
Abstract
Posaconazole is typically used for preventing invasive yeast and mold infections such as invasive aspergillosis in high-risk immunocompromised patients. The oral suspension was the first released formulation and many pharmacokinetic and pharmacodynamic studies of this formulation have been published. Erratic absorption profiles associated with this formulation were widely reported. Posaconazole exposure was found to be significantly influenced by food and many gastrointestinal conditions, including pH and motility. As a result, low posaconazole plasma concentrations were obtained in large groups of patients. These issues of erratic absorption urged the development of the subsequently marketed delayed-release tablet, which proved to be associated with higher and more stable exposure profiles. Shortly thereafter, an intravenous formulation was released for patients who are not able to take oral formulations. Both new formulations require a loading dose on day 1 to achieve high posaconazole concentrations more quickly, which was not possible with the oral suspension. So far, there appears to be no evidence of increased toxicity correlated to the higher posaconazole exposure achieved with the regimen for these formulations. The higher systemic availability of posaconazole for the delayed-release tablet and intravenous formulation have resulted in these two formulations being preferable for both prophylaxis and treatment of invasive fungal disease. This review aimed to integrate the current knowledge on posaconazole pharmacokinetics, pharmacodynamics, major toxicity, existing resistance, clinical experience in special populations, and new therapeutic strategies in order to get a clear understanding of the clinical use of this drug.
Similar content being viewed by others


Posaconazole is a systemic triazole antifungal drug that shows high variability in exposure within patients, but also between different patient populations and between the three available formulations, with the two most recent formulations (i.e., delayed-release tablet and intravenous formulation) providing higher and more stable exposure than the oral suspension. |
PK/PD targets for posaconazole are mostly derived from animal studies and quantified using conventional PK/PD indices based on MIC that do not take dynamic exposure patterns and mechanistic pharmacological knowledge into account. |
Posaconazole shows a low occurrence of hepatotoxicity and cardiotoxicity and no clear relationship between posaconazole exposure and treatment-related toxicity has been identified to date. |
1 Introduction
Posaconazole (Noxafil®) is a systemic triazole antifungal drug derived from itraconazole and exerts the same antifungal mechanism of action as other azole derivatives [1]. Three formulations are currently available, namely an oral suspension (40 mg/mL), a delayed-release tablet (100 mg), and an intravenous formulation (18 mg/mL). The posaconazole oral suspension and delayed-release tablet are approved for patients aged 13 years and older (USA) or adults aged 18 years and older (Europe), while the intravenous formulation is licensed only for patients aged 18 years and older. Posaconazole is mainly licensed for prophylaxis of invasive fungal diseases (IFD) in: (1) patients receiving remission-induction chemotherapy for acute myelogenous leukemia (AML) or myelodysplastic syndromes (MDS) expected to result in prolonged neutropenia and who are at high risk of developing IFD; (2) allogeneic hematopoietic stem cell transplant (HSCT) recipients who are undergoing high-dose immunosuppressive therapy for graft-versus-host disease and who are at high risk of developing IFD [2]. Additionally, posaconazole is approved for treatment of oropharyngeal candidiasis, for the treatment of patients with IFD who are intolerant to first-line therapy, and as salvage treatment of IFD caused by rare pathogens such as fusariosis, chromoblastomycosis, mycetoma, and coccidioidomycosis [3].
1.1 Dosing
The posaconazole suspension is indicated to be administered as 200 mg three times daily (TID) for prophylaxis or as 400 mg twice daily (BID), or 200 mg four times daily (QID) for treatment of refractory IFDs or for treatment of patients with IFD who are intolerant to first-line therapy. The delayed-release tablet and intravenous formulation are indicated to be given as a loading dose at 300 mg BID on the first day and a maintenance dose at 300 mg once daily (QD) thereafter.
1.2 Mechanism of Action
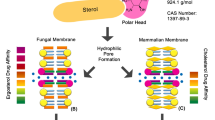
Similar to other azole derivatives, posaconazole inhibits the enzyme lanosterol 14α-demethylase and consequently inhibits the biosynthesis of ergosterol, which is an essential component of fungal cell membrane (see in Fig. 1). This results in an accumulation of methylated sterol precursors and depletion of ergosterol within the cell membrane, thereby weakening the structure and function of the fungal cell membrane, which is considered to be responsible for the antifungal activity of posaconazole [2].

Antifungal mechanism of action of posaconazole
1.3 In Vitro Antifungal Activity
Posaconazole shows broad activity against the majority of opportunistic pathogenic yeasts and molds in vitro, including the common pathogenic fungal species, such as Candida and Aspergillus species, but also against less common pathogens such as Mucorales and some Fusarium species [3]. According to European Committee on Antimicrobial Susceptibility Testing (EUCAST), the minimum inhibitory concentration (MIC) breakpoints for A. fumigatus are ≤ 0.12 mg/L for susceptible and > 0.25 mg/L for resistant strains, 0.25 mg/L for A. terreus and 0.5 mg/L for A. flavus, A. nidulans, and A. niger [4]. The breakpoints of posaconazole against C. albicans, C. dubliniensis, C. parapsilosis, and C. tropicalis are all defined as ≤ 0.06 mg/L for susceptible and > 0.06 mg/L for resistance. Higher resistant breakpoints of 0.25, 0.5, and 1.0 mg/L were set for C. guilliermondii, C. krusei, and C. glabrata, respectively [4].
1.4 Aspergillus Resistance
Posaconazole showed potent dose-dependent in vivo antifungal activity in many animal studies on prophylaxis and treatment against C. albicans, A. fumigatus, and other uncommon fungal infections [5,6,7,8,9,10,11,12,13]. The area under the concentration–time curve (AUC) versus MIC, i.e., AUC/MIC, showed the strongest correlation with therapeutic success. Despite the dose-dependent killing, some strains of A. fumigatus have become fully resistant against azoles, and this resistance has become of increasing clinical concern [14, 15]. The majority of isolates with azole-resistant phenotypes harbor mutations in the cyp51A gene, which codes for the enzyme lanosterol 14α-demethylase, or in the promotor region of this gene. Two routes of resistance development have been proposed [16]. Azole resistance can develop in-host during treatment (patient route) or alternatively through exposure to azole fungicides in the environment (environmental route). Generally, resistant mutations associated with these routes are different, as point mutations in locus G54, M220, G448, and P216 in the cyp51A gene and non-cyp51A-mediated mechanisms are mostly associated with in-host resistance development, while L98H mutations in combination with a 34 base pair tandem repeat in the promoter region (TR34/L98H) or Y121F/T289A in combination with a TR46 (TR46/Y121F/T289A) are associated with the environmental route. Importantly, resistant isolates with environmental mutations have been found in patients without prior antifungal exposure. Exceptions to the categorization in resistance development routes were recently described as isolates with cyp51A point mutations that have been recovered from the environment and azole-naive patients [17]. In addition, an isolate harboring a tandem repeat in the promotor region (TR120) was shown to have developed in-host through azole therapy [17, 18].
Case series indicate that azole resistance in A. fumigatus is associated with increased mortality rates [19,20,21]. Most resistance mutations affect the azole susceptibility of all the triazoles. However, as the triazoles are structurally different (e.g., long-tailed and short-tailed triazoles), different mutations may have various effects on the target binding of triazoles and thus variable azole resistance phenotypes [22]. For example, TR34/L98H often results in high itraconazole resistance (MIC typically ≥ 16 mg/L), with voriconazole, isavuconazole, and posaconazole MICs being variable, while isolates with TR46/Y121F/T289A have high resistance to voriconazole and isavuconazole (MICs typically ≥ 16 mg/L) with itraconazole and posaconazole being less affected. In most azole-resistant isolates, posaconazole retains the greatest in vitro activity, with MICs that are close to the resistance breakpoint, i.e., 0.5–1 mg/L. In vivo studies indicate that isolates with posaconazole MICs of 0.5–1 mg/L may still be treated with increased posaconazole exposure [7, 9]. As the azoles are the only drug class with activity against Aspergillus that can be administered orally, strategies are explored using higher than standard dosing to overcome resistance in selected patients and in infections by A. fumigatus isolates with azole MICs that are close to the resistance breakpoint [23]. An increasing number of studies on different formulations, together with an extended clinical use of posaconazole, have expanded our understanding regarding the pharmacology of this drug, but some discrepancies and controversial issues have also arisen. This review aimed to integrate the current knowledge on posaconazole pharmacokinetics, pharmacodynamics, major toxicity, existing resistance, new therapeutic strategies, and clinical experience in special populations, in order to get a clear understanding of the clinical use of this drug.
2 Clinical Pharmacokinetics
The posaconazole oral tablet—not the marketed delayed-release tablet, but a premarketing formulation used before the oral suspension became clinically available—showed dose-linearity in exposure up to a single dose of 800 mg, with saturation of absorption occurring above 800 mg in healthy volunteers [24]. Using simulation-based approaches, it has been proposed that the non-linear absorption might be attributable to the extensive precipitation of posaconazole in the small intestine due to incomplete gastric dissolution in the pH shift from the stomach to the intestine, caused by its high lipophilicity and weakly basic property [25, 26]. Hence, development of this oral tablet was not pursued and an oral suspension was brought to the market. Unfortunately, this suspension also demonstrated high inter-individual variability as typically patients who received the suspension did demonstrate dose-limited absorption above a daily dose of 800 mg with a highly variable and erratic absorption [27].
A gastric-resistant tablet formulation was subsequently designed for releasing posaconazole in the small intestine, in order to avoid the erratic absorption caused by the gastric conditions and to improve systemic absorption. Systemic exposure after administration of this delayed-release tablet showed dose-linearity between 200 and 400 mg, while higher doses were not explored [28]. Finally, an intravenous formulation was designed for patients who do not tolerate oral medication. Dose-linearity was observed between doses of 200 mg and 300 mg whereas non-linearities were observed below 200 mg [29, 30]. Intravenous doses above 300 mg were not investigated. The exposure of these two new formulations still shows substantial interpatient variability [31,32,33,34].
The published population pharmacokinetic findings on posaconazole are discussed below and are summarized in Table 1. Model-independent findings on the clinical pharmacokinetics of posaconazole in healthy volunteers and patients are also discussed and are summarized in Tables 2 and 3, respectively.
2.1 Absorption
The two relevant parameters for oral absorption are the absorption rate constant (ka), describing the rate of absorption, and bioavailability (F), describing the extent of absorption. The ka of the oral suspension was reported to be different in different patient groups and mostly ranged from 0.40 to 0.77 h−1, which corresponds to an absorption half-life (t1/2) between 0.90 and 1.7 h [35,36,37]. Both a slower absorption (absorption t1/2 of 17.5 h) and a faster absorption (absorption t1/2 of 0.55 h) with a delayed onset of absorption have been reported [38, 39]. High inter-individual variability (53.4%) was reported for the ka upon administration of the posaconazole suspension [39]. For the delayed-release tablet, similar ka values were reported (0.59 h−1 and 0.85 h−1) [40, 41], with inter-individual variability in ka (57.5%) being as high as for the oral suspension [41]. Food intake proved to be associated with an increase in ka, but was not expected to have a clinically relevant influence, because it had no impact on bioavailability or steady-state exposure parameters [41].
The mean value for F for the posaconazole suspension and delayed-release tablet were reported to be around 50% in healthy volunteers [42, 43], but was found to be about 2.6 times lower in patients receiving the posaconazole suspension [39]. It has been shown that food intake and nutritional supplements increase the F by improving solubility and delaying gastric emptying, thereby enhancing posaconazole exposure. Higher gastric pH and gastrointestinal motility decrease F of the oral suspension by reducing the solubility and shortening gastric residence time [44,45,46,47]. Additionally, administering the posaconazole suspension via nasogastric tube showed approximately 20% decreases in exposure compared to oral administration in healthy volunteers [47]. In immunocompromised patients, coadministration of proton pump inhibitors (PPIs) or metoclopramide or the occurrence of mucositis or diarrhea were proven to reduce the F of posaconazole by 45%, 35%, 58%, and 45%, respectively, while administration with nutritional supplements could increase F by 129% [39].
The systemic exposure of posaconazole upon dosing of the delayed-release tablet formulation is less susceptible to the aforementioned gastric conditions than the suspension. Coadministration with antacids, PPIs, H2-receptor functional antagonists, or metoclopramide proved to have a non-clinically relevant impact on the F of posaconazole in a healthy population receiving the delayed-release tablet [48]. A high-fat meal could only modestly increase the posaconazole AUC by 50%, in contrast to a 400% increase in similar conditions for the suspension, even though the high-fat meal postpones the median time to peak concentration (Tmax) by 1 h [49, 50].
