
Abstract
This study reviews recent research on volatile organic compound (VOC) emissions from motorized vehicle sources by means of online mass spectrometry. Chemical ionization is a powerful tool that usually permits soft ionization of chemical species and it allows the time-resolved measurement of multiple VOCs, even in complex samples where many kinds of VOCs coexist. The vehicular exhaust gasses are investigated using H3O+, NO+, Hg+, and CH3C(O)O− as a reagent ion in online chemical ionization mass spectrometry. The proton transfer using H3O+ as a reagent ion was used for the detection of nitro-organic compounds such as nitromethane and nitrophenol. The time-resolved measurement of the nitro-organic compounds in the laboratory experiments with a chassis dynamometer system revealed their emission properties, such as the dependence of the emissions as a function of vehicular velocity and acceleration/deceleration, as well as the effect of various types of exhaust gas treatment. The data regarding the nitromethane and nitrophenol emissions obtained in the field measurements were consistent with the results of the laboratory experiments done with a chassis dynamometer system. In the experiments involving evaporative emissions from gasoline-powered cars, NO+ was used as a reagent ion. Online measurements showed that the adsorption of hydrocarbons in a sealed housing evaporative determination unit could result in emissions being underestimated, if the concentrations are monitored only before and after a diurnal breathing loss test. The composition analysis gave an estimated ozone formation potential (OFP) approximately 20 % higher for breakthrough emissions and refueling emissions than for the gasoline that was tested, but the OFP for the permeation emissions was almost the same as the OFP for the test fuel.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chemical ionization usually permits the soft ionization of chemical species [1] and it allows the time-resolved measurement of multiple volatile organic compounds (VOCs) even in a complex sample where many kinds of VOCs coexist. Some techniques have been developed for the purpose: for example, proton transfer reaction mass spectrometry (PTR-MS) [2–5], selected ion flow tube mass spectrometry (SIFT-MS) [6], ion-molecule reaction mass spectrometry (IMR-MS) [7], ion attachment mass spectrometry (IA-MS) [8], and negative ion chemical ionization mass spectrometry (NI-CI-MS) [9]. Unlike gas chromatography, they do not usually require any sample treatment such as preconcentration. One of the main weaknesses of online chemical ionization mass spectrometry (CI-MS) is its reliance solely on mass spectrometry for discriminating between molecules, which means that isobaric and isomeric species cannot be distinguished. In addition to overlapping the ion signals of the isobaric and isomeric species, fragment ions from high-molecular weight species may interfere with the ion signal of a target molecule [10]. In order to overcome such an interference to apply CI-MS to the time-resolved measurement of VOCs in the complex sample, the reagent ion for the chemical ionization must be selected properly.
Automobiles are major sources of VOCs that are emitted not only from their tailpipes while running but also as evaporative emissions by gasoline vehicles [11]. Some reagent ions have been used for an online analysis of automotive exhaust gasses and are listed in Table 1. Hydronium ions (H3O+) and NO+ are used as a reagent ion in SIFT-MS [6] and resent proton transfer reaction plus switchable reagent ion mass spectrometry (PTR + SRI-MS) [18]. Hydronium ions react with VOCs that have a proton affinity (PA) higher than that of water to give a proton (i.e., proton transfer):
Most VOCs, including alkenes (except ethylene), aromatic hydrocarbons, oxygenated VOCs, and acetonitrile, can be ionized by reaction (1). However, it is known that reaction (1) does not occur for alkanes given that they have lower PAs than water [19]. A method to measure C12 − C18 n-alkanes using PTR-MS was demonstrated [20]. They were detected by a series of fragment ions with formula C n H2n+1. Similar results were reported for branched alkanes [21]. Since alkanes are one of the major components of fuel, the fact suggests that the ion signals of alkanes interfere with ion signals of alkenes in the PTR-MS detection.
Reactions of VOCs with NO+ have been extensively investigated by SIFT-MS [6]. Major reaction paths are reported as follows:
Alkanes can be detected as an [M − H]+ by using the NO+ ion as a reagent ion [22] and the ion signals of alkanes are detected separately from the ion signals of alkenes.
This paper provides a review of recent research on VOC emissions from vehicular sources by means of online mass spectrometry. As mentioned before, it is not easy to detect compounds without the interference of ion signals in automotive emissions. Recent research showed that nitro-organic compounds were detectable without substantial interference from other compounds by PTR-MS and the real-time measurement of the nitro-organic compounds such as nitromethane and nitrophenol emitted in diesel exhaust was carried out during a transient driving cycle tested on a chassis dynamometer system [23•] and in ambient air at a busy intersection [24•]. As pointed out later, an online measurement method is essential to measure nitro-organic compounds in automotive emissions. Another study found that the time-resolved measurement of C4 − C7 alkanes, C4 − C7 alkenes, and some aromatics such as benzene, toluene, C8-benzenes, C9-benzenes, and naphthalene was carried out using NO+ as the reagent ion for the first time, in order to determine evaporative and refueling emissions from gasoline-powered cars in Japan [25•, 26•]. The online measurement showed the adsorption of hydrocarbons in a sealed housing evaporative determination (SHED) unit, resulting in evaporative emissions being underestimated.
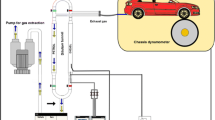
VOC tailpipe emissions are investigated by a chassis dynamometer system, equipped with a constant volume sampler (“dilution tunnel”) [27]. A schematic diagram is shown in Fig. 1. The sample from the dilution tunnel was introduced into a mass spectrometer through a 2.5-μm cut cyclone to remove particulate matter from the automotive exhaust. The regulated substances from automobiles such as CO, CO2, total hydrocarbon (THC) (or non-methane hydrocarbon (NMHC)), and nitrogen oxides (NO x ) are usually monitored, although it is not shown in Fig. 1. Evaporative emissions from gasoline-powered vehicles are investigated using a SHED unit [11]. A schematic diagram of the measurement of evaporative emissions and refueling emissions is shown in Fig. 2. A diurnal breathing loss (DBL) test lasts for 24 h and the temperature inside the SHED is raised from 20 to 35 °C and then reduced again to 20 °C in order to reproduce diurnal change on a sunny day [11]. For a refueling emission test, the test is started upon removing the fuel tank cap and then inserting an oil feed nozzle from a gasoline dispenser into the fuel tank of a gasoline-powered vehicle as quickly as possible. The test runs for approximately 5 min. Total hydrocarbon concentration is usually monitored using a total hydrocarbon gas analyzer.

A schematic diagram of the constant volume sampler and method for sampling the diluted exhaust gas

A schematic diagram of the measurement of evaporative emissions and refueling emissions
Nitro-Organic Compounds in Vehicular Exhaust
Diesel engines are an important part of the transportation sector and because of their superior fuel efficiency and high power output, their use is predicted to continue to grow. Despite these advantages, however, diesel exhaust largely contributes to air pollution. Combustion of diesel fuel results in substantial emissions consisting of NO x , VOCs, and diesel exhaust particles (DEPs) [28–31]. NO x and VOCs are thought as precursors of photochemical ozone and particulate matter (PM), resulting in urban and regional haze [32, 33]. DEPs contain various organic compounds, including polyaromatic hydrocarbons (PAHs), oxygenated PAHs (oxy-PAHs), and nitrated PAHs (nitro-PAHs) [23•, 34]. Some VOCs and DEPs themselves are toxic, causing adverse health effects in humans [35, 36]. To reduce NO x and DEP emissions, exhaust gas treatment (“aftertreatment”) have been extensively investigated besides fuel reformulation and engine redesign [23•, 28, 29, 37–39].
