
Abstract
It has been estimated by the World Health Organization (WHO) that over 71 million people were infected with the hepatitis C virus (HCV) in 2015. Since then, a number of highly effective direct-acting antiviral (DAA) regimens have been licensed for the treatment of chronic HCV infection: sofosbuvir/daclatasvir, sofosbuvir/ledipasvir, elbasvir/grazoprevir, sofosbuvir/velpatasvir, glecaprevir/pibrentasvir, and sofosbuvir/velpatasvir/voxilaprevir. With these treatment regimens, almost all chronic HCV-infected patients, even including prior DAA failures, can be treated effectively and safely. It is therefore likely that further development of DAAs will be limited. In this descriptive review we provide an overview of the clinical pharmacokinetic characteristics of currently available DAAs by describing their absorption, distribution, metabolism, and excretion. Potential drug–drug interactions with the DAAs are briefly discussed. Furthermore, we summarize what is known about the pharmacodynamics of the DAAs in terms of efficacy and safety. We briefly discuss the relationship between the pharmacokinetics of the DAAs and efficacy or toxicity in special populations, such as hard to cure patients and patients with liver cirrhosis, liver transplantation, renal impairment, hepatitis B virus or HIV co-infection, bleeding disorders, and children. The aim of this overview is to educate/update prescribers and pharmacists so that they are able to safely and effectively treat HCV-infected patients even in the presence of underlying co-infections or co-morbidities.
Similar content being viewed by others

The currently approved direct-acting antivirals (DAAs) can be used to treat a great majority of hepatitis C-infected patients. |
Some of the last remaining issues regarding DAA therapy are how to treat patients who have not responded to DAA therapy who have severe resistance associated-substitution, how to manage drug interactions with strong enzyme inducers, and how to treat patients with both decompensated cirrhosis and renal impairment. |
1 Introduction
Hepatitis C virus (HCV) infection is caused by a virus that replicates in the liver; this leads to scarring of the liver which can result in liver diseases such as cirrhosis and hepatocellular carcinoma. In 2015, it was estimated by the World Health Organization (WHO) that over 71 million people were infected with HCV (HCV-RNA positive) [1]. In addition, from 1988 to 2009, 59% of liver transplantations were caused by cirrhosis, of which 40% were virus related [2].
HCV is an RNA virus that can be divided in six genotypes with several subtypes. Recently, a case report of a seventh HCV genotype has been described [3]. Treatment success and development of liver disease differs per genotype. Another important aspect is the high replication rate and the error-prone nature of the HCV viral replication cycles. This results in a high prevalence of resistance-associated substitutions (RASs), which can occur with and without drug pressure [4].
The landscape of HCV therapy dramatically changed from 2015 onwards. Before 2015, the treatment of HCV was mainly with peg-interferon (peg-IFN) plus ribavirin (RBV) therapy, which was associated with suboptimal response rates and considerable short- and long-term toxicity. HCV cure rates markedly improved with the approval of boceprevir and telaprevir, the first direct-acting antivirals (DAAs), but toxicity was still high [5]. Since the approval of these drugs, several other DAAs have been licensed, with a further increase in response rates for the different HCV genotypes and special patient populations. The timeline of DAA development is presented in Fig. 1.

Timeline of approval of direct-acting antivirals for both the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA). 1Drugs are withdrawn or were not re-approved for the EMA and/or FDA markets
With the recent approval of the highly effective regimens of glecaprevir (GLE)/pibrentasvir (PIB) [6] and sofosbuvir (SOF)/velpatasvir (VEL) [7] with or without voxilaprevir (VOX) [8], more than 95% of chronic HCV-infected patients can be treated successfully and safely. A remaining challenge is drug pricing, which is still limiting HCV treatment access globally. Clinical challenges in HCV treatment only remain in a few special patient groups such as patients with decompensated cirrhosis, renal impairment, and drug–drug interactions (DDIs). Nevertheless, further DAA drug development has seemingly ended, with few DAAs left in the development pipelines of the major drug companies. The drugs licensed today are the regimens that we will use to reach the WHO goals to eliminate and eradicate HCV [1]. To accomplish this, the biggest challenge in resource-rich countries is to identify all patients who are HCV infected and lost from care or those who were never diagnosed. For the low-income countries, the availability of these drugs is often a major issue because of high pricing.
The aim of this descriptive review is to give an overview of the pharmacokinetics and pharmacodynamics (safety and efficacy) of the DAAs currently used for chronic HCV treatment. In addition, special populations are identified and separately discussed.
2 Methods
All phase II and III studies describing efficacy and/or safety of the following DAA combinations were selected for this review: SOF/daclatasvir (DCV); SOF/ledipasvir (LDV); elbasvir (EBR)/grazoprevir (GZR); SOF/VEL; GLE/PIB; and SOF/VEL/VOX. Boceprevir, telaprevir, simeprevir, and paritaprevir/ritonavir, ombitasvir, dasabuvir are not discussed as they have been withdrawn (licensed not extended) from the market and no generic versions of these DAAs will be produced. Retrospective studies and case reports/series were excluded. In addition, the Summaries of Product Characteristics (SmPCs) published by the European Medicines Agency (EMA), the prescribing information published by the US Food and Drug Administration (FDA), and poster presentations available online were used.
The overview of the search terms and results can be found in the Electronic Supplementary Material (ESM) (Table S1), as well as the summary of the efficacy and safety data found in the studies (ESM Table S3–S8).
3 Pharmacokinetics
Table 1 presents a summary of the product and dosing information of the DAAs approved by the FDA and EMA. Table 2 provides an overview of the pharmacokinetic parameters of the DAAs.
3.1 Daclatasvir (DCV)
3.1.1 Absorption
DCV is readily absorbed as the time to maximal plasma concentration (tmax) is 1–2 h. The maximal plasma concentration (Cmax), area under the concentration–time curve (AUC), and minimal plasma concentration (Cmin) increase in a dose-proportional matter. Exposure was comparable in HCV-infected patients and healthy volunteers after a dosage of 60 mg [9, 10]. A high-fat meal (950 kcal; 492 kcal fat, 312 kcal carbohydrates, 144 kcal protein) decreases absorption as both the Cmax and AUC decreased (28% and 23%, respectively). No effect on absorption of a low-fat meal was observed (277 kcal; 42 kcal fat, 190 kcal carbohydrates, and 44 kcal proteins). In vitro data have shown that DCV is a substrate of the transporter P-glycoprotein (P-gp) (Caco-2 cells) and the absolute bioavailability is 67% [10].
DCV is combined with SOF (Sovaldi®). SOF is quickly absorbed and the median Cmax was found after ~ 0.5–2 h. GS-331007 is the main circulating metabolite of SOF and frequently used to describe the pharmacokinetics of SOF. The Cmax of GS-331007 was found 2–4 h after administration. The exposure of SOF and GS-331007 were 57% higher and 39% lower when HCV-infected subjects were compared with healthy volunteers. Food increased SOF absorption by a factor of 1.8; however, there were no clinically relevant alterations in Cmax. GS-331007 was not affected by food. The AUC of SOF and GS-331007 show a near dose-proportional increase in the range of 200–1200 mg. SOF is a substrate of the transporters P-gp and breast cancer resistance protein (BCRP). This is not the case for GS-331007 [11, 12].
3.1.2 Distribution
DCV is highly bound to plasma proteins (~ 99%) and the apparent volume of distribution (Vd/L) is 47.1 L. DCV is passively and actively transported into hepatocytes. In vitro data have shown that DCV is actively transported by organic cation transporter (OCT) 1 and inhibits P-gp, organic anion transporting protein (OATP) 1B1, and BCRP. DCV also in vitro inhibits the renal transporters organic anion transporter (OAT) 1, OAT3, and OCT2 [9, 10]. OCT2 inhibition by DCV is not clinically relevant, as shown in a drug interaction study with metformin (an OCT1 and OCT2 substrate) [13].
SOF is 61–65% bound to plasma proteins and the binding of SOF is independent of drug concentrations (1–20 µg/mL). GS-331007 is minimally bound to plasma proteins [11, 12] .
3.1.3 Metabolism
DCV is a substrate of cytochrome P450 (CYP) 3A4; however, 97% of the circulating drug is the parent drug and < 5% of metabolites are found in plasma [9, 10].
SOF has a more complex metabolism (see Fig. 2) [14]. SOF is initially metabolized in the liver into the pharmacologically active nucleoside analog triphosphate GS-461203. This is followed by dephosphorylation to the main inactive metabolite GS-331007. GS-331007 accounts for over 90% of the systemic exposure. SOF only accounts for 4% of the systemic exposure [11, 12].

Metabolism of sofosbuvir (derived from Kirby et al. [14])
3.1.4 Excretion
DCV is primarily hepatically cleared, as 88% of a radioactive test dose was retrieved in the feces, of which 53% was the parent drug. Only 6.6% of the parent drug was excreted in the urine. The elimination half-life (t½) is 12–15 h and the clearance is 4.24 L/h [9, 10].
For SOF, the main route of excretion is via urine (80%); only 14% of a radioactive dose was recovered in feces. The majority was retrieved as GS-331007 (78%) and only 3.5% was recovered as the parent drug. The median t½ was 0.4 h for SOF and 27 h for GS-331007 [11, 12].
3.2 Sofosbuvir (SOF)/Ledipasvir (LDV)
When interpreting the SOF pharmacokinetic data it is important to recognize that the different formulations (e.g., Sovaldi®, Harvoni®, Epclusa®, Vosevi®) all have their own pharmacokinetic profiles and therefore there are slight differences in the described parameters within this review. See also Table 2 for a complete overview.
3.2.1 Absorption
The pharmacokinetics of the fixed-dose tablet SOF/LDV (Harvoni®) are described in this section. The LDV Cmax was reached after 4–4.5 h. The tmax of SOF was ~ 1 h after drug intake. The main inactive metabolite of SOF, GS-331007 (see Sect. 3.2.3) had a tmax of 4 h. The exposure of both GS-331007 and LDV were comparable between healthy volunteers and HCV-infected patients. The exposure of LDV and GS-331007 are not affected by moderate- (600 kcal; 30% fat) and high- (1000 kcal; 50% fat) fat meals. However, the AUC from time zero to infinity (AUC∞) of SOF when taken with food was increased ~ 2-fold but the Cmax of SOF was not affected [15, 16]. Despite these differences, response rates with and without food are comparable so SOF/LDV can be administered with or without food [16]. In addition, the solubility of LDV decreases with an increase of pH. Lastly, both SOF and LDV are substrates of the drug transporters P-gp and BCRP [15, 16].
3.2.2 Distribution
LDV and SOF are both bound to plasma proteins: > 99.8% and 61–65%, respectively. GS-331007 is not bound to plasma proteins. LDV is an inhibitor of the intestinal transporters P-gp and BCRP. In addition, OATP1B1/2 and bile salt export pump are inhibited [15, 16].
3.2.3 Metabolism
The metabolism of LDV is unknown, but is expected to be minimal as > 98% of the parent drug is responsible for systemic exposure [15, 16].
SOF has a more complex metabolism (see Fig. 2 and Sect. 3.1.3) [14]. SOF is metabolized in the liver into the pharmacologically active nucleoside analog triphosphate GS-461203. Dephosphorylation results in the main inactive metabolite, GS-331007. GS-331007 accounts for over 90% of the systemic exposure [15, 16].
3.2.4 Excretion
After a radioactive dose of LDV, 87% of the parent drug was retrieved in urine and feces, of which 86% was in feces. The median t½ was 47 h [15, 16].
For SOF the main route of excretion is via urine (80%); only 14% of a radioactive dose was recovered in feces. The majority was retrieved as GS-331007 (78%) and only 3.5% was found as the parent drug. The median t½ was 0.5 h for SOF and 27 h for GS-331007 [15, 16].
3.3 Elbasvir (EBR)/Grazoprevir (GZR)
3.3.1 Absorption
The median tmax of EBR is 3 h (range 3–6 h) and the estimated bioavailability is ~ 32%. A high-fat meal (900 kcal; 500 kcal fat) decreased absorption (AUC 11% and Cmax 15%). EBR is a substrate of P-gp.
The median tmax for GZR is 2 h (range 0.5–3 h) and the absolute bioavailability after a single dose varied from 15 to 27% and after multiple doses from 20 to 40%. A high-fat meal (900 kcal; 500 kcal fat) increased absorption (AUC 50% and Cmax 108%). When compared with healthy individuals, HCV-infected patients had increased exposure (~ 2-fold). GZR is a substrate of P-gp. Steady state is reached around the sixth day of administration [17, 18].
3.3.2 Distribution
EBR and GZR are highly bound to both albumin and α1-acid glycoprotein (> 99.9% and > 98.8%, respectively) [17, 18]. The estimated Vd/L values for EBR and GZR are 680 and 1250 L, respectively. GZR is actively transported by the hepatic transporter OATP1B1/3 [18]. Although EBR inhibits P-gp, it is not clinically relevant as shown in a drug interaction study with digoxin (11% increase of digoxin). Both drugs inhibit BCRP [17, 18].
3.3.3 Metabolism
Both EBR and GZR are substrates of CYP3A4; however, no circulating metabolites were detected in plasma. GZR is a weak inhibitor of CYP3A4 [17, 18].
3.3.4 Excretion
Both EBR and GZR are primary hepatically cleared as > 99% of a radioactive dose was retrieved in feces. The apparent t½ is 24 and 31 h for EBR and GZR, respectively [17, 18].
