
Abstract
Introduction
The phase II JO25567 study compared the efficacy and safety of erlotinib plus bevacizumab vs. erlotinib alone as first-line therapy for advanced epidermal growth factor receptor (EGFR) mutation-positive non-small-cell lung cancer (NSCLC).
Objective
Our objective is to provide updated analyses of safety and the assessment of manageability of specific adverse events.
Methods
Patients with stage IIIB/IV or recurrent, non-squamous, EGFR mutation-positive NSCLC were randomized to receive erlotinib plus bevacizumab or erlotinib. The primary endpoint was progression-free survival. Adverse event frequency rates, predictability and manageability, reasons for discontinuation, time to onset, and outcomes of specific adverse events were analyzed.
Results
The safety analysis population comprised 152 randomized patients (75 erlotinib plus bevacizumab; 77 erlotinib) who received at least one dose of study drug between February 2011 and March 2012. There was no difference in overall incidence of serious adverse events between arms, but more grade 3 or higher adverse events were reported with erlotinib plus bevacizumab (90.7%) than with erlotinib (53.2%), primarily due to grade 3 hypertension. Hypertension was controllable with antihypertensive medications in most cases. Proteinuria and bleeding were also more frequently reported with erlotinib plus bevacizumab than with erlotinib but were manageable and did not lead to early discontinuations.
Conclusions
The addition of bevacizumab to erlotinib prolonged progression-free survival in EGFR mutation-positive NSCLC. Follow-up safety data were consistent with the known safety profiles of both erlotinib and bevacizumab in NSCLC; this combination appeared to be manageable, and treatment was well tolerated.
JapicCTI-111390.
Similar content being viewed by others


The updated safety profile in JO25567 was largely consistent with the known safety profiles of both erlotinib and bevacizumab in a population with non-small-cell lung cancer (NSCLC). |
Adverse events observed more frequently in the erlotinib plus bevacizumab (EB) arm than in the erlotinib alone (E) arm included hypertension, proteinuria, bleeding events, and skin and nail disorders. These adverse events could be monitored, easily detected, and well managed in most cases. |
Although EB led to higher rates of bevacizumab discontinuation than previously reported (in the context of prolonged drug exposure), these were not early discontinuations and therefore did not prevent patients from deriving full benefit from treatment. |
1 Introduction
Epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs) have proven benefit for patients with non-small-cell lung cancer (NSCLC) with activating EGFR mutations (EGFR-positive [+] NSCLC) [1,2,3,4,5,6,7]. Current clinical guidelines recommend that patients with advanced inoperable EGFR+ NSCLC should receive treatment with an EGFR TKI in the first-line setting [8,9,10]. Although EGFR inhibitors prolong progression-free survival (PFS), treatment resistance often develops; most patients experience disease progression and almost inevitably revert to treatment with cytotoxic chemotherapy [1,2,3,4, 11, 12]. Novel therapeutic approaches are therefore needed to improve outcomes for these patients.
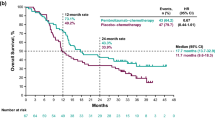
Clinical data suggesting activity of the EGFR TKI erlotinib plus the anti-angiogenic agent bevacizumab (EB) in NSCLC initially came from a small, randomized phase II study conducted in the second-line setting [13]. This study led to the initiation of further studies using the combination in advanced NSCLC (OSI3364g [BeTa-Lung] [14], BO20571 [TASK] [15], and AVF3671g [ATLAS]) [16]. These further studies were conducted in an EGFR-unselected patient population because, at the time of their initiation, the clinical relevance of EGFR-activating mutations in NSCLC had not been fully established. However, retrospective analyses of patients with EGFR+ tumors suggested greater benefit of the EB combination compared with the comparator arm. The phase II JO25567 study was undertaken to evaluate the efficacy and safety of EB vs. erlotinib alone (E) as first-line therapy in patients with advanced EGFR+ NSCLC in Japan [17]. In the primary analysis (data cut-off 30 June 2013), the study demonstrated a clinically meaningful and statistically significant improvement in terms of PFS (hazard ratio 0.54; 95% confidence interval 0.36–0.79; P = 0.0015; median 16.0 months for EB and 9.7 months for E); overall survival was immature at the time of the primary analysis [17]. Here, we report the safety and manageability analyses from the JO25567 study, with an additional 9 months of safety follow-up compared with the primary analysis, with a data cut-off date of 31 March 2014.