The posaconazole suspension exhibits a dose-dependent and saturable absorption profile, with more frequent dosing leading to higher exposure when the total daily dose is lower than 800 mg [46, 51]. This pattern was not observed in the delayed-release tablet [28] due to the distinct differences in the gastrointestinal drug-delivery features between these two oral formulations.
2.2 Distribution
Figure 2 shows posaconazole distribution in various human tissues and fluids after systemic administration [52,53,54,55,56,57,58,59,60,61]. This figure shows that posaconazole accumulates in peripheral tissues, especially in lungs, kidneys, liver, and heart [52, 62]. For instance, exposure in alveolar cells is about 32-fold higher than in plasma, although the exposure in the pulmonary epithelial lining fluid (ELF) is slightly lower than in plasma in healthy volunteers receiving the 400 mg posaconazole suspension BID [54]. The concentrations in skin are similar to those in blood [55]. Posaconazole showed inconsistent distribution profiles in the cerebral spinal fluid (CSF), with CSF/serum levels ranging from 0.4 to 237% [58, 59]. It is unclear how cerebral inflammation impacts the permeability of the blood–brain barrier to further influence posaconazole exposure in CSF [58, 59]. Posaconazole concentrations in brain tissue have not been reported in humans, but in two murine models these concentrations were reported to be about half those of serum concentrations [63, 64]. Based on the current evidence of posaconazole distribution in the central nervous system, there is no clear pharmacokinetic evidence to support the use of posaconazole for the treatment of cerebral infections.

Posaconazole distribution depicted as the ratios of tissue or fluid concentrations versus simultaneously measured plasma concentrations in different organs and tissues (tissue concentration unit: ng/g, fluid or plasma concentration unit: ng/mL). CSF cerebrospinal fluid, ELF pulmonary epithelial lining fluid
Posaconazole is bound to plasma proteins for more than 98%, predominantly to albumin [2], yet this does not limit extravascular distribution of posaconazole. With values of 61.6 L and 181 L for the central and peripheral volume of distribution (Vd) respectively, the Vd of posaconazole is relatively large [30]. When posaconazole is only administered orally, F cannot be estimated. In such studies apparent Vd (Vd/F) will be reported, which is inversely proportional to the value of F. Thus, the inter-individual variability in apparent Vd observed in patients receiving oral posaconazole is significantly affected by the F. In healthy volunteers, the Vd/F of the posaconazole suspension and the delayed-release tablet are about twice as high as the absolute Vd that was determined upon intravenous injection [29], which could be explained by the reported value of 50% for F. A compartmental pharmacokinetic model developed for patients with persistent febrile neutropenia or refractory IFD showed that the Vd/F of posaconazole suspension is 2447 L [27], which indicates a remarkably larger Vd/F than for the healthy population (427 L under fed and 1450 L under fasted conditions) [50]. Four population pharmacokinetic studies using non-linear mixed-effect modeling confirmed this finding in other hematological patients receiving posaconazole suspension [35, 37,38,39]. The markedly larger Vd/F in the patient population might be in part due to the lower F caused by concomitant medication and multiple clinical factors. Patients from the surgical intensive care unit (SICU) exhibited the largest Vd/F (5280 L, compared to 1100–2770 L in hematological patients), which might be mainly caused by poor absorption resulting from the application of nasogastric tubes and/or by increased distribution to peripheral tissue due to capillary leakage and edema [36].
Inter-individual variability in posaconazole Vd was reported to be high among AML/MDS/HSCT patients [30]. Disease status (patients vs. healthy volunteers) proved to increase both central and peripheral Vd; moreover peripheral Vd was found to increase with increasing body weight [30].
The delayed-release tablet formulation was found to exhibit a lower Vd/F than the suspension based on population pharmacokinetic analyses [35,36,37,38,39,40,41], but this is likely driven by the difference in F rather than by a true difference in Vd. In patients with AML/MDS receiving the oral suspension, ethnicity (non-White vs. White), higher weight, PPI use, occurrence of diarrhea, and high gamma-glutamyl transpeptidase or bilirubin levels (≥ 2 times the upper limit of normal) proved to significantly increase the Vd/F [37, 38], among which the impact of diarrhea and PPI use are likely driven by the decrease in F. In contrast, coadministration of chemotherapy has been shown to decrease the Vd/F [37]. In patients receiving allogeneic HSCT, increasing age proved to be associated with decreases in Vd/F [35]. No variable was associated with inter-individual variability in Vd/F for the delayed-release tablet [40, 41], which might be partly due to the weak influence of gastric conditions on the extent of absorption.
2.3 Biotransformation and Elimination
After administration of the posaconazole suspension, 77% of the dose is excreted in feces, of which > 66% is unchanged, while 13% of the dose is eliminated in urine, of which < 0.2% is unchanged [2]. Unlike other triazole antifungal agents, posaconazole is barely metabolized by the cytochrome P450 (CYP) pathway. About 17% is glucuronidated by UGT1A4 and the remainder is eliminated unchanged [65, 66]. There are no major circulating metabolites. Nevertheless, posaconazole may still be impacted as a victim drug by interactions with drugs that interact with UGT enzymes, like phenytoin, rifampin, and fosamprenavir [2]. In addition, posaconazole is a potent inhibitor of CYP3A4 [2]. Clinicians and pharmacists should remember that the inhibitory potency of posaconazole is concentration, and thus formulation, dependent [67]. Several clinically relevant drug-drug interactions have been identified that require substantial empirical dose reductions of victim drugs (i.e., 30–50%), like cyclosporine A or tacrolimus. Adding to these examples are the interactions of posaconazole with new targeted therapies such as ibrutinib, venetoclax, and ruxolitinib, which make optimal management with these combinations challenging [68].
The posaconzole intravenous injection showed a decrease in clearance (CL) when increasing a single dose from 50 to 200 mg, while the CL remained stable for doses of 200 mg and 300 mg [29]. This may be attributable to saturation of for instance enzyme or transporter involved in the elimination of posaconazole, which leads to the observed more-than-dose-proportional increase in exposure. Posaconazole CL reported in a population pharmacokinetic analysis using combined data from both healthy volunteers and patients with AML/MDS/HSCT receiving an intravenous infusion appeared to be in line with these results reported from a clinical pharmacokinetic study in healthy volunteers (7.8 vs. 6.5—6.9 L/h) [29, 30]. The apparent CL/F observed upon administration of the posaconazole suspension in patients is significantly higher than in healthy volunteers and differs among different patient populations. Patients with persistent febrile neutropenia or refractory IFD, patients from ICU, and cystic fibrosis patients after lung transplantation appear to have high CL/F values (283.0, 195.0 and 143.2 L/h, respectively) [27, 36, 69], compared with those suffering from AML/MDS/HSCT (42.5–67.0 L/h) [35, 37, 38]. In general, the difference in F plays an important role in the substantial differences of posaconazole reported absolute clearance with the intravenous formulation and apparent clearance with the oral suspension.
The posaconazole CL upon administration of the delayed-release tablet showed a similar CL profile in both healthy volunteers and patient populations [28, 40, 41]. The CL/F observed for the delayed-release tablet is twice as high as the CL of the intravenous formulation in healthy volunteers (15.4 vs. 7.6 L/h), which is also in line with F being estimated around 50% [30]. Two population pharmacokinetic models developed on data obtained upon administration of the posaconazole delayed-release tablet demonstrated that CL/F is slightly lower, with values of 7.3 and 9.7 L/h in patients with hematological malignancies [40, 41]. Generally, the CL/F after administration of the oral suspension is higher than the CL/F after administration of the delayed-release tablet, which could be explained by the lower F caused by the lower F of the suspension.
In patients receiving the posaconazole suspension, occurrence of diarrhea and coadministration of PPI or phenytoin/rifampin was associated with increases in posaconazole CL/F [35, 37, 39]. No clinically relevant covariate was identified with a significant impact on CL/F or CL of posaconazole delayed-release tablet or intravenous formulation [30, 40, 41, 70].
Since posaconazole is metabolized by UGT and is a substrate for P-glycoprotein, inhibitors (e.g., verapamil, ciclosporin, quinidine, clarithromycin, erythromycin, etc.) or inducers (e.g. rifampicin, rifabutin, certain anticonvulsants, etc.) of these proteins may increase or decrease posaconazole plasma concentrations, respectively [3]. On the other hand, as a potent CYP3A4 inhibitor, posaconazole can induce large increases in exposure of CYP3A4 substrates as exemplified before. More details about drug-drug interactions for posaconazole can be found in previously published reviews [71,72,73,74].
2.4 Posaconazole Descriptive Pharmacokinetics
The AUC and peak concentration (Cmax) after a single 100 mg dose of the posaconazole delayed-release tablet to healthy volunteers under fasting conditions were found to be similar to the oral suspension under fed conditions using the same dosage. This concentration is three times higher compared to the suspension under fasted conditions [42], which could be explained by the great impact of food and formulation on F for the oral suspension. The AUC and Cmax of posaconazole upon intravenous administration are twofold and sevenfold higher, respectively, compared to the delayed-release tablet after a single dose of 300 mg [30]. Posaconazole exposure after administration of the oral suspension in healthy volunteers is about two to three times higher compared to hematological patients [42]. The steady-state exposures of posaconazole after administration of the delayed-release tablet or intravenous formulation are similar in patients with AML/MDS/HSCT, but are significantly higher than exposures achieved through administration of the suspension [32, 34, 75,76,77]. The variability in posaconazole average concentration (Cavg) upon administration of the oral suspension in patients with AML/MDS/HSCT is relatively high, ranging from 57 to 68% [75, 76]. As the variability in exposure (i.e., AUC or Cavg) upon dosing with the posaconazole delayed-release tablet and intravenous formulation in patients with AML/MDS/HSCT is smaller, i.e., 40% and 35%, respectively [32, 34], it seems that absorption-related factors are attributable to the variation. A higher steady-state concentration was reported in HSCT patients compared to AML/MDS patients receiving posaconazole suspension and delayed-release tablet (1.47 vs. 0.58 mg/L for suspension, 1.87 vs. 1.44 mg/L for delayed-release tablet) [32, 75, 76], but not for the intravenous administration (1.56 vs. 1.47 mg/L) [34]. The accumulation ratio upon dosing of the posaconazole suspension in patients is similar to the other two formulations (2.4–3.9 for suspension, 2.2–2.5 for delayed-release tablet, 2.8–3.6 for intravenous solution) based on the magnitude of AUC [31, 33, 78].
The mean Tmax observed after administration of the posaconazole suspension ranged from 5.0 to 6.0 h in healthy subjects under fed conditions and 4.0 h under fasted conditions [50], which is similar to the mean Tmax of the delayed-release tablet (4.0–5.0 h) under fasted conditions [24, 48]. The Tmax of an intravenous dose is attained around the time of termination of infusion [28, 29, 32, 34]. The mean elimination t1/2 of the posaconazole suspension is 25.1–29.2 h, which is comparable to the delayed-release tablet (27.0–28.1 h) in healthy volunteers [48, 50]. However, the mean t1/2 of the intravenous injection in healthy volunteers showed a dose-dependent prolongation from a single dose of 50 mg (18.7 h) to 200 mg (23.6 h), which can be explained by the aforementioned decreased CL [29]. When giving a single dose of 250–300 mg, the elimination t1/2 of posaconazole intravenous formulation is similar to that of the other two oral formulations (24.6–28.8 h) [29].
3 Pharmacodynamics
Since neither one single dose nor one single target concentration may be appropriate for all patients, researchers integrate the in vivo drug exposure and the in vitro antimicrobial susceptibility of the pathogen (MIC) as a pharmacokinetic/pharmacodynamic (PK/PD) predictor for the in vivo antimicrobial efficacy. The relationship between the exposure to posaconazole and the corresponding antifungal response (PD) in relation to the pathogen susceptibility (MIC) has been verified in many preclinical studies.