3.4 SOF/Velpatasvir (VEL)
3.4.1 Absorption
The pharmacokinetics of the fixed-dose tablet SOF/VEL (Epclusa®) is described in this section.
The SOF Cmax was found 0.5–1 h after administration and the GS-331007 Cmax was found 3 h after administration. The exposures of SOF and GS-331007 were comparable in healthy volunteers and HCV-infected patients. A moderate- and high-fat meal increased the AUC∞ of SOF by 60% and 78%, respectively. However, the SOF Cmax was not affected by food and the GS-331007 AUC∞ was decreased by 25% and 37%, respectively [7, 19].
The median tmax of VEL is 3 h and the AUC and Cmax values were 41% and 37% lower in healthy volunteers than in HCV-infected patients. The VEL AUC was increased 34% and 21% after intake of moderate- (600 kcal; 30% fat) and high-fat (800 kcal; 50% fat) meals; the Cmax was only increased by 34% and 5%, respectively. In addition, VEL has pH-dependent solubility and the solubility (and thus absorption) decreases with increasing pH [7, 19].
3.4.2 Distribution
VEL is highly protein bound (> 99.5%), which is independent of the concentration range of 0.09–1.8 µg/mL. SOF is a substrate of P-gp and BCRP and VEL is a substrate of P-gp, OATP1B, and BCRP [7, 19]. SOF is bound 61–65% to plasma proteins, which is dose independent (1–20 µg/mL) [7, 19].
3.4.3 Metabolism
VEL is metabolized by CYP2B6, CYP2C8, and CYP3A4. However, after a single dose > 98% of the parent drug was found in the blood. VEL is an inhibitor of P-gp, BCRP, and OATP1B1/3 [7, 19].
SOF metabolism is discussed in Sect. 3.1.3.
3.4.4 Excretion
As > 94% of VEL was retrieved in feces and 0.4% in urine, clearance of VEL is mainly hepatic. 77% of VEL was recovered in feces as the parent drug. The t½ of VEL is around 15 h [7, 19]. SOF is mainly renally excreted (80%) and the majority of the dose found was GS-331007 (78%); only 3.5% was found as SOF. The t½ of SOF was 0.5 h and for GS-331007 was 25 h [7, 19].
3.5 Glecaprevir (GLE)/Pibrentasvir (PIB)
3.5.1 Absorption
The tmax of GLE/PIB is approximately 5 h and food increases absorption (both moderate- and high-fat meals). GLE exposure was increased 83–163% and PIB 40–53% when taken with a meal. Both drugs are P-gp substrates [6, 20].
3.5.2 Distribution
GLE and PIB are highly bound to plasma proteins (GLE 97.5%; PIB > 99.9%) and actively transported by BCRP. GLE is also a substrate for the hepatic transporter OATP1B1/3 [6, 20].
3.5.3 Metabolism
GLE is metabolized by CYP3A4 and PIB is not subject to biotransformation. In vivo GLE and PIB weakly inhibit CYP3A4 and uridine 5′-diphospho-glucuronosyltransferase (UGT) 1A1 [6, 20].
3.5.4 Excretion
GLE is primarily hepatically excreted as 92.1% of a radioactive dose was retrieved in feces. The t½ at steady state is 6–9 h. PIB is also primarily excreted in feces (96.6%) and the t½ is 23–29 h [6, 20].
3.6 SOF/Velpatasvir (VEL)/Voxilaprevir (VOX)
3.6.1 Absorption
The pharmacokinetics of the combination tablet SOF/VEL/VOX (Vosesi®) is described in this section. VOX, VEL, and GS-331007 reach Cmax after approximately 4 h and SOF after 2 h. Compared with healthy individuals, SOF and GS-331007 pharmacokinetics were not altered in HCV-infected patients. For VEL, the AUC and Cmax were 41% and 39% decreased in patients, respectively. For VOX, the AUC and Cmax were both elevated by 260% when HCV-infected individuals and healthy volunteers were compared [8, 21].
When taken with food (type of meal not defined), the SOF AUC∞ and Cmax increased from 64 to 114% and 9 to 76%, respectively. For GS-331007, the Cmax was decreased, ranging from 19 to 35%. For VEL, the AUC∞ and Cmax were increased, ranging from 40 to 166% and 37 to 187%, respectively. The VOX AUC and Cmax increased, ranging from 112 to 435% and 147 to 680%, respectively. Therefore, it is recommended that SOF/VEL/VOX is taken together with a meal [8, 21].
3.6.2 Distribution
SOF, VEL, and VOX are highly bound to plasma proteins (61–65%, > 99%, and > 99%, respectively). For SOF and VEL this was concentration independent in the ranges of 1–20 and 0.09–1.8 µg/mL, respectively. GS-331007 is not bound to plasma proteins. SOF is a substrate of P-gp and BCRP, VEL is a substrate of P-gp, OATP1B1/3, and BCRP, and VOX is a substrate of P-gp and BCRP [8, 21].
3.6.3 Metabolism
VOX is a CYP3A4 substrate; however, after a single radioactive dose, approximately 91% of the circulating drug was the parent drug. VOX is an inhibitor of P-gp, BCRP, and OATP1B1/3 [8, 21].
See Sects. 3.1.3 and 3.4.3 for the metabolism of SOF and VEL, respectively.
3.6.4 Excretion
SOF is mainly renally excreted (80%) and the majority of the dose was GS-331007 (78%); only 3.5% was found as SOF. The t½ of SOF was 0.5 h and for GS-331007 this was 29 h [8, 21].
As > 94% of VEL was retrieved in feces and 0.4% in urine, clearance of VEL is mainly hepatic. In feces, 77% of VEL was recovered as the parent drug. The t½ of VEL is around 17 h [7, 19].
The major route of excretion is biliary, as 94% of VOX was recovered in the feces. After a single dose, almost 40% was found as the parent drug and 22.1% as the metabolite des-[methylcyclopropylsulfonamide]-voxilaprevir and three other metabolites (< 10%) [8, 21].
4 Drug–Drug Interactions
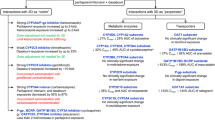
As presented in Fig. 3, most DAAs are substrates and inhibitors of drug transporters and CYP enzymes and, therefore, have the potential for DDIs. In this section the most important classes of drugs with interactions with DAAs are briefly discussed. For more information, and for help in clinical decision-making, we recommend the HEP Drug Interactions website (www.hep-druginteractions.org) [22].

Overview of the drug metabolism enzymes and drug transporters involved in the metabolism and distribution of the several direct-acting antivirals (DAAs). Only enzymes and drug transporters involved in the metabolism/transport of DAAs are included. Information obtained from the relevant Summaries of Product Characteristics (SmPCs) and from Chu et al. [205]. 1See substrates and inhibitors relating to hepatocyte. 2See substrates and inhibitors relating to intestine. 3Minor substrate. 4Weak inhibitor. BCRP breast cancer resistance protein, CYP cytochrome P450, EBR elbasvir, GLE glecaprevir, I inhibitor of drug transporter and/or enzyme, MRP multidrug resistance protein, OATP organic anion transporting polypeptide, OCT organic cation transporter, P-gp P-glycoprotein, PIB pibrentasvir, S substrate of drug transporter and/or enzyme, SOF sofosbuvir, UGT uridine 5′-diphospho-glucuronosyltransferase, VEL velpatasvir, VOX voxilaprevir
4.1 HIV Drugs
Antiretroviral drugs have a high potential for DDIs due to induction or inhibition of CYP3A and drug transporters. Non-nucleoside reverse transcriptase inhibitors (NNRTIs) have inducing effects, predominantly on CYP3A4. For example, after coadministration with efavirenz, the EBR and GZR AUC decreased by 54% and 87% [17], respectively, and the VEL AUC by ~ 50% [7]. The preferred NNRTIs for coadministration with GLE/PIB, SOF/VEL, and SOF/VEL/VOX are doravirine and rilpivirine [23, 24]. Concomitant use of DCV with a CYP3A4 inducer, such as efavirenz, ertravirine, and nevirapine, requires an increase in dose from 60 to 90 mg due to lower DCV exposure [25].
Boosted protease inhibitors (PIs) are inhibitors of CYP3A, OATP, and P-gp. Concomitant use of CYP3A4 inhibitors, such as atazanavir/ritonavir and cobicistat-boosted regimens, requires a decrease in DCV dose from 60 to 30 mg because of decreased CYP3A4 metabolism resulting in elevated DCV plasma concentrations [26, 27]. Use of GLE/PIB with ritonavir-boosted atazanavir is, however, contraindicated due to extreme elevations in GLE and PIB plasma concentrations. The Cmin of GLE and PIB increased 14.0-fold and ~ 1.3-fold, respectively [6]. As all boosted PIs are strong CYP3A4 inhibitors, they are not recommended with the GLE and PIB combination [17, 28].
SOF/VEL may be coadministered with any antiretroviral regimen including the boosted PIs atazanavir, darunavir, and lopinavir (boosted with ritonavir or cobicistat) without dose adjustment as no clinically relevant differences in pharmacokinetics of SOF/VEL were observed [29]. SOF/VEL/VOX with darunavir/ritonavir and atazanavir/ritonavir resulted in a 2.4- and 4.3-fold increase in the VOX AUC, respectively, due to OATP1B, P-gp, and CYP3A inhibition and is therefore not recommended [8]. Pharmacokinetic data with other boosted PIs are lacking, but they are not recommended for use together with SOF/VEL/VOX either.
SOF/LDV increases tenofovir exposure due to inhibition of P-gp. This is particularly relevant when LDV is combined with tenofovir disoproxil fumarate (TDF) because of the high tenofovir plasma concentrations [15, 30]. When combined with tenofovir alafenamide this drug interaction is not clinically relevant due to lower circulating tenofovir concentrations [31]. A comparable increase in the tenofovir plasma concentration is observed when TDF is combined with VEL [7, 8]. This increases the risk of tenofovir-related renal toxicity. Therefore, additional monitoring of renal function is advised.
The integrase inhibitors raltegravir, dolutegravir, and bictegravir have favorable interaction profiles and can be safely combined with DAAs [32]. The exception is cobicistat-boosted [31] elvitegravir as this combination also strongly inhibits CYP3A4 and P-gp.
4.2 Immunosuppressive Drugs
Since HCV is a major cause of cirrhosis, which could result in liver transplantation, immunosuppressant agents are commonly used by HCV-infected patients. Therefore, potential DDIs should be considered when treating these patients with both DAAs and immunosuppressive agents.
The immunosuppressive agent cyclosporine (ciclosporin) is a perpetrator of DDIs as it is a strong inhibitor of OATP1B1. Coadministration with EBR/GZR resulted in a 15-fold increase of the GZR AUC. Higher GZR exposure potentially leads to hepatotoxicity and coadministration is thus not recommended [17]. Similarly, combining cyclosporine 100 mg with GLE resulted in an increased AUC (37%) and the GLE AUC increased by 451% after a dose of 400 mg. Since cyclosporine seemingly has a dose-dependent influence on OATP, a maximum of 100 mg/day is recommended [6]. For VOX, the AUC and Cmax increased by 19- and 9.4-fold when combined with cyclosporine, respectively. Therefore, SOF/VEL/VOX is not recommended in subjects using cyclosporine [8]. However, to overcome this drug interaction, therapeutic drug monitoring (TDM) of the DAA could be used when no other DAA treatment options are available.
Tacrolimus is a CYP3A4 substrate and its AUC increased by 43% and 1.45-fold after coadministration with EBR/GZR and GLE/PIB, respectively, due to CYP3A4 inhibition [6, 17]. Additional monitoring or dose alterations may be necessary as tacrolimus has a narrow therapeutic range. This DDI can be overcome by using frequent TDM when starting DAA therapy. We strongly recommend frequent monitoring of the tacrolimus plasma concentration when HCV-infected patients are treated, not only to overcome possible DDIs but also because in patients recovering from an HCV infection the tacrolimus plasma concentration can be altered due to altered CYP3A4 metabolism [33]. Significant pharmacokinetic DDIs are not expected with SOF/DCV, SOF/LDV, SOF/VEL, and SOF/VEL/VOX as they do not influence CYP3A4 and tacrolimus.
4.3 Cardiovascular Drugs
DDIs with cardiovascular drugs have been previously discussed in a paper published in this journal [34]. In short, statins (HMG-CoA reductase inhibitors) are substrates for several drug transporters, such as P-gp, BCRP, OATP1B1, and CYP enzymes. Generally, coadministration of statins with DAAs results in an increase of the statin AUC, causing higher risk of toxicities such as myopathies. Therefore, caution is needed, and at least adjustment and close monitoring for statin adverse effects are needed. Given the short treatment duration of HCV therapy (only 8–12 weeks), temporary discontinuation of the lipid-lowering drug may be considered as well.
The antiarrhythmic agent amiodarone causes clinically relevant DDIs with SOF-containing regimens: events of severe bradycardia occurred after concomitant use of amiodarone and SOF [35,36,37]. The mechanism behind it, however, remains uncertain [38], although it is thought to reflect a pharmacodynamic interaction enhancing the bradycardic effect of amiodarone. Because of the long and variable t½ (20–100 days) of amiodarone, the timing of starting or discontinuing amiodarone should be kept in mind as DDIs can have a prolonged effect.
