli, ClinicalTrials.gov COVID-19 Trials, Coronavirus Clinical Trials Explorer, and STAT’s COVID-19 Drugs and Vaccines Tracker. These strains had full-length genomes and were isolated between 1941 and 2020 and included SARS-CoV-2 strains.
Selection of antigenic epitope peptides
The specificity of peptides was confirmed by Basic Local Alignment Search Tool (BLAST) search against the UniProtKB/Swiss-Prot database, which required that the peptide length should be not more than ~ 10–13 amino acids according to the human MHC I and II epitope-recognized compatibility. The specific search criteria were the protein–protein algorithm, and no organism was excluded. According to the hit number, all antigenic peptide results were based on three grades: high (H, hit number = 0), medium (M, 1 ≤ hit number ≤ 10) and low (L, hit number ≥ 11), and grade “high” indicated the best peptide specificity. Antigenic epitope peptide region predictions were based on amino acid properties, including antigenicity [26], secondary structure [27], solvent accessibility [28], flexibility [29] and hydrophilicity [30]. Furthermore, based on the epitope refined to databases such as IEDB [31], FIMM [32] and Bcipep, there are also some methods using machine learning approaches, such as the support vector machine and hidden Markov model (HMM) [33], to locate linear epitopes, such as BEPITOPE [34], BayesB [35], BepiPred [33], ABCPred [36], BEOracle/BROracle [37] and BEST [38].
Identification of T-cell- and β-cell-recognized epitopes
T cell epitope predictions established the shorter peptides of ~ 10–13 amino acids in the antigen that can stimulate either CD4 or CD8 T cells [39]. This propensity to induce T cell immunogenicity has been verified in experiments requiring synthetic antigen-derived peptides [40]. Several different peptides among antigens and T cell prediction methods are structured to classify those that are immunogenic. The full novel coronavirus of the antigenic epitope regions was precisely described by BepiPred-2.0 [41] that was strongly immune-recognized. T cell epitope immunogenicity was contingent on three basics: (1) antigen processing, (2) peptide binding to pMHC molecules and (3) cognate TCR identification. Of such three bases, pMHC–peptide binding was the most selective in determining T cell epitopes [42]. The essential basis for estimating T cell epitopes was, therefore, the prediction of peptide–pMHC binding. Besides, β-cell epitope predictions often promote β-cell epitopes by substituting the antigen for antibody development. Antibodies can recognize any solvent-exposed area of the antigen. β-cell epitopes consist of peptide amino acids ~ 10–13, while conformational β-cell epitopes consist of patches of solvent-exposed atoms of residues that were not necessarily sequential. The majority of β-cell epitopes were approximately 90% conformational, and in fact, only a minority of native antigens generated linear β-cell epitopes.
Epitope-based vaccine construction and validation
Synthetic peptide vaccine candidates for antigenic epitope fragment peptide sequences were used for alignment accuracy, fold recognition and structure prediction by using several benchmarks that were determined by SPARKS-X [43]: protein fold recognition, and ORION: optimized protein fold recognition [44, 45]. In addition, the PAComplex server was used to predict peptide antigens and search the template-based homologous specific peptide antigens of our query short vaccine candidate peptide amino acid sequence by the following methods [46]. The server initially divides the vaccine candidate peptide sequence into fixed lengths ranging from ~ 8 to 13 peptides based on selected pMHC class I–II alleles. Each selected epitope peptide was aligned to the bound peptide of TCR–pMHC templates gathered from PDB. Next, the peptide antigen was examined by utilizing the template-based scoring function to statistically evaluate the complex similarity (Jz ≥ 4.0) between TCR–pMHC and TCR–pMHC. The binding interacting residues based on hydrogen bonds and VDW forces of pMHC and antigenic peptide–TCR interface for each antigen. Finally, the hit homolog identified a template vaccine candidate of antigenic conserved amino acid peptides with a ratio of Jz ≥ 4.0, which was more recognized and interacts with TCR-pMHC. A validation of peptide measures by ProSA-web [47–49] exploits interactive scores and energy plots that highlight potential problems spotted in peptide structures. In particular, the quality scores of peptides determined whether problematic parts of a structure are shown and highlighted in a structure model. Furthermore, constructing vaccine candidate peptides does not allow highly polar and charged molecules to enter cells without causing irreversible lipid membrane damage, which was a novel approach determined in CELLPM [50] cell-penetrating peptides (CPPs) and the Positioning of Proteins in Membrane (PPM) server [51] to understand mechanisms of direct peptide translocation across the lipid bilayer.
Immune response for vaccine efficiency
Immune simulations were determined by the C-ImmSim server [52] to evaluate the immunogenic characteristic of multi-epitope vaccines in real life. C-ImmSim seems to be an agent-based dynamic immune response simulator that utilizes a condition score matrix (PSSM), and predictive analytics techniques predict epitope and immune interactions. The conditions were determined in the C-ImmSim immune simulator four weeks apart at ~ 1, 84 and ~ 168 time steps at each time step relating to ~ 8 h in real-life time step ~ 1 injection time = 0 with a total of ~ 1050 simulation fixed asset. Also, ~ 12 small scattered epitope injections of the developed peptide were provided four weeks apart to imitate the antigen’s repetitive exposure in the standard endemic region, i.e., the clonal probe collection. The Simpson Index, D (Measure of Diversity), was perceived from the narrative.
Peptide molecular dynamics (MD) simulations
The program PEPstrMOD [53] allows extended molecular dynamics (MD) simulation of predicted peptides that enables all-atom simulations of biomolecules. The initial structure was treated for energy minimization and molecular dynamics by using both AMBER11 [54] and GROMACS [55, 56] to generate the final peptide structure. Systems were being minimized for ~ 10,000 phases followed by ~ 200 ps of balancing, followed by a molecular dynamics output run of ~ 200 ns at a temperature of ~ 300 K using a time step of ~ 2 fs. Long-range ion interactions were measured using the Ewald (PME) particle mesh [57]. After simulations, a short energy minimization of all peptides was performed, and the final tertiary structure was predicted. Further analysis of the molecular docking of vaccine peptide candidates revealed interactions between TCR and pMHC II. The generated peptide–docking complex was visualized in Molecular Operating Environment (MOE) [58].