2 Methods
Methods are reported in brief, as a detailed description has been published previously [17]. The study is registered with the Japan Pharmaceutical Information Center (JapicCTI-111390).
2.1 Study Design
JO25567 was a randomized, open-label, multicenter phase II study comparing the efficacy and safety of oral erlotinib 150 mg/day plus intravenous bevacizumab 15 mg/kg every 3 weeks (EB) vs. erlotinib 150 mg/day (E) as first-line therapy for patients with stage IIIB/IV EGFR+ NSCLC. Stepwise modification of the dosing of erlotinib (to 100 or 50 mg/day) due to adverse events (AEs) was allowed. Dose reductions due to toxicities were not allowed for bevacizumab; however, temporary dose interruptions were allowed to manage toxicities (see the Electronic Supplementary Material). As per protocol, patients in the combination arm could continue with one component of study medication if the other drug was discontinued. All patients provided informed consent, and the study was approved by all relevant ethics committees.
2.2 Patients
Eligible patients were aged ≥20 years with histologically or cytologically confirmed stage IIIB/IV or post-operative, recurrent, non-squamous NSCLC with an activating EGFR mutation (exon 19 deletion or L858R point mutation) without concurrent T790M mutation. Other eligibility criteria included an Eastern Cooperative Oncology Group performance status 0 or 1; adequate hematological, hepatic, and renal function; and no previous chemotherapy for advanced disease. Previous radiotherapy was allowed for lesions not located in the lungs. Key exclusion criteria included patients with brain metastases at baseline; those who had received previous treatment with an EGFR or vascular EGFR inhibitor; patients with a confirmed T790M mutation; and patients with a history of interstitial lung disease [17].
2.3 Study Endpoints
The primary endpoint was PFS assessed by an independent review committee [17]. Secondary endpoints included overall survival, tumor response, PFS according to investigator assessment, quality of life (QoL), and safety.
2.4 Assessments
Patients were monitored for AEs throughout the study period and graded according to the National Cancer Institute Common Terminology Criteria for AEs v4.03 [18]. AEs were classified using the Medical Dictionary for Regulatory Activities (MedDRA®), version 14.0. Serious AEs were classified using the Japanese Pharmaceuticals and Medical Devices Act [19]. The causal relationship between the AE and the investigational drugs was classed as not related, unlikely to be related, possibly related, or probably related [20, 21]. Action taken for the treatment of AEs and action taken regarding the investigational drug were recorded, as were the outcomes of all AEs. All randomized patients were included to assess the time to and incidence of hypertension, proteinuria, and bleeding events. Patients who received at least one dose of study treatment were included to assess the incidence of all other safety events.
3 Results
3.1 Patients
Between February 2011 and March 2012, a total of 154 patients were randomized to receive EB (n = 77) or E (n = 77). Of the 154 patients randomized, two withdrew due to concomitant illness before treatment started, so were excluded from the safety analysis population (n = 152; 75 in the EB arm, 77 in the E arm). Patient characteristics were generally well balanced between the two treatment arms. As previously mentioned, the study met its primary efficacy endpoint [17].
3.2 Drug Exposure
Patients received a median 16 cycles of bevacizumab, and median duration of exposure was 325 days (range 1–1053). The median dose intensity of bevacizumab in the EB arm was 93.7% (range 68.9–99.7). With respect to erlotinib, the median dose intensity of erlotinib was similar in both arms: 95.3% (range 34.0–100) in the EB arm compared with 98% (range 33.3–100) in the E arm. Median duration of exposure to erlotinib was much longer in the EB arm at 431 days (range 21–1065) compared with 254 days (range 18–1075) in the E arm.
3.3 Updated Safety
The most common AEs (≥50% frequency, any grade, regardless of relation to treatment) in the EB and E arms, respectively, were diarrhea (81.3 vs. 79.2%), paronychia (78.7 vs. 64.9%), hypertension (78.7 vs. 14.3%), dry skin (74.7 vs. 58.4%), stomatitis (62.7 vs. 59.7%), and acneiform dermatitis (61.3 vs. 53.2%). AEs with the most substantial difference in incidence between the EB and E arms were attributed to known bevacizumab-related AEs of special interest, including all-grade hypertension, bleeding events, and proteinuria. All-grade and grade 3 or higher AEs of special interest are shown in Table 1.