Since all of the new oral anticoagulants (NOACs) are substrates for P-gp, and apixaban and rivaroxaban are also substrates for BCRP and CYP3A, there could be an increased risk of toxicity when these drugs are combined with DAAs. This possibly includes bleeding. For both GLE/PIB and SOF/VEL/VOX, the dabigatran etexilate AUC and Cmax values were approximately 2- to 3-fold increased after concomitant use [6, 8]. Pharmacokinetic data after co-administration of anticoagulants with other DAAs are not available, but similar results are expected. Therefore, these drugs should either not be used with DAAs or used with caution and careful monitoring when no other anticoagulant is possible [23].
4.4 Anticonvulsant Drugs
The first-generation anticonvulsant drugs (e.g., carbamazepine, phenytoin, phenobarbital) are strong inducers of CYP3A4 and P-gp. These drugs decrease DAA exposure significantly after coadministration, which could result in decreased virological effectiveness. Accordingly, these drugs are either not recommended or contraindicated with all HCV regimens [23]. If no other antiepileptic treatment is possible, the most optimal anti-HCV therapy seems to be SOF with an increased dose of DCV [39].
4.5 Tuberculosis Drugs
In some parts of the world, for instance Eastern Europe, tuberculosis (TB) disease is a burden among HCV or HIV/HCV co-infected patients. This is mainly caused by the fact that these diseases (including hepatitis B virus [HBV]) are prevalent in the same (vulnerable) groups, such as men who have sex with men, prisoners, people who inject drugs, and immigrants and refugees from high endemic countries [40].
TB disease can be treated with combination therapy, but compliance to therapy is very important due to resistance issues. The first-line drugs for the treatment of TB disease include isoniazid, rifampicin (rifampin), rifabutin, rifapentine, ethambutol, and pyrazinamide [41, 42]. All current DAA regimens are contraindicated with rifampicin, rifabutin, and rifapentine as all these drugs are strong inducers of CYP3A4. For example, the AUCs of both SOF and VEL were decreased with 72% and 82% and the Cmax by 77% and 71% when combined with rifampicin 600 mg once daily. Comparable results are found with the other DAAs and it is expected that rifabutin and rifapentine also reduce plasma concentrations in the same manner as they are also strong CYP3A4 inducers. Thus, it is not recommended to treat patients with DAAs combined with rifampicin, rifabutin, or rifapentine (similar to anticonvulsant drugs, as noted in Sect. 3.7.4) [7, 19], making it almost impossible to treat a patient for HCV and TB disease at the same time as rifampicin is the cornerstone of current TB disease treatment [41].
Isoniazid, ethambutol, and pyrazinamide are not expected to have any clinically relevant drug interactions with the DAA regimens as the metabolic pathways do not interfere with each other [22].
4.6 Acid-Reducing Agents
Acid-reducing agents such as proton pump inhibitors (PPIs) and histamine H2-receptor antagonists are drugs that influence gastric pH. Both LDV and VEL have pH-dependent absorption. VEL is a weak base which is insoluble in water (pH 7; 0.003 mg/mL) and solubility increases at pH 2 (> 2 mg/mL) [43]. Clinically, this results in decreased exposure of VEL when combined with PPIs such as omeprazole. When omeprazole (20 mg) was combined with SOF/VEL in fasted subjects, the AUC and Cmax of VEL decreased (36% and 37%). The VEL AUC and Cmax decreased even more when omeprazole 20 mg once daily (fasted) was taken 12 h before SOF/VEL intake (55% and 57%). The same dose of omeprazole administered 2 h before SOF/VEL, with food, also resulted in decreased AUC and Cmax values of 38% and 48%. The most promising results were found when omeprazole 20 mg was taken 4 h after SOF/VEL was taken with food; the AUC and Cmax only dropped 26% and 33% [7, 19]. Our recommendation for VEL is not to combine it with acid-reducing agents except when it is really clinically necessary and the PPI cannot be discontinued. Based on the results mentioned here, we advise use of omeprazole 20 mg once daily and administration of SOF/VEL with food 4 h before the PPI is used. H2-receptor antagonists (famotidine 40 mg twice daily or equivalent) should be taken together with SOF/VEL or separated by at least 12 h.
LDV solubility is also pH dependent as it is slightly soluble at pH 2.3 but practically insoluble at pH 4–7.5 [44]. When SOF/LDV was coadministered with omeprazole 20 mg once daily simultaneously, the AUC and Cmax were decreased by 4% and 11%. When the intake was separated by 2 h, the LDV AUC and Cmax were decreased by 42% and 48% [15, 16]. As for VEL, we do not recommend SOF/LDV be combined with high-dose acid-reducing agents. However, if necessary it can be combined with omeprazole 20 mg once daily or famotidine 40 mg twice daily if taken at the same time as SOF/LDV. It is important to note that these recommendations are based on pharmacokinetic changes of the DAAs and not on efficacy endpoints.
5 Efficacy and Toxicity
In this section the pharmacokinetic–pharmacodynamic (PK-PD) relationship of the DAAs and the viral inhibition are described. Pharmacodynamics is defined as reduction of HCV-RNA virus when a patient is treated with the DAAs. To determine the optimal dose, there is a balance between the effect (HCV-RNA reduction) and toxicity (adverse effects, adverse events [AEs]).
ESM Table S2 provides an overview of the dose–response relationships of the DAAs. For most DAAs the approved dose is one of the highest dosages tested. This shows that all the DAAs were well-tolerated during the dose-finding and PK-PD studies. However, this also means that these dosages are likely in the upper part of the s-curve (plateau) where the effect is maximal (Fig. 4). When these thoughts are translated to, for instance, drug interactions, this helps explain why the LDV + PPI drug interaction is marked from a pharmacokinetic perspective (absorption and exposure decreases) but may not be clinically relevant (since in most patients at least, you are still in the upper part of the PK-PD curve with no effect on SVR). It is also important when interpreting these PK-PD relationships to note that there is always variability between individual patients (inter-subject variation). This variation could have several causes such as physiology (age, sex), genetic factors influencing metabolism (e.g., CYP polymorphisms) and drug transport (e.g., OCT or OATP polymorphisms), or environmental factors (smoking, nutrition, alcohol use).

Pharmacodynamic–pharmacokinetic relationship and factors influencing pharmacodynamics and pharmacokinetics. Every drug has a pharmacodynamic (= effect and toxicity) and pharmacokinetic (= exposure) relationship
5.1 SOF/DCV
5.1.1 Efficacy
The combination of SOF/DCV is licensed to treat patients with all HCV genotypes; however, DCV is mostly used to treat HCV in patients infected with HCV genotypes 1, 2, 3, and 4 [23]. In a dose-finding study, DCV was administered in doses of 1, 10, 30, 60, and 100 mg once daily. Geometric mean AUCs showed dose-proportional pharmacokinetics. After 1 day of dosing, the HCV-RNA was decreased by ~ 3 log10 IU/mL for all study groups (except 1 mg). The exposure–response analysis suggested a dose varying from 3 to 60 mg once daily [45].
Similarly, single and multiple doses of SOF 50, 100, 200, and 400 mg once daily were administered to HCV genotype 1-infected patients for 3 days. The decline in HCV-RNA viral load was dose dependent and for 400 mg a HCV-RNA reduction of ~ 2 log10 IU/mL was seen (200 mg ~ 1 log10 IU/mL) [46].
Due to good results in phase II studies, SOF/DCV received early approval. These data are not described in this review as it mainly consists of data in combination with peg-IFN and RBV. Later on, efficacy and safety were studied to a greater extent. The ANRS (France REcherche Nord&Sud Sida-hiv Hépatites) CO22 HEPATHER trial included DAA-naïve HCV genotype 1-infected patients with and without cirrhosis. Overall, 92–99% of the patients achieved sustained virological response (SVR) 12 weeks after treatment (SVR12). The SVR12 rates were 98% and 94% in those without and with cirrhosis, respectively [47]. In a real-world study in DAA-naïve HCV genotype 2-infected patients with and without cirrhosis, all 32 (100%) reached SVR12 [48]. In the ALLY-3 study (phase III), the overall SVR12 rate in HCV genotype 3-infected patients was 89%. Patients with and without cirrhosis yielded SVR12 rates of 63% and 96%, respectively. So, SOF/DCV is not the ideal regimen for HCV genotype 3-infected patients with cirrhosis [49]. Another study showed that patients with advanced fibrosis or compensated cirrhosis obtained an overall SVR12 rate of 90%: 88% and 92% following 12 and 16 weeks of treatment, respectively [50]. HCV genotype 4-infected patients yielded SVR12 rates of 92% in patients with cirrhosis or treatment-experienced patients without cirrhosis [51] (see ESM Table S3).
5.1.2 Toxicity
The most commonly reported adverse effects for the SOF/DCV combination were fatigue, headache, and gastrointestinal complaints such as diarrhea and nausea. In several studies, concomitant therapy with RBV resulted, as previously described [52], in anemia and leukopenia [47, 51, 53,54,55,56,57]. Following an AE, ≤ 7% of the patients discontinued treatment with SOF/DCV, with the exception of patients with a life expectancy of less than 12 months due to decompensated cirrhosis or recurrence of HCV following a liver transplant, in whom 10% and 18%, respectively, discontinued therapy due to an AE [54].
5.2 SOF/LDV
5.2.1 Efficacy
A dose-ranging study using LDV 1, 3, 10, 30, and 90 mg for the duration of 3 days was performed in HCV-infected subjects. On day 3, the AUC during a dosing interval (AUCτ) varied from 34.0 to 3815.5 ng/mL for these dose ranges, showing dose-proportional pharmacokinetics. For all doses the median HCV-RNA reduction was > 3 log10 IU/mL, showing comparable viral suppression over the dose range. In the same study the exposure–response relationship of LDV was simulated. The AUC and maximal HCV-RNA decline were used and for genotype 1a patients it was found that a dose of 30 mg would be optimal to have a > 95% antiviral response [58]. However, no formal dose/concentration–effect relationship has been established for the combination of SOF plus LDV.
SOF/LDV is an effective treatment in the dose of 90 mg/400 mg once daily. The regimen can be used in patients infected with HCV genotypes 1a, 1b, 4, 5, and 6 (12 weeks) in treatment-naïve patients with or without compensated (Child–Pugh [CP] score A) cirrhosis. Additionally, SOF/LDV (12 weeks) is also effective in treatment-experienced HCV genotype 1b-infected patients [23]. In patients with an HCV-RNA viral load of < 6,000,000 IU/mL, the SOF/LDV treatment duration can be decreased to 8 weeks when patients are infected with genotype 1b [59, 60]. Boerekamps et al. [61] even showed that 8 weeks of SOF/LDV can be used to treat both HCV and HIV/HCV co-infected patients with genotype 4-infected patients. The efficacy and safety of SOF/LDV were studied in the ION-1 study, where treatment-naïve HCV genotype 1a-infected patients yielded SVR12 in 99% of individuals after 12 weeks of treatment without RBV. In patients with HCV genotype 1b infection, SVR12 rates of 100% were obtained after the same treatment regimen [62]. In the ION-2 study, 94% of treatment-experienced HCV genotype 1-infected patients, including those with cirrhosis, achieved SVR12 [63]. In a phase III study, treatment-naïve and treatment-experienced patients infected with HCV genotype 1 were treated for 12 weeks without RBV: 100% achieved SVR12, including those with compensated cirrhosis [64]. After 12 weeks of therapy with SOF/LDV, 95% of HCV genotype 4-infected patients achieved SVR12 in the SYNERGY trial [65]. Results were confirmed with SVR12 rates of 96% in treatment-naïve and 91% in treatment-experienced patients with HCV genotype 4 [66]. HCV genotype 5-infected patients were treated in an open-label phase II study, in which 95% achieved SVR [67]. SVR rates of 64% and 96% were achieved in patients with HCV genotypes 3 and 6, respectively, in an open-label study [68]. In conclusion, the regimen containing LDV and SOF is effective in patients with genotypes 1, 4, 5, and 6 and has reduced efficacy to HCV genotype 3 (ESM Table S4).
5.2.2 Toxicity
The most frequently occurring AEs reported from studies with SOF/LDV were mild, being fatigue, headache, and nausea. In multiple studies no adverse effects at all were reported to occur in over 10% of the study population [69,70,71]. Less than 5% of the patients discontinued treatment due to an AE in all studies.
5.3 EBR/GZR
5.3.1 Efficacy
EBR/GZR is an effective regimen when used for 12 weeks against HCV genotypes 1 and 4. It is approved for patients with renal insufficiency and compensated cirrhosis [23]. In a dose-finding study, patients with HCV genotype 1 or 3 infection were treated with EBR (5–100 mg) for 5 days and GZR (10–800 mg) for 7 days (both monotherapy). For HCV genotype 1-infected patients treated with GZR with doses > 30 mg, the mean maximum HCV-RNA reduction was > 4.0 log10 IU/mL. For HCV genotype 3, this amount of HCV-RNA reduction was achieved following doses > 400 mg. These data suggest that for GZR the dose–response plateau is reached at a dose of 50 mg once daily; however, this cannot be confirmed for HCV genotype 3-infected patients. For genotypes 1a and 1b the mean maximum HCV-RNA reduction was > 4.0 log10 IU/mL at a dose of EBR 50 mg. For genotype 3 the doses of EBR 50 and 100 mg had a mean maximum HCV-RNA reduction of > 3.0 log10 IU/mL. These data suggest a dose of 50 mg of EBR is sufficient [72].