New technologies for aftertreatments have been developed to decrease NO x and DEP emissions from diesel-powered vehicles [29, 37–39]. Selective catalytic reduction (SCR) and lean NO x traps (LNTs) can be used to control NO x emissions. SCR works by reducing NO x on a selective catalyst using an ammonia-based reductant (e.g., urea, (NH2)2CO). LNTs store NO x as nitrates on alkaline earth materials under lean conditions and allow the nitrates to dissociate under slightly rich conditions every minute or so, releasing the NO x , which are then converted by an integrated three-way catalyst [38]. A diesel particulate filter (DPF) has been developed to control PM, and all new diesel-powered vehicles in Europe, Japan, and the USA have, or will shortly have, DPFs [38]. A DPF is usually coupled with a diesel oxidation catalyst (DOC), which oxidizes hydrocarbons (HCs) to CO2, burning the fuel to actively regenerate the filter and generating NO2 to passively regenerate the DPF [38]. It has been reported that DPFs are very effective at converting gaseous HCs and PM-associated PAHs [40]. However, the secondary production of some nitro-PAHs during catalytic aftertreatments has been found in several recent studies [41, 42]. Locating a DOC upstream of a DPF allows the intentional conversion of NO to NO2, to accelerate the oxidation of accumulated PM in the DPF [43].
It is thought that nitro-PAHs, which account for a major portion of direct acting mutagens contained in DEPs [44, 45], are produced during fuel combustion, by nitration of the corresponding PAHs [46, 47]. As mentioned above, some nitro-PAHs are produced secondarily during catalytic aftertreatment [41, 42]. In addition to nitro-PAHs, several kinds of nitrophenols have been observed in DEP emissions [48, 49], which show vasodilatory activity and estrogenic and anti-androgenic activity [50]. Speciation and quantification of the nitro-organic compounds in DEPs have been extensively conducted via the collection of DEP samples on filters followed by gas chromatography (GC) or liquid chromatography analyses [29]. However, there are few studies in which emissions of gaseous nitro-organic compounds in diesel exhaust are investigated [23•]. When quantifying nitro-organic compounds in diesel vehicle exhaust, it should be noted that organic compounds could be easily nitrated on the filter during sampling, which results in “artifacts” [51]. An online measurement method is therefore essential to avoid these artifacts, and time-resolved measurements are helpful for revealing in which driving mode each nitro-organic compound is generated [23•]. In addition, such information will be useful for improving engine design and aftertreatment in order to reduce nitro-organic compound emissions [23•].
Laboratory Experiments Using a Chassis Dynamometer System
A recent experiment used PTR-MS for online analysis of gaseous nitro-organic compounds in diesel vehicle exhaust [23•]. The time-resolved measurement of the nitro-organic compounds revealed their emission properties, including the dependence of the emission as a function of vehicular velocity and acceleration/deceleration as well as the effect of various types of aftertreatment [52•, 53•].
When detecting nitro-organic compounds by PTR-MS, protonated species (M·H+) usually give ion signals at odd m/z values. But protonated mono-nitro-organic compounds usually give ion signals at even m/z values as Inomata et al. (2013) [23•] pointed out. Protonated mono-nitro-organic compounds observed at even m/z tend to be buried, due to an overlap of 13C isotopologue signals of protonated organic compounds, also observed at even m/z. The 13C isotopologue signal can easily be calculated from the protonated organic compound signal. By subtracting the contributions of the 13C isotopologue signals from the ion signals at even m/z values, more accurate emission properties of the nitro-organic compounds can be obtained [23•].
Nitro-organic compound emissions were investigated from the two light-duty trucks with either a diesel oxidation catalyst (vehicle-DOC) or a diesel PM-NO x reduction system (vehicle-DPNR), and a heavy-duty diesel truck fitted with a urea-selective catalytic reduction system (vehicle-SCR) [23•]. In addition, a compact gasoline-powered passenger car, which has a three-way catalyst as the aftertreatment (vehicle-GASOLINE) was also tested [52•, 53•]. The emission levels of regulated substances, such as CO, THC (or NMHC), NO x , and PM in diesel exhaust is related to the installed aftertreatment system type. For example, a DOC-only aftertreatment is an older system meeting 2003 Japanese regulations. DPNR and urea SCR are newer systems, which were developed to meet 2005 Japanese regulations.
The PTR mass spectrum obtained for diesel exhaust gasses consists of many signals attributable to a series of alkenes and/or unsaturated aldehydes/ketones (m/z 43, 57, 71, 85, 99, 113, etc.), saturated aldehydes/ketones (m/z 31, 45, 59, 73, 87, 101, 115, etc.), and aromatics (m/z 79, 93, 107, 121, 135, 149, etc.) [6, 13, 23•]. To identify the mono-nitrogen containing compounds from the ion signals in the spectrum, the ratio of an ion signal at an even m/z [M] to the adjacent ion signal at an odd m/z [M − 1], against even m/z values (I even/I odd) was introduced as an indicator of mono-nitro-organic compounds by Inomata et al. (2013) [23•]. If there is no signal from mono-nitrogen containing compounds at any even m/z [M] and the ion signals at the even m/z [M] are attributed only to 13C isotopologue signals of the compounds at m/z [M − 1], then the I even/I odd ratios should range from 0.01 to 0.18, depending on the number of carbons from C1 to C16. However, high ratios were observed at some m/z values highlighted in the figure, suggesting that there were substantial contributions from mono-nitrogen containing compounds when the 13C isotopologue contributions of the adjacent odd ion signals were subtracted. In the exhaust gasses of vehicle-DOC, the residual signals at the m/z values with high ratios were assigned as follows: acetonitrile (CH3CN) at m/z 42, NO2 + (fragment from alkyl nitrates [54]) at m/z 46, acrylonitrile (C2H3CN) at m/z 54, nitromethane (CH3NO2) at m/z 62, methyl nitrate (CH3ONO2) at m/z 78), ethyl nitrate (C2H5ONO2) at m/z 92, nitrophenol at m/z 240, C7-, C8-, C9-, and C10-nitrophenols at m/z 154, m/z 168, m/z 182, and m/z 196, respectively, and dihydroxynitrobenzenes at m/z 156. The result indicated the presence of nitro-organic compounds, such as nitromethane, nitrophenols, and dihydroxynitrobenzenes in diesel exhaust, along with hydrocarbons, aromatics, and oxygenated VOCs. The presence of acetonitrile in diesel-powered engine exhaust has been shown by means of PTR-MS [13, 14], and vehicular exhaust has been shown to emit nitromethane [55] and gaseous nitrophenols by means of GC/MS [49, 56].