Time-to-event analyses revealed that, in the EB arm, the number of patients who experienced first onset of hypertension, bleeding, and proteinuria continued to increase until 12–14 months after study initiation (Fig. 1). Skin and nail disorders, which are commonly seen with erlotinib, were also observed in both arms; the addition of bevacizumab to erlotinib appeared to increase their incidence (paronychia [all grades]: 78.7% with EB and 64.9% with E; grade 3 or higher: 2.7% for EB, 3.9% for E).

Time to first onset of (a) hypertension, (b) proteinuria, and (c) bleeding events in all randomized patients. E erlotinib alone, EB erlotinib plus bevacizumab
The incidence of any grade 3 or higher AEs (any relationship to treatment) was higher in the EB arm than in the E arm. Grade 3 or higher AEs were reported by 90.7% of EB patients and 53.2% of E patients (grade 3: 89.3% with EB and 42.9% with E; grade 4: 1.3% with EB and 9.1% with E; grade 5 AE: one patient in the E arm [drowning; assessed by the investigator as unlikely to be causally related to study treatment]) [17]. The increase in grade 3 AEs in the EB arm was primarily driven by the higher incidence of grade 3 hypertension (no grade 4 hypertension was reported). No nephrotic syndrome or permanent organ dysfunction was seen in the study. The proportion of patients experiencing serious AEs (SAEs) was similar between arms: 25.3% for EB and 24.7% for E.
The reason for discontinuation from any component of treatment in the EB arm was due to an AE in 45.3% of patients (mostly discontinuation of bevacizumab due to associated AEs in 42.7% of EB patients); most patients continued to receive erlotinib after discontinuation of bevacizumab. The incidence of AEs leading to discontinuation of erlotinib was similar in the EB and E arms (17.3 and 18.2%, respectively). These data demonstrate that discontinuation of bevacizumab due to AEs did not prevent continuation of erlotinib. The higher proportion of patients discontinuing in the EB arm compared with the E arm was primarily driven by proteinuria (16.0%) and bleeding events (epistaxis 4.0%; hemorrhoidal hemorrhage 2.7%) (Table 2). Discontinuations tended to occur later in the study, with the median duration of bevacizumab exposure being 8.2 months in those who discontinued. A total of 43.8% of patients who discontinued had ≥10 months of bevacizumab exposure.
3.4 Predictability and Manageability of Specific Adverse Events in the Erlotinib plus Bevacizumab Arm
3.4.1 Hypertension
Hypertension (any grade) events (comprising hypertension and increased blood pressure) were reported in 59 EB patients (78.7%; 58 patients with hypertension, one patient with increased blood pressure) compared with 11 E patients (14.3%). Hypertension events occurred in patients with prior or concurrent hypertension at baseline at a lower rate than in those without (65.6 vs. 86.0%, respectively). Most patients developed hypertension within 1–2 months, with the incidence steadily increasing until 14 months (Fig. 1).
Although 45 EB patients experienced grade 3 hypertension, these events were controllable with one or two antihypertensive medications in most cases. A total of 33 EB patients initiated anti-hypertensive medication, 18 of these patients needed a second medication to control blood pressure and five needed a third medication. None of these events was assessed as serious, and hypertension only led to discontinuation of bevacizumab in two patients (of 58), who both responded to antihypertensive medication and received more than the median number of bevacizumab cycles. Of the 58 patients with hypertension, 15 had recovered or were recovering at the last visit (most of these patients stopped antihypertensive treatment at the last visit). Cardiovascular or cerebrovascular events occurred in three patients (4%) in the EB arm (two patients with cerebral infarction events and one patient with cerebral ischemia) compared with one patient in the E arm (1.3%; arterial thrombosis). Arterial thromboembolism is a known adverse reaction to bevacizumab. Despite prolonged exposure to bevacizumab treatment and an increased incidence of hypertension, the incidence of arterial thromboembolism observed in this study was within the range described in the previous studies.