Results
Detection of β-cell- and T-cell-recognized epitopes and antigenic analysis
Antigenic epitope peptide selection from SARS-nCoV-2 based on the peptide quantification method for peptide selection; we challenged screen peptides via specificity, digestibility, recovery and stability to determine the prototypic peptides and investigated the presence of predicted peptides from the UniProt SARS-nCoV-2 pre-release (Jun 23, 2020). MHC T cells and β-cells are highly recognized antigens that are prioritized based on the consensus score, binding affinity and antigenicity. Data-dependent acquisition analysis revealed that the presence of more than ~ 20 unique antigenic epitope peptides ranked at the top of the list would correlate with the highest antigenicity Frank score of 0, and a Frank score of ~ 0.5 would correspond to a random prediction. It was suggested that the consensus approach would improve the specificity and accuracy of the epitope prediction, as it can reduce false positives (Fig. 1a). That were predicted, but overall construct ~ 12 synthetic small antigenic peptides of range among ~ 13 amino acids out of the ~ 12 different proteins (S, E, M, N and ~ 8 other ORFs) in each predicted by UniProt SARS-nCoV-2 (Fig. 1b). That was more interrelating between histocompatibility complex (pMHC) and T cell receptor (TCR). T cells and β-cells recognize small peptide fragments of optimal amino acid residues for pMHC binding, produce a more stable pMHC complex and improve the recognition of antigenic epitope peptides. Small peptides can significantly alter TCR binding in ways that are difficult to predict and thereby prime T cells with altered TCR repertoires. These repertoire effects have clinical relevance, as it was found that vaccination with contact residue-altered peptides was less effective than vaccination with natural peptides at priming SARS-nCoV-2-specific T cells in patients. The peptide backbone was a fixed amino acid, and all other positions were degenerate, with degenerate positions containing any one of the photogenic amino acids within the range (~ 1–13) excluded to reduce disulfide bond formation within the compound mixture.

a Systematic structures of SARS-nCoV-2 genome organization and identified antigenic epitopes. A ~ 72 nucleotides of long chain flank the SARS-nCoV-2 genome at the 5′ end and a poly(A) tail at the 3′ end. Many other open reading frames (ORFs) relating to viral structural components (S, E, M and N proteins) and companion genes (ORF 1a, 1b, 3a, 6, 7a, 7b, 8 and 10) were identified, all of which reflect different colors. The antigen-presenting epitope peptide sequence regions and ranges of each protein have been identified in various colors. b Library design of antigenic epitope peptides for β-cell/T cell recognition. SARS-nCoV-2 antigen epitope model selection based on score feature to infer peptide antigens and homologous peptide antigens through structural templates based on score Z value (Jz) < v3, along with experimental peptides and full pathogen genome databases. The preference of epitopes is known as a β-cell/T cell displaying various colors through prioritization
Vaccine construction and validation by MD simulation
Different selected epitope sequences were designed, as described in the Materials and Methods. We further estimated the effect of linker, epitope location and sequence continuity on peptide structural characteristics. The range of linkers varies from more than ~ 2 to ~ 30 amino acids, optimized for each condition so that the linker does not impose any constraints on the conformation or interactions of the linked partners. The IBIVU analysis results showed that the linker between the epitopes, which used the flexible linker “AVKELF” lyase, was found to connect every single epitope to form a vaccine candidate structure for the therapeutic index. Every epitopes joined by “GPGPG” linkers prevent the formation of junctional epitopes and also facilitate the immune processing of antigens. The peptides adjuvant was attached to the N-terminal of the construct via “EAAAK” linker in order to boost a long-lasting immune response. The final vaccine construct consisted of ~ 156 amino acids with a molecular weight of ~ 18.72 kDa (Fig. 2A). However, once linkers linked with peptides, the structural parameters changed greatly, especially indexes of α-, β-, turn and coil regions that are indications of protein secondary structure. In particular, “AVKELF” linkers reduced the number of β-, turn and coil regions but increased the number of α-regions. This provides a strategy of combinatorial biosynthesis, in which modules are the building blocks of genetic manipulation. Epitope peptides’ connection with a linker makes highly polar and charged molecules incapable to enter cells without causing irreversible membrane damage to passively penetrate across the lipid bilayer.

A Constructed vaccine candidates by linker and quality assessment. a Tertiary structural constructs of ~ 1–12 P′ small peptides in each connect to ~ 1–7(L) linkers (GPGPG and EAAAK) of the vaccine model were determined by (ORION) peptide fold recognition. b The structural quality estimation of vaccine model was DOPE Energy: − 181.9, and Z-score of Energy: − 0.961, and c overall vaccine model quality has Z-score: − 1.99 determined by ProSA and arranged all residues show knowledge-based energy plot. B Energy minimizations of SARS-nCoV-2 proteins construct peptides. Energy graph of SARS-nCoV-2-designed peptide-based vaccine region range (~ 1–12 P′) and simulation of RMSD graph
The structure of each SARS-nCoV-2 protein epitope peptide (P′1–P′13) was further refined with energy minimization and molecular dynamics simulations. The duration of MD simulations was from ~ 100 ps to ~ 1 ns (ns), and the performances determined by PEPstrMOD and ORION at both of these time steps were compared (Fig. 2B). The results were slightly better for the first time compared to the quality of each peptide based on the ab initio method. The performance was further improved from ~ 3.97 to ~ 3.82 Å (CA-RMSD: PEPstrMOD) by extending the duration of MD runs from ~ 100 ps to ~ 1 ns in a hydrophilic environment, even in the case of very short peptides. The potential energy minimization on the initial peptides to obtain the minimized structures was computed based on RMSD values. The results improved to average CA-RMSD: PEPstrMOD and B-RMSD: ORION of ~ 4.12 Å and ~ 3.85 Å, respectively. The initial and minimized structure achieved an average RMSD: ORION of ~ 3.76 Å and ~ 3.64 Å, respectively. Finally, the minimized structure of peptides and structure after ~ 100 ps MD simulations improved to ~ 4.78 Å and ~ 4.31 Å, respectively. Overall, no improvement in the results was observed by extending MD simulation from 100 ps to 1 ns by ORION and PEPstrMOD. Performing MD simulations in a hydrophilic environment also produced similar results with an average CA-RMSD of ~ 4.35 Å compared to ~ 4.31 Å in vacuum.