Temporal variations of the mixing ratios for three nitro-organic compounds (nitromethane, nitrophenol, and dihydroxynitrobenzenes) along with CO2, CO, NO x , CH4, N2O, and the selected NMVOCs, including benzene, toluene, acetone, acetic acid, phenol, and acetonitrile during a transit cycle (JE05 for diesel-powered vehicles and JC08 for a gasoline-run car) were measured for vehicle-DOC [23•]. Two types of emission peaks, sharp and broad, were observed in the temporal profiles [23•]. Sharp peaks were typically observed for light molecules such as CO, NO x , and hydrocarbons (CH4 and benzene); these sharp peaks were synchronized with acceleration processes. In contrast, broad peaks were observed, particularly for oxidized NMVOCs, including acetic acid and phenol, and were located after the sharp peaks. The nitromethane peaks fell into the sharp peak category, whereas the peaks of nitrophenol and dihydroxynitrobenzenes fell into the broad peak category [23•].
It was found that nitromethane emission was strongly correlated with emissions of CO, benzene, and acetone [23•]. Nitrophenol emissions were moderately correlated with phenol emissions, multiplied by NO2 emissions, suggesting that both phenol and NO2 are related to the production of nitrophenol [23•]. The effects of the oxidation catalyst on nitro-organic compound emissions were investigated by performing the transit cycle experiments with the catalytic converter of vehicle-DOC, replaced with a straight pipe. Nitromethane emissions did not change while nitrophenol emissions were suppressed, in addition to acetic acid, phenol, and acetonitrile [52•, 53•]. This suggests that the oxidation catalyst played an important role in the production of gaseous nitrophenol.
Nitromethane emissions were commonly observed for vehicles-DPNR and -SCR; however, obvious emissions of other nitro-organic compounds, including nitrophenol, were not. It was concluded that nitrophenol emissions depend on the vehicle, possibly due to the type of aftertreatment installed, while nitromethane emissions do not [23•, 52•]. Nitromethane is likely produced near the engine [23•, 52•]. Higher emission ratios of nitromethane to CO and benzene relative to those for vehicle-DOC were observed for vehicle-SCR. This was probably caused by the suppression of CO and benzene emissions in vehicle-SCR [23•, 52•]. For vehicle-DPNR, CO and benzene emissions were completely suppressed [23•, 52•]. It was also found that CO and VOC emissions appear to be suppressed by the newer aftertreatment systems, urea-SCR and DPNR but that nitromethane emissions were not suppressed for vehicle-SCR and were only partially suppressed for vehicle-DPNR [23•, 52•].
While strong nitromethane emissions were observed from the first moment of the first stage of the cold-start experiments for vehicle-GASOLINE, nitromethane emissions were weak throughout the hot-start experiment. This result suggests that nitromethane was produced within the gasoline engine and that aftertreatment (i.e., the three-way catalyst) substantially reduced the nitromethane emissions. It was suggested that the reduction function in the three-way catalyst played a role in reducing nitromethane emissions. Indeed, the emission factor of nitromethane for vehicle-DPNR was the smallest among those of the diesel trucks, probably because the PM-NO x reduction system was improved through use of a three-way catalyst [52•].
From the dependence of nitromethane emissions on vehicular velocity and acceleration/deceleration, the following properties of nitromethane emissions from vehicle-DOC and vehicle-SCR were observed: (1) Emissions were strong at low vehicle velocities (0–20 km h−1). (2) Emissions generally increased with increasing acceleration. (3) At high vehicle velocities (70–90 km h−1), emissions were slightly greater than those at moderate vehicle velocities (50–70 km h−1) [52•].
Field Measurement of Nitro-Organic Compounds
A field study was conducted at a busy intersection in Kawasaki, a large city in Japan, where nitro-organic compounds were measured in wintertime from February 26 to March 6, 2011 [24•]. Nitromethane, nitrophenols, nitrocresols, difhydroxynitrobenzenes, nitrobenzene, nitrotoluenes, and nitronaphthalenes along with some related VOCs by PTR-MS were continuously monitored.
Nitromethane was usually observed among air pollutants emitted from vehicles; temporal variation of the mixing ratios of nitromethane was similar to that of NO x [24•]. The mixing ratios of nitromethane varied substantially and sometimes clearly varied at an approximately constant interval [24•]. The interval corresponded to the traffic signal cycle at the intersection, and it was found that fast-moving diesel-powered trucks emitted nitromethane, and that air with high mixing ratios of nitromethane reached the measurement site [24•]. This was consistent with the laboratory experiment results using a chassis dynamometer system, where emissions of nitromethane were observed at high vehicular velocity [52•]. In addition to the regular peaks of nitromethane, sharp increases of the nitromethane mixing ratios were irregularly observed, a result of emissions released from vehicles turning around the corner in front of the observation site [24•]. This was also consistent with the results of the laboratory experiments, i.e., that nitromethane emissions were strong at acceleration with low vehicular velocity [52•]. During the measurement, the average and the maximum of the nitromethane mixing ratios were 0.16 and 4.6 ppbv, respectively [24•]. Interestingly, an investigation of the relationship between the nitromethane emissions and the CO and benzene emissions showed that the observed data was located in the range between the emission ratios previously obtained from vehicle-DOC and vehicle-GASOLINE, suggesting that the major sources of nitromethane emissions at the intersection were relatively old diesel-run vehicles [24•].
For other nitro-VOCs, such as nitrophenol, nitrocresol, DHNB, nitrobenzene, nitrotoluene, and nitronaphthalene, most of the data fluctuated within the detection limit, with occasional emission peaks of these compounds observed [24•]. Indeed, the maximum mixing ratios of these compounds were higher than the detection limits [24•]. Further improvements of the detection sensitivity and/or the S/N ratios are necessary in order to obtain meaningful data for these nitro-organic compounds, even at a busy intersection.
Very recently, nitromethane was measured as a tracer for diesel exhaust in the Kathmandu Valley in Nepal in the winter of 2012–2013 by means of high-mass resolution proton transfer reaction time-of-flight mass spectrometry (PTR-TOF-MS), which makes it possible to differentiate ion signals of protonated nitromethane (m/z 62.024) from those of 13C isotopologue signals of protonated acetic acid (m/z 62.032) [57]. The nitromethane mixing ratios observed in the Kathmandu Valley were similar to those at the busy Japanese intersection [24•].
Evaporative Emissions from Gasoline-Powered Vehicles
Evaporative emissions from gasoline-powered vehicles are substantial among the total VOC emissions in Japan according to the recent estimation [11]; however, recent VOC reduction strategies in Japan have focused on tailpipe emissions rather than evaporative emissions. Alkanes, alkenes, and aromatics are major components of gasoline [20]. Evaporative emissions consist of “permeation” (the leakage of fuel from the fuel tank and piping) and “breakthrough” (the overflow of fuel vapor from the carbon canister). These components have been well-studied in the USA [58–61] and regulated since the 1970s. Current US regulations are the most strict, requiring a running loss test is administered, which examines one of the major VOC emissions from light-duty gasoline-powered vehicle [62, 63]; this is in addition to two- or three-day diurnal breathing loss (DBL) and hot soak loss (HSL) tests. Regulations in Europe and Japan still only require one day DBL and HSL tests, which stopped being used in the USA decades ago. In addition, Japan is one of the countries where refueling emissions have not been controlled, even though prevention techniques have been already established and introduced in many other countries. Refueling gasoline emissions are also a serious problem in countries where refueling emissions have not been controlled, including Japan.