3.4.2 Proteinuria
Proteinuria (any grade) events (comprising proteinuria or protein urine present) were reported in 41 patients (54.7%; 35 patients with proteinuria, six with protein in the urine) in the EB arm compared with three (3.9%) in the E arm. Six patients (8%) had grade 3 events in the EB arm. Of these patients, three had a history of hypertension, a known risk factor for proteinuria; no history of renal disorder or diabetes was reported in these patients. Proteinuria appeared to occur at any time during treatment, with 40–50% of patients who experienced proteinuria reporting it within the first 10 months of treatment. Proteinuria led to a total of 12 bevacizumab discontinuations (16% of the EB arm). These 12 patients received a median of 15.5 cycles of bevacizumab before discontinuation due to proteinuria, which was similar to the overall median cycles of bevacizumab received in the EB arm (16 cycles). After discontinuation of bevacizumab, proteinuria events were reported as resolved in seven patients and unresolved in five patients. No associated renal events or relevant laboratory events were reported for any of these patients.
3.4.3 Bleeding
A total of 56 patients (74.7%) experienced overall bleeding events of any grade in the EB arm, compared with 23 patients (29.9%) in the E arm. Mucocutaneous bleeding accounted for most of these events in the EB arm, with epistaxis being the main bleeding event contributing to the higher incidence in the EB arm (46.7 vs. 9.1%, respectively). Pulmonary bleeding (hemoptysis) occurred in seven patients in the EB arm and one patient in the E arm, all of which were grade 1 or 2. Two patients experienced serious bleeding events (grade 3 epistaxis and grade 2 duodenal ulcer hemorrhage) in the EB arm.
All hemoptysis events resolved without sequelae and did not lead to discontinuation of erlotinib or bevacizumab. The majority of non-pulmonary bleeding events were either resolved or resolving at the clinical data cut-off (31 March 2014). Unresolved events reported in more than one patient were grade 1 epistaxis (eight patients, all in the EB arm), grade 1 blood in the urine (two patients, both in the EB arm), and grade 1 subcutaneous hemorrhage (two patients, both in the E arm).
Non-pulmonary bleeding events led to a total of nine bevacizumab discontinuations, representing 12% of the EB arm. Among these nine patients, latency to event onset ranged from 1.5 to 11.8 months, with a median latency of 4.2 months (approximately 6 cycles). No events secondary to bleeding were reported in the nine patients who discontinued bevacizumab. Treatment to control bleeding included nil by mouth for duodenal hemorrhage and medications such as carbazochrome and tranexamic acid. All but one patient (with unresolved unspecified hemorrhage), including those with serious and grade 3 events, reported favorable outcomes after bevacizumab discontinuation.
3.4.4 Skin and Nail Conditions
All-grade skin and nail events (dry skin and paronychia) were reported with a >10% increase in the EB arm compared with the E arm; grade 3 skin events (rash, dermatitis acneiform, and dry skin) were reported with a >2% increase. No grade 4 or 5 skin or nail events were reported. The rates of grade 3 rash, all-grade dermatitis acneiform, and grade 3 dermatitis acneiform were similar when adjusted for treatment exposure. The exposure-adjusted incidence of all-grade dermatitis acneiform (AE rate per 100 patient-years) was 118 vs. 107 and of grade ≥3 dermatitis acneiform was 7.1 vs. 8.8 (E vs. EB, respectively). All-grade paronychia occurred with a higher incidence in the EB arm than in the E arm (78.7 vs. 64.9%, respectively); however, the frequency of grade 3 paronychia was similar in both arms (3.9 and 2.7%, respectively). For all-grade paronychia, exposure-adjusted analysis revealed a trend towards an increased incidence rate in the EB arm vs. the E arm (213 vs. 171 events per patient-year, respectively).
Overall, the reported skin and nail conditions were mild in severity (mainly grade 1–2), could be well managed, and infrequently led to study drug discontinuation (n = 2; EB). It is of note that the two patients who discontinued only did so after a long duration of study treatment, having received 15 and 16 cycles of treatment, respectively.
4 Discussion
Safety results from the JO25567 study, with an additional 9 months of follow-up from the primary data analysis are presented here (data cut-off, 31 March 2014; median follow-up 25.9 months for EB, 27 months for E). This analysis supports the finding that the EB regimen has a manageable safety profile, in addition to providing a PFS advantage compared with E alone [17].