Peptide across bilayer and membrane–peptide interactions
The molecular dynamics simulations were the direct translocation of cell-penetrating peptides (CPPs)-constructed ~ 1–12 P′ antigenic peptides of SARS-nCoV-2 across the lipid membrane driven by the membrane electrostatic potential interaction. The interactions between CPPs and lipid membranes have been observed for a wide spectrum of simple biomimetic models of the plasma membrane assembled from phospholipids using a mixture of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) bilayers. The local membrane potential is produced by the ion–concentration imbalance across the lipid membrane, which is ubiquitous in biological environments. A produced charge of cell-penetrating antigenic peptides was adsorbed on the lipid membrane, which further enhances the classical nucleation theory that was applied to estimate the translocation time by calculating the changes in the free energy upon transferring peptides across the lipid membrane. Overall, the results of SARS-nCoV-2 for each peptide were used to calculate the parameters of a lipid membrane to bound peptide (binding affinity), the position of the predicted alpha-helix in the amino acid peptide sequence, alpha-helix stability, membrane penetration depth and tilt angle, which are provided in a table, and the addition of structural stability with an interaction between the lipid membrane and each peptide, as shown in Fig. 3 and Table 1. The accuracy of the membrane-binding energy, which was assessed as the root-mean-square error (RMSE) between the experimental and calculated values, was found to be ~ 0.74 kcal/mol for small molecules and ~ 1.13 kcal/mol for peripheral antigenic peptides. The hydrophobic thickness of each peptide was approximately ~ 5 °C for the tilt angle, but the constructs of ~ 1–12 P′ small peptides connected by ~ 1–7 L linker adaptors were used to construct large single peripheral peptides, especially conformations of the membrane to interacting loops, where the uncertainty in tilt angle may reach 50°. Large differences in orientations may be observed for alternative conformations of peptide. Moreover, distinct conformations of Ca2 + -ATPase, a TM α-helical protein, differ in protein tilt by ~ 17° and membrane thickness by an average of ~ 3 Å, which might be of other functional importance.

Short peptides of SARS-nCoV-2 and penetrated cellular lipid bilayers. The fragmentation of (~ 1–12 P′) peptides interacting with the lipid membrane is shown in electrostatic potential color in molecular surface representation, the lipid tail spears are shown in white, and the stability and energy minimization were calculated for each peptide
Peptide bindings to MHC class I–II in an extended conformation with TCR
A crystal structure with a resolution of 1.61 Å of the TCR class I–II antigen (PDB ID: 2AK4) as Swiss-model and PAComplex identified the best template. The sequence identity template was 92%. The best TCR complex model was then selected, and templates were evaluated based on the accuracy of scoring functions on variant conditions, including single templates, multiple templates, and single and both sides. The model-selected TCR complex prototype with a binding antigenic peptide score was ~ 1.77 to ~ 4.22 for these validation approaches. The induced dataset consisted of ≥ 108 peptide candidates (JZ ≥ 1.645) derived from (IEDB) ~ 864 628 sequences of ~ 389 pathogens and had been used to test the reliability of SARS-nCoV-2 homologous peptide antigens. The homologous peptide antigens were tested for > 1010 peptides obtained from ~ 864 628 protein sequences of ~ 389 pathogens. More than 108 JZ ≥ 1645 peptide candidates were chosen for analysis of the relationship between JZ values and the number of positive homologous peptide antigens. When the JZ ratio was more significant than ~ 4.0, the accuracy of > 0.8 and the number of positive antigens reached ~ 1600 by both positive and negative data sets. The JZ threshold was set at ~ 4.0, and the total number of potential inferring peptide antigens ~ 4 000 000, reportedly obtained from ~ 12 antigenic peptides recognized by TCR–pMHC complexes. PMHC class II peptide binding was complicated compared to pMHC class I peptide binding due to their limited duration. Our pMHC class II peptide binding numbers were ~ 10–13 long amino acids with a binding base of ~ 4–6 amino acids containing primary binding residues. The docked allele from each epitope out of ~ 1–12 P′ complex showed the same residue of antigenic peptide interactions found in the epitope bound crystal structures of TCR–pMHC. Additional details can be found in Fig. 4 and Table 2.

MHC class I and II T cell allele and epitope modeling and docking. The T cell receptor consists of four binding domains in different colors (Cα: green; Cβ: violet; Vα: green; and Vβ: violet). MHC class II and I consist of four binding domains in different colors (α1, α2, α3, and β2). Vaccine candidates of SARS-nCoV-2 antigenic peptides consist of a ~ 1–12 P′ binding complex between pMHC and TCR. MHC–TCR epitope complex minimized structure at the end of simulations t = 0 and t = 200 ns antigenic peptide, as shown in between the middle of complex (yellow)
Simulation of vaccine and immune response to pathogens
The immune response created by the C-ImmSim immune simulator was based on the actual immune response, as shown in Fig. 5a, b. The various potential responses were substantially higher than in the first response. The typical high levels of immunoglobulin activity characteristic of IgG1 + IgG2, IgM and IgG + IgM epitopes known as antibodies were apparent in both various potential responses with a corresponding decrease in specific antigen concentrations. In contrast, multiple β-cell isotopes with long-lasting activity were observed, suggesting possible swapping potential isotopes and memory generation. Similarly, a strong response was seen in the T-helper (Th) and cytotoxic T cell populations with corresponding memory growth, and it was most important to complement the immune response. Increased macrophage activity was shown during exposure, while dendritic cell activity was observed to be stable. High levels of interferon-gamma (IFN-γ) and interleukin-2 (L-2) were also apparent. In comparison, the lower Simpson index suggests a broader range. A designed vaccine was injected at ~ 12 doses throughout ~ 12 months. Repeated exposure to several ~ 12 small fragmented antigenic epitopes caused a rise in IgG1 and a decrease in IgM levels, thus retaining IFN-γ and T-helper cell populations at a high level during exposure, as illustrated in Fig. 5. This profile reveals the event of immune memory and, subsequently, the antigen’s increased clearance at subsequent exposures. Subsequent exposures result in a secondary reaction that was quicker and greater as a result of the first exposure memory of the body.