3.1 Posaconazole PK/PD in Preclinical Studies
3.1.1 Prophylaxis
Posaconazole given as prophylactic therapy against pulmonary aspergillosis showed a dose-(and concentration)-dependent response in a neutropenic rabbit model and a neutropenic murine model [6, 11]. In the rabbit model, posaconazole was administered orally with three dosing levels of 2, 6, and 20 mg/kg/day 4 h before endotracheal inoculation with A. fumigatus. Rabbits receiving prophylactic posaconazole at all dosages showed a significant reduction in infarct scores, total lung weights, and organism clearance from lung tissue in comparison to those of untreated controls. A dose-dependent microbiological clearance of A. fumigatus from lung tissue in response to posaconazole was observed [6]. In the murine model, oral posaconazole was administered once daily with five dosing levels of 1, 4, 8, 16, and 32 mg/kg and mice were infected through instillation of the inoculum in the nares. A 24 h-AUC/MIC ratio (AUC0-24/MIC) of 37.4 (95% confidence interval, 7.1–196) was able to achieve half-maximal survival for preventing invasive pulmonary aspergillosis caused by azole-resistant A. fumigatus for which the MIC against posaconazole was 0.5 mg/L [11]. Table 4 shows the posaconazole exposure–response relationships in various murine models.
3.1.2 Treatment
In addition to prophylaxis models, many preclinical PK/PD models have been established for the treatment of invasive candidiasis and aspergillosis [5,6,7,8,9,10]. The posaconazole exposure–response relationship was described using an inhibitory sigmoid Emax model based on an in vitro human alveolus model consisting of a bilayer of human alveolar epithelial and endothelial cells [8, 79]. EC50 with an AUC/MIC ratio of 2.2 and 11.6 was observed in endothelial and alveolar compartments of an in vitro model infected with A. fumigatus, respectively, and an AUC/MIC ratio of 100 was able to achieve near a maximal decrease of galactomannan concentrations in both endothelial and alveolar compartments [8].
The relationship between AUC/MIC and response to posaconazole therapy were confirmed in three neutropenic murine models of invasive pulmonary aspergillosis and one non-neutropenic murine model of disseminated aspergillosis, all infected with A. fumigatus strains [7,8,9,10]. The AUC0-24/MIC target associated with half-maximal antifungal response differs from model to model, with a ratio of the AUC/MIC of 321 when using mice mortality as endpoints [7] versus an AUC/MIC ratio of 167 when using the decline in serum galactomannan concentrations as end point [8], or an AUC/MIC of 179 and 53 when the fungal burden in the mouse lung were used as PD endpoint [9, 10]. The difference in PD endpoints, number and variety of fungal strains, inoculum size, and data analysis method as well as drug source might contribute to the observed differences among these PK/PD targets. EUCAST accepted a PK/PD target of 167–178 AUC0-24/MIC for Aspergillus infections. Using the licensed dose of 400 mg BID of the posaconazole oral suspension an AUC0-24 of 17.2 ± 14.8 mg·h/L (mean ± standard deviation [SD]) was achieved [27], suggesting 98% probability of target attainment for aspergillosis when the MIC is ≤ 0.015 mg/L [4]. If the delayed-release tablet or intravenous formulation is used in the licensed dose of 300 mg QD, an AUC0-24 of 34.3 ± 12.4 mg·h/L [31] and of 34.3 ± 14.4 mg·h/L (mean ± SD) [33] are achieved, respectively, yielding 100% probability of target attainment for a pathogen with an MIC ≤ 0.06 mg/L [4].
3.1.3 Posaconazole PK/PD in Treating Mucormycosis
Apart from the promising in vitro activity against Mucorales species, posaconazole also showed potential for preventing neutropenic mice from pulmonary mucormycosis by Rhizopus delemar [80], and disseminated mucormycosis by Lichtheimia corymbifera or R. arrhizus [81]. When posaconazole is used for treatment of mucormycosis, an AUC0-24/MIC ratio of 63 proved to be the target that was associated with half-maximal effect of lung fungal burden based on a neutropenic murine model of pulmonary mucormycosis infected with R. arrhizus [10]. Unfortunately, no controlled, adequately powered clinical efficacy trial is available to confirm this finding in humans. In clinical practice, the posaconazole suspension has been used as salvage therapy of mucormycosis and showed satisfactory efficacy in many cases [82, 83], which also indicates an encouraging prospect for the new formulations with higher drug exposures [84, 85]. Similar to the treatment of invasive aspergillosis, the delayed-release tablet or intravenous formulation of posaconazole are preferred for the treatment of invasive mucormycosis due to the more favorable exposure attained with these formulations.
3.2 Posaconazole PK/PD in Clinical Studies
Although controversial, some studies suggest an exposure–response relationship for both prophylaxis and treatment of IFD in patients. As a proportion of patients receiving the oral suspension showed low plasma concentrations [2, 77, 86,87,88,89], therapeutic drug monitoring (TDM) may be needed to ensure adequate exposure [87, 88, 90,91,92].
3.2.1 Prophylaxis
In general it can be stated that target concentrations for posaconazole prophylaxis are still under debate [86, 93]. A lower boundary of steady-state Cavg of 0.7 mg/L for posaconazole is accepted as a target for prophylaxis by the US Food and Drug Administration (FDA) and in European guidelines [94, 95], which was supported by the analysis from two randomized, active-controlled clinical studies [86]. Posaconazole trough concentrations (Cmin) proved to be well correlated with Cavg or AUC0-24 [32, 96]. Thus, Cmin is also frequently used for TDM measures in practice and considered as a more conservative and practicable index [30, 97]. A recent meta-analysis indicated that a Cmin of 0.5 mg/L could represent a clear margin separating successful from failed prophylaxis [98].
3.2.2 Treatment
For treatment purposes, posaconazole plasma Cavg ≥ 1.25 mg/L at steady-state proved to be associated with 75% successful response rates in patients with invasive aspergillosis and other mycoses, and therefore was considered as a cut-off value for IFD treatment [77]. The 2017 ESCMID-ECMM-ERS guidelines for management of Aspergillus disease recommended a slightly lower target trough concentration of 1.0 mg/L for treatment [99]. However, both targets lack validation in a larger cohort.
3.3 Challenges of Conventional PK/PD Indices
Although PK/PD indices based on MICs are widely used for target exposures, there are some inherent drawbacks of these indices. Firstly, the PK/PD indices are mostly based on animal studies that used various rodents, but differences in pharmacokinetics in various rodent animals are not taken into account. Secondly, the in vitro MIC is a static threshold value often established with limited precision that is obtained in experiments with static antifungal concentrations, while it is not known how fungal susceptibility towards the antifungals is impacted by the dynamics in the exposure in vivo, nor how this impacts the development of resistance. By not considering the concentration–time course in a dosing interval, these indices are basically assumed to be independent of the drug pharmacokinetics. Finally, the indices do not take the hosts’ immune response to the fungal infection into account, which may decrease the required in vivo drug exposure needed to obtain the same antifungal effect as in an in vitro setting.
Figure 3 illustrates how the currently applied PK/PD indices for antifungals relate to the pharmacological and physiological processes that occur in vivo. Upon antifungal administration, a dynamic concentration-effect profile is obtained. Subsequently, it is the combination of the antifungal effect of the dynamic drug exposure as well as the immune system of the host that will determine the fungal burden. The fungal burden then drives the responses that are observed in preclinical or clinical studies. The PK/PD indices ignore most of this mechanistic information by summarizing the dynamic exposure into a single value and empirically establishing which of the available exposure metrics best correlates with the observed responses, using the MIC value obtained in in vitro experiments with static exposure and in the absence of host immune response. In the field of antibacterial drugs, more mechanism-based PK/PD models that do take this mechanistic information into account have been established to overcome the weaknesses associated with the use of the PK/PD indices [100,101,102,103]. Unfortunately, this approach has not yet been applied in the antifungal field. This should yield better target exposure values as well as improved between-species (i.e., animal to human) scaling of findings.

Schematic illustration of the pharmacological and physiological processes driving antifungal drug response and how they link to the currently used PK/PD indices. Cmax peak concentration, Cmin trough concentration, AUC area under the concentration–time curve, MIC minimum inhibitory concentration, GM test detection of galactomannan, G test detection of (1–3)-β-D-glucan, IFD invasive fungal disease
3.4 Toxicity
No clear relationship between posaconazole exposure and treatment-related toxicity has been identified to date [32, 86]. During the development process of the delayed-release tablet and the intravenous formulation, an upper plasma toxicity limit of 3.75 mg/L was selected, which was derived from the 90th percentile of the exposure achieved from previous clinical studies that characterized safety for approval of the posaconazole oral suspension [32]. The most frequently reported adverse events (AEs) during posaconazole treatment included gastrointestinal disorders—such as diarrhea, nausea, vomiting—and also hypokalemia, pyrexia, which are manageable from a clinical perspective [2, 75, 76]. In the following sections we summarize the two posaconazole-related toxicities that are of most clinical concern, namely hepatotoxicity and cardiotoxicity.
3.4.1 Hepatotoxicity
Hepatotoxicity is a common AE of azole antifungal drugs. The occurrence of treatment-related increases in hepatic enzymes was 1–3% reported in 605 patients receiving the posaconazole suspension in two prophylaxis studies [75, 76]. Other treatment-related serious hepatotoxicities, such as hepatic failure and hepatocellular damage, appeared to be very rare (≤ 1%) among these hematological patients [75, 76]. The incidence of treatment-related abnormal liver function test (LFT) in 447 hematological patients receiving delayed-release tablets or intravenous injections was ≤ 2%, which is similar to the suspension despite significantly higher exposure [32, 34]. It was also reported that switching from suspension to delayed-release tablets can significantly increase posaconazole concentrations more than twofold without worsening its hepatotoxicity [104]. Apart from hematological patients, posaconazole also showed a low occurrence of hepatotoxicity in patients with chronic pulmonary aspergillosis, refractory IFD, and lung transplantation [105,106,107].
Some studies indicated that the incidences of LFT abnormalities are generally transient and reversible for long-term posaconazole use [2, 108, 109]. Most studies found no correlation between posaconazole exposure and hepatotoxicity occurrence [107, 110,111,112]. Nevertheless, in 343 hematological patients receiving delayed-release tablets or intravenous injections, a posaconazole concentration of > 1.83 mg/L was proven to be correlated with grade 3/4 hepatotoxicity using classification and regression-tree analysis, although no association was found using logistic regression [113]. In general, even though the incidence is low, monitoring LFT is necessary and TDM together with dose adjustments or discontinuation and alternative medication should be considered when treatment-related liver toxicity is assessed.
3.4.2 Cardiotoxicity
QT interval prolongation is another a class effect of the azoles. Posaconazole was reported to be associated with prolonged QT interval and other cardiac AEs, such as atrial fibrillation and torsades de pointes [75]. Treatment-related prolongation of the QT interval or corrected QT (QTc) interval occurred in 4% of 304 neutropenic patients receiving posaconazole suspension in one active-controlled prophylaxis study [75]. However, QT prolongation was not observed in healthy volunteers [2]. The incidences of treatment-related atrial fibrillation and torsades de pointes are less than 1% [75]. There is no evidence of an increased risk of cardiotoxicity in hematological patients receiving posaconazole delayed-release tablets or intravenous injections. Surprisingly, the incidence rate of the treatment-related prolonged QT interval is slightly lower for these two new formulations (≤ 1%) [34].
Coadministration with CYP3A4 substrates, such as pimozide and quinidine, can increase the exposure of these drugs and result in a higher risk of cardiotoxicity, including QTc prolongation and torsades de pointes [113], therefore these drugs are contraindicated with posaconazole. Posaconazole is also contraindicated in patients receiving drugs that are known to prolong the QTc interval or those identified with potentially proarrhythmic conditions such as cardiomyopathy and QTc prolongation. Potassium, magnesium, and calcium should be corrected before posaconazole administration, in order to reduce the risk of posaconazole-related cardiotoxicity [2]. There are less safety concerns with respect to prolonged QT or QTc in patients with persistent febrile neutropenia or refractory IFD, patients with chronic pulmonary aspergillosis, and lung transplant patients [105,106,107]. No discernable correlation between posaconazole exposure and cardiotoxicity has been found to date [30, 110].