It is well-known that different VOC species have different impacts on the formation of ozone. Therefore, it is important to assess the effects of VOC emissions using an ozone formation potential (OFP) parameter [62, 64–66], such as the maximum incremental reactivity (MIR) [67, 68]. Because of this, automobile exhaust evaluations using an OFP have often been performed not only on tailpipe exhaust [69–77] but also on evaporative emissions [78–80]. In those studies, the VOC compositions were analyzed using traditional analytical chemistry techniques such as GC with flame ionization detection (GC/FID) or GC/MS. While these methods are excellent for identifying and quantifying VOC species, samples of vehicular exhaust gas have to be stored for analysis in Teflon bags or air sampling canisters, where sample hydrocarbons with relatively high-molecular weights have been found to be lost when stored in air sampling canisters [81]. These losses have a serious impact on DBL tests using a sealed housing evaporative determination (SHED) unit as such tests last for 24 h and measurements are usually only conducted before and after the test.
Permeation and Breakthrough Emissions During Parking on a Sunny Day
In order to estimate the OFPs for permeation and breakthrough emissions produced during the DBL tests, an online measurement of evaporative VOCs from gasoline-run vehicles was carried out using a PTR + SRI-MS and NO+ as a reagent ion [25•]. Emissions that occur during a DBL test are usually estimated by subtracting the initial analyte concentration from the final concentration. However, there is a risk of underestimating emissions using this approach because the analytes can be adsorbed onto the walls of the SHED, due to the DBL tests being conducted for long periods (up to 24 h). The PTR + SRI-MS method can be used to perform real-time measurements, and by assessing the emission profiles over time, the effects of adsorption on the emission measurements could be estimated.
From the time profiles of the concentrations of some aromatic and alkane compounds over time during the DBL test, the concentration of every analyzed species increased during the rising temperature phase, but the time profile for the aromatic compound with a molecular mass of 120 (trimethylbenzens and/or ethylmethylbenzenes and/or propylbenzenes) clearly decreased after 15 h. The concentrations of the aromatic compound with a molecular mass of 106 (xylenes and/or ethylbenzene) and the alkane with a molecular mass of 100 (heptanes) also decreased in the latter part of the DBL test. These results indicate that adsorption can occur during DBL tests in a SHED. The relatively heavy compounds, which have low volatilities, were particularly affected by adsorption. Thanks to real-time measurement, it was found that determining the concentrations of the analytes only before and after a DBL test, would have led to an underestimation of these compound emissions. The evaporative emissions of each component were derived by subtracting the initial concentrations from the maximum concentrations (not the final concentrations) to avoid the possibility of adsorption by the SHED. It should be noted that values obtained by subtracting the initial concentrations from the maximum concentrations may still be underestimated, because emissions only reach the maximum when the desorption rate and the permeation rate are balanced.
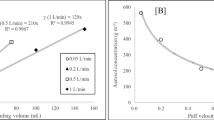
The species that were detected by PTR + SRI-MS were the aromatic compounds benzene, toluene, xylene/ethylbenzene (C8-benzenes), trimethylbenzene/other isomers (C9-benzenes), naphthalene, and the aliphatic compounds C4–C7 alkanes and C4–C7 alkenes. In Fig. 3a, mass ratios of three components, aromatics, alkanes, and alkenes, obtained for the test fuel, the permeation emissions, and the breakthrough emissions are shown. In the test fuel, aromatics, alkenes, and alkenes were contained with a ratio 0.34:0.48:0.18. In the permeation emissions, it was found that aromatics, alkanes, and alkenes were emitted and that the mass ratios were accidentally similar to those of the test fuel, although the C3 species present in the test fuel were not measured. In addition, almost no C8 branched-chain alkanes were detected in the permeation emissions even though they were present in the test fuel. In contrast, only the emissions of alkanes and alkenes were observed in the breakthrough emissions, and the emissions of aromatics were not observed. This is probably because aromatic hydrocarbons have lower volatility than alkanes and alkenes observed in the breakthrough emissions. Indeed, the alkanes and alkenes were both dominated by C4 and C5 species.

a Mass ratios of three components, aromatics, alkanes, and alkenes, obtained for the test fuel, the permeation emission, and the breakthrough emission. b Ozone formation potential of the test fuel, the permeation emission, and the breakthrough emission
The OFP of the test fuel, the permeation emissions, and the breakthrough emissions were calculated as the sum of the individual MIR values:
The MIR values shown were taken from a report by the California Environmental Protection Agency (2009) [82]. The derived OFPs for the test fuel, the permeation emissions, and the breakthrough emissions are shown in Fig. 3b. The OFP of the permeation emissions was 3.31, which was similar to the OFP of the test fuel (3.33). The OFP of the breakthrough emissions was 3.89, which was 17 % higher than the OFP of the test fuel. This is probably because the breakthrough emissions contained large amounts of relatively small alkenes, which have higher MIR values than do aromatic compounds.
Refueling Emissions
Prevention techniques regarding refueling emissions have already been established and introduced in many countries. The strategy for preventing refueling emissions in the USA is to employ an onboard-refueling vapor recovery (ORVR) system, which traps the vapor displaced by the liquid fuel entering into the tank, by using a carbon canister installed in the car. Unlike the USA, in Europe, the refueling emissions are vacuumed by a stage II system installed on a gasoline dispenser [83]. These two are the commonly used techniques, and often countries wishing to control refueling emissions choose one of them after considering their merits and disadvantages. The ORVR system not only exhibits high trapping efficiency (95 %) but also reduces evaporative emissions dramatically. The benefits of ORVR will be fully delivered after most vehicles on the road are replaced with ORVR vehicles, and this may take more than 10 years. In contrast, although the real world trapping efficiency of stage II systems is only 70 %, they can be implemented more quickly [84]. The theoretical efficiency of stage II system is similar to that of ORVR; in reality, however, they are often not well-maintained, which results in lowered efficiency. Japan is one of the countries where refueling emissions have not been controlled and there is also no data on the effectiveness of control techniques for refueling emissions for the country. There were many studies discussing the effectiveness of these techniques in the USA in the 1980s. However, the current situation in Japan is quite different from the situation in the USA in the 1980s, such as car specifications, fuel composition, and fuel dispenser specifications. The results of these evaluations will be useful not only to Japan but also to many other countries that have not yet introduced refueling emission controls.
Alkanes, alkenes, and diene species concentrations from carbon numbers 3 to 11 were monitored in the SHED with PTR + SRI-MS using NO+ as a reagent ion [26•]. Benzene, alkyl benzenes, naphthalene, and alkyl naphthalenes were also monitored. Mass ratios of three components, aromatics, alkanes, and alkenes, obtained for the test fuel and the refueling emission are shown in Fig. 4a. The SHED temperature and the fuel temperature were set to 20 °C. It was found that the detailed composition of the refueling emissions was quite different from that of the fuel. The ratio of aromatic compounds in the refueling emissions was less than 10 % and the main components were C4–C6 alkanes and alkenes. These features were similar to the breakthrough emissions of evaporative emissions [25•]. The estimated OFP of the refueling emissions was 4.21 ± 0.35 higher than that of the test fuel (3.78) (Fig. 4b). It was suggested that the difference in MIR between the breakthrough emissions and the test fuel was almost the same as that between the refueling emissions and the test fuel. It was proposed that the OFPs of breakthrough emissions (OFP breakthrough ), refueling emissions (OFP refueling), and permeation emissions (OFP permeation ) are related by:
where OFP fuel is the OFP of the test fuel [26•].