Immune simulation response to pathogens. The vaccine was administered over ~ 12 doses throughout ~ 12 months. The number of cells was displayed. Legend: Act (active), Intern (internalized) Ag, Press II posing MHC II, Dup (duplicating) in the mitotic cycle, Anergic residing was not active. a; 1 B-total count lymphocytes, memory cells and subdivided into IgM, IgG1 and IgG2 isotypes. a; 2 Plasma B-lymphocytes are subdivided by isotypes (IgM, IgG1 and IgG2). a; 3 B-lymphocytes population per entity state (i.e., displaying counts for involved, presenting on class II, internalizing the Ag, duplicating and anergic. a; 4 CD4 T-helper lymphocytes count. The plot compares the average number, and the memory counts. a; 5 CD4 T-helper lymphocytes count subdivided by entity state (i.e., involved, sleeping, anergic and duplicating). b Same symbols, as seen above. b; 1 CD8 T-cytotoxic count of lymphocytes and general information is seen. b; 2 CD8 T-cytotoxic count of lymphocytes as shown in the entity state. b; 3 Natural killer cells in total shown. b; 4 Complete count macrophages were internalized, indicating active and rested macrophages in MHC class II. b; 5 Dendritic cells. DC can present antigenic peptides for both class I and class II MHC molecules. Curves display the overall amount broken down to find, rest, internalizing, and addressing the Ag. b; 6 Complete epithelial cells broken down to acgive, virus-infected, and present in class I MHC molecules. c The virus, immunoglobulins and immunocomplexes. Antigen and immunoglobulin antibodies are subdivided by isotype. d Cytokine and interleukin concentrations. The inset plot displays the threat signal along with the leukocyte growth factor IL-2. Cytokines and related signals were seen for the concentration of cytokines and interleukins. D was a danger signal in the inset plot
Peptide vaccine produced antibodies against a specific antigen
Immunization effectors seem to be effectively antibodies produced by β-cells responsible for binding, specifically to a pathogen. Other potential effectors are cytotoxic CD8 + T lymphocytes that may prevent pathogen agents’ transmission by detecting and destroying invading pathogens or cells that secrete unique antiviral cytokines and CD4 + T-helper lymphocytes. This T helper cell leads to resistance by developing cytokines and supports the initiation and progression of β-cell and CD8 + T cell responses. Adaptive immune CD4 + T-helper cells are reliant on their crucial cytokine production of interferon-γ or interleukin [IL]-4, meanwhile. SARS-nCoV-2 peptide-based vaccine antigens penetrated the lipid membrane and translocated into the β-cell via pMHC class I–II. The β-cells filled with surface β-cell receptors responsible for binding to vaccine antigens have been activated and moved to the interface between the β-cell and the T cell. Here, β-cells engage T cells and begin to increase. The accumulated number of activation signals obtained by the β-cells affects their ultimate fate.
SARC-nCoV-2 protein antigens have been used as small peptides on the substrate of particular antigen-presenting cells (APCs) to activate T cells. Such causes a massively influential β-cell differentiation process through complex mechanisms wherein the antigen-specific β-cells proliferate and divide into antibody-secreting plasma cells or memory β-cells against SARS-nCoV-2. They detect an antigen of its SARC-nCoV-2 protein over which their unique substrate IgM receptor binds. Antigen binding initiates β-cell activation and activates the up-regulation of the chemokine receptor (CCR7). This chemokine receptor induces antigen-specific β-cells toward the outer T cell region of the lymph nodes. At such sites, peptide vaccine antigen-specific β-cells are revealed to (< 24 h) stimulate dendritic cells (DCs) and T cells that have uncontrolled unique surface receptors and therefore have β-cell activating signals (Fig. 6). This T cell allows to effectively trigger β-cell proliferation into Ig-secreting plasma cells that contain low-affinity germline antibodies toward different follicular reactions.

Systematic overview of antibodies developed from specific epitopes of SARS-nCoV-2. This figure consists of six steps. (1) The linker adaptor attached to each small antigenic peptide of SARS-Cov-2 ordered by each protein (ORF 1a and ORF1b)—structural protein [Spike—S, Envelope—E, Membrane—M, Nucleocapsid—N)-3′, and some of the additional accessory proteins, such as ORF 3a, 6, 7a, 7b, 8 and 10, are inserted into the genes of structural proteins. Full vaccine candidates of whole peptides with linker adaptors to inclusion through cell penetration inside the membrane and the exit of the peptide from the bilayer. (2 and 3) Transduce signals to downstream kinase complexes that activate IFN regulatory factor-3 (IRF-3), protein kinase kB (NF-kB) and ATF-2/c-jun. So these signaling pathways regulate the activation of type I interferons (IFN-α and β). Type I IFNs trigger the stimulation of STAT expression factors that influence the activation of hundreds of IFN-stimulated genes that create antiviral immunity in cellular components, thus restricting viral replication and spread. Also, T-helper-2 subsets rely on their crucial output of cytokines (interferon-Δ or interleukin [IL]-4 and 12). (4) Lymphocytes that were applied to both β-cells and T cells. We shall see that each lymphocyte is committed to respond to a specific antigen and that its response during its first encounter with an antigen ensures that a more rapid and effective response occurs on subsequent encounters with the same antigen. (5) Β-cells translate our peptide vaccine antigens into small peptides ~ 1–13 amino acids seen on the surface by primary MHC class I–II molecules. Thus, MHC peptide complexes are made accessible for binding by a tiny segment of CD4 + T-helper cells. (6) This peptide vaccine is based on SARS-nCoV-2 protein antigens as well-targeted β-cells and the types of T cells that support the production of specific antibodies, such as IgM, IgD, IgG, IgA and IgE, against SARS-nCoV-2. In such cases, this peptide vaccine can also be developed to promote cytotoxic T cell activity or perhaps a combination of both antibody and cytotoxic T cells immune responses
Also, β-cell affinity was its outcome of a comprehensive somatic hypermutation mechanism within Ig genes variable area segments. In a tiny proportion of β-cells, the initiation of mutations in their Ig genes increases the affinity of the IgG surface to that same antigen, which allows such β-cells to defend effectively for binding to small quantities of peptide vaccine antigens interacting with the surface of DCs. Such peptide vaccine antigens are transcribed by β-cells into small surface peptides by significant class molecules I–II MHC. Thus, MHC–peptide complexes are made available for binding by a particular subset of CD4 + T cells. These T cells, which carry CXCR5, migrate to the CXCL13-expressing FDCs. Differentiating from T-helper ~ 1 and ~ 2 cells by their chemokine receptors, transcription factors, surface markers and interleukins, they are ideally suited to provide effective β-cell aid through a sequence of co-stimulation molecules, which include CD40L, ICOS intracellular T cell co-stimulator, IL-10 β-cell growth factor and IL-21; these were antigen-specific anti-SARS-nCoV-2 antibodies which further arise in the serum and therefore are another hallmark of peripheral antibody responses.