The combination of EBR/GZR is approved in the fixed-dose combination of 50 mg/100 mg once daily for genotypes 1 and 4. In the C-WORTHY trial, HCV genotype 1-infected patients were effectively treated (with or without cirrhosis, 12–18 weeks) and SVR12 rates varied from 91 to 100% [73]. C-CORAL (treatment-naive ± cirrhosis) showed SVR12 of 94% in patients with genotypes 1, 4, or 6 [74]. Jacobson et al. [75] performed an integrated analysis showing SVR rates varying from 89 to 100% when treating patients with CP-A (treatment-naive and treatment-experienced). In addition, genotype 1a patients with HCV-RNA > 800,000 IU/mL had lower SVR rates than genotype 1a patients with HCV-RNA < 800,000 IU/mL (12 weeks: 91% vs. 98%; 16 weeks + RBV: 94% vs. 100%). It was also shown that patients with non-structural protein (NS)5A baseline RASs had an SVR12 of 53% (16/30) after 12 weeks of treatment with EBR/GZR; this was increased to 100% (4/4) when treated for 16 weeks [75]. Therefore, in patients with a high viral load and patients with NS5A RASs, physicians should consider treating for 16 weeks with EBR/GZR.
The C-SALVAGE phase II study was a hypothesis-generating trial to study EBR/GZR + RBV for 12 weeks as a salvage therapy for genotype 1a/1b. Patients who did not respond to a licensed DAA-containing therapy were included, and overall an SVR of 96% was achieved [76].
Low SVR rates were found, varying from 45 to 57%, when treating genotype 3 patients for 12–18 weeks, respectively (C-WORTHY), which fits the previous PK-PD results [77]. Therefore, SOF was added in the C-ISLE trial. This combination was highly effective as SVR12 rates over 94% were reported [78] (ESM Table S5).
5.3.2 Toxicity
Overall, EBR/GZR has a favorable safety profile with low discontinuation rates (≤ 5%). The exception was the ANRS HC34 REVENGE trial where patients with advanced fibrosis or compensated cirrhosis were treated for 24 weeks in combination with RBV, in which 15% (2/13) of the patients discontinued due to AEs [79]. The most frequently reported AEs were fatigue, headache, asthenia, nausea, rash, alanine aminotransferase (ALT)/aspartate aminotransferase (AST) increase, and alkaline phosphatase increase. These increased liver enzymes mostly recovered after treatment with EBR/GZR and are related to the plasma concentration of GZR. The presence of cirrhosis is not a risk factor for this ALT/AST increase [17].
5.4 SOF/VEL
5.4.1 Efficacy
The PK-PD relationship for VEL was established in a dose-ranging study with ascending doses of 5, 25, 50, 100, and 150 mg once daily given for 3 days to HCV genotype 1a-infected patients. The reported AUCτ values after 3 days varied from 86.4 to 5003.0 ng∙h/mL for the doses of VEL 5–50 mg, showing dose-proportional pharmacokinetics. For all dose ranges in HCV genotype 1a-infected patients, a maximal HCV-RNA decline of ≥ 3 log10 IU/mL was established. For genotype 3, only the 150 mg dose had a maximal HCV-RNA decline > 3 log10 IU/mL [80].
Treatment with SOF/VEL for 12 weeks is highly effective in both treatment-experienced and treatment-naïve patients with all genotypes of HCV infection. The phase III ASTRAL-1 trial in patients with HCV genotype 1a, 1b, 2, 4, 5, and 6 infections yielded SVR12 rates varying from 97 to 100% [81]. These results were confirmed by the ASTRAL-2 trial (99% in genotype 2) [82] and multiple real-world studies in patients with all HCV genotypes [83, 84]. ASTRAL-3 showed that patients with genotype 3 infection achieved SVR12 rates of 98% (treatment naïve without cirrhosis). Therefore, this regimen can be used for the treatment of HCV genotype 3-infected patients. However, lower rates were observed in patients with genotype 3 plus cirrhosis or treatment experience (91% and 90%, respectively). All these patients received 12 weeks of SOF/VEL without RBV [82]. Additionally, suboptimal results in HCV genotype 3 patients with compensated cirrhosis were reported (SVR12 78% and 88%, respectively) [85, 86]. For patients with HCV genotype 3 infection and cirrhosis, addition of a third drug to this regimen may be necessary, justifying the triple therapy of SOF/VEL/VOX in this subpopulation (ESM Table S6).
5.4.2 Toxicity
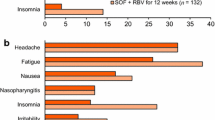
The most frequently observed adverse effects from the phases II and III trials of SOF/VEL were headache, fatigue, nausea, and insomnia. As expected, combination therapy with RBV led to anemia in over 10% of the patients in two studies [87, 88]. Discontinuation rates due to an AE were low (≤ 5%) in all studies.
5.5 GLE/PIB
5.5.1 Efficacy
The dose–response relationship of GLE and PIB (both as monotherapy) was assessed in HCV genotype 1-infected patients for 3 days. The GLE dose ranged from 100 to 700 mg and PIB from 15 to 400 mg. Both GLE and PIB have more than dose proportional pharmacokinetics. After a dose of GLE 1200 mg once daily, the AUC was 516-fold higher than with 200 mg once daily. In comparison, PIB 120 mg once daily resulted in a 10-fold increased AUC compared with 30 mg once daily. Both increases are probably caused by saturation of efflux transporters. When PIB is combined with GLE (which is always the case), the exposure is 3-fold higher than with PIB monotherapy [6, 20]. For GLE, the mean maximal decrease in HCV-RNA ranged from 4.1 to 4.3 log10 IU/mL. For PIB, HCV-RNA declines of > 3.4 log10 IU/mL were seen. The 15 mg dose of PIB resulted in a smaller decline in HCV-RNA than with the doses of 40, 120, and 400 mg [89].
This pan-genotypic regimen is highly effective when administered for 8 or 12 weeks in doses of 100 mg/40 mg once daily (treatment-naïve and treatment-experienced patients, with and without cirrhosis). Multiple phases II and III studies also yielded high rates of SVR12 in patients treated with GLE/PIB in the harder to treat HCV genotypes 1b and 3. In the SURVEYOR phase II study, DAA-naïve patients without cirrhosis received GLE/PIB for 8 or 12 weeks and SVR12 rates varied from 97 to 100% [90]. The ENDURANCE studies reported SVR12 rates of 95% in non-cirrhotic patients with HCV genotype 3. Shorter treatment is also possible, as SVR12 rates of 99% and 100% in HCV genotype 1-infected patients without cirrhosis after 8 and 12 weeks of treatment, respectively, were reported [91]. The EXPEDITION-1 trial in HCV genotype 1–6 patients with compensated cirrhosis resulted in SVR12 rates of 99% [92]. In conclusion, GLE/PIB is a pan-genotypic regimen with high efficacy rates in all HCV genotypes, including treatment-experienced patients and those with cirrhosis (ESM Table S7).
5.5.2 Toxicity
In general, GLE/PIB has a mild toxicity profile. The most commonly reported AEs were headache, fatigue, nasopharyngitis, and nausea. Some regimens comprising HCV PIs have been associated with hepatotoxicity (e.g., GZR) [93]. In contrast to these findings, clinically significant laboratory abnormalities for liver function were rare. Elevated ALT, AST, or alkaline phosphatase levels as common AEs were not reported in any of the studies. In addition, increased blood bilirubin and a total bilirubin of more than 1.5–3.0 × the upper limit of normal were reported in only one study [94]. Low rates of discontinuation due to an AE (≤ 6%) were found with and without concomitant use of RBV in both cirrhotic and non-cirrhotic patients.
5.6 SOF/VEL/VOX
5.6.1 Efficacy
The PK-PD relationship of VOX was established in a study in HCV patients infected with all genotypes. Dependent on the genotype, doses of 50–300 mg were administered once daily for 3 days. The AUCτ values at day 3 of administration for all genotypes were 372.3, 1357.6, and 3926.9 ng∙h/mL for the doses of 50, 100, and 300 mg, respectively [95]. This means that VOX has more than dose-proportional pharmacokinetics [21]. For all genotypes, the doses ranging from 100 to 300 mg once daily resulted in a mean maximal HCV-RNA reduction of > 3 log10 IU/mL [95].
This pan-genotypic combination is highly effective and licensed to treat patients with and without compensated cirrhosis and patients that previously failed to respond on both peg-IFN/RBV and DAAs. Phase II studies showed high efficacy in treatment-naive patients with compensated cirrhosis and all genotypes (including genotype 3). SVR12 rates varied from 93 to 97% in patients with cirrhosis and rates of 88 to 100% without cirrhosis with varying treatment durations of 6–12 weeks were achieved in patients with genotypes 2, 3, 4, or 6 [96]. Therefore, the phase III trials (POLARIS) continued with a treatment duration of 8 and 12 weeks. The POLARIS 2/3 analysis showed an SVR12 rate of 95% after treating a diverse group of 501 patients (treatment-naive, treatment-experienced, with and without compensated cirrhosis) [97].
In the POLARIS 1 trial where, among others, treatment-experienced, HCV genotype 3-infected patients were treated for 12 weeks, an SVR12 rate of 100% was achieved. In the POLARIS 4 study an SVR of 99% was achieved in a comparable group of patients [98]. Taking these results into account, the current recommendation is to treat DAA treatment-experienced and/or compensated cirrhotic patients with this combination for 12 weeks and to treat treatment-naïve patients for 8 weeks. However, in our opinion, this regimen should be used as salvage therapy due to the efficacy of double medication regimens (ESM Table S8).
5.6.2 Toxicity
The toxicity profile of this combination regimen is mild. In the different trials the most frequently reported AEs were headache, diarrhea, fatigue, nausea, and constipation. In addition, the discontinuation rate due to AEs was low (≤ 3%) in all trials. As VOX is a PI, this combination should not be used in patients with CP-B/C cirrhosis as exposure increases, which could potentially cause safety issues (see Sect. 5.2).
6 Special Populations
The overall ranges of SVR12 for the combination treatment regimens in different study populations are presented in Table 3.
6.1 Hard to Cure Patients
In the era of DAA therapy, patients with genotype 3, cirrhosis, or who are treatment experienced were initially considered hard to cure (or hard to treat) as SVR rates were lower than with other patient populations. To increase the chance of achieving SVR, RBV was added to the first DAA regimens or treatment durations were extended. In this section we discuss the current treatment options for those patients.
6.1.1 Genotype 3
Genotype 3 patients were easier to cure with peg-IFN/RBV than genotype 1 patients. Nevertheless, response rates were higher with the first approved DAA regimens; however, the SVR rates were lower than with genotype 1 or 4 patients. For example, a real-world study showed an SVR12 rate of 83% in genotype 3 patients with advanced fibrosis treated with SOF/DCV [99]. Additionally, 90% SVR12 was achieved in genotype 3 patients (with advanced fibrosis/compensated cirrhosis) treated with SOF/DCV/RBV for 12–16 weeks [50]. Due to good response rates, the preferred regimens, at this time, are SOF/VEL [82, 84,85,86,87, 100,101,102,103,104], GLE/PIB [90, 91, 105,106,107], or SOF/VEL/VOX [96,97,98, 108] for at least 12 weeks of treatment. With the currently available pan-genotypic treatment options, there no longer seems to be the need to approach HCV genotype 3-infected patients as hard to cure patients.
6.1.2 Treatment-Experienced Patients
Treatment-experienced patients can be divided into two categories: patients who failed to respond to peg-IFN/RBV and those who failed to respond to DAA therapy. Both groups are considered hard to cure, but DAA-experienced patients are a higher risk due to the appearance of RASs. However, with the current treatment options, these treatment-experienced patients also obtain high SVR12 rates when DAA regimens are used that combine DAAs from the three currently available HCV drug classes.
Importantly, patients previously treated with peg-IFN/RBV without cirrhosis can be treated as ‘normal’ patients. For all treatment regimens the SVR12 rates are up to 100%. The most successful results were achieved with treatment regimens consisting of SOF/DCV [47, 48, 53, 56, 109,110,111,112,113,114], GLE/PIB [94, 105, 106, 115,116,117], and SOF/VEL/VOX [96, 98, 108, 118]. Although efficacious, triple combination therapy is not indicated since double treatment regimens are also effective.
Limited data are available for the DAA-experienced patients. Excellent results were obtained in these patients with SOF/VEL/VOX [98] and GLE/PIB also showed good results for re-treatment of patients who previously failed to respond to NS5A inhibitors [116]. In patients with NS5A RASs, triple therapy with either SOF/VEL/VOX or the experimental combination SOF/GLE/PIB could be options [119].
With the DAA regimens currently available, peg-IFN/RBV treatment-experienced patients have numerous options to attain successful virological eradication. The remaining hard to cure population is those patients who have previously failed to respond to both PI and NS5A inhibitor-containing regimens, with the possibility of NS5A RASs.
6.1.3 Patients with Resistance-Associated Substitutions
Resistance-associated substitutions (RASs) to DAAs, due to the error-prone nature of the HCV RNA polymerase, may be present at baseline and can significantly affect treatment outcomes and the achievement of SVR [120].