a Mass ratios of three components, aromatics, alkanes, and alkenes, obtained for the test fuel and the refueling emissions. b Ozone formation potential of the test fuel and the refueling emissions
By changing the SHED temperature and the fuel temperature from 5 to 35 °C, the temperature dependence of the composition was also measured [26•]. Aromatics (summed) showed a gradual increase with increasing temperature. The emissions of alkanes (summed), alkenes (summed), and dienes (summed) seemed to be constant, having no dependence on the temperature. However, the detailed emissions of individual species indicated that the compositions changed as the temperature changed, even in alkanes, alkenes and dienes. However, there was no significant temperature dependence of the MIR of the refueling emissions.
Concluding Remarks
This study reviews recent research on VOC emissions from motorized vehicle sources by means of online mass spectrometry. Chemical ionization is a powerful tool that usually permits soft ionization of chemical species and it allows the time-resolved measurement of multiple VOCs, even in a complex sample where many kinds of VOCs coexist. An online CI-MS was used to investigate the automotive exhaust gasses using an H3O+, NO+, Hg+, and CH3C(O)O− reagent ion. The reagent ion for the chemical ionization must be selected properly so as to overcome interference from other compounds when applying CI-MS to the time-resolved measurement of VOCs in a complex sample.
The proton transfer using H3O+ as a reagent ion was used for the detection of nitro-organic compounds such as nitromethane and nitrophenol. It is known that substantial fragmentation occurs during photoionization of nitro-organic compounds [85]. Therefore, it is useless to detect nitro-organic compounds by mass spectrometry coupled with photoionization. This suggests that parent ions (M+) of nitro-organic compounds are fragile and ionizations other than proton transfer may be not effective for the detection of nitro-organic compounds. To identify the compounds containing mono-nitrogen oxides from many ion signals in the mass spectrum, the ratio of an ion signal at an even m/z [M] to the adjacent ion signal at an odd m/z [M − 1] against even m/z values (I even/I odd) was introduced as an indicator of organic mono-nitrogen compounds. Owing to a recent advance of mass spectrometry, a high-mass resolution TOFMS makes it possible to differentiate ion signals of protonated organic mono-nitrogen compounds from those of 13C isotopologue signals of the adjacent odd ion signals. The time-resolved measurement of the nitro-organic compounds in the laboratory experiments using a chassis dynamometer system revealed emission properties, such as the dependence of the emissions as a function of vehicular velocity and acceleration/deceleration as well as the effect of various types of aftertreatment. The data regarding the nitromethane and nitrophenol emissions obtained in the field measurements were consistent with the laboratory results from experiments conducted using a chassis dynamometer system.
In the experiments examining evaporative emissions from gasoline-run cars, NO+ was used as a reagent ion given that besides alkenes and aromatic hydrocarbons, alkanes can be detectable as a sole ion, [M–H]+ [22]. The ion was not overlapped with the product ion of alkenes and aromatic hydrocarbons, which are usually detected as M+; note that aldehydes give [M–H]+ if they are. Fortunately, there is no aldehyde in the evaporative emissions. The high-mass-resolution TOFMS, however, does make it possible to differentiate ion signals of alkanes from those of aldehydes. The online measurements by means of NO+ ionization PTR + SRI-MS showed that the adsorption of hydrocarbons in a SHED unit could result in emissions being underestimated if the concentrations are monitored only before and after a DBL test. Based on the composition analysis, OFP was estimated at approximately 20 % higher for the breakthrough emissions and the refueling emissions than for the gasoline that was tested, but the OFP for the permeation emissions was almost the same as the OFP for the test fuel. In the papers [25•, 26•], cycloalkanes were not quantified because cycloalkanes were relatively minor components compared with alkanes, alkenes, and aromatics in fuels. Cycloalkanes can also be detectable as a sole ion, [M–H]+, in which case the ion signal does not overlap with those of alkanes, alkenes, nor aromatics (Inomata S., unpublished data).
In IMR-MS, Hg+, Xe+, and Kr+ are used as a reagent ion [7, 15]. The ionization is based on the charge transfer from the detective molecule (M) to the reagent ion (R+).
Reaction (6) can occur only for the compounds that have an ionization potential (IP) lower than that of the primary ion (Hg 10.44 eV; Xe 12.13 eV; Kr 14.00 eV) [86]. Since the IPs of benzene, toluene, ethylbenzene, p-xylene, o-xylene, and m-xylene are 9.24, 8.83, 8.77, 8.44, 8.56, and 8.55 eV, respectively [86], these compounds can be detected using Hg+ as the reagent ion [15]. The reaction channel (2b) using NO+ as the reagent ion is the same charge transfer reaction as reaction (6). Since the IP of NO is 9.26 eV [86] that is lower than those of Hg, Xe, and Kr, the excess energy in reaction (2b) is smaller than those in reaction (6), resulting in less fragmentation when using NO+ as the reagent ion. However, it should be noted that the reaction is switched to reaction (2c) when the IP of the analyte is higher than that of NO. For example, 3-hexene (IP ∼9 eV [86]) is detected by the M+ ion while the [M + NO]+ ion is produced for 1-hexene (IP = 9.44 eV [86]) by NO+ ionization PTR + SRI-MS (Inomata S., unpublished data).
IA-MS is known as fragmentation-free mass spectrometry, in which ionization can be performed because a neutral molecule becomes an adduct ion by the attachment of Li+ (in most cases) with a small amount of energy by the Coulomb force [8, 87].
However, there is little research on the direct analysis of automotive exhaust gasses by IA-MS. This is probably because the Li+ ion as the reagent ion would be substantially consumed by water vapor that is present in the automotive exhaust gasses. IA-MS works well only under dry conditions and the direct analysis of DEPs was demonstrated by IA-MS [87]. IA-MS might be suitable for the analysis of the evaporative and refueling emissions.
NI-CI-MS was recently applied for online measurements of isocyanic acid (HNCO) besides organic acids, HONO, HNO3 [16]. CH3C(O)O− was used as a reagent ion. The ionization is based on a proton transfer from the detective molecule (M) to CH3C(O)O−.
Reaction (8) occurs for acids that are stronger than acetic acid (CH3C(O)OH). Therefore, many acids such as formic acid, acrylic acid, propionic acid, glycolic acid, methacrylic acid, butyric acid, pyruvic acid, lactic acid, benzonic acid, nitrophenols, and pentafluoropropionic acid can become detectable by this ionization [88]. It was demonstrated that isocyanic acid could be measured as the protonated molecule in ambient air using a PTR-TOF-MS instrument in the Kathmandu Valley [57].
Online CI-MS is useful for the analysis of not only the automotive exhaust gasses but also emitted gasses from biomass burning. Biomass burning is the largest source of primary fine carbonaceous particles and the second largest source of trace gasses in the global atmosphere [89]. PTR-MS is extensively used for the analysis [90–94] and NI-CI-MS is also used [95, 96]. Recently, Inomata et al. (2015) [94] pointed out the importance of online measurements in the analysis of emitted gasses, especially oxygenated VOCs, from biomass burning, given that they found that emission factors for oxygenated VOCs obtained by using online PTR-MS tended to be higher than those measured using cartridges to collect gaseous NMVOCs. It should be again noted, however, that one of the main weaknesses of CI-MS is its reliance solely on mass spectrometry for discriminating between molecules, which means that isomeric species cannot be distinguished. Instead, GC/FID and GC/MS are excellent methods for identifying VOC species.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Harrison AG. Chemical ionization mass spectrometry 2nd edition. Boca Raton: CRC Press; 1992.