Hypertension, proteinuria, and bleeding are known AEs associated with bevacizumab. However, in the EB arm of our study, their incidences were higher than in previous experience [20]. This was most likely due to the extended exposure to bevacizumab (median of 16 cycles) as compared with most previous studies of bevacizumab across indications that used 4–6 cycles [20]. In the EB arm of JO25567, the number of patients who experienced first onset of hypertension, proteinuria, and bleeding continued to increase until 12–14 months after study treatment initiation, highlighting the role of the extended exposure as a contributing factor to the increased incidence of these AEs. The skin and nail events reported in this study (e.g., paronychia) are known to be associated with erlotinib treatment and are well characterized [20, 21]. Dry skin is also linked with bevacizumab treatment [20], so an increased incidence with combination therapy was expected. These conditions were generally mild to moderate in severity and are considered of low clinical importance for patients with NSCLC.
The incidence of AEs leading to bevacizumab discontinuation was higher than in previous EB studies, mainly driven by proteinuria and bleeding events. Patients discontinued bevacizumab due to AEs from 0 to 32.5 months after initiation of study treatment, with the median duration of bevacizumab exposure for these patients being 8.2 months. Of the 32 patients who discontinued bevacizumab, 14 (44%) did so at least 10 months after initiation of study treatment, which was longer than the median PFS in the E arm. Incidences of AEs leading to erlotinib discontinuation and SAEs were consistent with previous trials [15, 16]. As per the protocol, discontinuation of bevacizumab due to AEs did not prevent the patients from continuing erlotinib and vice versa. Therefore, discontinuation of bevacizumab did not adversely impact patients’ ability to receive treatment with erlotinib, as only seven patients discontinued both treatments simultaneously.
In JO25567, the high incidence of grade ≥3 AEs (90.7%) seen in the EB arm was primarily due to increased grade 3 hypertension (60.0%). The high incidence of hypertension did not lead to a correspondingly high level of discontinuations (n = 2). In addition, none of the hypertension events were assessed as serious, and they did not lead to significantly more clinical complications such as cardiovascular or cerebrovascular events. A history of hypertension did not appear to predispose patients to the development of hypertension, nor did treatment with the EB combination appear to exacerbate existing conditions. However, regardless of baseline hypertension, patients should be monitored for worsening of hypertension, and, if necessary, modifications should be made to existing hypertension medication during treatment with EB. Hypertension is a known risk associated with bevacizumab but was manageable with antihypertensive medication and routine blood pressure monitoring. Therefore, hypertension should not be considered a major limitation when deciding on bevacizumab treatment.
Although caution in making direct comparisons between studies is recommended due to differing patient populations and study designs, safety data associated with this combination from previous studies [14,15,16, 22] suggest that AEs reported with the EB combination are tolerable. The increase in AEs seen in this study for EB may be mainly due to extended exposure to the combination. The primary analysis results (cut-off 30 June 2013) demonstrated that adding bevacizumab to erlotinib can improve efficacy outcomes, and the safety profile of this combination is manageable in this population [17].
Potential limitations of this safety analysis of the JO25567 study include the relatively small sample size and enrollment of only Japanese patients. However, there is no known clinical evidence to suggest that the safety profile of these drugs would differ by ethnicity, and the pharmacokinetics are similar for both erlotinib and bevacizumab in Japanese and Caucasian patients.
5 Conclusions
JO25567 has previously shown a clinically meaningful and statistically significant efficacy benefit for EB vs. E in patients with EGFR+ NSCLC. The updated safety profile in JO25567 was largely consistent with the known safety profiles of both erlotinib and bevacizumab in a population with NSCLC. AEs observed more frequently in the EB arm than in the E arm included hypertension, proteinuria, bleeding events, and skin and nail disorders; these were likely due to the extended exposure to bevacizumab and erlotinib. They could be easily detected and well managed in most cases. No clinically significant complications of these events were noted. Although the EB combination led to higher rates of bevacizumab discontinuation than previously reported (in the context of prolonged drug exposure), these were not early discontinuations and therefore did not prevent patients from deriving the full benefit from treatment. This is reflected in the 6.3-month prolongation in PFS with the addition of bevacizumab.
References
Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–8.
Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–8.
Rosell R, Carcereny E, Gervais R, Spanish Lung Cancer Group in collaboration with Groupe Français de Pneumo-Cancérologie and Associazione Italiana Oncologia Toracica, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–46.
Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–42.
Wu YL, Zhou C, Liam CK, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label. ENSURE study. Ann Oncol. 2015;26:1883–9.
Yang J, Wu YL, Schuler M, et al. Afatinib versus cisplatin-based chemotherapy for EGFR mutation-positive lung adenocarcinoma (LUX-Lung 3 and LUX-Lung 6): analysis of overall survival data from two randomised, phase 3 trials. Lancet Oncol. 2015;16:141–51.