RASs in NS3 may affect virological results in patients treated with an NS3 inhibitor, such as GZR, GLE, and VOX. NS3 RASs at baseline were associated with lower rates of SVR12 after 12 weeks of treatment with EBR/GZR than in HCV-infected patients without this type of RAS [77, 78, 121,122,123]. This pattern was also observed after treatment with GLE/PIB [91]. On the contrary, in a phase III trial 12 weeks of treatment resulted in SVR12 rates of 100% (6/6) and 92% (34/37) in patients with and without baseline NS3 RASs, respectively [107]. These findings may, however, be a result of small sample sizes. Similar patterns were observed for SOF/VEL/VOX, which implicates that NS3 RASs indeed are associated with lower virological efficacy rates [97, 98].
DCV, EBR, LDV, PBR, and VEL are NS5A inhibitors and are therefore expected to alter SVR rates in subjects with baseline NS5A RASs [55]. Similar results were achieved in studies with SOF/LDV [66, 70, 124,125,126,127], EBR/GZR [75, 78, 121,122,123, 128,129,130], SOF/VEL [82,83,84, 86,87,88, 103], GLE/PIB [91, 107, 131], and SOF/VEL/VOX [97, 98].
Because of its interference with NS5B, a SOF-containing regimen could have decreased efficacy in patients with baseline NS5B RASs. However, this does not seem to be such an issue as it is with the other RASs. Many patients are treated with SOF and only a few incidental reports of these kinds of NS5B RASs are presented. SVR12 rates in patients with NS5B RASs varied from 88 to 100% with all SOF-containing regimens [66, 70, 82, 84, 104, 126, 130, 132,133,134,135,136,137,138].
Although the SVR in patients with baseline RASs is somewhat lower than in those without RASs at baseline, overall rates for efficacy remain high even in this population. Additionally, patients with baseline RASs for one protein have other treatment options in reserve that encounter another HCV protein.
6.2 Liver Cirrhosis
Chronic hepatitis may lead to progressive liver fibrosis and subsequently result in cirrhosis, which could alter the pharmacokinetics and pharmacodynamics of a drug. We have previously described the possible issues with liver cirrhosis in more detail [139].
Total DCV Cmax and AUC values were lower in subjects with cirrhosis than in subjects with normal liver function after a single 30 mg dose of DCV. The DCV AUC was 43, 38, and 36% lower in patients with mild (CP-A), moderate (CP-B), and severe (CP-C) hepatic impairment, respectively. For Cmax, the geometric mean is estimated to be at least 45% lower in patients with hepatic dysfunction. However, hepatic impairment has no clinically significant effect on unbound DCV concentrations and thus on exposure to the active fraction [9, 140].
The steady-state AUC for SOF increased 126% and 143% in CP-B and CP-C patients, respectively, compared with controls after 7 days dosing of SOF 400 mg. The GS-331007 AUC was increased by 18% and 9%, respectively [11]. After treatment with SOF/DCV combination therapy for 12 or 24 weeks (according to disease severity), SVR12 rates of 50–100% [47,48,49,50, 53,54,55,56,57, 112] were reached in CP-A/B/C patients. Lower rates of SVR12 (< 90%) were obtained in the studies with small sample sizes [54]. Accordingly, SOF/DCV is a treatment option for CP-A/B/C patients without any dose adjustments needed (see Table 4).
Following a single dose of LDV 90 mg, HCV-uninfected patients with CP-C had no clinically relevant changes in LDV pharmacokinetics [141]. The effect of hepatic impairment on the pharmacokinetics of a fixed-dose combination of SOF/LDV is expected to be similar to when SOF and LDV were administered separately. The safety and efficacy of 12 weeks of treatment with SOF/LDV with and without RBV was evaluated in HCV-infected patients with compensated cirrhosis. Treatment resulted in high efficacy, irrespective of transplantation status [65, 67, 71, 126, 142,143,144,145,146]. In studies including CP-C decompensated cirrhotic patients, SVR12 rates were lower (40–92%), although retrieved from small sample sizes [144,145,146,147]. These patients seem to benefit from the addition of RBV.
Following administration of EBR 50 mg, the AUC decreased by 39, 28, and 12% and Cmax by 42, 31, and 42% in patients with CP-A, CP-B, and CP-C, respectively, compared with healthy controls [148]. For GZR, however, the steady-state GZR exposure was 1.66-, 4.82-, and 11.67-fold higher following varying doses of GZR (200, 100, and 50 mg, respectively) based on their hepatic function (CP-A, CP-B, and CP-C, respectively). Correspondingly, Cmax values increased, although AEs did not [148]. Based on these findings, GZR is contraindicated in patients with CP-B or CP-C hepatic impairment (see Table 4). EBR/GZR in patients with liver CP-A cirrhosis obtained high SVR12 rates (90–100%) in all studies [73, 74, 78, 79, 122, 129]. Increased values of ALT, AST, and alkaline phosphatase were reported in some studies [73, 74, 78, 121, 129, 149, 150]. The presence of cirrhosis was, however, not thought to be a risk factor for this elevation [17]. In summary, EBR/GZR can be used in patients with CP-A with monitoring of liver function.
After a single dose of VEL 100 mg, the AUC was 17% lower and 14% higher in non-HCV patients with CP-B and CP-C cirrhosis, respectively [151]. Additionally, subjects with compensated cirrhosis (CP-B) received either SOF/VEL for 12 weeks with or without RBV or SOF/VEL for 24 weeks. The VEL AUCτ was similar and SOF exposure was higher (~ 100%) [152]. Efficacy of SOF/VEL in CP-A patients was high, with SVR12 rates up to 100% after treatment for 12 or 24 weeks [81,82,83, 86, 101,102,103]. In CP-B patients, SVR rates increased with use of RBV: from 83 to 94% [87]. Efficacy seems to decrease with increasing liver impairment based on lower SVR rates in CP-C patients [88]. Therefore, SOF/VEL is not the therapy of choice in CP-C and CP-B patients and therefore RBV should be added (12 weeks of treatment).
A single dose of PIB 120 mg resulted in an AUC increase of 51%, 31%, and 5.2-fold in patients with CP-A, CP-B, and CP-C, respectively. For the fixed-dose combination of GLE/PIB, the GLE AUC increased by 33%, 2.0-fold, and 11-fold in patients with CP-A, CP-B, and CP-C, respectively, compared with normal subjects. GLE is therefore contraindicated in CP-C patients. The PIB AUC differed by 26% or less for patients with CP-A or CP-B cirrhosis, and increased to 2.1-fold for those with CP-C [153]. The results for efficacy of GLE/PIB in patients with CP-A cirrhosis are promising. Overall, 96–100% of patients achieve SVR12 following 12 weeks of treatment without [92, 105, 107, 154] or with RBV [105]. GLE/PIB is not recommended in patients with decompensated cirrhosis, but may be effective in treating CP-A patients.
No pharmacokinetic studies of the triple combination therapy SOF/VEL/VOX for HCV-infected patients with cirrhosis were performed. Since VOX is a PI, it is expected that it will increase hepatotoxicity and should therefore not be used in patients with decompensated cirrhosis (see Table 4). The treatment regimen was found to be highly effective in patients with CP-A cirrhosis, with SVR12 rates of 80–100% [96, 97, 108, 118, 155]. However, none of these studies included patients with decompensated cirrhosis, which makes implementation to all types of cirrhotic patients difficult.
6.3 Transplant Patients
HCV infection has a negative impact on both patient and graft survival in kidney transplant recipients compared to those without HCV infection [156]. Additionally, DDIs with immunosuppressants also make this population prone to negative treatment outcomes (see Sect. 4.2).
Efficacy and safety have not been studied thoroughly in transplant patients, but, to date, results seem promising that DAA treatment is safe and effective. For kidney transplant recipients, SOF/LDV therapy resulted in an SVR12 of 100% following treatment for 12 or 24 weeks without RBV. No significant changes in estimated glomerular filtration rate (eGFR) were observed during and after treatment [157]. These successful virological results were confirmed in several clinical trials and real-world studies [158,159,160,161]. High efficacy rates with favorable safety profiles in kidney transplant recipients were also retrieved with SOF/DCV [159,160,161] and GLE/PIB [131]. DAA therapy was well-tolerated in all studies, with mild AEs and laboratory abnormalities being infrequent.
Liver transplant recipients with HCV recurrence obtained an SVR rate of 94% following SOF/DCV/RBV for 12 weeks. Similar results were achieved in other studies with liver transplant patients [54, 110, 162]. Efficacy rates for SOF/LDV varying from 50 to 100% were seen in HCV recurrent liver transplant recipients following the fixed-dose combination of SOF/LDV [69, 124, 146]. SVR12 rates lower than 85% were only achieved in studies with low numbers of included patients (n < 10) [124]. Comparably, 12 weeks of treatment with SOF/VEL and GLE/PIB resulted in SVR12 rates of 96% and 89%, respectively, in liver transplant recipients [104, 131]. Therapies were generally well-tolerated with no serious adverse effects, laboratory abnormalities, or graft transplant rejection in relation to the treatment.
In conclusion, DAA therapy seems to be effective and safe in transplant patients with no therapy alterations being necessary.
6.4 Renal Impairment
Two SOF-free regimens are available: EBR/GZR and GLE/PIB. EBR and GZR are both minimally (< 1%) renally excreted; it is therefore not expected that overall clearance of either drug will be affected. After normal doses, EBR/GZR exposure is increased by 50–86% in both advanced chronic kidney disease (CKD) patients with and without HCV infection compared with healthy controls. For patients with end-stage renal disease (ESRD), EBR and GZR exposure was not significantly affected. Also, the steady-state pharmacokinetics of EBR and GZR were not influenced by dialysis [17, 18]. The efficacy and safety of EBR/GZR in patients with renal impairment were studied in multiple trials and SVR12 varied from 94 to 100% in various populations [149, 163,164,165]. Therefore, this regimen is safe and effective for patients with renal impairment, requiring no dose adjustment (see Table 5).
Renal excretion of both GLE and PIB are minimal (< 4%). For both drugs, the AUC was increased up to 13, 30, 45, and 56% in subjects with mild, moderate, and severe renal impairment or ESRD, respectively, compared with normal subjects [166]. The efficacy and safety of GLE/PIB were studied in patients with severe renal impairment (CKD stage 4 or 5): the SVR12 rate was 98% after treatment with the fixed-dose combination of GLE/PIB for 12 weeks [105]. These results were confirmed with an SVR12 rate of 100% in patients with severe renal dysfunction and prior DAA treatment [106]. This treatment, therefore, is considered to be effective and safe in patients with severe renal insufficiency and no dose adjustments are required (see Table 5).
SOF is mainly renally excreted and reduced clearance can be expected in patients with renal dysfunction [11]. Concerns have risen due to substantially higher concentrations of the primary metabolite of SOF, GS-331007, in these patients (see Fig. 2). Patients with mild, moderate, and severe renal insufficiency obtained increased SOF AUC values of 61, 107, and 171%, respectively, compared with controls. GS-331007 AUC values were elevated by 55, 88, and 451%, respectively, so accumulation could occur [167]. Therefore, SOF-free regimens are generally preferred in patients with severe renal impairment (eGFR < 30 mL/min/1.73 m2). In all other patients, restrictions regarding SOF are unnecessary [23].
However, increased exposure of GS-331007 is not associated with increased toxicity [12]. Due to the metabolism of SOF, it makes pharmacological sense to give a full dose of 400 mg to patients with impaired renal function. The reason for this is that we need to achieve high enough active metabolite (GS-461203) concentrations in the liver for the antiviral action of SOF. Lower doses possibly result in lower active metabolite concentrations and response rates (see Sect. 4). It has been shown that SOF in standard doses is well-tolerated in patients with renal impairment [160, 168, 169] and that alternate dosing of LDV/SOF in hemodialysis patients yielded SVR12 rates of 91% [170]. In patients undergoing dialysis, a 95% SVR12 rate was achieved after SOF/VEL use for 12 weeks. The safety of this combination regimen was consistent with advanced renal disease and was well-tolerated [171]. In case no safer DAA options are available, e.g., in patients with other co-morbidities, treatment with SOF may therefore be acceptable.
6.5 Hepatitis C Virus (HCV)/HIV Co-Infection and Hepatitis B Virus/HCV Co-Infection
Historically, HCV/HIV co-infected patients were harder to treat patients, as SVR rates were lower in the peg-IFN/RBV era than in HCV mono-infected patients. However, SVR12 rates after treatment with DAA regimens in HCV/HIV co-infected patients are comparable with HCV mono-infected patients and this is extensively shown in the literature [52, 56, 111, 114, 123, 125, 128, 137, 172,173,174].
HBV co-infected patients have a potential risk of HBV reactivation during or after HCV clearance, although this remains unpredictable [175]. The efficacy of HCV therapy in HBV/HCV co-infected patients was high (SVR12 100%) with SOF/LDV therapy for 12 weeks. Approximately two-thirds of the patients had an increased HBV DNA level during treatment, although this was not associated with any signs or symptoms. Only 2 of the 111 included patients required HBV therapy during SOF/LDV treatment. No (serious) AEs were reported and no patients discontinued treatment [70]. Based on this study, there seems to be no reason to treat HBV co-infected patients differently to mono-infected patients. Therefore, HBV/HCV co-infected patients should be treated with the same anti-HCV treatment regimens as HCV mono-infected patients, keeping the HBV reactivation in mind—meaning that HBV DNA and ALT flares should be monitored at least every 4–8 weeks and 3 months post treatment according to American Association for the Study of Liver Diseases (AALSD) guidance [176]. Patients that are hepatitis B surface antigen (HBsAg)-negative, antibody to hepatitis B core antigen (anti-HBc)-positive have a low reactivation risk and ALT should only be monitored at baseline, end of treatment, and during follow-up [176].