3.5 Posaconazole Resistance
Although the use of azole monotherapy is precluded in most patients with azole-resistant Aspergillus disease, a modest role of azole therapy may remain in infections caused by isolates with azole MICs that are close to the resistance breakpoint. In such cases dose escalation might be a feasible strategy provided that drug toxicity is avoided. The posaconazole MICs of azole-resistant A. fumigatus often remain close to the wild-type MIC distribution (i.e., MIC 0.5–1 mg/L) [114, 115]. Preclinical studies indicated that isolates with a posaconazole MIC of 0.5 mg/L can be treated successfully with increased exposure [7, 9]. The required AUC/MIC to treat isolates with increased posaconazole MICs was calculated based on preclinical experiments and bridged to human infections. Thus for each posaconazole MIC the required exposure was calculated. As the posaconazole AUC is linearly correlated with Cmin, target Cmin values could be extracted from this correlation [96]. Thus, it is postulated that A. fumigatus isolates with posaconazole MICs of 0.5 mg/L may be treated with augmented posaconazole dosing in order to achieve plasma levels that exceed 3 mg/L [23]. One should bear in mind that evidence regarding the clinical efficacy of this strategy is absent. A major concern of a strategy using augmented dosing is the revelation of AEs. One study evaluated the AE in patients with posaconazole high-dosing regimens and incidental high posaconazole serum concentrations. This study concluded that the number of AEs in these patients was comparable to previous reports that evaluated standard dosing. A direct comparison between high dosing and standard dosing has not been reported [23].
3.6 New Strategies for Posaconazole Targeted Therapy
The finding that posaconazole accumulates in human peripheral blood mononuclear cells and polymorphonuclear leukocytes triggered an investigation on the impact of posaconazole-loaded leukocytes on the antifungal activity and functional capacity of different leukocytes [116,117,118,119]. High posaconazole intracellular concentrations did not show a significant impact on the functional capacities of human neutrophils and macrophages in vitro [117]. Natural killer cells also have proven to still be viable and they maintained their capacity under therapeutic concentrations of posaconazole [119]. Similar results were also found in neutrophil-like leukocyte cells. Furthermore, an improved antifungal activity was observed both in vitro and in an in vivo invasive pulmonary aspergillosis mouse model, which indicates the potential of posaconazole-loaded leukocytes as a novel antifungal strategy, in which leukocytes serve as a vehicle to target the infection site and further increase the antifungal effect [118]. Apart from this, these endogenous vehicles are supposed to be associated with less AE problems and are considered as a promising strategy for the prophylaxis and treatment of IFD.
4 Special Populations
4.1 Patients with Hepatic or Renal Impairment
Posaconazole showed slightly lower CL/F in patients with mild, moderate, and severe hepatic impairment (corresponding to Child–Pugh class A, B, and C, respectively) in comparison with healthy subjects after a single 400 mg dose of the oral suspension [120], which might be attributable to decreased metabolism by UGT1A4. The AUC was increased by 36% in patients with hepatic dysfunction compared to patients with normal hepatic function. Due to this minor change in the pharmacokinetics and the observed safety in patients with hepatic impairment, no dose adjustments are proposed for the posaconazole suspension in patients with hepatic impairment. This recommendation was directly applied to the later released formulations, without clear evidence on the influence of liver function on posaconazole pharmacokinetics or on the safety profile with these formulations in this population [2]. Future studies may still be needed to investigate the long-term pharmacokinetics and safety of all posaconazole formulations in patients with hepatic impairment.
No clinically significant difference in posaconazole CL/F or the exposure was observed between patients with mild, moderate, and severe chronic renal disease (corresponding to creatinine clearance levels at 50–80, 20–49, and < 20 mL/min, respectively) and healthy subjects after a 400 mg single dose of oral suspension [121]. Posaconazole suspension also appears to be effective and well tolerated in patients with refractory IFD and renal impairment (creatinine clearance < 50 mL/min or serum creatinine level > 2 mg/dL) [122]. Therefore, no dose adjustment was suggested in patients with mild and moderate renal impairment receiving the posaconazole suspension. Despite the fact that no dose adjustments are needed in patients with renal impairment, there is still a necessity for monitoring of the symptoms of IFD just like other patients with IFD. This is due to the high variability in exposure of the oral suspension [3]. The recommendation not to adjust the dose in patients with renal impairment was also directly applied to posaconazole delayed-release tablets without support by a clinical study [3]. The posaconazole intravenous formulation is not recommended for patients with moderate or severe renal impairment, because of the expected accumulation of the sulfobutylether-β-cyclodextrin excipient in the kidneys. However, from the experience with voriconazole, also containing sulfobutylether-β-cyclodextrin, we have learned that the benefits of efficacious treatment may outweigh the risk associated with accumulation of sulfobutylether-β-cyclodextrin. In addition, sulfobutylether-β-cyclodextrin appeared to accumulate by about sixfold in the kidney, but was not nephrotoxic itself [123,124,125]. Data on pharmacokinetics, efficacy, and safety upon long-term posaconazole use are lacking in this special population, for which future studies are expected to fill the gap.
4.2 Obesity
According to the label, patients weighing ≥ 120 kg are at increased risk of lower posaconazole exposure [3]. Additionally, in patients with hematological malignancies, significantly lower trough concentrations were also observed in patients weighing ≥ 90 kg compared to those weighing < 90 kg (0.65 vs. 1.31 mg/L), as well as between patients with a body mass index ≥ 30 and those with a body mass index < 30 (0.89 vs. 1.29 mg/L) receiving posaconazole delayed-release tablets [126]. The delayed-release tablet administration showed a significantly lower exposure and longer washout half-life in healthy obese subjects [weighing of 116.8 ± 19.6 kg and 140.4 ± 32 kg (mean ± SD)] compared to healthy normal-weight subjects [weighing of 71.2 ± 8.2 kg and 67.9 ± 9.1 kg (mean ± SD)] [127, 128]. The lower exposure can be attributed to an increased clearance and distribution volume [128]. In addition to this, the washout half-life is further prolonged by an increase in the already large distribution volume resulting from the extensive distribution of posaconazole into adipose tissue, which can also lead to a prolonged drug-drug interaction with CYP3A4 substrates in obese patients [127, 128].
A recent population pharmacokinetic study in 16 obese patients receiving posaconazole by peripheral venous catheter showed that a maintenance dose of 300 mg QD can only ensure target attainment in patients weighing less than 180 kg for prophylactic purposes (using Cmin > 0.7 mg/L as target). For patients with greater weights, 400 mg is required. For treatment purpose (using a Cmin > 1.0 mg/L), the maintenance dose needs to be increased to 400 mg and 500 mg for patients weighing between 120 and 170 kg, and more than 170 kg, respectively [129].
4.3 ICU Patients
A limited number of studies were performed on the use of posaconazole in patients admitted to the intensive care unit (ICU). The posaconazole oral suspension given via nasogastric tube showed very low systemic exposure in 27 ICU patients with only 17% of the cohort achieving a steady-state Cmin above 0.25 mg/L after a treatment of 400 mg BID or 200 mg QID, which indicates the posaconazole oral suspension to be unsuitable in this population and the use of intravenous formulations may be preferred [130].
A recent study reported the pharmacokinetic profiles of a single intravenous dose of posaconazole in eight ICU patients [131]. Clearance and Vd were more than twice the value reported in healthy volunteers (16.8 L/h vs. 6.9 L/h and 529 L vs. 236 L, respectively) [29]. This could result from hypoalbuminemia increasing the unbound posaconazole, which can then distribute into the tissue and be eliminated by clearing organs, but unfortunately there are no studies available on the influence of hypoalbuminemia on the pharmacokinetics of posaconazole. The AUC and Cmax in these patients are comparable to patients with AML/MDS, but lower than in healthy volunteers [29, 33, 131].
In brief, intravenous posaconazole displays encouraging pharmacokinetic characteristics in ICU patients and further studies with larger cohorts are required to demonstrate the efficacy and safety of this formulation in this special population.
4.4 Pediatrics
While the posaconazole oral formulations are approved in patients older than 13 years (USA) or 18 years (Europe), the intravenous form is only labeled for patients older than 18 years, due to potential toxicity to brain ventricle development observed in juvenile dogs [2, 30]. However, many studies have reported its off-label use in pediatric patients, which could be attributed to the promising efficacy and safety profile in adults [132,133,134]. A recent population pharmacokinetic model was developed for 171 pediatric immunocompromised patients aged between 5 month and 18 years receiving one of the oral formulations, with nearly 96% of the samples being obtained after administration of the suspension [70]. The estimated values of CL/F and V/F related to the delayed-release tablet formulation and standardized to a 70 kg individual are comparable to those reported in adults [40, 41]. These children showed a higher inter-individual variability on CL/F compared to that of adults (63.0% vs. 24.2% or 37.9%) [40, 41]. This might be partly attributable to the age-associated maturation of hepatic UGT1A4 [135].
A twice-daily allometric dosing algorithm based on body-weight (index at 0.75) resulted in adequate posaconazole concentrations at day 10 in 12 children aged 3–16 years with chronic granulomatous disease [136]. In children aged ≤ 13 years, a bodyweight-based dosing regimen of the oral suspension of 4 mg/kg TID or body surface area-based regimen of 120 mg/m2 TID showed a considerable proportion of hematologic children to reach < 0.7 mg/L steady-state plasma concentrations [137,138,139,140]. Therefore, higher initial dosing strategies of ≥ 20 mg/kg/day were recommended and are expected to ensure adequate concentrations [141, 142]. Experience with the posaconazole delayed-release tablet in pediatric patients is limited. A model-derived dosing strategy was applied in 34 children and adolescents (range 5–17 years) receiving the posaconazole delayed-release tablet, and more than 90% of the patients were reported to have steady-state trough concentrations above the target of 0.7 mg/L [134]. However, to implement such size-based dosing approaches in younger children, the delayed-release tablet displays an unattractive prospect as it is indivisible and large in size. A new delayed-release tablet formulation of smaller dosage and size or a new oral suspension formulation with better bioavailability might benefit young children.
High variability in posaconazole concentrations was also reported in the pediatric population as a result of the erratic bioavailability for which TDM was recommended [138,139,140,141, 143]. Consistent with the previous findings in adults [37,38,39], diarrhea and concomitant PPI use also had a negative impact on the bioavailability of the suspension in children [70]. A population pharmacokinetic analysis in children illustrated the insufficient therapeutic target attainment even on the highest feasible dose of oral suspension in children with diarrhea and/or PPI administration [70]. Based on the model-based simulations, this study recommended different dosing regimens for different age groups for both prophylactic and treatment purposes in children patients aged < 13 years. Due to the poor and saturable bioavailability of the suspension, the delayed-release tablet formulation is considered a superior choice compared to the oral suspension once the children are able to take it [70, 99, 134].
The establishment of pediatric target exposure is currently based on the concentration targets recommended in adults, which assumes that the same exposure will result in the same effect in adults and children. Although the susceptibility of fungi to antifungals can reasonably be expected to be the same in adult and pediatric patients, it still remains to be established whether differences in the developmental status of the immune system result in different required target concentrations in vivo. Differences in target concentrations could be likely, because despite the fact that the proportion of the target attainment was not high in children, the posaconazole oral suspension was demonstrated to be effective, safe, and well tolerated in preventing and treating IFD in immunocompromised children [137, 138, 140, 144,145,146,147].
4.5 Patients with Cystic Fibrosis
As the steady-state trough concentration for posaconazole delayed-release tablet is significantly higher than for the suspension both in cystic fibrosis (CF) (1.1 mg/L vs. 0.19 mg/L) and in non-CF lung transplant patients (1.9 mg/L vs. 0.47 mg/L) [69, 148], the delayed-release tablet form is considered a promising alternative to the suspension with satisfactory drug exposure and good tolerance. In lung transplant patients, patients with CF showed significantly lower posaconazole concentrations compared to non-CF patients with both oral formulations [69, 148, 149], which can increase the risk of subtherapeutic concentration in this subgroup, especially for the suspension.
Higher posaconazole concentrations were found to be correlated with lower Aspergillus Immunoglobulin E levels [150]. Posaconazole oral formulations, especially the delayed-release tablet, exhibited satisfactory exposure in children (median age 13 years, range 3–17 years) with CF and was proven to be generally safe and well tolerated [151]. Overall, the posaconazole delayed-release tablet appears to be a suitable antifungal agent in patients with CF due to the improved absorption and wide intrinsic distribution into the lung tissue. Further studies are still needed to confirm the efficacy of posaconazole in CF patients.