Discussion
The novel coronavirus disease 2019 (COVID-19) pandemic has created a worldwide crisis and inspired an urgent search for the prevention and treatment of severe acute respiratory syndrome coronavirus 2 (SARS-nCoV-2) infection [4–7, 25]. The world around researchers is currently developing more than ~ 140 vaccines against coronavirus by different target proteins each. An effective vaccine is the only necessary solution to the COVID-19 pandemic [59]. Although the vaccine development process typically requires years of research and testing before reaching the clinic, scientists are racing to produce a safe and effective vaccine. The first vaccine safety trials in humans started in March 2019, but the road ahead remains uncertain. Some trials fail, and others may end without clear results [60]. However, a few may succeed in stimulating the better immune system to produce effective antibodies against the virus. Here is the current status of all the vaccines that have reached trials in humans, along with a selection of promising vaccines still being in a pipeline tested in cells or animals, as shown in Fig. 6 [61].
The S-protein is considered an attractive therapeutic target given its location in Fig. 7, and it is therefore targetable using antibodies [62]. Immunization of animals with S-protein-based vaccines has been shown to induce the formation of neutralizing antibodies that are effective in preventing infection by homologous coronaviruses [63], but the phase III clinical trial was not successful in humans. Although S-protein may elicit an immune reaction, it is not yet known whether this would be sufficient to mount the sustained immune response needed to fight COVID-19 infection [64]. As for a recent report among all SARS-nCoV antigens, the S protein stimulates the highest level of antibody production. The epitopes of this protein that bound to CD4 and CD8 T cells were identified. Two of these epitopes binding to CD8 cells were presented by the MHC II molecule HAL-A*02:01 and stimulated a specific response in patients who had recovered from SARS-nCoV-2 but not healthy individuals. Many other T cell epitopes in the M and N proteins of the virus have also been identified. However, we are not targeted vaccines based on a single antigen, such as S-protein [65]. Here, we designed epitopes of peptide-based vaccines based on the complete genome of the SARS-CoV-2 Wuhan-Hu-1 isolate (GenBank accession number: MN908947.3) [48, 49] and constructed ~ 12 small fragmented antigenic epitope peptides by ~ 13 amino acids in different proteins (S, E, M and N) and other necessary proteins (ORFs 1a, 1b, 3a, 6, 7a, 7b, 8 and 10) of SARS-nCoV-2. We converted the ~ 1–12 P′ antigenic epitopes into a single vaccine candidate, using the “AVKELF” peptide linker adaptor, which was penetratingly translocated to the cell or lipid membrane [66]. These epitopes can be used to make immunogenic multi-epitopes peptide vaccines against SARS-nCoV-2.

Coronavirus vaccine tracker tested in cells or animals
Human MHC I–II molecule antigen appearances continue through two separate pathways. MHC II molecules contain peptide antigens obtained from endocyted antigens that are damaged and activated to the MHC II molecule in endosome compartments. In contrast, other class I MHC molecule introduces peptides obtained predominantly from antigens degraded to the cytosol [67]. Our corresponding peptide antigens are then carried by TAP to the endoplasmic reticulum, where they have been loaded onto latent MHC class I–II molecules. Vaccines most successfully activate the innate immune system’s stimulation by several pathogen-associated signals, enabling them to be identified by pattern recognition receptors [68, 69]. Although many antigens and vaccines stimulate β-cell and T cell responses, there is no reason for rejecting vaccines to favor antibody development and T cell responses. Also, CD4 + T cells are needed for most antibody reactions, while antibodies exert major fluencies on T cell responses to intracellular pathogens [70]. Further, toll-like receptors (TLRs) might also modulate innate immune activities and the production of new therapeutic molecules by targeting TLR5 inhibition against SARS-CoV-2 [4–7]. Interleukin (IL) receptor has also been caused by antagonist therapies [4–7]. Cytokine storm syndrome–extreme acute respiratory syndrome (SARS-CoV-2) treatment affected people to improve clinical possibilities for cytokine storms.
Our vaccine candidates of antigenic peptides that were recognized by MHC class I–II antigens, providing valuable insight into the detection of specific pMHC–peptide complexes, rely on the string induction strong activation of T cells bearing relevant CD8 + T cell responses. Therefore, our vaccine candidates of antigenic immune complexes are taken up by macrophages and DCs, dissociate into their acidic phagolysosome compartment, and are processed into fragmented small peptides. These peptides are exposed at the surface of APCs and are bound by CD4 + and CD8 + T cells. Overall, most protective antibody responses are dependent on CD4 + T cell help. Therefore, we assessed SARS-nCoV-2-collotion of antigens from different proteins that all specific CD4 + T cell responses were associated with production of higher specific antibody against SARS-nCoV-2.
Conclusion
We are addressing and challenging the antigenic immune response, the potential of structural peptide-based vaccines against coronaviruses and the observed phenomenon of long-term immunity to SARS-nCoV-2 likely stemming from T cells and β-cells activity. The identification of such peptides would indeed help to better understand the immune response to SARS-nCoV-2 and promote the development of peptide-based specific vaccines. This vaccine produces specific antibodies for specific antigens that are functionally non-allergenic, antigenic and more effective, which might be strong against SARS-nCoV-2. Further clinical trials are required to check the efficacy of this vaccine.
Significance statement
This study provides information on the COVID-19 of SARS-nCoV-2. The developed peptide-based vaccine elicitation epitope triggers specific antibodies against the novel coronavirus to prevent infection.