6.6 Bleeding Disorders
Since treatment with interferon and RBV are associated with anemia, anti-HCV therapy has often been withheld in patients with bleeding disorders. Several phases II and III studies have examined the safety and efficacy of DAA treatment regimens in this special population. Results in patients with bleeding disorders do not seem to differ from those in patients without these conditions [122, 135, 177,178,179,180,181]. Recommendations for HCV treatment in this special population are therefore equal to those without a bleeding disorder. However, as several RBV-free regimens are available, the preferred option is to treat these patients with the RBV-free regimens of SOF/VEL or GLE/PIB.
6.7 Children and Adolescents
Mother-to-child transmission of HCV is the most common source of HCV infection in children; however, treatment of HCV-infected newborns is not yet possible [182, 183]. Therefore, children can be treated (see below) or HCV treatment during pregnancy is an option, not only to cure maternal HCV but also to reduce the incidence of pediatric HCV cases. With the introduction of RBV-free DAA treatment combinations, which are highly effective and show a favorable safety profile in preclinical animal studies, treatment during pregnancy seems realistic in the nearby future [184].
Children and adolescents may require treatment alterations compared with adults due to differences in pharmacokinetics that potentially lead to efficacy and/or safety issues in this population. Administration of half of the adult fixed-dose of SOF/LDV in children (6 to < 12 years) resulted in comparable exposure to that in adults exposed to the fixed-dose combination: the AUC was 13% lower, 16% higher, and 34% lower for LDV, SOF, and GS-331007, respectively. The Cmax was 16, 39, and 6% higher for these three compounds. An SVR of 99% was achieved in these DAA-naïve children with genotypes 1, 3, or 4 HCV infection [134].
Exposure in adolescents (12 to < 18 years) was comparable with that in adults following the adult fixed-dose combination of SOF/LDV: SOF, GS-331007, and LDV AUCτ increased by 60, 5, and 27%, respectively, and Cmax was 56, 39, and 62% higher, respectively [132]. Efficacy was also comparable with that in adults, as all 40 adolescents (treatment-naïve, treatment-experienced, genotype 4) achieved SVR when treated with LDV/SOF [185]. The SVR12 rate was 98% in HCV genotype 1-infected adolescents (12–17 years) with and without compensated cirrhosis [132]. No serious AEs were reported in any of the studies.
Weight-based (≥ 45 kg: 400 mg/60 mg; 17–45 kg: 200 mg/30 mg) SOF/DCV resulted in SVR12 rates of 97% in HCV genotype 4-infected adolescents (12–17 years) [186]. In a study with patients aged 8–18 years, SVR12 was 98% after SOF/DCV 400 mg/60 mg per 1.7 m2 [187].
At present, DAA therapy seems to be effective and safe (probably after dose adjustments based on bodyweight) in children and adolescents; however, additional studies may be necessary to confirm these statements.
7 Conclusion
In this descriptive review, we have provided an overview of the clinical pharmacokinetics and pharmacodynamics, in terms of efficacy and toxicity, of the DAA combination regimens for the treatment of chronic HCV. Although we have listed all available treatment DAA regimens and options, no differences have been discussed in pricing and/or availability of the regimens. One should consider that in some counties only the pan-genotypic regimens SOF/VEL and GLE/PIB are available as first-line agents. In our opinion, SOF/VEL/VOX should be preserved as the salvage regimen for DAA failures.
We have also discussed the relationship between the pharmacokinetics of the DAAs and efficacy or toxicity in patients with liver cirrhosis, liver transplantation, renal impairment, HBV or HIV co-infection, and bleeding disorders, and in children and adolescents.
As the development of treatments for HCV therapy is thought to have come to an end, this review gives a complete overview of all possible treatment options for chronic hepatitis C in well-resourced countries. This aids in ensuring that making informed decisions in clinical practice is feasible and physicians, pharmacists, and nurse practitioners are educated to effectively and safely treat HCV-infected patients.
References
World Health Organization. Hepatitis C fact sheet. 2018. https://www.who.int/news-room/fact-sheets/detail/hepatitis-c. Accessed 19 Feb 2019.
European Association for the Study of the Liver. The burden of liver disease in Europe: a review of available epidemiological data. 2013. http://www.easl.eu/medias/EASLimg/Discover/EU/54ae845caec619f_file.pdf.
Schreiber J, McNally J, Chodavarapu K, Svarovskaia E, Moreno C. Treatment of a patient with genotype 7 hepatitis C virus infection with sofosbuvir and velpatasvir. Hepatology. 2016;64(3):983–5.
Esposito I, Trinks J, Soriano V. Hepatitis C virus resistance to the new direct-acting antivirals. Expert Opin Drug Metab Toxicol. 2016;12(10):1197–209.
European Medicines Agency. Summary of product characteristics: Victrelis. 2019. https://www.ema.europa.eu/documents/product-information/victrelis-epar-product-information_en.pdf. Accessed 2 Feb 2019.
European Medicines Agency. Summary of product characteristics: Maviret [in Dutch]. 2019. https://www.ema.europa.eu/documents/product-information/maviret-epar-product-information_nl.pdf. Accessed 2 Feb 2019.
European Medicines Agency. Summary of product characteristics: Epclusa [in Dutch]. 2019. https://www.ema.europa.eu/documents/product-information/epclusa-epar-product-information_nl.pdf. Accessed 2 Feb 2019.
European Medicines Agency. Summary of product characteristics: Vosevi. 2019. https://www.ema.europa.eu/documents/product-information/vosevi-epar-product-information_en.pdf. Accessed 2 Feb 2019.
European Medicines Agency. Summary of product characteristics: Daklinza. 2019. https://www.ema.europa.eu/documents/product-information/daklinza-epar-product-information_en.pdf. Accessed 2 Feb 2019.
US Food and Drug Administration. Highlights of prescribing information: Daklinza. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/206843s006lbl.pdf. Accessed 19 Feb 2019.
European Medicines Agency. Summary of product characteristics: Sovaldi. 2019. https://www.ema.europa.eu/documents/product-information/sovaldi-epar-product-information_en.pdf. Accessed 2 Feb 2019.
US Food and Drug Administration. Highlights of prescribing information: Sovaldi. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/204671s002lbl.pdf. Accessed 19 Feb 2019.
Smolders EJ, Colbers A, de Kanter C, Velthoven-Graafland K, Wolberink LT, van Ewijk-Beneken Kolmer N, et al. Metformin and daclatasvir: absence of a pharmacokinetic-pharmacodynamic drug interaction in healthy volunteers. Br J Clin Pharmacol. 2017;83(10):2225–34.
Kirby BJ, Symonds WT, Kearney BP, Mathias AA. Pharmacokinetic, pharmacodynamic, and drug-interaction profile of the hepatitis C virus NS5B polymerase inhibitor sofosbuvir. Clin Pharmacokinet. 2015;54(7):677–90.
European Medicines Agency. Summary of product characteristics: Harvoni. 2019. https://www.ema.europa.eu/documents/product-information/harvoni-epar-product-information_en.pdf. Accessed 2 Feb 2019.
US Food and Drug Administration. Highlights of prescribing information: Harvoni. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/205834s017lbl.pdf. Accessed 19 Feb 2019.
European Medicines Agency. Summary of product characteristics: Zepatier. 2019. https://www.ema.europa.eu/documents/product-information/zepatier-epar-product-information_en.pdf. Accessed 2 Feb 2019.
US Food and Drug Administration. Highlights of prescribing information: Zepatier. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208261s002lbl.pdf. Accessed 19 Feb 2019.
US Food and Drug Administration. Highlights of prescribing information: Epclusa. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208341s007lbl.pdf. Accessed 19 Feb 2019.
US Food and Drug Administration. Highlights of prescribing information: Maviret. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209394s000lbl.pdf. Accessed 19 Feb 2019.
US Food and Drug Administration. Highlights of prescribing information: Vosevi. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/209195s000lbl.pdf. Accessed 19 Feb 2019.
Univerisity of Liverpool. Liverpool HEP Drug Interactions. 2019. https://www.hep-druginteractions.org/. Accessed 19 Mar 2019.
European Association for the Study of the Liver. EASL recommendations on treatment of hepatitis C 2018. J Hepatol. 2018;69(2):461–511.
European Medicines Agency. Summary of product characteristics: Pifeltro. 2019. https://www.ema.europa.eu/en/documents/product-information/pifeltro-epar-product-information_en.pdf. Accessed 18 Mar 2019.
Bifano M, Hwang C, Oosterhuis B, Hartstra J, Grasela D, Tiessen R, et al. Assessment of pharmacokinetic interactions of the HCV NS5A replication complex inhibitor daclatasvir with antiretroviral agents: ritonavir-boosted atazanavir, efavirenz and tenofovir. Antivir Ther. 2013;18(7):931–40.
Smolders EJ, Colbers EP, de Kanter CT, Velthoven-Graafland K, Drenth JP, Burger DM. Daclatasvir 30 mg/day is the correct dose for patients taking atazanavir/cobicistat. J Antimicrob Chemother. 2017;72(2):486–9.
Wyles DL, Ruane PJ, Sulkowski MS, Dieterich D, Luetkemeyer A, Morgan TR, et al. Daclatasvir plus sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med. 2015;373(8):714–25.
Feng HP, Caro L, Fandozzi C, Chu X, Guo Z, Talaty J, et al. Pharmacokinetic interactions between the HCV inhibitors elbasvir and grazoprevir and HIV protease inhibitors ritonavir, atazanavir, lopinavir, or darunavir in healthy participants. Antimicrob Agents Chemother. 2019;63(4):e02142-18.
Mogalian E, Stamm LM, Osinusi A, Brainard DM, Shen G, Ling KHJ, et al. Drug-drug interaction studies between hepatitis C virus antivirals sofosbuvir/velpatasvir and boosted and unboosted human immunodeficiency virus antiretroviral regimens in healthy volunteers. Clin Infect Dis. 2018;67(6):934–40.
Solas C, Bregigeon S, Faucher-Zaegel O, Quaranta S, Obry-Roguet V, Tamalet C, et al. Ledipasvir and tenofovir drug interaction in human immunodeficiency virus-hepatitis C virus coinfected patients: Impact on tenofovir trough concentrations and renal safety. Br J Clin Pharmacol. 2018;84(2):404–9.
Pozniak A, Arribas JR, Gathe J, Gupta SK, Post FA, Bloch M, et al. Switching to tenofovir alafenamide, coformulated with elvitegravir, cobicistat, and emtricitabine, in HIV-infected patients with renal impairment: 48-week results from a single-arm, multicenter, open-label phase 3 study. J Acquir Immune Defic Syndr. 2016;71(5):530–7.
Feng HP, Guo Z, Ross LL, Fraser I, Panebianco D, Jumes P, et al. Assessment of drug interaction potential between the HCV direct-acting antiviral agents elbasvir/grazoprevir and the HIV integrase inhibitors raltegravir and dolutegravir. J Antimicrob Chemother. 2019;74(3):710–7.
Smolders EJ, Pape S, de Kanter CT, van den Berg AP, Drenth JP, Burger DM. Decreased tacrolimus plasma concentrations during HCV therapy: a drug–drug interaction or is there an alternative explanation? Int J Antimicrob Agents. 2017;49(3):379–82.
Smolders EJ, Ter Horst PJG, Wolters S, Burger DM. Cardiovascular risk management and hepatitis C: combining drugs. Clin Pharmacokinet. 2019;58(5):565–92.
Renet S, Chaumais MC, Antonini T, Zhao A, Thomas L, Savoure A, et al. Extreme bradycardia after first doses of sofosbuvir and daclatasvir in patients receiving amiodarone: 2 cases including a rechallenge. Gastroenterology. 2015;149(6):1378.e1–1380.e1.
Regan CP, Morissette P, Regan HK, Travis JJ, Gerenser P, Wen J, et al. Assessment of the clinical cardiac drug-drug interaction associated with the combination of hepatitis C virus nucleotide inhibitors and amiodarone in guinea pigs and rhesus monkeys. Hepatology. 2016;64(5):1430–41.
FDA Drug Safety Communication. FDA warns of serious slowing of the heart rate when the antiarrhytmic drug amiodarone is used with hepatitis C treatments containing sofosbuvir (Harvoni) of Sovaldi in combination with another direct acting antiviral drug. 2015. https://www.fda.gov/downloads/Drugs/DrugSafety/UCM439492.pdf.
Caldeira D, Rodrigues FB, Duarte MM, Sterrantino C, Barra M, Goncalves N, et al. Cardiac harms of sofosbuvir: systematic review and meta-analysis. Drug Saf. 2018;41(1):77–86.
van Seyen M, Smolders EJ, van Wijngaarden P, Drenth JPH, Wouthuyzen-Bakker M, de Knegt RJ, et al. Successful HCV treatment of patients on contraindicated anti-epileptic drugs: Role of drug level monitoring. J Hepatol. 2019;70(3):552–4.
European Commission. Commission staff working document on combatting HIV/AIDS, viral hepatitis and tuberculosis in the European Union and neighbouring countries—state of play, policy instruments and good practices. 2018. https://ec.europa.eu/health/sites/health/files/communicable_diseases/docs/swd_2018_387_en.pdf. Accessed 19 Mar 2019.
PanACEA (Pan-African Consortium for the Evaluation of Antituberculosis Antibiotics). 2019. http://panacea-tb.net/. Accessed 19 Mar 2019.