5 Conclusions
Posaconazole is widely used for the prevention and treatment of IFD. As this drug is going off patent, new generic formulations are expected to enter the European market in the beginning of 2020, which will likely result in increased clinical use due to anticipated price drops. The current review will help those that are less familiar with the use of posaconazole to better understand the pharmacological behavior of this drug. We want to alert clinicians that especially the absorption profile and bioavailability of posaconazole appear to be highly dependent on the formulation, meaning that proposed dosages may not always be directly translatable from one formulation to another.
There is a plethora of pharmacokinetic information available for the oral suspension, while new information on the pharmacokinetics of both the intravenous formulation and the delayed-release tablet is emerging rapidly. These studies are predominantly performed in healthy volunteers and hematological patients. There is therefore an urgent remaining need for more (population) pharmacokinetic knowledge in other patient groups including critically ill patients and the pediatric population. For all populations three distinct pharmacological issues should be further explored:
- 1.
Differences in oral absorption profiles, bioavailability, and exposure of the three pharmaceutical formulations need to be clarified for each special patient population.
- 2.
Protein binding, the variability in protein binding, and its relation to PD must be investigated. This is typically relevant for populations with a high likelihood of altered protein binding such as critically ill patients, (pediatric) leukemic patients, and patients with renal failure.
- 3.
More information on site-specific penetration of posaconazole, specifically brain tissue, is needed. Now that higher and more predictable plasma concentrations are attained with the new formulations, it might be possible to achieve detectable brain concentrations—thereby opening up treatment strategies, but also toxicological risks. Some neurological side effects have been described pointing towards an increased exposure in the brain [152], but this is yet to be confirmed.
There is a paucity of data related to the PD of posaconazole, especially on a mechanistic level. Past work on exposure–response relationships needs to be revisited using unbound concentrations and taking into account dynamic exposure profiles. Simultaneously, the scientific community could invest in detecting new biomarkers that could provide useful information on the efficacy of treatment. Such markers should perform better than current measures of outcome that leave room for interpretation such as mycological response. These biomarkers should be subsequently linked to the dynamic pharmacokinetic profiles to define the PK-PD relations. Finally, knowledge should be gained on how to treat fungal disease with pathogens with attenuated MICs. Adaptive targets, i.e., targets based on the pathogens’ MIC, have been investigated in animal models, but their clinical utility needs to be validated. Ultimately, information on the hosts’ immune response should also be utilized to complete the understanding of the interplay between pathogen, host, and drug to predict treatment outcome.
References
Hof H. A new, broad-spectrum azole antifungal: posaconazole–mechanisms of action and resistance, spectrum of activity. Mycoses. 2006;49(Suppl 1):2–6. https://doi.org/10.1111/j.1439-0507.2006.01295.x.
U.S FDA. Noxafil instruction. 2015. https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/205053s1lbl.pdf. Accessed 15 Apr 2020.
European Medicines Agency. Summary of posaconazole characteristics. 2010. https://www.ema.europa.eu/en/documents/product-information/noxafil-epar-product-information_en.pdf. Accessed 15 Apr 2020.
The European Committee on Antimicrobial Susceptibility Testing. Posaconazole: Rationale for the EUCAST clinical breakpoints, version 2.0. 2017. https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/AFST/Files/Posaconazole_Yeast_Molds_RD_V.2_Apr__2017.pdf. Accessed 15 Apr 2020.
Andes D, Marchillo K, Conklin R, Krishna G, Ezzet F, Cacciapuoti A, et al. Pharmacodynamics of a new triazole, posaconazole, in a murine model of disseminated candidiasis. Antimicrob Agents Chemother. 2004;48(1):137–42.
Petraitiene R, Petraitis V, Groll AH, Sein T, Piscitelli S, Candelario M, et al. Antifungal activity and pharmacokinetics of posaconazole (SCH 56592) in treatment and prevention of experimental invasive pulmonary aspergillosis: correlation with galactomannan antigenemia. Antimicrob Agents Chemother. 2001;45(3):857–69. https://doi.org/10.1128/AAC.45.3.857-869.2001.
Mavridou E, Bruggemann RJ, Melchers WJ, Mouton JW, Verweij PE. Efficacy of posaconazole against three clinical Aspergillus fumigatus isolates with mutations in the cyp51A gene. Antimicrob Agents Chemother. 2010;54(2):860–5. https://doi.org/10.1128/AAC.00931-09.
Howard SJ, Lestner JM, Sharp A, Gregson L, Goodwin J, Slater J, et al. Pharmacokinetics and pharmacodynamics of posaconazole for invasive pulmonary Aspergillosis: clinical implications for antifungal therapy. J Infect Dis. 2011;203(9):1324–32. https://doi.org/10.1093/infdis/jir023.
Lepak AJ, Marchillo K, Vanhecker J, Andes DR. Posaconazole pharmacodynamic target determination against wild-type and Cyp51 mutant isolates of Aspergillus fumigatus in an in vivo model of invasive pulmonary aspergillosis. Antimicrob Agents Chemother. 2013;57(1):579–85. https://doi.org/10.1128/AAC.01279-12.
Lewis RE, Albert ND, Kontoyiannis DP. Comparative pharmacodynamics of posaconazole in neutropenic murine models of invasive pulmonary aspergillosis and mucormycosis. Antimicrob Agents Chemother. 2014;58(11):6767–72. https://doi.org/10.1128/AAC.03569-14.
Seyedmousavi S, Mouton JW, Melchers WJ, Verweij PE. Posaconazole prophylaxis in experimental azole-resistant invasive pulmonary aspergillosis. Antimicrob Agents Chemother. 2015;59(3):1487–94. https://doi.org/10.1128/AAC.03850-14.
Rodriguez MM, Pastor FJ, Sutton DA, Calvo E, Fothergill AW, Salas V, et al. Correlation between in vitro activity of posaconazole and in vivo efficacy against Rhizopus oryzae infection in mice. Antimicrob Agents Chemother. 2010;54(5):1665–9. https://doi.org/10.1128/AAC.01463-09.
Wiederhold NP, Najvar LK, Bocanegra R, Graybill JR, Patterson TF. Efficacy of posaconazole as treatment and prophylaxis against Fusarium solani. Antimicrob Agents Chemother. 2010;54(3):1055–9. https://doi.org/10.1128/AAC.01445-09.
Verweij PE, Ananda-Rajah M, Andes D, Arendrup MC, Brüggemann RJ, Chowdhary A, et al. International expert opinion on the management of infection caused by azole-resistant Aspergillus fumigatus. Drug Resist Updates. 2015;21–22:30–40. https://doi.org/10.1016/j.drup.2015.08.001.
Meis JF, Chowdhary A, Rhodes JL, Fisher MC, Verweij PE. Clinical implications of globally emerging azole resistance in Aspergillus fumigatus. Philos Trans R Soc Lond B Biol Sci. 2016;371(1709):20150460. https://doi.org/10.1098/rstb.2015.0460.
Verweij PE, Zhang J, Debets AJM, Meis JF, van de Veerdonk FL, Schoustra SE, et al. In-host adaptation and acquired triazole resistance in Aspergillus fumigatus: a dilemma for clinical management. Lancet Infect Dis. 2016;16(11):e251–e260260. https://doi.org/10.1016/S1473-3099(16)30138-4.
Buil JB, Hare RK, Zwaan BJ, Arendrup MC, Melchers WJG, Verweij PE. The fading boundaries between patient and environmental routes of triazole resistance selection in Aspergillus fumigatus. PLoS Pathogens. 2019;15(8):e1007858-e. https://doi.org/10.1371/journal.ppat.1007858.
Hare RK, Gertsen JB, Astvad KMT, Degn KB, Løkke A, Stegger M, et al. In vivo selection of a unique tandem repeat mediated azole resistance mechanism (TR(120)) in Aspergillus fumigatus cyp51A, Denmark. Emerg Infect Dis. 2019;25(3):577–80. https://doi.org/10.3201/eid2503.180297.
Verweij PE, van de Sande-Bruisma N, Kema GHJ, Melchers WJG. Azole resistance in Aspergillus fumigatus in the Netherlands--increase due to environmental fungicides? Nederlands tijdschrift voor geneeskunde. 2012;156(25):A4458-A.
Resendiz-Sharpe A, Mercier T, Lestrade PPA, van der Beek MT, von dem Borne PA, Cornelissen JJ, et al. Prevalence of voriconazole-resistant invasive aspergillosis and its impact on mortality in haematology patients. J Antimicrob Chemother. 2019;74(9):2759–66. https://doi.org/10.1093/jac/dkz258.
Lestrade PP, Bentvelsen RG, Schauwvlieghe AFAD, Schalekamp S, van der Velden WJFM, Kuiper EJ, et al. Voriconazole resistance and mortality in invasive aspergillosis: a multicenter retrospective cohort study. Clin Infect Dis. 2019;68(9):1463–71. https://doi.org/10.1093/cid/ciy859.
Snelders E, Karawajczyk A, Schaftenaar G, Verweij PE, Melchers WJ. Azole resistance profile of amino acid changes in Aspergillus fumigatus CYP51A based on protein homology modeling. Antimicrob Agents Chemother. 2010;54(6):2425–30. https://doi.org/10.1128/aac.01599-09.
Schauwvlieghe AFAD, Buil JB, Verweij PE, Hoek RAS, Cornelissen JJ, Blijlevens NMA et al. High-dose posaconazole for azole-resistant aspergillosis and other difficult-to-treat mould infections. Mycoses. 2019. https://doi.org/10.1111/myc.13028.
Courtney R, Pai S, Laughlin M, Lim J, Batra V. Pharmacokinetics, safety, and tolerability of oral posaconazole administered in single and multiple doses in healthy adults. Antimicrob Agents Chemother. 2003;47(9):2788–95.
Hens B, Brouwers J, Corsetti M, Augustijns P. Supersaturation and precipitation of posaconazole upon entry in the upper small intestine in humans. J Pharm Sci. 2016;105(9):2677–84. https://doi.org/10.1002/jps.24690.
Hens B, Pathak SM. In silico modeling approach for the evaluation of gastrointestinal dissolution, supersaturation, and precipitation of posaconazole. Mol Pharm. 2017. https://doi.org/10.1021/acs.molpharmaceut.7b00396.
Ullmann AJ, Cornely OA, Burchardt A, Hachem R, Kontoyiannis DP, Topelt K, et al. Pharmacokinetics, safety, and efficacy of posaconazole in patients with persistent febrile neutropenia or refractory invasive fungal infection. Antimicrob Agents Chemother. 2006;50(2):658–66. https://doi.org/10.1128/AAC.50.2.658-666.2006.
Krishna G, Ma L, Martinho M, Preston RA, O'Mara E. A new solid oral tablet formulation of posaconazole: a randomized clinical trial to investigate rising single- and multiple-dose pharmacokinetics and safety in healthy volunteers. J Antimicrob Chemother. 2012;67(11):2725–30. https://doi.org/10.1093/jac/dks268.
Kersemaekers WM, van Iersel T, Nassander U, O'Mara E, Waskin H, Caceres M, et al. Pharmacokinetics and safety study of posaconazole intravenous solution administered peripherally to healthy subjects. Antimicrob Agents Chemother. 2015;59(2):1246–51. https://doi.org/10.1128/aac.04223-14.
European Medicines Agency. Posaconazole injection assessment report: EPAR-assessment report-Variation. 2014. https://www.ema.europa.eu/en/documents/variation-report/noxafil-h-c-610-x-0033-epar-assessment-report-variation_en.pdf. Accessed 15 Apr 2020.
Duarte RF, Lopez-Jimenez J, Cornely OA, Laverdiere M, Helfgott D, Haider S, et al. Phase 1b study of new posaconazole tablet for prevention of invasive fungal infections in high-risk patients with neutropenia. Antimicrob Agents Chemother. 2014;58(10):5758–65. https://doi.org/10.1128/aac.03050-14.
Cornely OA, Duarte RF, Haider S, Chandrasekar P, Helfgott D, Jimenez JL, et al. Phase 3 pharmacokinetics and safety study of a posaconazole tablet formulation in patients at risk for invasive fungal disease. J Antimicrob Chemother. 2016;71(3):718–26. https://doi.org/10.1093/jac/dkv380.
Maertens J, Cornely OA, Ullmann AJ, Heinz WJ, Krishna G, Patino H, et al. Phase 1B study of the pharmacokinetics and safety of posaconazole intravenous solution in patients at risk for invasive fungal disease. Antimicrob Agents Chemother. 2014;58(7):3610–7.