Lindinger W, Hansel A, Jordan A. Proton-transfer-reaction mass spectrometry (PTR-MS): on-line monitoring of volatile organic compounds at pptv levels. Chem Soc Rev. 1998;27:347–54.
de Gouw J, Warneke C. Measurements of volatile organic compounds in the earth’s atmosphere using proton-transfer-reaction mass spectrometry. Mass Spectrom Rev. 2007;26:223–57.
Blake RS, Monks PS, Ellis AM. Proton-transfer reaction mass spectrometry. Chem Rev. 2009;109:861–96.
Ellis AM, Mayhew CA. Proton transfer reaction mass spectrometry, Wiley, 2014.
Smith D, Španěl P. Selected ion flow tube mass spectrometry (SIFT-MS) for on-line trace gas analysis. Mass Spectrom Rev. 2005;24:661–700.
Schubert H, Guntow U, Hofmann K, Schlögl R. Performance and application potential of ion-molecule reaction mass spectrometry (IMR-MS) in the analysis of complex gas mixtures. Fresenius J Anal Chem. 1996;356:127–37.
Selvin PC, Iwase K, Fujii T. Design and performance of an atmospheric pressure inlet system for lithium ion attachment mass spectrometry. Anal Chem. 2002;74:2053–7.
Huey LG. Measurement of trace atmospheric species by chemical ionization mass spectrometry: speciation of reactive nitrogen and future directions. Mass Spectrom Rev. 2007;26:166–84.
Warneke C, de Gouw JA, Kuster WC, Goldan PD, Fall R. Validation of atmospheric VOC measurements by proton-transfer-reaction mass spectrometry using a gas-chromatographic preseperation method. Environ Sci Technol. 2003;37:2494–501.
Yamada H. Contribution of evaporative emissions from gasoline vehicles toward total VOC emissions in Japan. Sci Total Environ. 2013;449:143–9.
Smith D, Španěl P, Dabill D, Cocker J, Rajan B. On-line analysis of diesel engine exhaust gases by selected ion flow tube mass spectrometry. Rapid Commun Mass Spectrom. 2004;18:2830–8.
Jobson BT, Alexander ML, Maupin GD, Muntean GG. On-line analysis of organic compounds in diesel exhaust using a proton transfer reaction mass spectrometer (PTR-MS). Int J Mass Spectrom. 2005;245:78–89.
Holizinger R, Jordan A, Hansel A, Lindinger W. Automobile emissions of acetonitrile: assessment of its contribution to the global source. J Atmos Chem. 2001;38:187–93.
Heeb NV, Forss A-M, Bach C. Fast and quantitative measurement of benzene, toluene and C2-benzenes in automotive exhaust during transient engine operation with and without catalytic exhaust gas treatment. Atmos Environ. 1999;33:205–15.
Wentzell JJB, Liggo J, Li S-M, Vlasenko A, Staebler R, Lu G, et al. Measurements of gas phase acids in diesel exhaust: a relevant source of HNCO? Environ Sci Technol. 2013;47:7663–71.
Brady JM, Crisp TA, Collier S, Kuwayama T, Forestieri SD, Perraud V, et al. Real-time emission factor measurements of isocyanic acid from light duty gasoline vehicles. Environ Sci Technol. 2014;48:11405–12.
Jordan A, Haidacher S, Hanel G, Hartungen E, Herbig J, Märk L, et al. An online ultra-high sensitivity proton-transfer-reaction mass-spectrometer combined with switchable reagent ion capability (PTR + SRI-MS). Int J Mass Spectrom. 2009;286:32–8.
Hunter KC, East ALL. Properties of C-C bonds in n-alkanes: relevance to cracking mechanisms. J Phys Chem A. 2002;106:1345–56.
Erickson MH, Gueneron M, Jobson BT. Measuring long chain alkanes in diesel engine exhaust by thermal desorption PTR-MS. Atmos Meas Tech. 2014;7:225–39.
Gueneron M, Erickson ME, VanderSchelden GS, Jobson BT. PTR-MS fragmentation patterns of gasoline hydrocarbons. Int J Mass Spectrom. 2015;379:97–109.
Inomata S, Tanimoto H, Yamada H. Mass spectrometric detection of alkanes using NO+ chemical ionization in proton-transfer-reaction plus switchable reagent ion mass spectrometry. Chem Lett. 2014;43:538–40.
Inomata S, Tanimoto H, Fujitani Y, Sekimoto K, Sato K, Fushimi A, et al. On-line measurements of gaseous nitro-organic compounds in diesel vehicle exhaust by proton-transfer-reaction mass spectrometry. Atmos Environ. 2013;73:195–203. The real-time measurement of the nitro-organic compounds by PTR-MS was carried out during a transient driving cycle tested on a chassis dynamometer system for the first time.
Inomata S, Fujitani Y, Fushimi A, Tanimoto H, Sekimoto K, Yamada H. Field measurement of nitromethane from automotive emissions at a busy intersection using proton-transfer-reaction mass spectrometry. Atmos Environ. 2014;96:301–9. The real-time measurement of the nitro-organic compounds by PTR-MS in ambient air was carried out at a busy intersection for the first time.
Yamada H, Inomata S, Tanimoto H. Evaporative emissions in three-day diurnal breathing loss tests on passenger cars for the Japanese market. Atmos. Environ. 2015; 107, 166–173. The time-resolved measurements by NO + ionization CI-MS showed that the adsorption of hydrocarbons in a sealed housing evaporative determination unit could result in evaporative emissions being underestimated for the first time.
Yamada H, Inomata S, Tanimoto H. Refueling emissions from cars in Japan: Compositions, temperature dependence and effect of vapor liquefied collection system. Atmos. Environ. 2015; 120, 455–462. NO + ionization CI-MS was applied to the analysis of the refueling emissions for the first time.
Yamada H, Misawa K, Suzuki D, Tanaka K, Matsumoto J, Fujii M, et al. Detailed analysis of diesel vehicle exhaust emissions: nitrogen oxides, hydrocarbons and particulate size distribution. Proc Combust Inst. 2011;33:2895–902.
Lloyd AC, Cackette TA. Diesel engines: environmental impact and control. J Air Waste Manage Assoc. 2001;51:809–47.
Maricq MM. Chemical characterization of particulate emissions from diesel engines: a review. J Aerosol Sci. 2007;38:1079–118.
Schauer JJ, Kleeman MJ, Cass GR, Simoneit BRT. Measurement of emissions from air pollution sources. 2. C1 through C30 organic compounds from medium duty diesel trucks. Environ Sci Technol. 1999;33:1578–87.
Siegl WO, Hammerle RH, Herrmann HM, Wenclawiak BW, Luers-Jongen B. Organic emissions profile for a light-duty diesel vehicle. Atmos Environ. 1999;33:797–805.
Atkinson R. Atmospheric chemistry of VOCs and NO x . Atmos Environ. 2000;34:2063–101.