Abbreviations
- SARS-nCoV-2:
-
Severe acute respiratory syndrome coronavirus 2
- MHC:
-
Major histocompatibility complex
- TCR:
-
T cell receptor
- APC:
-
Antigen-presenting cell
- ICTV:
-
The International Committee on Taxonomy of Viruses
- HMM:
-
Hidden Markov model
- TCR:
-
T cell receptor
- APCs:
-
Antigen-presenting cells
- RMSD:
-
Root-mean-square deviations
- RMSE:
-
Root-mean-square error
- PDB:
-
Protein data bank
- CPPs:
-
Cell-penetrating peptides
- MD:
-
Molecular dynamics
- MOE:
-
Molecular operating environment
- IEDB:
-
The immune epitope database and analysis resource
- hACE:
-
Human angiotensin-converting enzyme 2
References
Lai CC, Shih TP, Ko WC, Tang HJ, Hsueh PR (2020) Severe acute respiratory syndrome coronavirus 2 (Sars-Cov-2) and coronavirus disease-2019 (Covid-19): the epidemic and the challenges. Int J Antimicrob Agents 55(3):105924. https://doi.org/10.1016/j.ijantimicag.2020.105924
Li K, Li Z, Wohlford-Lenane C, Meyerholz DK, Channappanavar R, An D, Perlman S, McCray PB, He B (2020) Single-dose, intranasal immunization with recombinant parainfluenza virus 5 expressing middle east respiratory syndrome coronavirus (Mers-Cov) spike protein protects mice from fatal Mers-Cov infection. mBio. https://doi.org/10.1128/mBio.00554-20
Lim YX, Ng YL, Tam JP, Liu DX (2016) Human coronaviruses: a review of virus-host interactions. Diseases 4(3):26. https://doi.org/10.3390/diseases4030026
Chakraborty C, Sharma AR, Bhattacharya M, Sharma G, Lee SS, Agoramoorthy G (2020) Consider Tlr5 for new therapeutic development against Covid-19. J Med Virol. https://doi.org/10.1002/jmv.25997
Chakraborty C, Sharma AR, Bhattacharya M, Sharma G, Lee SS, Agoramoorthy G (2020) Covid-19: consider Il-6 receptor antagonist for the therapy of cytokine storm syndrome in Sars-Cov-2 infected patients. J Med Virol. https://doi.org/10.1002/jmv.26078
Chakraborty C, Sharma AR, Sharma G, Bhattacharya M, Lee SS (2020) Sars-Cov-2 causing pneumonia-associated respiratory disorder (Covid-19): diagnostic and proposed therapeutic options. Eur Rev Med Pharmacol Sci 24(7):4016–4026. https://doi.org/10.26355/eurrev_202004_20871
Chakraborty C, Sharma AR, Sharma G, Bhattacharya M, Saha RP, Lee SS (2020) Extensive partnership, collaboration, and teamwork is required to stop the Covid-19 outbreak. Arch Med Res. https://doi.org/10.1016/j.arcmed.2020.05.021
Adachi S, Koma T, Doi N, Nomaguchi M, Adachi A (2020) Commentary: origin and evolution of pathogenic coronaviruses. Front Immunol 11:811. https://doi.org/10.3389/fimmu.2020.00811
Fontanet A, Cauchemez S (2020) COVID-19 herd immunity: where are we? Nat Rev Immunol 20(10):583–584. https://doi.org/10.1038/s41577-020-00451-5
Viruses, Coronaviridae Study Group of the International Committee on Taxonomy of Viruses (2020) The species severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat Microbiol 5(4):536–544. https://doi.org/10.1038/s41564-020-0695-z
Battegay M, Kuehl R, Tschudin-Sutter S, Hirsch HH, Widmer AF, Neher RA (2020) 2019-novel coronavirus (2019-nCoV): estimating the case fatality rate—a word of caution. Swiss Med Wkly 150:w20203. https://doi.org/10.4414/smw.2020.20203
Saha A, Sharma AR, Bhattacharya M, Sharma G, Lee SS, Chakraborty C (2020) Probable molecular mechanism of remdesivir for the treatment of Covid-19: need to know more. Arch Med Res 51(6):585–586. https://doi.org/10.1016/j.arcmed.2020.05.001
Saha A, Sharma AR, Bhattacharya M, Sharma G, Lee SS, Chakraborty C (2020) Tocilizumab: a therapeutic option for the treatment of cytokine storm syndrome in Covid-19. Arch Med Res 51(6):595–597. https://doi.org/10.1016/j.arcmed.2020.05.009
Saha RP, Sharma AR, Singh MK, Samanta S, Bhakta S, Mandal S, Bhattacharya M, Lee SS, Chakraborty C (2020) Repurposing drugs, ongoing vaccine, and new therapeutic development initiatives against Covid-19. Front Pharmacol 11:1258. https://doi.org/10.3389/fphar.2020.01258
Stockton J, Kyle-Sidell C (2020) Dexmedetomidine andworsening hypoxemia in the setting of Covid-19: a case report. Am J Emerg Med. https://doi.org/10.1016/j.ajem.2020.05.066
Tang W, Cao Z, Han M, Wang Z, Chen J, Sun W, Wu Y, Xiao W, Liu S, Chen E, Chen W, Wang X, Yang J, Lin J, Zhao Q, Yan Y, Xie Z, Li D, Yang Y, Liu L, Qu J, Ning G, Shi G, Xie Q (2020) Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: open label, randomised controlled trial. BMJ 369:m1849. https://doi.org/10.1136/bmj.m1849
Zhang H, Baker A (2017) Recombinant human Ace2: acing out angiotensin ii in ards therapy. Crit Care 21(1):305. https://doi.org/10.1186/s13054-017-1882-z
Bérubé K, Prytherch Z, Job C, Hughes T (2010) Human primary bronchial lung cell constructs: the new respiratory models. Toxicology 278(3):311–318. https://doi.org/10.1016/j.tox.2010.04.004
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu NH, Nitsche A, Müller MA, Drosten C, Pöhlmann S (2020) Sars-Cov-2 cell entry depends on Ace2 and Tmprss2 and is blocked by a clinically proven protease inhibitor. Cell 181(2):271–280.e8. https://doi.org/10.1016/j.cell.2020.02.052
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Abiona O, Graham BS, McLellan JS (2020) Cryo-em structure of the 2019-nCoV spike in the prefusion conformation. bioRxiv. https://doi.org/10.1101/2020.02.11.944462
Iwata-Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N (2019) Tmprss2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol. https://doi.org/10.1128/JVI.01815-18