Centers for Disease Control and Prevention. Treatment for TB disease. 2019. https://www.cdc.gov/tb/topic/treatment/tbdisease.htm. Accessed 19 Mar 2019.
Mogalian E, German P, Kearney BP, Yang CY, Brainard D, Link J, et al. Preclinical pharmacokinetics and first-in-human pharmacokinetics, safety, and tolerability of velpatasvir, a pangenotypic hepatitis C virus NS5A inhibitor, in healthy subjects. Antimicrob Agents Chemother. 2017;61(5):e02084-16.
European Medicines Agency. Harvoni: assessment report. 2019. https://www.ema.europa.eu/en/documents/assessment-report/harvoni-epar-public-assessment-report_en.pdf. Accessed 19 Mar 2019.
Nettles RE, Gao M, Bifano M, Chung E, Persson A, Marbury TC, et al. Multiple ascending dose study of BMS-790052, a nonstructural protein 5A replication complex inhibitor, in patients infected with hepatitis C virus genotype 1. Hepatology. 2011;54(6):1956–65.
Lawitz E, Rodriguez-Torres M, Denning JM, Albanis E, Cornpropst M, Berrey MM, et al. Pharmacokinetics, pharmacodynamics, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor, following multiple ascending doses in patients with chronic hepatitis C infection. Antimicrob Agents Chemother. 2013;57(3):1209–17.
Pol S, Bourliere M, Lucier S, Hezode C, Dorival C, Larrey D, et al. Safety and efficacy of daclatasvir-sofosbuvir in HCV genotype 1-mono-infected patients. J Hepatol. 2017;66(1):39–47.
Cheng PN, Chiu YC, Chien SC, Chiu HC. Real-world effectiveness and safety of sofosbuvir plus daclatasvir with or without ribavirin for genotype 2 chronic hepatitis C in Taiwan. J Formos Med Assoc. 2019;118(5):907–13.
Nelson DR, Cooper JN, Lalezari JP, Lawitz E, Pockros PJ, Gitlin N, et al. All-oral 12-week treatment with daclatasvir plus sofosbuvir in patients with hepatitis C virus genotype 3 infection: ALLY-3 phase III study. Hepatology. 2015;61(4):1127–35.
Leroy V, Angus P, Bronowicki JP, Dore GJ, Hezode C, Pianko S, et al. Daclatasvir, sofosbuvir, and ribavirin for hepatitis C virus genotype 3 and advanced liver disease: a randomized phase III study (ALLY-3+). Hepatology. 2016;63(5):1430–41.
Abdel-Moneim A, Aboud A, Abdel-Gabaar M, Zanaty MI, Ramadan M. Efficacy and safety of sofosbuvir plus daclatasvir with or without ribavirin: large real-life results of patients with chronic hepatitis C genotype 4. Hepatol Int. 2018;12(4):348–55.
de Kanter CT, Drenth JP, Arends JE, Reesink HW, van der Valk M, de Knegt RJ, et al. Viral hepatitis C therapy: pharmacokinetic and pharmacodynamic considerations. Clin Pharmacokinet. 2014;53(5):409–27.
El-Khayat H, Fouad Y, Mohamed HI, El-Amin H, Kamal EM, Maher M, et al. Sofosbuvir plus daclatasvir with or without ribavirin in 551 patients with hepatitis C-related cirrhosis, genotype 4. Aliment Pharmacol Ther. 2018;47(5):674–9.
Kwo P, Fried MW, Reddy KR, Soldevila-Pico C, Khemichian S, Darling J, et al. Daclatasvir and sofosbuvir treatment of decompensated liver disease or post-liver transplant hepatitis C virus recurrence in patients with advanced liver disease/cirrhosis in a real-world cohort. Hepatol Commun. 2018;2(4):354–63.
Poordad F, Schiff ER, Vierling JM, Landis C, Fontana RJ, Yang R, et al. Daclatasvir with sofosbuvir and ribavirin for hepatitis C virus infection with advanced cirrhosis or post-liver transplantation recurrence. Hepatology. 2016;63(5):1493–505.
Rockstroh JK, Ingiliz P, Petersen J, Peck-Radosavljevic M, Welzel TM, Van der Valk M, et al. Daclatasvir plus sofosbuvir, with or without ribavirin, in real-world patients with HIV-HCV coinfection and advanced liver disease. Antivir Ther. 2017;22(3):225–36.
Shiha G, Soliman R, ElBasiony M, Hassan AA, Mikhail NNH. Sofosbuvir plus daclatasvir with or without ribavirin for treatment of chronic HCV genotype 4 patients: real-life experience. Hepatol Int. 2018;12(4):339–47.
Lawitz EJ, Gruener D, Hill JM, Marbury T, Moorehead L, Mathias A, et al. A phase 1, randomized, placebo-controlled, 3-day, dose-ranging study of GS-5885, an NS5A inhibitor, in patients with genotype 1 hepatitis C. J Hepatol. 2012;57(1):24–31.
Wilson E, Kattakuzhy S, Kwo P, Davitkov P, Qureshi K, Sundaram V, et al. Real world effectiveness of 8 and 12 weeks of ledipasvir/sofosbuvir (LDV/SOF) in Blacks with HCV: a comparative analysis of clinical trials with real world cohorts. AASLD: The Liver Meeting; 20–24 Oct 2017; Washington, DC.
Buggisch P, Vermehren J, Mauss S, Gunther R, Schott E, Pathil A, et al. Real-world effectiveness of 8-week treatment with ledipasvir/sofosbuvir in chronic hepatitis C. J Hepatol. 2018;68(4):663–71.
Boerekamps A, Vanwolleghem T, van der Valk M, van den Berk GE, van Kasteren M, Posthouwer D, et al. 8 weeks of sofosbuvir/ledipasvir is effective in DAA-naive non-cirrhotic HCV genotype 4 infected patients (HEPNED-001 study). J Hepatol. 2019;70(3):554–7.
Afdhal N, Zeuzem S, Kwo P, Chojkier M, Gitlin N, Puoti M, et al. Ledipasvir and sofosbuvir for untreated HCV genotype 1 infection. N Engl J Med. 2014;370(20):1889–98.
Afdhal N, Reddy KR, Nelson DR, Lawitz E, Gordon SC, Schiff E, et al. Ledipasvir and sofosbuvir for previously treated HCV genotype 1 infection. N Engl J Med. 2014;370(16):1483–93.
Mizokami M, Yokosuka O, Takehara T, Sakamoto N, Korenaga M, Mochizuki H, et al. Ledipasvir and sofosbuvir fixed-dose combination with and without ribavirin for 12 weeks in treatment-naive and previously treated Japanese patients with genotype 1 hepatitis C: an open-label, randomised, phase 3 trial. Lancet Infect Dis. 2015;15(6):645–53.
Kohli A, Kapoor R, Sims Z, Nelson A, Sidharthan S, Lam B, et al. Ledipasvir and sofosbuvir for hepatitis C genotype 4: a proof-of-concept, single-centre, open-label phase 2a cohort study. Lancet Infect Dis. 2015;15(9):1049–54.
Abergel A, Metivier S, Samuel D, Jiang D, Kersey K, Pang PS, et al. Ledipasvir plus sofosbuvir for 12 weeks in patients with hepatitis C genotype 4 infection. Hepatology. 2016;64(4):1049–56.
Abergel A, Asselah T, Metivier S, Kersey K, Jiang D, Mo H, et al. Ledipasvir-sofosbuvir in patients with hepatitis C virus genotype 5 infection: an open-label, multicentre, single-arm, phase 2 study. Lancet Infect Dis. 2016;16(4):459–64.
Gane EJ, Hyland RH, An D, Svarovskaia E, Pang PS, Brainard D, et al. Efficacy of ledipasvir and sofosbuvir, with or without ribavirin, for 12 weeks in patients with HCV genotype 3 or 6 infection. Gastroenterology. 2015;149(6):1454–1461.e1.
Ueda Y, Ikegami T, Akamatsu N, Soyama A, Shinoda M, Goto R, et al. Treatment with sofosbuvir and ledipasvir without ribavirin for 12 weeks is highly effective for recurrent hepatitis C virus genotype 1b infection after living donor liver transplantation: a Japanese multicenter experience. J Gastroenterol. 2017;52(8):986–91.
Liu CJ, Chuang WL, Sheen IS, Wang HY, Chen CY, Tseng KC, et al. Efficacy of ledipasvir and sofosbuvir treatment of HCV infection in patients coinfected with HBV. Gastroenterology. 2018;154(4):989–97.
Lim YS, Ahn SH, Lee KS, Paik SW, Lee YJ, Jeong SH, et al. A phase IIIb study of ledipasvir/sofosbuvir fixed-dose combination tablet in treatment-naive and treatment-experienced Korean patients chronically infected with genotype 1 hepatitis C virus. Hepatol Int. 2016;10(6):947–55.
Yeh WW, Fraser IP, Jumes P, Petry A, Lepeleire I, Robberechts M, et al. Antiviral activity, safety, and tolerability of multiple ascending doses of elbasvir or grazoprevir in participants infected with hepatitis C virus genotype-1 or -3. Clin Ther. 2018;40(5):704–718.e6.
Lawitz E, Gane E, Pearlman B, Tam E, Ghesquiere W, Guyader D, et al. Efficacy and safety of 12 weeks versus 18 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin for hepatitis C virus genotype 1 infection in previously untreated patients with cirrhosis and patients with previous null response with or without cirrhosis (C-WORTHY): a randomised, open-label phase 2 trial. Lancet. 2015;385(9973):1075–86.
Wei L, Jia JD, Wang FS, Niu JQ, Zhao XM, Mu S, et al. Efficacy and safety of elbasvir/grazoprevir in participants with HCV genotype 1, 4, or 6 infection from the Asia-Pacific region and Russia: final results from the randomized C-CORAL study. J Gastroenterol Hepatol. 2019;34(1):12–21.
Jacobson IM, Lawitz E, Kwo PY, Hezode C, Peng CY, Howe AYM, et al. Safety and efficacy of elbasvir/grazoprevir in patients with hepatitis C virus infection and compensated cirrhosis: an integrated analysis. Gastroenterology. 2017;152(6):1372–1382.e2.
Forns X, Gordon SC, Zuckerman E, Lawitz E, Calleja JL, Hofer H, et al. Grazoprevir and elbasvir plus ribavirin for chronic HCV genotype-1 infection after failure of combination therapy containing a direct-acting antiviral agent. J Hepatol. 2015;63(3):564–72.
Gane E, Nahass R, Luketic V, Asante-Appiah E, Hwang P, Robertson M, et al. Efficacy of 12 or 18 weeks of elbasvir plus grazoprevir with ribavirin in treatment-naive, noncirrhotic HCV genotype 3-infected patients. J Viral Hepat. 2017;24(10):895–9.
Foster GR, Agarwal K, Cramp ME, Moreea S, Barclay S, Collier J, et al. Elbasvir/grazoprevir and sofosbuvir for hepatitis C virus genotype 3 infection with compensated cirrhosis: a randomized trial. Hepatology. 2018;67(6):2113–26.
de Ledinghen V, Laforest C, Hezode C, Pol S, Renault A, Alric L, et al. Retreatment with sofosbuvir plus grazoprevir/elbasvir plus ribavirin of patients with hepatitis C virus genotype 1 or 4 who previously failed an NS5A- or NS3-containing regimen: the ANRS HC34 REVENGE study. Clin Infect Dis. 2018;66(7):1013–8.
Lawitz E, Freilich B, Link J, German P, Mo H, Han L, et al. A phase 1, randomized, dose-ranging study of GS-5816, a once-daily NS5A inhibitor, in patients with genotype 1–4 hepatitis C virus. J Viral Hepat. 2015;22(12):1011–9.
Feld JJ, Jacobson IM, Hezode C, Asselah T, Ruane PJ, Gruener N, et al. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N Engl J Med. 2015;373(27):2599–607.
Foster GR, Afdhal N, Roberts SK, Brau N, Gane EJ, Pianko S, et al. Sofosbuvir and velpatasvir for HCV genotype 2 and 3 infection. N Engl J Med. 2015;373(27):2608–17.
Liu CH, Sun HY, Liu CJ, Sheng WH, Hsieh SM, Lo YC, et al. Generic velpatasvir plus sofosbuvir for hepatitis C virus infection in patients with or without human immunodeficiency virus coinfection. Aliment Pharmacol Ther. 2018;47(12):1690–8.
Everson GT, Towner WJ, Davis MN, Wyles DL, Nahass RG, Thuluvath PJ, et al. Sofosbuvir with velpatasvir in treatment-naive noncirrhotic patients with genotype 1 to 6 hepatitis C virus infection: a randomized trial. Ann Intern Med. 2015;163(11):818–26.
Gane EJ, Shiffman ML, Etzkorn K, Morelli G, Stedman CAM, Davis MN, et al. Sofosbuvir-velpatasvir with ribavirin for 24 weeks in hepatitis C virus patients previously treated with a direct-acting antiviral regimen. Hepatology. 2017;66(4):1083–9.
Pianko S, Flamm SL, Shiffman ML, Kumar S, Strasser SI, Dore GJ, et al. Sofosbuvir plus velpatasvir combination therapy for treatment-experienced patients with genotype 1 or 3 hepatitis C virus infection: a randomized trial. Ann Intern Med. 2015;163(11):809–17.