Kato T, Yoshioka H, Okamoto I, et al. Afatinib versus cisplatin plus pemetrexed in Japanese patients with advanced non-small-cell lung cancer harboring activating EGFR mutations: subgroup analysis of LUX-Lung 3. Cancer Sci. 2015;106:1202–11.
Kobayashi K, Hagiwara K. Treatment guideline for advanced NSCLC based on driver gene mutations. J Genet Syndr Gene Ther. 2014;5:212.
Reck M, Popat S, Reinmuth N, et al. Metastatic non-small-cell lung cancer (NSCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2014;25(Suppl 3):iii27–39.
National Comprehensive Cancer Network. NCCN guidelines for treatment of cancer by site. Fort Washington, PA: NCCN; 2017. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp#site. Accessed 17 Jan 2017.
Fukuoka M, Wu YL, Thongprasert S, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non–small-cell lung cancer in Asia (IPASS). J Clin Oncol. 2011;29:2866–74.
Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–34.
Herbst R, O’Neill V, Fehrenbacher L, et al. Phase II study of efficacy and safety of bevacizumab in combination with chemotherapy or erlotinib compared with chemotherapy alone for treatment of recurrent or refractory non-small-cell lung cancer. J Clin Oncol. 2007;25:4743–50.
Herbst RS, Ansari R, Bustin F, et al. Efficacy of bevacizumab plus erlotinib versus erlotinib alone in advanced non-small-cell lung cancer after failure of standard first-line chemotherapy (BeTa): a double-blind, placebo-controlled, phase 3 trial. Lancet. 2011;377:1846–54.
Ciuleanu T, Tsai C, Tsao C, et al. A phase II study of erlotinib in combination with bevacizumab versus chemotherapy plus bevacizumab in the first-line treatment of advanced non-squamous non-small cell lung cancer. Lung Cancer. 2013;82:276–81.
Johnson B, Kabbinavar F, Fehrenbacher L, et al. ATLAS: randomized, double-blind, placebo-controlled, phase IIIB trial comparing bevacizumab therapy with or without erlotinib, after completion of chemotherapy, with bevacizumab for first-line treatment of advanced non-small-cell lung cancer. J Clin Oncol. 2013;31:3926–34.
Seto T, Kato T, Nishio M, et al. Erlotinib alone or with bevacizumab as first-line therapy in patients with advanced non-squamous non-small-cell lung cancer harbouring EGFR mutations (JO25567): an open-label, randomized, multicenter, phase 2 study. Lancet Oncol. 2014;15:1236–44.
National Cancer Institute. NCI Common Terminology Criteria for Adverse Events (CTCAE), v.4 data files. NCI; 2010. https://evs.nci.nih.gov/ftp1/CTCAE/About.html. Accessed 27 Jun 2017.
Japan Pharmaceutical Manufacturers Association. Pharmaceutical administration and regulations in Japan. Tokyo: JPMA; 2010. http://apps.who.int/medicinedocs/documents/s18577en/s18577en.pdf. Accessed 24 July 2017.
Melosky B, Hirsh V. Management of common toxicities in metastatic NSCLC related to anti-lung cancer therapies with EGFR-TKIs. Front Oncol. 2014;4:238.
Yamazaki N, Kiyohara Y, Kudoh S, et al. Optimal strength and timing of steroids in the management of erlotinib-related skin toxicities in a post-marketing surveillance study (POLARSTAR) of 9909 non-small-cell lung cancer patients. Int J Clin Oncol. 2016;21:248–53.
Rosell R, Dafni U, Felip E, et al. Erlotinib and bevacizumab in patients with advanced non-small-cell lung cancer and activating EGFR mutations (BELIEF): an international, multicentre, single-arm, phase 2 trial. Lancet Respir Med. 2017;5:435–44.
Acknowledgements
The authors thank the investigators, the patients, and their families for their contribution to this clinical study. The authors also thank Naohito Inagaki of Chugai Pharmaceutical Co. Ltd for useful discussions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The JO25567 study was sponsored by Chugai Pharmaceutical Co. Ltd. Support for third-party writing assistance under the direction of the authors for this manuscript was provided by Joanna Musgrove MRes of Gardiner-Caldwell Communications and was funded by F. Hoffmann-La Roche Ltd.