Deng X, Zou W, Xiong M, Wang Z, Engelhardt JF, Ye SQ, Yan Z, Qiu J (2017) Human parvovirus infection of human airway epithelia induces pyroptotic cell death by inhibiting apoptosis. J Virol. https://doi.org/10.1128/JVI.01533-17
Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJL (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25(11):1422–1423. https://doi.org/10.1093/bioinformatics/btp163
Qiagen (2020) Hilden, Germany
Zhao WM, Song SH, Chen ML, Zou D, Ma LN, Ma YK, Li RJ, Hao LL, Li CP, Tian DM, Tang BX, Wang YQ, Zhu JW, Chen HX, Zhang Z, Xue YB, Bao YM (2020) The 2019 novel coronavirus resource. Yi Chuan 42(2):212–221. https://doi.org/10.16288/j.yczz.20-030
Kolaskar AS, Tongaonkar PC (1990) A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett 276(1–2):172–174. https://doi.org/10.1016/0014-5793(90)80535-q
Pellequer JL, Westhof E, Van Regenmortel MH (1993) Correlation between the location of antigenic sites and the prediction of turns in proteins. Immunol Lett 36(1):83–99. https://doi.org/10.1016/0165-2478(93)90072-a
Emini EA, Hughes JV, Perlow DS, Boger J (1985) Induction of hepatitis a virus-neutralizing antibody by a virus-specific synthetic peptide. J Virol 55(3):836–839
Karplus PA, Schulz GE (1985) Prediction of chain flexibility in proteins: a tool for the selection of peptide antigen. Naturwiss 72:13–221
Parker JM, Guo D, Hodges RS (1986) New hydrophilicity scale derived from high-performance liquid chromatography peptide retention data: correlation of predicted surface residues with antigenicity and x-ray-derived accessible sites. Biochemistry 25(19):5425–5432. https://doi.org/10.1021/bi00367a013
Vita R, Zarebski L, Greenbaum JA, Emami H, Hoof I, Salimi N, Damle R, Sette A, Peters B (2010) The immune epitope database 2.0. Nucl Acids Res 38(Database issue):D854–D862. https://doi.org/10.1093/nar/gkp1004
Schönbach C, Koh JL, Sheng X, Wong L, Brusic V (2000) Fimm, a database of functional molecular immunology. Nucl Acids Res 28(1):222–224. https://doi.org/10.1093/nar/28.1.222
Larsen JE, Lund O, Nielsen M (2006) Improved method for predicting linear B-cell epitopes. Immunome Res 2:2. https://doi.org/10.1186/1745-7580-2-2
Odorico M, Pellequer JL (2003) Bepitope: predicting the location of continuous epitopes and patterns in proteins. J Mol Recognit 16(1):20–22. https://doi.org/10.1002/jmr.602
El-Manzalawy Y, Dobbs D, Honavar V (2008) Predicting linear b-cell epitopes using string kernels. J Mol Recognit 21(4):243–255. https://doi.org/10.1002/jmr.893
Saha S, Raghava GP (2006) Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins 65(1):40–48. https://doi.org/10.1002/prot.21078
Wang Y, Wu W, Negre NN, White KP, Li C, Shah PK (2011) Determinants of antigenicity and specificity in immune response for protein sequences. BMC Bioinform 12:251. https://doi.org/10.1186/1471-2105-12-251
Gao J, Faraggi E, Zhou Y, Ruan J, Kurgan L (2012) Best: improved prediction of B-cell epitopes from antigen sequences. PLoS ONE 7(6):e40104. https://doi.org/10.1371/journal.pone.0040104
Ahmed RK, Maeurer MJ (2009) T-cell epitope mapping. Methods Mol Biol 524:427–438. https://doi.org/10.1007/978-1-59745-450-6_31
Ahmad TA, Eweida AE, El-Sayed LH (2016) T-cell epitope mapping for the design of powerful vaccines. Vaccine Rep 6:13–22. https://doi.org/10.1016/j.vacrep.2016.07.002
Jespersen MC, Peters B, Nielsen M, Marcatili P (2017) Bepipred-2.0: improving sequence-based B-cell epitope prediction using conformational epitopes. Nucl Acids Res 45(W1):W24–W29. https://doi.org/10.1093/nar/gkx346
Lafuente EM, Reche PA (2009) Prediction of Mhc-peptide binding: a systematic and comprehensive overview. Curr Pharm Des 15(28):3209–3220. https://doi.org/10.2174/138161209789105162
Yang Y, Faraggi E, Zhao H, Zhou Y (2011) Improving protein fold recognition and template-based modeling by employing probabilistic-based matching between predicted one-dimensional structural properties of query and corresponding native properties of templates. Bioinformatics 27(15):2076–2082. https://doi.org/10.1093/bioinformatics/btr350
Ghouzam Y, Postic G, de Brevern AG, Gelly JC (2015) Improving protein fold recognition with hybrid profiles combining sequence and structure evolution. Bioinformatics 31(23):3782–3789. https://doi.org/10.1093/bioinformatics/btv462
Ghouzam Y, Postic G, Guerin PE, de Brevern AG, Gelly JC (2016) Orion: a web server for protein fold recognition and structure prediction using evolutionary hybrid profiles. Sci Rep 6:28268. https://doi.org/10.1038/srep28268
Liu IH, Lo YS, Yang JM (2011) Pacomplex: a web server to infer peptide antigen families and binding models from Tcr-Pmhc complexes. Nucl Acids Res 39(Web Server issue):W254–W260. https://doi.org/10.1093/nar/gkr434
Wiederstein M, Sippl MJ (2007) Prosa-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucl Acids Res 35(Web Server issue):W407–W410. https://doi.org/10.1093/nar/gkm290
Bhattacharya M, Sharma AR, Patra P, Ghosh P, Sharma G, Patra BC, Lee SS, Chakraborty C (2020) Development of epitope-based peptide vaccine against novel coronavirus 2019 (Sars-Cov-2): immunoinformatics approach. J Med Virol 92(6):618–631. https://doi.org/10.1002/jmv.25736
Bhattacharya M, Sharma AR, Patra P, Ghosh P, Sharma G, Patra BC, Saha RP, Lee SS, Chakraborty C (2020) A Sars-Cov-2 vaccine candidate. Inform Med Unlocked 20:100394. https://doi.org/10.1016/j.imu.2020.100394
Lomize AL, Pogozheva ID (2018) Modeling of peptide folding and translocation across membranes. Biophys J 114(3):267a. https://doi.org/10.1016/j.bpj.2017.11.1545