Finlayson-Pitts BJ, Pitts Jr JM. Chemistry of the upper and lower atmosphere. New York: Academic Press; 2005.
Levsen K. The analysis of diesel particulate. Fresenius’ Z Anal Chem. 1988;331:467–78.
International Agency for Research on Cancer. Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. IARC monographs on the evaluation of carcinogenic risks to humans, 2010; 2.
International Agency for Research on Cancer, Diesel and gasoline engine exhausts and some nitroarenes. IARC monographs on the evaluation of carcinogenic risks to humans 2013; 105.
Hammerle R, Schuetzle D, Adams W. A perspective on the potential development of environmentally acceptable light-duty diesel vehicles. Environ Health Perspect. 1994;102 Suppl 4:25–30.
Johnson TV. Review of diesel emissions and control. Int J Engine Res. 2009;10:275–85.
Knecht W. Diesel engine development in view of reduced emission standards. Energy. 2008;33:264–71.
Ratclift MA, Dane AJ, Williams A, Ireland J, Luecke J, Mccorhees RL, et al. Diesel particulate filter and fuel effects on heavy-duty diesel engine emission. Environ Sci Technol. 2010;42:8343–9.
Heeb N, Schmid P, Kohler M, Gujer E, Zennegg M, Wenger D, et al. Secondary effects of catalytic diesel particulate filters: conversion of PAHs versus formation of nitro-PAHs. Environ Sci Technol. 2008;42:3773–9.
Heeb NV, Schmid P, Kohker M, Gujer E, Zennegg M, Wenger D, et al. Impact of low- and high-oxidation diesel particulate filters on genotoxic exhaust constituents. Environ Sci Technol. 2010;44:1078–84.
Kobayashi S, Hasegawa S, Kondo Y, Fushimi A, Tanabe K. Nitrogen dioxide emission from diesel vehicles equipped with exhaust aftertreatment systems. Rev Automot Eng. 2008;29:229–35.
Schuetzle D. Sampling of vehicle emissions for chemical analysis and biological testing. Environ Health Perspect. 1983;47:65–80.
Hayakawa K, Nakamura A, Terai N, Kizu R, Ando K. Nitroarene concentrations and direst-acting mutagenicity of diesel exhaust particulates fractionated by silica-gel column chromatography. Chem Pharm Bull. 1997;45:1820–2.
Paputa-Peck MC, Marano RS, Schuetzle D, Riley TL, Hampton CV, Prater TJ, et al. Determination of nitrated polynuclear aromatic hydrocarbons in particulate extracts by capillary column gas chromatography with nitrogen selective detection. Anal Chem. 1983;55:1946–54.
Zielinska B, Sagebiel J, McDonald JD, Whitney K, Lawson DR. Emission rates and comparative chemical composition from selected in-use diesel and gasoline-fueled vehicles. J Air Waste Manage Assoc. 2004;54:1138–50.
Taneda S, Kamata K, Hayashi H, Toda N, Seki K, Sakushima A, et al. Investigation of vasodilatory substances in diesel exhaust particles (DEP): isolation and identification of nitrophenol derivatives. J Health Sci. 2004;50:133–41.
Nojima K, Kawaguchi A, Ohya T, Kanno S, Hirobe M. Studies on photochemical reaction of air pollutants X identification of nitrophenols in suspended particulates. Chem Pharm Bull. 1983;31:1047–51.
Taneda S, Mori Y, Kamata K, Hayashi H, Furuta C, Li C, et al. Estrogenic and anti-androgenic activity of nitrophenols in diesel exhaust particles (DEP). Biol Pharm Bull. 2004;27:835–7.
Levsen K, Puttins U, Schilhabel J, Prieß B. Artifacts during the sampling of nitro PAHs of diesel exhaust. Fresenius’ Z Anal Chem. 1988;330:527–8.
Sekimoto K, Inomata S, Tanimoto H, Fushimi A, Fujitani Y, Sato K, et al. Characterization of nitromethane emission from automotive exhaust. Atmos Environ. 2013;81:523–31. The effect of various types of aftertreatment on the nitromethane emissions in diesel exhaust gasses was investigated by on-line PTR-MS.
Inomata S, Fushimi A, Sato K, Fujitani Y, Yamada H. 4-Nitrophenol, 1-nitropyrene, and 9-nitroanthracene emissions in exhaust particles from diesel vehicles with different exhaust gas treatments. Atmos Environ. 2015;110:93–102. The effect of various types of aftertreatment on the emissions of nitro-organic compounds in gas and aerosol phases was summarized.
Aoki N, Inomata S, Tanimoto H. Detection of C1–C5 alkyl nitrates by proton transfer reaction time-of-flight mass spectrometry. Int J Mass Spectrom. 2007;263:12–21.
Seizinger DE, Dimitriades B. Oxygenates in exhaust from simple hydrocarbon fuels. J Air Pollut Control Assoc. 1972;22:47–51.
Tremp J, Mattrel P, Fingler S, Eawag WG. Phenols and nitrophenols as tropospheric pollutants: emissions from automobile exhausts and phase transfer in the atmosphere. Water Air Soil Pollut. 1993;68:113–23.
Sarkar C, Sinha V, Kumar V, Rupakheti M, Panday A, Mahata KS, et al. Overview of VOC emissions and chemistry from PTR-TOF-MS measurements during the SusKat-ABC campaign: high acetaldehyde, isoprene and isocyanic acid in wintertime air of the Kathmandu Valley. Atmos Chem Phys. 2016;16:3979–4003.
Furey R.L., Perry K.L. Composition and reactivity of fuel vapor emissions from gasoline—oxygenated blends. SAE Technical Paper 1991; 912429.
Pierson WR, Schorran DE, Fujita EM, Sagebiel JC, Lawson DR, Tanner RL. Assessment of nontailpipe hydrocarbon emissions from motor vehicles. J Air Waste Manage Assoc. 1999;49:498–519.
Rubin JI, Kean AJ, Harley RA, Millet DB, Goldstein AH. Temperature dependence of volatile organic compound evaporative emissions from motor vehicles. J Geophys Res. 2006;111:D03305.
Gentner DR, Harley RA, Miller AM, Goldstein AH. Diurnal and seasonal variability of gasoline-related volatile organic compound emissions in Riverside, California. Environ Sci Technol. 2009;43:4247–52.
Kirchstetter TW, Singer BC, Harley RA, Kendall GR, Chan W. Impact of oxygenated gasoline use on California light-duty vehicle emissions. Environ Sci Technol. 1996;30:661–70.
Lough GC, Schauer JJ, Lonneman WA, Allen MK. Summer and winter nonmethane hydrocarbon emissions form on-road motor vehicles in the Midwestern United States. J Air Waste Manage Assoc. 2005;55:629–46.
Dimitriades B. Scientific basis for the VOC reactivity issues raised by section 183(e) of the Clean Air Act Amendments of 1990. J Air Waste Manage Assoc. 1996;46:963–70.
Martien PT, Harley RA, Milford JB, Russell AG. Evaluation of incremental reactivity and its uncertainty in southern California. Environ Sci Technol. 2003;37:1598–608.
Chen J, Luo D. Ozone formation potentials of organic compounds from different emission sources in the South Coast Air Basin of California. Atmos Environ. 2012;55:448–55.
Carter WPL. Development of the SAPRC-07 chemical mechanism. Atmos Environ. 2010;44:5324–35.