Cornely OA, Robertson MN, Haider S, Grigg A, Geddes M, Aoun M, et al. Pharmacokinetics and safety results from the Phase 3 randomized, open-label, study of intravenous posaconazole in patients at risk of invasive fungal disease. J Antimicrob Chemother. 2017;72(12):3406–13. https://doi.org/10.1093/jac/dkx263.
Kohl V, Muller C, Cornely OA, Abduljalil K, Fuhr U, Vehreschild JJ, et al. Factors influencing pharmacokinetics of prophylactic posaconazole in patients undergoing allogeneic stem cell transplantation. Antimicrob Agents Chemother. 2010;54(1):207–12. https://doi.org/10.1128/AAC.01027-09.
Storzinger D, Borghorst S, Hofer S, Busch CJ, Lichtenstern C, Hempel G, et al. Plasma concentrations of posaconazole administered via nasogastric tube in patients in a surgical intensive care unit. Antimicrob Agents Chemother. 2012;56(8):4468–70. https://doi.org/10.1128/AAC.06167-11.
Vehreschild JJ, Muller C, Farowski F, Vehreschild MJ, Cornely OA, Fuhr U, et al. Factors influencing the pharmacokinetics of prophylactic posaconazole oral suspension in patients with acute myeloid leukemia or myelodysplastic syndrome. Eur J Clin Pharmacol. 2012;68(6):987–95. https://doi.org/10.1007/s00228-012-1212-y.
AbuTarif MA, Krishna G, Statkevich P. Population pharmacokinetics of posaconazole in neutropenic patients receiving chemotherapy for acute myelogenous leukemia or myelodysplastic syndrome. Curr Med Res Opin. 2010;26(2):397–405. https://doi.org/10.1185/03007990903485056.
Dolton MJ, Bruggemann RJ, Burger DM, McLachlan AJ. Understanding variability in posaconazole exposure using an integrated population pharmacokinetic analysis. Antimicrob Agents Chemother. 2014;58(11):6879–85. https://doi.org/10.1128/aac.03777-14.
Petitcollin A, Boglione-Kerrien C, Tron C, Picard S, Lalanne S, Nimubona S, et al. Population pharmacokinetics and monte-carlo simulations of posaconazole administered as tablets in a real-life cohort of patients with hematological malignancies: Towards dose reduction? Fundam Clin Pharmacol. 2017;31:19.
van Iersel M, Rossenu S, de Greef R, Waskin H. A population pharmacokinetic model for a solid oral tablet formulation of posaconazole. Antimicrob Agents Chemother. 2018. https://doi.org/10.1128/aac.02465-17.
European Medicines Agency. Posaconazole tablet assessment report. Committee for Medicinal Products for Human Use (CHMP). 2014. https://www.ema.europa.eu/en/documents/variation-report/noxafil-h-c-610-x-0028-epar-scientific-discussion-extension_en.pdf. Accessed 15 Apr 2020.
Chen L, Brüggemann RJM, Knibbe CAJ, Krekels EHJ. Bioavailability and the variability of posaconazole exposure in healthy volunteers using a population pharmacokinetic analysis. Population Approach Group Europe; 2019. https://www.page-meeting.org/default.asp?abstract=8958. Accessed 15 Apr 2020.
Courtney R, Wexler D, Radwanski E, Lim J, Laughlin M. Effect of food on the relative bioavailability of two oral formulations of posaconazole in healthy adults. Br J Clin Pharmacol. 2004;57(2):218–22.
Krishna G, Ma L, Vickery D, Yu X, Wu I, Power E, et al. Effect of varying amounts of a liquid nutritional supplement on the pharmacokinetics of posaconazole in healthy volunteers. Antimicrob Agents Chemother. 2009;53(11):4749–52.
Krishna G, Moton A, Ma L, Medlock MM, McLeod J. Pharmacokinetics and absorption of posaconazole oral suspension under various gastric conditions in healthy volunteers. Antimicrob Agents Chemother. 2009;53(3):958–66. https://doi.org/10.1128/AAC.01034-08.
Dodds Ashley ES, Varkey JB, Krishna G, Vickery D, Ma L, Yu X, et al. Pharmacokinetics of posaconazole administered orally or by nasogastric tube in healthy volunteers. Antimicrob Agents Chemother. 2009;53(7):2960–4. https://doi.org/10.1128/aac.01178-08.
Kraft WK, Chang PS, van Iersel ML, Waskin H, Krishna G, Kersemaekers WM. Posaconazole tablet pharmacokinetics: lack of effect of concomitant medications altering gastric pH and gastric motility in healthy subjects. Antimicrob Agents Chemother. 2014;58(7):4020–5. https://doi.org/10.1128/AAC.02448-13.
Kersemaekers WM, Dogterom P, Xu J, Marcantonio EE, de Greef R, Waskin H, et al. Effect of a high-fat meal on the pharmacokinetics of 300-milligram posaconazole in a solid oral tablet formulation. Antimicrob Agents Chemother. 2015;59(6):3385–9. https://doi.org/10.1128/AAC.05000-14.
Krishna G, Ma L, Martinho M, O'Mara E. Single-dose phase I study to evaluate the pharmacokinetics of posaconazole in new tablet and capsule formulations relative to oral suspension. Antimicrob Agents Chemother. 2012;56(8):4196–201.
Park WB, Cho JY, Park SI, Kim EJ, Yoon S, Yoon SH, et al. Effectiveness of increasing the frequency of posaconazole syrup administration to achieve optimal plasma concentrations in patients with haematological malignancy. Int J Antimicrob Agents. 2016;48(1):106–10. https://doi.org/10.1016/j.ijantimicag.2016.04.013.
Blennow O, Eliasson E, Pettersson T, Pohanka A, Szakos A, El-Serafi I, et al. Posaconazole concentrations in human tissues after allogeneic stem cell transplantation. Antimicrob Agents Chemother. 2014;58(8):4941–3. https://doi.org/10.1128/AAC.03252-14.
Conte JE Jr, DeVoe C, Little E, Golden JA. Steady-state intrapulmonary pharmacokinetics and pharmacodynamics of posaconazole in lung transplant recipients. Antimicrob Agents Chemother. 2010;54(9):3609–13. https://doi.org/10.1128/aac.01396-09.
Conte JE Jr, Golden JA, Krishna G, McIver M, Little E, Zurlinden E. Intrapulmonary pharmacokinetics and pharmacodynamics of posaconazole at steady state in healthy subjects. Antimicrob Agents Chemother. 2009;53(2):703–7. https://doi.org/10.1128/aac.00663-08.
Krishna G, Beresford E, Ma L, Vickery D, Martinho M, Yu X, et al. Skin concentrations and pharmacokinetics of posaconazole after oral administration. Antimicrob Agents Chemother. 2010;54(5):1807–10. https://doi.org/10.1128/aac.01616-09.
Krishna G, Ma L, Martinho M, Prasad P, Wahl J, Tavakkol A. Determination of posaconazole levels in toenails of adults with onychomycosis following oral treatment with four regimens of posaconazole for 12 or 24 weeks. Antimicrob Agents Chemother. 2011;55(9):4424–6. https://doi.org/10.1128/aac.01302-10.
Kuipers S, Brüggemann RJM, De Sévaux RGL, Heesakkers JPFA, Melchers WJG, Mouton JW, et al. Failure of posaconazole therapy in a renal transplant patient with invasive aspergillosis due to Aspergillus fumigatus with attenuated susceptibility to posaconazole. Antimicrob Agents Chemother. 2011;55(7):3564–6. https://doi.org/10.1128/AAC.01544-10.
Reinwald M, Uharek L, Lampe D, Grobosch T, Thiel E, Schwartz S. Limited penetration of posaconazole into cerebrospinal fluid in an allogeneic stem cell recipient with invasive pulmonary aspergillosis. Bone Marrow Transpl. 2009;44(4):269–70.
Rüping MJGT, Albermann N, Ebinger F, Burckhardt I, Beisel C, Müller C, et al. Posaconazole concentrations in the central nervous system. J Antimicrob Chemother. 2008;62(6):1468–70.
Sponsel WE, Graybill JR, Nevarez HL, Dang D. Ocular and systemic posaconazole(SCH-56592) treatment of invasive Fusarium solani keratitis and endophthalmitis. Br J Ophthalmol. 2002;86(7):829–30. https://doi.org/10.1136/bjo.86.7.829-a.
Taesotikul S, Dilokpattanamongkol P, Nosoongnoen W, Panusitthikorn P, Rotjanapan P. Pharmacokinetic study of intravenous posaconaozle in a critically ill patient with multiple organ failure: a case report. Aust Med J. 2017;10(8):734–42. https://doi.org/10.21767/AMJ.2017.3137.
Felton T, Troke PF, Hope WW. Tissue penetration of antifungal agents. Clin Microbiol Rev. 2014;27(1):68–88. https://doi.org/10.1128/cmr.00046-13.
Calvo E, Pastor FJ, Rodriguez MM, Mayayo E, Salas V, Guarro J. Murine model of a disseminated infection by the novel fungus Fonsecaea monophora and successful treatment with posaconazole. Antimicrob Agents Chemother. 2010;54(2):919–23. https://doi.org/10.1128/AAC.01284-09.
Calvo E, Pastor FJ, Rodriguez MM, Pujol I, Guarro J. Antifungal therapy in a murine model of disseminated infection by Cryptococcus gattii. Antimicrob Agents Chemother. 2010;54(10):4074–7. https://doi.org/10.1128/AAC.00172-10.
Ghosal A, Hapangama N, Yuan Y, Achanfuo-Yeboah J, Iannucci R, Chowdhury S, et al. Identification of human UDP-glucuronosyltransferase enzyme(s) responsible for the glucuronidation of posaconazole (Noxafil). Drug Metab Dispos. 2004;32(2):267–71. https://doi.org/10.1124/dmd.32.2.267.
Krieter P, Flannery B, Musick T, Gohdes M, Martinho M, Courtney R. Disposition of posaconazole following single-dose oral administration in healthy subjects. Antimicrob Agents Chemother. 2004;48(9):3543–51. https://doi.org/10.1128/AAC.48.9.3543-3551.2004.
Petitcollin A, Crochette R, Tron C, Verdier MC, Boglione-Kerrien C, Vigneau C, et al. Increased inhibition of cytochrome P450 3A4 with the tablet formulation of posaconazole. Drug Metab Pharmacokinet. 2016;31(5):389–93. https://doi.org/10.1016/j.dmpk.2016.05.001.
Maschmeyer G, De Greef J, Mellinghoff SC, Nosari A, Thiebaut-Bertrand A, Bergeron A, et al. Infections associated with immunotherapeutic and molecular targeted agents in hematology and oncology. A position paper by the European Conference on Infections in Leukemia (ECIL). Leukemia. 2019;33(4):844–62. https://doi.org/10.1038/s41375-019-0388-x.
Zhang H, Nguyen MH, Clancy CJ, Joshi R, Zhao W, Ensor C, et al. Pharmacokinetics of posaconazole suspension in lung transplant patients with and without cystic fibrosis. Antimicrob Agents Chemother. 2016;60(6):3558–622. https://doi.org/10.1128/aac.00424-16.
Boonsathorn S, Cheng I, Kloprogge F, Alonso C, Lee C, Doncheva B, et al. Clinical pharmacokinetics and dose recommendations for posaconazole in infants and children. Clin Pharmacokinet. 2019;58(1):53–61. https://doi.org/10.1007/s40262-018-0658-1.
Katragkou A, Tsikopoulou F, Roilides E, Zaoutis TE. Posaconazole: when and how? The clinician's view. Mycoses. 2012;55(2):110–22. https://doi.org/10.1111/j.1439-0507.2011.02061.x.
Rachwalski EJ, Wieczorkiewicz JT, Scheetz MH. Posaconazole: an oral triazole with an extended spectrum of activity. Ann Pharmacother. 2008;42(10):1429–38. https://doi.org/10.1345/aph.1L005.
Frampton JE, Scott LJ. Posaconazole. Drugs. 2008;68(7):993–1016. https://doi.org/10.2165/00003495-200868070-00008.
Sandherr M, Maschmeyer G. Pharmacology and metabolism of voriconazole and posaconazole in the treatment of invasive aspergillosis-review of the literature. Eur J Med Res. 2011;16(4):139. https://doi.org/10.1186/2047-783X-16-4-139.
Cornely OA, Maertens J, Winston DJ, Perfect J, Ullmann AJ, Walsh TJ et al. Posaconazole vs. fluconazole or itraconazole prophylaxis in patients with neutropenia. N Engl J Med. 2007;356(4):348–59. https://doi.org/10.1056/NEJMoa061094.