Lomize MA, Pogozheva ID, Joo H, Mosberg HI, Lomize AL (2012) OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res 40:D370–D376. https://doi.org/10.1093/nar/gkr703
Rapin N, Lund O, Bernaschi M, Castiglione F (2010) Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One 5(4):e9862. https://doi.org/10.1371/journal.pone.0009862
Singh S, Singh H, Tuknait A, Chaudhary K, Singh B, Kumaran S, Raghava GP (2015) Pepstrmod: structure prediction of peptides containing natural, non-natural and modified residues. Biol Direct 10:73. https://doi.org/10.1186/s13062-015-0103-4
Salomon-Ferrer R, Case DA, Walker RC (2013) An overview of the Amber biomolecular simulation package. WIREs Comput Mol Sci 3:198–210. https://doi.org/10.1002/wcms.1121
Jo S, Kim T, Iyer VG, Im W (2008) Charmm-Gui: a web-based graphical user interface for Charmm. J Comput Chem 29(11):1859–1865. https://doi.org/10.1002/jcc.20945
Jo S, Cheng X, Lee J, Kim S, Park SJ, Patel DS, Beaven AH et al (2017) Charmm-Gui 10 years for biomolecular modeling and simulation. J Comput Chem 38(15):1114–1124. https://doi.org/10.1002/jcc.24660
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG (1995) A smooth particle mesh Ewald method. J Chem Phys 103(19):8577. https://doi.org/10.1063/1.470117
Wilson GL, Lill MA (2011) Integrating structure-based and ligand based approaches for computational drug design. Future Med Chem 3(6):735–750. https://doi.org/10.4155/fmc.11.18
Corey L, Mascola JR, Fauci AS, Collins FS (2020) A strategic approach to Covid-19 vaccine R&D. Science 368(6494):948–950. https://doi.org/10.1126/science.abc5312
Callaway E (2020) Coronavirus vaccine trials have delivered their first results—but their promise is still unclear. Nature 581(7809):363–364. https://doi.org/10.1038/d41586-020-01092-3
Feng W, Newbigging AM, Le C, Pang B, Peng H, Cao Y, Wu J et al (2020) Molecular diagnosis of Covid-19: challenges and research needs. Anal Chem 92(15):10196–10209. https://doi.org/10.1021/acs.analchem.0c02060
Marovich M, Mascola JR, Cohen MS (2020) Monoclonal antibodies for prevention and treatment of Covid-19. JAMA. https://doi.org/10.1001/jama.2020.10245
Padron-Regalado E (2020) Correction to: Vaccines for sars-Cov-2: Lessons from Other Coronavirus Strains. Infect Dis Ther. https://doi.org/10.1007/s40121-020-00371-w
Catanzaro M, Fagiani F, Racchi M, Corsini E, Govoni S, Lanni C (2020) Immune response in Covid-19: addressing a pharmacological challenge by targeting pathways triggered by Sars-Cov-2. Signal Transduct Target Ther 5(1):84. https://doi.org/10.1038/s41392-020-0191-1
Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR, Rawlings SA, Sutherland A, Premkumar L, Jadi RS, Marrama D, de Silva AM, Frazier A, Carlin AF, Greenbaum JA, Peters B, Krammer F, Smith DM, Crotty S, Sette A (2020) Targets of T cell responses to Sars-Cov-2 coronavirus in humans with Covid-19 disease and unexposed individuals. Cell 181(7):1489–1501.e15. https://doi.org/10.1016/j.cell.2020.05.015
Khan M, Khan S, Ali A, Akbar H, Sayaf AM, Khan A, Wei DQ (2019) Immunoinformatics approaches to explore helicobacter pylori proteome (virulence factors) to design B and T cell multi-epitope subunit vaccine. Sci Rep 9(1):13321. https://doi.org/10.1038/s41598-019-49354-z
Blum JS, Wearsch PA, Cresswell P (2013) Pathways of antigen processing. Annu Rev Immunol 31:443–473. https://doi.org/10.1146/annurev-immunol-032712-095910
Querec T, Bennouna S, Alkan S, Laouar Y, Gorden K, Flavell R, Akira S, Ahmed R, Pulendran B (2006) Yellow fever vaccine Yf-17d activates multiple dendritic cell subsets via Tlr2, 7, 8, and 9 to stimulate polyvalent immunity. J Exp Med 203(2):413–424. https://doi.org/10.1084/jem.20051720
PrabhuDas M, Adkins B, Gans H, King C, Levy O, Ramilo O, Siegrist CA (2011) Challenges in infant immunity: implications for responses to infection and vaccines. Nat Immunol 12(3):189–194. https://doi.org/10.1038/ni0311-189
Ishikawa H, Barber GN (2008) Sting is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455(7213):674–678. https://doi.org/10.1038/nature07317
Acknowledgements
The authors would like to thank the Deanship of Scientific Research at Umm Al-Qura University for supporting this work by Grant Code: (20UQU0064DSR).
Author information
Authors and Affiliations
Contributions
Z.A. and F.A. designed the research and contributed to new reagents. S.A. and Z.A. analyzed the analytic tools data. F.A. analyzed the data and S.A. wrote the paper. Z.A. is the principal investigator.
Corresponding author
Ethics declarations
Conflict of interest
All authors: No reported conflicts of interest. All authors have submitted form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
Ethical approval
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Disclaimer: The research sponsor did not have a role to play in the design, collection of data, review of data, presentation of the findings or writing of the paper. And the first and corresponding author had complete access to all research data and were often directly responsible for the decision to apply for publication.
Rights and permissions
About this article

Cite this article
Abduljaleel, Z., Al-Allaf, F.A. & Aziz, S.A. Peptides-based vaccine against SARS-nCoV-2 antigenic fragmented synthetic epitopes recognized by T cell and β-cell initiation of specific antibodies to fight the infection. Bio-des. Manuf. 4, 490–505 (2021). https://doi.org/10.1007/s42242-020-00114-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42242-020-00114-3