Role of the funding source
This trial was designed, funded, and monitored by Chugai Pharmaceutical Co. Ltd. Data were gathered, analyzed, and interpreted by Chugai Pharmaceutical Co. Ltd and F. Hoffmann-La Roche Ltd, with input from all authors. The drugs were supplied by Chugai Pharmaceutical Co. Ltd. The corresponding author had full access to the relevant data and took full responsibility for the final decision to submit the report for publication.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Conflicts of interest
Terufumi Kato has received grants and personal fees from Chugai Pharmaceutical Co. Ltd, F. Hoffmann-La Roche Ltd, Eli Lilly, AstraZeneca, Taiho Pharmaceutical, Pfizer, and Kyowa Hakko Kirin and grants from Takeda Pharmaceutical, Daiichi Sankyo Co Ltd, Yacult Honsha, Bristol-Myers Squibb, Abbvie, Astellas Pharma, Quintiles, and Parexel International. Takashi Seto has received grants and honoraria from AstraZeneca, Chugai Pharmaceutical Co. Ltd, Daiicho Sankyo, Eli Lilly, and Eisai; grants from Astellas Pharma, Bayer, Merck, Serono, Novartis, Pfizer, and Verastam; and honoraria from Bristol-Myers Squibb, Fuju Pharma, Hisamitu Pharmaceutical, Kissei Pharmaceutical, Kyowa Hakko Kirin, Nippon Kayaku, Mochida Pharmaceutical, Ono Pharmaceutical, F. Hoffmann-La Roche Ltd, Sanofi, Showa Yakuhin Kako, Sumitomo Dainippon Pharma, Taiho Pharmaceutical, and Takeda Pharmaceutical. Makoto Nishio has received grants and personal fees from Pfizer, Bristol-Myers Squibb, Ono Pharmaceutical, Chugai Pharmaceutical Co. Ltd, Eli Lilly, Taiho Pharmaceutical, Astellas Pharma, AstraZeneca, and Boehringer Ingelheim. Koichi Goto has received grants from Chugai Pharmaceutical Co. Ltd, MSD, Taiho Pharmaceutical, Boehringer Ingelheim, Ono Pharmaceutical, GlaxoSmithKline, OxOnc, Pfizer, Kyowa Hakko Kirin, Eli Lilly, Sumitomo, Dainippon Pharma, Takeda Pharmaceutical, Novartis, Daiichi Sankyo, Astellas Pharma, Eisai, Amgen Astellas Biopharma, AstraZeneca, Merck, Serono, Riken, and Genesis; and personal fees from Chugai Pharmaceutical Co. Ltd, MSD, Taiho Pharmaceutical, Boehringer Ingelheim, Ono Pharmaceutical, Pfizer, Kyowa Hakko Kirin, Eli Lilly, Novartis, Daiichi Sankyo, Bristol-Myers Squibb, AstraZeneca, Nippon, Kayaku, Life Technologies Japan, SRL, Riken, and Genesis. Noboru Yamamoto has received grants from Chugai Pharmaceutical Co. Ltd, Eli Lilly, Taiho Pharmaceutical, Eisai, Quintiles, Astellas Pharma, Bristol-Myers Squibb, Novartis, Daiichi Sankyo, Pfizer, Boehringer Ingelheim, Kyowa Hakko Kirin, Bayer, Takeda, and Ono Pharmaceutical. Isamu Okamoto has received personal fees from Chugai Pharmaceutical Co. Ltd. Liang Tao and Tarik Khaznadar are employees of F. Hoffmann-La Roche Ltd. Wei Yu is an employee of Genentech Inc. Kosei Tajima, Masahiko Shibata, and Akihiro Seki are employees of Chugai Pharmaceutical Co. Ltd. Nobuyuki Yamamoto has received grants and personal fees from Chugai Pharmaceutical Co. Ltd, Boehringer Ingelheim, and Ono Pharmaceutical, and personal fees from AstraZeneca, Eli Lilly, MSD, and Pfizer.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kato, T., Seto, T., Nishio, M. et al. Erlotinib Plus Bevacizumab Phase ll Study in Patients with Advanced Non-small-Cell Lung Cancer (JO25567): Updated Safety Results. Drug Saf 41, 229–237 (2018). https://doi.org/10.1007/s40264-017-0596-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-017-0596-0