Ullmann AJ, Lipton JH, Vesole DH, Chandrasekar P, Langston A, Tarantolo SR, et al. Posaconazole or fluconazole for prophylaxis in severe graft-versus-host disease. N Engl J Med. 2007;356(4):335–47. https://doi.org/10.1056/NEJMoa061098.
Walsh TJ, Raad I, Patterson TF, Chandrasekar P, Donowitz GR, Graybill R, et al. Treatment of invasive aspergillosis with posaconazole in patients who are refractory to or intolerant of conventional therapy: an externally controlled trial. Clin Infect Dis. 2007;44(1):2–12. https://doi.org/10.1086/508774.
Gubbins PO, Krishna G, Sansone-Parsons A, Penzak SR, Dong L, Martinho M, et al. Pharmacokinetics and safety of oral posaconazole in neutropenic stem cell transplant recipients. Antimicrob Agents Chemother. 2006;50(6):1993–9. https://doi.org/10.1128/aac.00157-06.
Hope WW, Kruhlak MJ, Lyman CA, Petraitiene R, Petraitis V, Francesconi A, et al. Pathogenesis of Aspergillus fumigatus and the kinetics of galactomannan in an in vitro model of early invasive pulmonary aspergillosis: implications for antifungal therapy. J Infect Dis. 2007;195(3):455–66. https://doi.org/10.1086/510535.
Gebremariam T, Alkhazraji S, Baldin C, Kovanda L. Prophylaxis with isavuconazole or posaconazole protects immunosuppressed mice from pulmonary mucormycosis. Antimicrob Agents Chemother. 2017;61(5):5. https://doi.org/10.1128/aac.02589-16.
Barchiesi F, Spreghini E, Santinelli A, Fothergill AW, Pisa E, Giannini D, et al. Posaconazole prophylaxis in experimental systemic zygomycosis. Antimicrob Agents Chemother. 2007;51(1):73–7. https://doi.org/10.1128/AAC.00969-06.
van Burik JA, Hare RS, Solomon HF, Corrado ML, Kontoyiannis DP. Posaconazole is effective as salvage therapy in zygomycosis: a retrospective summary of 91 cases. Clin Infect Dis. 2006;42(7):e61–e6565. https://doi.org/10.1086/500212.
Vehreschild JJ, Birtel A, Vehreschild MJ, Liss B, Farowski F, Kochanek M, et al. Mucormycosis treated with posaconazole: review of 96 case reports. Crit Rev Microbiol. 2013;39(3):310–24. https://doi.org/10.3109/1040841X.2012.711741.
Cornely OA, Alastruey-Izquierdo A, Arenz D, Chen SCA, Dannaoui E, Hochhegger B, et al. Global guideline for the diagnosis and management of mucormycosis: an initiative of the European Confederation of Medical Mycology in cooperation with the Mycoses Study Group Education and Research Consortium. Lancet Infect Dis. 2019;19(12):e405–21. https://doi.org/10.1016/s1473-3099(19)30312-3.
Salmanton-García J, Seidel D, Koehler P, Mellinghoff SC, Herbrecht R, Klimko N, et al. Matched-paired analysis of patients treated for invasive mucormycosis: standard treatment versus posaconazole new formulations (MoveOn). J Antimicrob Chemother. 2019;74(11):3315–27. https://doi.org/10.1093/jac/dkz344.
Jang SH, Colangelo PM, Gobburu JV. Exposure-response of posaconazole used for prophylaxis against invasive fungal infections: evaluating the need to adjust doses based on drug concentrations in plasma. Clin Pharmacol Ther. 2010;88(1):115–9. https://doi.org/10.1038/clpt.2010.64.
Dolton MJ, Ray JE, Chen SC, Ng K, Pont L, McLachlan AJ. Multicenter study of posaconazole therapeutic drug monitoring: exposure-response relationship and factors affecting concentration. Antimicrob Agents Chemother. 2012;56(11):5503–10. https://doi.org/10.1128/AAC.00802-12.
Dolton MJ, Ray JE, Marriott D, McLachlan AJ. Posaconazole exposure-response relationship: evaluating the utility of therapeutic drug monitoring. Antimicrob Agents Chemother. 2012;56(6):2806–13. https://doi.org/10.1128/AAC.05900-11.
Eiden C, Meniane JC, Peyriere H, Eymard-Duvernay S, Le Falher G, Ceballos P, et al. Therapeutic drug monitoring of posaconazole in hematology adults under posaconazole prophylaxis: influence of food intake. Eur J Clin Microbiol Infect Dis. 2012;31(2):161–7. https://doi.org/10.1007/s10096-011-1288-9.
Lebeaux D, Lanternier F, Elie C, Suarez F, Buzyn A, Viard JP, et al. Therapeutic drug monitoring of posaconazole: a monocentric study with 54 adults. Antimicrob Agents Chemother. 2009;53(12):5224–9. https://doi.org/10.1128/AAC.00939-09.
Neubauer WC, Engelhardt M, Konig A, Hieke S, Jung M, Bertz H, et al. Therapeutic drug monitoring of posaconazole in hematology patients: experience with a new high-performance liquid chromatography-based method. Antimicrob Agents Chemother. 2010;54(9):4029–32. https://doi.org/10.1128/AAC.00150-10.
Patterson TF, Thompson GR 3rd, Denning DW, Fishman JA, Hadley S, Herbrecht R, et al. Practice guidelines for the diagnosis and management of Aspergillosis: 2016 update by the Infectious Diseases Society of America. Clin Infect Dis. 2016;63(4):e1–e60. https://doi.org/10.1093/cid/ciw326.
Cornely OA, Ullmann AJ. Lack of evidence for exposure-response relationship in the use of posaconazole as prophylaxis against invasive fungal infections. Clin Pharmacol Ther. 2011;89(3):351–2.
U.S FDA. Medical review of posaconazole. Center for Drug Evaluation and Research; 2006. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2006/022003s000_noxafil_medr.pdf. Accessed 15 Apr 2020.
Maertens JA, Girmenia C, Bruggemann RJ, Duarte RF, Kibbler CC, Ljungman P, et al. European guidelines for primary antifungal prophylaxis in adult haematology patients: summary of the updated recommendations from the European Conference on Infections in Leukaemia. J Antimicrob Chemother. 2018;73(12):3221–30. https://doi.org/10.1093/jac/dky286.
Seyedmousavi S, Mouton JW, Melchers WJ, Bruggemann RJ, Verweij PE. The role of azoles in the management of azole-resistant aspergillosis: from the bench to the bedside. Drug Resist Updates. 2014;17(3):37–50. https://doi.org/10.1016/j.drup.2014.06.001.
Groll AH, Castagnola E, Cesaro S, Dalle JH, Engelhard D, Hope W et al. Fourth European Conference on Infections in Leukaemia (ECIL-4): guidelines for diagnosis, prevention, and treatment of invasive fungal diseases in paediatric patients with cancer or allogeneic haemopoietic stem-cell transplantation. Lancet Oncol. 2014;15(8):e327–40. https://doi.org/10.1016/s1470-2045(14)70017-8.
Chen L, Wang Y, Zhang T, Li Y, Meng T, Liu L, et al. Utility of posaconazole therapeutic drug monitoring and assessment of plasma concentration threshold for effective prophylaxis of invasive fungal infections: a meta-analysis with trial sequential analysis. BMC Infect Dis. 2018;18(1):155. https://doi.org/10.1186/s12879-018-3055-3.
Ullmann AJ, Aguado JM, Arikan-Akdagli S, Denning DW, Groll AH, Lagrou K, et al. Diagnosis and management of Aspergillus diseases: executive summary of the 2017 ESCMID-ECMM-ERS guideline. Clin Microbiol Infect. 2018;24(Suppl 1):e1–e38. https://doi.org/10.1016/j.cmi.2018.01.002.
Nielsen EI, Cars O, Friberg LE. Pharmacokinetic/pharmacodynamic (PK/PD) indices of antibiotics predicted by a semimechanistic PKPD model: a step toward model-based dose optimization. Antimicrob Agents Chemother. 2011;55(10):4619–30. https://doi.org/10.1128/aac.00182-11.
Mohamed AF, Nielsen EI, Cars O, Friberg LE. Pharmacokinetic-pharmacodynamic model for gentamicin and its adaptive resistance with predictions of dosing schedules in newborn infants. Antimicrob Agents Chemother. 2012;56(1):179–88. https://doi.org/10.1128/aac.00694-11.
Clewe O, Aulin L, Hu Y, Coates ARM, Simonsson USH. A multistate tuberculosis pharmacometric model: a framework for studying anti-tubercular drug effects in vitro. J Antimicrob Chemother. 2015;71(4):964–74. https://doi.org/10.1093/jac/dkv416.
Khan DD, Lagerback P, Cao S, Lustig U, Nielsen EI, Cars O, et al. A mechanism-based pharmacokinetic/pharmacodynamic model allows prediction of antibiotic killing from MIC values for WT and mutants. J Antimicrob Chemother. 2015;70(11):3051–60. https://doi.org/10.1093/jac/dkv233.
Jung DS, Tverdek FP, Kontoyiannis DP. Switching from posaconazole suspension to tablets increases serum drug levels in leukemia patients without clinically relevant hepatotoxicity. Antimicrob Agents Chemother. 2014;58(11):6993–5. https://doi.org/10.1128/AAC.04035-14.
Raad II, Graybill JR, Bustamante AB, Cornely OA, Gaona-Flores V, Afif C, et al. Safety of long-term oral posaconazole use in the treatment of refractory invasive fungal infections. Clin Infect Dis. 2006;42(12):1726–34. https://doi.org/10.1086/504328.
Felton TW, Baxter C, Moore CB, Roberts SA, Hope WW, Denning DW. Efficacy and safety of posaconazole for chronic pulmonary aspergillosis. Clin Infect Dis. 2010;51(12):1383–91. https://doi.org/10.1086/657306.
Stelzer D, Weber A, Ihle F, Matthes S, Ceelen F, Zimmermann G, et al. Posaconazole liquid vs tablet formulation in lung transplant recipients. Mycoses. 2018;61(3):186–94. https://doi.org/10.1111/myc.12724.
Cumpston A, Caddell R, Shillingburg A, Lu X, Wen S, Hamadani M, et al. Superior serum concentrations with posaconazole delayed-release tablets compared to suspension formulation in hematological malignancies. Antimicrob Agents Chemother. 2015;59(8):4424–8. https://doi.org/10.1128/AAC.00581-15.
Perissinotti AJ, Marini BL. Managing liver dysfunction in haematology patients: Switch antifungals, or use the tincture of time? Mycoses. 2019;62(3):214–6. https://doi.org/10.1111/myc.12871.
Pettit NN, Miceli MH, Rivera CG, Narayanan PP, Perissinotti AJ, Hsu M, et al. Multicentre study of posaconazole delayed-release tablet serum level and association with hepatotoxicity and QTc prolongation. J Antimicrob Chemother. 2017;72(8):2355–8. https://doi.org/10.1093/jac/dkx122.
Nickless JR, Bridger KE, Vora SB, Brothers AW. Evaluation of intravenous posaconazole dosing and pharmacokinetic target attainment in pediatric patients. J Pediatric Infect Dis Soc. 2018. https://doi.org/10.1093/jpids/piy094.
DiPippo AJ, Rausch CR, Kontoyiannis DP. Tolerability of isavuconazole after posaconazole toxicity in leukaemia patients. Mycoses. 2019;62(1):81–6. https://doi.org/10.1111/myc.12851.
Tverdek FP, Heo ST, Aitken SL, Granwehr B, Kontoyiannis DP. Real-life assessment of the safety and effectiveness of the new tablet and intravenous formulations of posaconazole in the prophylaxis of invasive fungal infections via analysis of 343 courses. Antimicrob Agents Chemother. 2017. https://doi.org/10.1128/aac.00188-17.
van Ingen J, van der Lee HA, Rijs TAJ, Zoll J, Leenstra T, Melchers WJG, et al. Azole, polyene and echinocandin MIC distributions for wild-type, TR34/L98H and TR46/Y121F/T289A Aspergillus fumigatus isolates in the Netherlands. J Antimicrob Chemother. 2015;70(1):178–81. https://doi.org/10.1093/jac/dku364.