Carter WPL, Heo G. Development of revised SAPRC aromatics mechanisms. Atmos Environ. 2013;77:404–14.
Yang Y, Milford JB. Quantification of uncertainty in reactivity adjustment factors from reformulated gasolines and methanol fuels. Environ Sci Technol. 1996;30:196–203.
Olson K.L., Loo J.F., Mulawa P.A. Rapid hydrocarbon speciation and exhaust reactivity measurements using high speed high resolution gas chromatography. SAE Technical Paper 2000; 2000-01-2950.
Schmitz T, Hassel D, Weber F. Determination of VOC-components in the exhaust of gasoline and diesel passenger cars. Atmos Environ. 2000;34:4639–47.
Farrugia J., Olson K. Evaluation of a high speed, high resolution gas chromatography instrument for exhaust hydrocarbon speciation, SAE Technical Paper 2005; 2005-01-0683.
Nakashima Y, Kamei N, Kobayashi S, Kajii Y. Total OH reactivity and VOC analyses for gasoline vehicular exhaust with a chassis dynamometer. Atmos Environ. 2010;44:468–75.
Adam TW, Astorga C, Clairotte M, Duane M, Elsasser M, Kransenbrink A, et al. Chemical analysis and ozone formation potential of exhaust from dual-fuel (liquefied petroleum gas/gasoline) light duty vehicles. Atmos Environ. 2011;45:2842–8.
Tsai J, Chang S, Chiang H. Volatile organic compounds from the exhaust of light-duty diesel vehicles. Atmos Environ. 2012;61:499–506.
Wang J, Jin L, Gao J, Shi J, Zhao Y, Liu S, et al. Investigation of speciated VOC in gasoline vehicular exhaust under ECE and EUDC test cycles. Sci Total Environ. 2013;445–446:110–6.
Costagliola MA, Murena F, Prati MV. Exhaust emissions of volatile organic compounds of powered two-wheelers: Effect of cold start and vehicle speed. Contribution to greenhouse effect and tropospheric ozone formation. Sci Total Environ. 2014;468–469:1043–9.
Burns V.R., Gorse R.A., Reuter R.M., Hochhauser A.M., Benson J.D., Koehl W.J., Painter L.J. Effects of gasoline composition on evaporative and running loss emissions—auto/oil air quality improvement research program. SAE Technical Paper 1992; 920323.
Harley RA, Coulter-Burke SC, Yeung TS. Relating liquid fuel and headspace vapor composition for California reformulated gasoline samples containing ethanol. Environ Sci Technol. 2000;34:4088–94.
Gentner DR, Worton DR, Isaacman G, Davis LC, Dallmann TR, Wood EC, et al. Chemical composition of gas-phase organic carbon emissions from motor vehicles and implications for ozone production. Environ Sci Technol. 2013;47:11837–48.
Zielinska B, Sagebiel JC, Harshfield G, Gertler AW, Pierson WR. Volatile organic compounds up to C20 emitted from motor vehicles; measurement methods. Atmos Environ. 1996;30:2269–86.
California Environmental Protection Agency. Tables of maximum incremental reactivity (MIR) values. 2009; (http://www.arb.ca.gov/regact/2009/mir2009/mir2009.htm).
Lin M-F, Lee YT, Ni C-K. Photodissociation dynamics of nitrobenzene and o-nitrotoluene. J Chem Phys. 2007;126:064310.
European Parliament. Council Directive 2009/126/EC of the European Parliament and of the Council of 21 October 2009 on stage II petrol vapour recovery during refuelling of motor vehicles at service stations; 2009.
Fung F. Maxwell B. Onboard refueling vapor recovery: evaluation of the ORVR program in the United States. The International Council on Clean Transportation Working Paper 2011–12, 2011; (http://www.theicct.org/onboard-refueling-vapor-recovery-evaluation-orvr-program-united-states).
Lias SG. Ionization energy data in NIST Chemistry Web-Book, NIST Standard Reference Database, 2015; vol. 69 edited by P.J. Linstrom, National Institute of Standards and Technology, Gaithersburg, MD; (http://webbook.nist.gov/).
Masaki H, Chen L, Korenaga T. Direct analysis of diesel exhaust particles by fragmentation-free mass spectrometry using ion attachment mass spectrometry. Environ Sci. 2006;13:347–52.
Veres P, Roberts JM, Warneke C, Welsh-Bon D, Zahniser M, Herndon S, et al. Development of negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS) for the measurement of gas-phase organic acids in the atmosphere. Int J Mass Spectrom. 2008;274:48–55.
Akagi SK, Yokelson RJ, Wiedinmyer C, Alvarado MJ, Reid JS, Karl T, et al. Emission factors for open and domestic biomass burning for use in atmospheric models. Atmos Chem Phys. 2011;11:4039–72.
Karl TG, Christian TJ, Yokelson RJ, Artaxo P, Hao WM, Guenther A. The tropical forest and fire emissions experiment: method evaluation of volatile organic compound emissions measured by PTR-MS, FTIR, and GC from tropical biomass burning. Atmos Chem Phys. 2007;7:5883–97.
Yokelson RJ, Christian TJ, Karl TG, Guenther A. The tropical forest and fire emissions experiment: laboratory fire measurements and synthesis of campaign data. Atmos Chem Phys. 2008;8:3509–27.
Warneke C, Roberts JM, Veres P, Gilman J, Kuster WC, Burling I, et al. VOC identification and inter-comparison from laboratory biomass burning using PTR-MS and PIT-MS. Int J Mass Spectrom. 2011;303:6–14.
Stockwell CE, Veres PR, Williams J, Yokelson RJ. Characterization of biomass burning emissions from cooking fires, peat, crop residue, and other fuels with high-resolution proton-transfer-reaction time-of-flight mass spectrometry. Atmos Chem Phys. 2015;15:845–65.
Inomata S, Tanimoto H, Pan X-L, Taketani F, Komazaki Y, Miyakawa T, et al. Laboratory measurements of emission factors of nonmethane volatile organic compounds from burning of Chinese crop residues. J Geophys Res Atmos. 2015;120:5237–52.
Veres P, Roberts JM, Burling IR, Warneke C, de Gouw J, Yokelson RJ. Measurements of gas-phase inorganic and organic acids from biomass fires by negative-ion proton-transfer chemical-ionization mass spectrometry. J Geophys Res. 2010;115:D23302.
Roberts JM, Veres P, Warneke C, Neuman JA, Washenfelder RA, Brown SS, et al. Measurement of HONO, HNCO, and other inorganic acids by negative-ion proton-transfer chemical-ionization mass spectrometry (NI-PT-CIMS): application to biomass burning emissions. Atmos Meas Tech. 2010;3:981–90.
Acknowledgments
This study was partly supported by the Environmental Research and Technology Development Fund (S2-05, S2-06, and 2–1505) of the Ministry of the Environment, Japan.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
This article is part of the Topical Collection on Air Pollution
Rights and permissions
About this article

Cite this article
Inomata, S., Yamada, H. & Tanimoto, H. Investigation on VOC Emissions from Automobile Sources by Means of Online Mass Spectrometry. Curr Pollution Rep 2, 188–199 (2016). https://doi.org/10.1007/s40726-016-0032-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40726-016-0032-6