Buil JB, Hagen F, Chowdhary A, Verweij PE, Meis JF. Itraconazole, voriconazole, and posaconazole CLSI MIC distributions for wild-type and azole-resistant Aspergillus fumigatus isolates. J Fungi. 2018;4(3):103. https://doi.org/10.3390/jof4030103.
Farowski F, Cornely OA, Vehreschild JJ, Hartmann P, Bauer T, Steinbach A, et al. Intracellular concentrations of posaconazole in different compartments of peripheral blood. Antimicrob Agents Chemother. 2010;54(7):2928–31. https://doi.org/10.1128/AAC.01407-09.
Farowski F, Cornely OA, Hartmann P. High intracellular concentrations of posaconazole do not impact on functional capacities of human polymorphonuclear neutrophils and monocyte derived macrophages in vitro. Antimicrob Agents Chemother. 2016. https://doi.org/10.1128/aac.02060-15.
Baistrocchi SR, Lee MJ, Lehoux M, Ralph B, Snarr BD, Robitaille R, et al. Posaconazole-loaded leukocytes as a novel treatment strategy targeting invasive pulmonary Aspergillosis. J Infect Dis. 2017;215(11):1734–41. https://doi.org/10.1093/infdis/jiw513.
Schmidt S, Schubert R, Tramsen L, Lehrnbecher T. Impact of antifungal compounds on viability and anti-aspergillus activity of human natural killer cells. Antimicrob Agents Chemother. 2019;63(2):5. https://doi.org/10.1128/aac.01993-18.
Moton A, Krishna G, Ma L, O'Mara E, Prasad P, McLeod J, et al. Pharmacokinetics of a single dose of the antifungal posaconazole as oral suspension in subjects with hepatic impairment. Curr Med Res Opin. 2010;26(1):1–7.
Courtney R, Sansone A, Smith W, Marbury T, Statkevich P, Martinho M, et al. Posaconazole pharmacokinetics, safety, and tolerability in subjects with varying degrees of chronic renal disease. J Clin Pharmacol. 2005;45(2):185–92. https://doi.org/10.1177/0091270004271402.
Hachem RY, Langston AA, Graybill JR, Perfect JR, Pedicone LD, Patino H, et al. Posaconazole as salvage treatment of invasive fungal infections in patients with underlying renal impairment. J Antimicrob Chemother. 2008;62(6):1386–91. https://doi.org/10.1093/jac/dkn401.
Kim S-H, Kwon J-C, Park C, Han S, Yim D-S, Choi J-K, et al. Therapeutic drug monitoring and safety of intravenous voriconazole formulated with sulfobutylether β-cyclodextrin in haematological patients with renal impairment. Mycoses. 2016;59(10):644–51. https://doi.org/10.1111/myc.12517.
Kiser TH, Fish DN, Aquilante CL, Rower JE, Wempe MF, MacLaren R, et al. Evaluation of sulfobutylether-β-cyclodextrin (SBECD) accumulation and voriconazole pharmacokinetics in critically ill patients undergoing continuous renal replacement therapy. Crit Care. 2015;19(1):32. https://doi.org/10.1186/s13054-015-0753-8.
Oude Lashof AML, Sobel JD, Ruhnke M, Pappas PG, Viscoli C, Schlamm HT, et al. Safety and tolerability of voriconazole in patients with baseline renal insufficiency and candidemia. Antimicrob Agents Chemother. 2012;56(6):3133–7. https://doi.org/10.1128/AAC.05841-11.
Miceli MH, Perissinotti AJ, Kauffman CA, Couriel DR. Serum posaconazole levels among haematological cancer patients taking extended release tablets is affected by body weight and diarrhoea: single centre retrospective analysis. Mycoses. 2015;58(7):432–6. https://doi.org/10.1111/myc.12339.
Chow CR, Harmatz JS, Ryan MJ, Greenblatt DJ. Persistence of a posaconazole-mediated drug-drug interaction with ranolazine after cessation of posaconazole administration: impact of obesity and implications for patient safety. J Clin Pharmacol. 2018;58(11):1436–42. https://doi.org/10.1002/jcph.1257.
Greenblatt DJ, Harmatz JS, Ryan MJ, Chow CR. Sustained impairment of lurasidone clearance after discontinuation of posaconazole: impact of obesity, and implications for patient safety. J Clin Psychopharmacol. 2018;38(4):289–95. https://doi.org/10.1097/jcp.0000000000000892.
Wasmann RE, Smit C, van Donselaar MH, van Dongen EPA, Wiezer RMJ, Verweij PE, et al. Implications for IV posaconazole dosing in the era of obesity. J Antimicrob Chemother. 2020;75(4):1006–13. https://doi.org/10.1093/jac/dkz546.
Ray J, Campbell L, Rudham S, Nguyen Q, Marriott D. Posaconazole plasma concentrations in critically ill patients. Ther Drug Monit. 2011;33(4):387–92. https://doi.org/10.1097/FTD.0b013e31821fb197.
Sime FB, Stuart J, Butler J, Starr T, Wallis SC, Pandey S, et al. Pharmacokinetics of intravenous posaconazole in critically ill patients. Antimicrob Agents Chemother. 2018. https://doi.org/10.1128/aac.00242-18.
Vicenzi EB, Cesaro S. Posaconazole in immunocompromised pediatric patients. Expert Rev Anti Infect Therapy. 2018;16(7):543–53. https://doi.org/10.1080/14787210.2018.1490177.
Arrieta AC, Sung L, Bradley JS, Zwaan CM, Gates D, Waskin H, et al. A non-randomized trial to assess the safety, tolerability, and pharmacokinetics of posaconazole oral suspension in immunocompromised children with neutropenia. PLoS One. 2019;14(3):e0212837-e. https://doi.org/10.1371/journal.pone.0212837.
Tragiannidis A, Herbrüggen H, Ahlmann M, Vasileiou E, Gastine S, Thorer H, et al. Plasma exposures following posaconazole delayed-release tablets in immunocompromised children and adolescents. J Antimicrob Chemother. 2019;74(12):3573–8. https://doi.org/10.1093/jac/dkz359.
Badee J, Qiu N, Collier AC, Takahashi RH, Forrest WF, Parrott N, et al. Characterization of the ontogeny of hepatic UDP-Glucuronosyltransferase enzymes based on glucuronidation activity measured in human liver microsomes. J Clin Pharmacol. 2019;59(Suppl 1):S42–s55. https://doi.org/10.1002/jcph.1493.
Welzen ME, Bruggemann RJ, Van Den Berg JM, Voogt HW, Gilissen JH, Pajkrt D, et al. A twice daily posaconazole dosing algorithm for children with chronic granulomatous disease. Pediatr Infect Dis J. 2011;30(9):794–7. https://doi.org/10.1097/INF.0b013e3182195808.
Doring M, Muller C, Johann PD, Erbacher A, Kimmig A, Schwarze CP, et al. Analysis of posaconazole as oral antifungal prophylaxis in pediatric patients under 12 years of age following allogeneic stem cell transplantation. BMC Infect Dis. 2012;12:263. https://doi.org/10.1186/1471-2334-12-263.
Vanstraelen K, Colita A, Bica AM, Mols R, Augustijns P, Peersman N, et al. Pharmacokinetics of posaconazole oral suspension in children dosed according to body surface area. Pediatr Infect Dis J. 2016;35(2):183–8. https://doi.org/10.1097/inf.0000000000000963.
McMahon J, Theoret Y, Autmizguine J, Bittencourt H, Tapiero B, Ovetchkine P. Posaconazole plasma monitoring in immunocompromised children. J Pediatric Infect Dis Soc. 2017. https://doi.org/10.1093/jpids/piw087.
Heinz WJ, Cabanillas Stanchi KM, Klinker H, Blume O, Feucht J, Hartmann U, et al. Posaconazole plasma concentration in pediatric patients receiving antifungal prophylaxis after allogeneic hematopoietic stem cell transplantation. Med Mycol. 2016;54(2):128–37. https://doi.org/10.1093/mmy/myv087.
Bernardo VA, Cross SJ, Crews KR, Flynn PM, Hoffman JM, Knapp KM, et al. Posaconazole therapeutic drug monitoring in pediatric patients and young adults with cancer. Ann Pharmacother. 2013;47(7–8):976–83. https://doi.org/10.1345/aph.1R775.
Mathew S, Kussin ML, Liu D, Pozotrigo M, Seyboth B, Thackray J, et al. Retrospective analysis of posaconazole suspension dosing strategies in a pediatric oncology population: single-center experience. Mol Pharm. 2017;6(3):e149–e151151. https://doi.org/10.1093/jpids/pix058.
Jancel T, Shaw PA, Hallahan CW, Kim T, Freeman AF, Holland SM, et al. Therapeutic drug monitoring of posaconazole oral suspension in paediatric patients younger than 13 years of age: a retrospective analysis and literature review. J Clin Pharm Ther. 2017;42(1):75–9. https://doi.org/10.1111/jcpt.12483.
Doring M, Blume O, Haufe S, Hartmann U, Kimmig A, Schwarze CP, et al. Comparison of itraconazole, voriconazole, and posaconazole as oral antifungal prophylaxis in pediatric patients following allogeneic hematopoietic stem cell transplantation. Eur J Clin Microbiol Infect Dis. 2014;33(4):629–38. https://doi.org/10.1007/s10096-013-1998-2.
Döring M, Cabanillas Stanchi KM, Queudeville M, Feucht J, Blaeschke F, Schlegel P, et al. Efficacy, safety and feasibility of antifungal prophylaxis with posaconazole tablet in paediatric patients after haematopoietic stem cell transplantation. J Cancer Res Clin Oncol. 2017;143(7):1281–92. https://doi.org/10.1007/s00432-017-2369-7.
Lehrnbecher T, Attarbaschi A, Duerken M, Garbino J, Gruhn B, Kontny U, et al. Posaconazole salvage treatment in paediatric patients: a multicentre survey. Eur J Clin Microbiol Infect Dis. 2010;29(8):1043–5.
Vicenzi EB, Calore E, Decembrino N, Berger M, Perruccio K, Carraro F, et al. Posaconazole oral dose and plasma levels in pediatric hematology-oncology patients. Eur J Haematol. 2018;100(3):315–22. https://doi.org/10.1111/ejh.13017.
Launay M, Roux A, Beaumont L, Douvry B, Lecuyer L, Douez E, et al. Posaconazole tablets in real-life lung transplantation: impact on exposure, drug–drug interactions, and drug management in lung transplant patients, including those with cystic fibrosis. Antimicrob Agents Chemother. 2018. https://doi.org/10.1128/aac.02061-17.
Stelzer D, Weber A, Ihle F, Matthes S, Ceelen F, Zimmermann G, et al. Comparing azole plasma trough levels in lung transplant recipients: percentage of therapeutic levels and intrapatient variability. Ther Drug Monit. 2017;39(2):93–101. https://doi.org/10.1097/ftd.0000000000000371.
Periselneris J, Nwankwo L, Schelenz S, Shah A, Armstrong-James D. Posaconazole for the treatment of allergic bronchopulmonary aspergillosis in patients with cystic fibrosis. J Antimicrob Chemother. 2019. https://doi.org/10.1093/jac/dkz075.
Shearin S, Bell T. Treatment of Aspergillus fumigatus infection with posaconazole delayed-release tablets. Am J Health Syst Pharm. 2018;75(13):958–61. https://doi.org/10.2146/ajhp170534.
Parkes LO, Cheng MP, Sheppard DC. Visual hallucinations associated with high posaconazole concentrations in serum. Antimicrob Agents Chemother. 2016;60(2):1170–1. https://doi.org/10.1128/aac.02739-15.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
No disclosures are applicable for this work. Disclosures outside of this work: R.J.B. and P.V have served as consultants to Astellas Pharma, Inc., F2G, Amplyx, Gilead Sciences, Merck Sharp & Dohme Corp., and Pfizer, Inc., and have received unrestricted and research grants from Astellas Pharma, Inc., Gilead Sciences, Merck Sharp & Dohme Corp., and Pfizer, Inc. All contracts were through Radboudumc, and all payments were invoiced by Radboudumc. None of the other authors have a conflict to declare.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Chen, L., Krekels, E.H.J., Verweij, P.E. et al. Pharmacokinetics and Pharmacodynamics of Posaconazole. Drugs 80, 671–695 (2020). https://doi.org/10.1007/s40265-020-01306-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01306-y