Curry MP, O’Leary JG, Bzowej N, Muir AJ, Korenblat KM, Fenkel JM, et al. Sofosbuvir and velpatasvir for HCV in patients with decompensated cirrhosis. N Engl J Med. 2015;373(27):2618–28.
Takehara T, Sakamoto N, Nishiguchi S, Ikeda F, Tatsumi T, Ueno Y, et al. Efficacy and safety of sofosbuvir-velpatasvir with or without ribavirin in HCV-infected Japanese patients with decompensated cirrhosis: an open-label phase 3 trial. J Gastroenterol. 2019;54(1):87–95.
Lawitz EJ, O’Riordan WD, Asatryan A, Freilich BL, Box TD, Overcash JS, et al. Potent antiviral activities of the direct-acting antivirals ABT-493 and ABT-530 with three-day monotherapy for hepatitis C virus genotype 1 infection. Antimicrob Agents Chemother. 2015;60(3):1546–55.
Kwo PY, Poordad F, Asatryan A, Wang S, Wyles DL, Hassanein T, et al. Glecaprevir and pibrentasvir yield high response rates in patients with HCV genotype 1–6 without cirrhosis. J Hepatol. 2017;67(2):263–71.
Zeuzem S, Foster GR, Wang S, Asatryan A, Gane E, Feld JJ, et al. Glecaprevir-pibrentasvir for 8 or 12 weeks in HCV genotype 1 or 3 infection. N Engl J Med. 2018;378(4):354–69.
Forns X, Lee SS, Valdes J, Lens S, Ghalib R, Aguilar H, et al. Glecaprevir plus pibrentasvir for chronic hepatitis C virus genotype 1, 2, 4, 5, or 6 infection in adults with compensated cirrhosis (EXPEDITION-1): a single-arm, open-label, multicentre phase 3 trial. Lancet Infect Dis. 2017;17(10):1062–8.
Kwo PY, Vinayek R. The therapeutic approaches for hepatitis C virus: protease inhibitors and polymerase inhibitors. Gut Liver. 2011;5(4):406–17.
Toyoda H, Chayama K, Suzuki F, Sato K, Atarashi T, Watanabe T, et al. Efficacy and safety of glecaprevir/pibrentasvir in Japanese patients with chronic genotype 2 hepatitis C virus infection. Hepatology. 2017. https://doi.org/10.1002/hep.29510 (Epub 2017 Sep 2).
Rodriguez-Torres M, Glass S, Hill J, Freilich B, Hassman D, Di Bisceglie AM, et al. GS-9857 in patients with chronic hepatitis C virus genotype 1–4 infection: a randomized, double-blind, dose-ranging phase 1 study. J Viral Hepat. 2016;23(8):614–22.
Gane EJ, Kowdley KV, Pound D, Stedman CA, Davis M, Etzkorn K, et al. Efficacy of sofosbuvir, velpatasvir, and GS-9857 in patients with hepatitis C virus genotype 2, 3, 4, or 6 infections in an open-label, phase 2 trial. Gastroenterology. 2016;151(5):902–9.
Jacobson IM, Lawitz E, Gane EJ, Willems BE, Ruane PJ, Nahass RG, et al. Efficacy of 8 weeks of sofosbuvir, velpatasvir, and voxilaprevir in patients with chronic HCV infection: 2 phase 3 randomized trials. Gastroenterology. 2017;153(1):113–22.
Bourliere M, Gordon SC, Flamm SL, Cooper CL, Ramji A, Tong M, et al. Sofosbuvir, velpatasvir, and voxilaprevir for previously treated HCV infection. N Engl J Med. 2017;376(22):2134–46.
Hezode C, Lebray P, De Ledinghen V, Zoulim F, Di Martino V, Boyer N, et al. Daclatasvir plus sofosbuvir, with or without ribavirin, for hepatitis C virus genotype 3 in a French early access programme. Liver Int. 2017;37(9):1314–24.
Grebely J, Dore GJ, Zeuzem S, Aspinall RJ, Fox R, Han L, et al. Efficacy and safety of sofosbuvir/velpatasvir in patients with chronic hepatitis C virus infection receiving opioid substitution therapy: analysis of phase 3 ASTRAL trials. Clin Infect Dis. 2016;63(11):1479–81.
von Felden J, Vermehren J, Ingiliz P, Mauss S, Lutz T, Simon KG, et al. High efficacy of sofosbuvir/velpatasvir and impact of baseline resistance-associated substitutions in hepatitis C genotype 3 infection. Aliment Pharmacol Ther. 2018;47(9):1288–95.
Wyles D, Brau N, Kottilil S, Daar ES, Ruane P, Workowski K, et al. Sofosbuvir and velpatasvir for the treatment of hepatitis C virus in patients coinfected with human immunodeficiency virus type 1: an open-label, phase 3 study. Clin Infect Dis. 2017;65(1):6–12.
Esteban R, Pineda JA, Calleja JL, Casado M, Rodriguez M, Turnes J, et al. Efficacy of sofosbuvir and velpatasvir, with and without ribavirin, in patients with hepatitis C virus genotype 3 infection and cirrhosis. Gastroenterology. 2018;155(4):1120–1127.e4.
Agarwal K, Castells L, Mullhaupt B, Rosenberg WMC, McNabb B, Arterburn S, et al. Sofosbuvir/velpatasvir for 12 weeks in genotype 1–4 HCV-infected liver transplant recipients. J Hepatol. 2018;69(3):603–7.
Gane E, Lawitz E, Pugatch D, Papatheodoridis G, Brau N, Brown A, et al. Glecaprevir and pibrentasvir in patients with HCV and severe renal impairment. N Engl J Med. 2017;377(15):1448–55.
Kumada H, Watanabe T, Suzuki F, Ikeda K, Sato K, Toyoda H, et al. Efficacy and safety of glecaprevir/pibrentasvir in HCV-infected Japanese patients with prior DAA experience, severe renal impairment, or genotype 3 infection. J Gastroenterol. 2018;53(4):566–75.
Wyles D, Poordad F, Wang S, Alric L, Felizarta F, Kwo PY, et al. Glecaprevir/pibrentasvir for hepatitis C virus genotype 3 patients with cirrhosis and/or prior treatment experience: a partially randomized phase 3 clinical trial. Hepatology. 2017. https://doi.org/10.1002/hep.29541 (Epub 2017 Sep 19).
Gane EJ, Schwabe C, Hyland RH, Yang Y, Svarovskaia E, Stamm LM, et al. Efficacy of the combination of sofosbuvir, velpatasvir, and the NS3/4A protease inhibitor GS-9857 in treatment-naive or previously treated patients with hepatitis C virus genotype 1 or 3 infections. Gastroenterology. 2016;151(3):448–56.e1.
Babatin MA, Alghamdi AS, Albenmousa A, Alaseeri A, Aljarodi M, Albiladi H, et al. Efficacy and safety of simeprevir or daclatasvir in combination with sofosbuvir for the treatment of hepatitis C genotype 4 infection. J Clin Gastroenterol. 2018;52(5):452–7.
Coilly A, Fougerou-Leurent C, de Ledinghen V, Houssel-Debry P, Duvoux C, Di Martino V, ANRS C023 CUPILT study group, et al. Multicentre experience using daclatasvir and sofosbuvir to treat hepatitis C recurrence—the ANRS CUPILT study. J Hepatol. 2016;65(4):711–8.
Luetkemeyer AF, McDonald C, Ramgopal M, Noviello S, Bhore R, Ackerman P. 12 weeks of daclatasvir in combination with sofosbuvir for HIV-HCV coinfection (ALLY-2 study): efficacy and safety by HIV combination antiretroviral regimens. Clin Infect Dis. 2016;62(12):1489–96.
Mangia A, Arleo A, Copetti M, Miscio M, Piazzolla V, Santoro R, et al. The combination of daclatasvir and sofosbuvir for curing genotype 2 patients who cannot tolerate ribavirin. Liver Int. 2016;36(7):971–6.
Omar H, El Akel W, Elbaz T, El Kassas M, Elsaeed K, El Shazly H, et al. Generic daclatasvir plus sofosbuvir, with or without ribavirin, in treatment of chronic hepatitis C: real-world results from 18 378 patients in Egypt. Aliment Pharmacol Ther. 2018;47(3):421–31.
Wyles DL, Ruane PJ, Sulkowski MS, Dieterich D, Luetkemeyer A, Morgan TR, et al. Daclatasvir plus sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med. 2015;373(8):714–25.
Gane E, Poordad F, Wang S, Asatryan A, Kwo PY, Lalezari J, et al. High efficacy of ABT-493 and ABT-530 treatment in patients with HCV genotype 1 or 3 infection and compensated cirrhosis. Gastroenterology. 2016;151(4):651–659.e1.
Poordad F, Felizarta F, Asatryan A, Sulkowski MS, Reindollar RW, Landis CS, et al. Glecaprevir and pibrentasvir for 12 weeks for hepatitis C virus genotype 1 infection and prior direct-acting antiviral treatment. Hepatology. 2017;66(2):389–97.
Rockstroh JK, Lacombe K, Viani RM, Orkin C, Wyles D, Luetkemeyer AF, et al. Efficacy and safety of glecaprevir/pibrentasvir in patients coinfected with hepatitis C virus and human immunodeficiency virus type 1: the EXPEDITION-2 study. Clin Infect Dis. 2018;67(7):1010–7.
Lawitz E, Reau N, Hinestrosa F, Rabinovitz M, Schiff E, Sheikh A, et al. Efficacy of sofosbuvir, velpatasvir, and GS-9857 in patients with genotype 1 hepatitis C virus infection in an open-label, phase 2 trial. Gastroenterology. 2016;151(5):893–901.e1.
Wyles D, Weiland O, Yao B, Weilert F, Gordon F, Poordad F, et al. Retreatment of hepatitis C infection in patients who failed glecaprevir/pibrentasvir [abstract no. 127]. Conference on Retroviruses and Opportunistic Infections (CROI); 4–7 Mar 2018; Boston.
Li DK, Chung RT. Overview of direct-acting antiviral drugs and drug resistance of hepatitis C virus. Methods Mol Biol. 2019;1911:3–32.
George J, Burnevich E, Sheen IS, Heo J, Kinh NV, Tanwandee T, et al. Elbasvir/grazoprevir in Asia-Pacific/Russian participants with chronic hepatitis C virus genotype 1, 4, or 6 infection. Hepatol Commun. 2018;2(5):595–606.
Hezode C, Colombo M, Bourliere M, Spengler U, Ben-Ari Z, Strasser SI, et al. Elbasvir/grazoprevir for patients with hepatitis C virus infection and inherited blood disorders: a phase III study. Hepatology. 2017;66(3):736–45.
Sulkowski M, Hezode C, Gerstoft J, Vierling JM, Mallolas J, Pol S, et al. Efficacy and safety of 8 weeks versus 12 weeks of treatment with grazoprevir (MK-5172) and elbasvir (MK-8742) with or without ribavirin in patients with hepatitis C virus genotype 1 mono-infection and HIV/hepatitis C virus co-infection (C-WORTHY): a randomised, open-label phase 2 trial. Lancet. 2015;385(9973):1087–97.
Manns M, Samuel D, Gane EJ, Mutimer D, McCaughan G, Buti M, et al. Ledipasvir and sofosbuvir plus ribavirin in patients with genotype 1 or 4 hepatitis C virus infection and advanced liver disease: a multicentre, open-label, randomised, phase 2 trial. Lancet Infect Dis. 2016;16(6):685–97.
Naggie S, Cooper C, Saag M, Workowski K, Ruane P, Towner WJ, et al. Ledipasvir and sofosbuvir for HCV in patients coinfected with HIV-1. N Engl J Med. 2015;373(8):705–13.
Tam E, Luetkemeyer AF, Mantry PS, Satapathy SK, Ghali P, Kang M, et al. Ledipasvir/sofosbuvir for treatment of hepatitis C virus in sofosbuvir-experienced, NS5A treatment-naive patients: findings from two randomized trials. Liver Int. 2018;38(6):1010–21.
Rockstroh JK, Bhagani S, Hyland RH, Yun C, Dvory-Sobol H, Zheng W, et al. Ledipasvir-sofosbuvir for 6 weeks to treat acute hepatitis C virus genotype 1 or 4 infection in patients with HIV coinfection: an open-label, single-arm trial. Lancet Gastroenterol Hepatol. 2017;2(5):347–53.
Braun DL, Hampel B, Kouyos R, Nguyen H, Shah C, Flepp M, et al. High cure rates with grazoprevir-elbasvir with or without ribavirin guided by genotypic resistance testing among human immunodeficiency virus/hepatitis C virus-coinfected men who have sex with men. Clin Infect Dis. 2019;68(4):569–76.
Kumada H, Suzuki Y, Karino Y, Chayama K, Kawada N, Okanoue T, et al. The combination of elbasvir and grazoprevir for the treatment of chronic HCV infection in Japanese patients: a randomized phase II/III study. J Gastroenterol. 2017;52(4):520–33.
Sperl J, Horvath G, Halota W, Ruiz-Tapiador JA, Streinu-Cercel A, Jancoriene L, et al. Efficacy and safety of elbasvir/grazoprevir and sofosbuvir/pegylated interferon/ribavirin: a phase III randomized controlled trial. J Hepatol. 2016;65(6):1112–9.
Reau N, Kwo PY, Rhee S, Brown RS Jr, Agarwal K, Angus P, et al. Glecaprevir/pibrentasvir treatment in liver or kidney transplant patients with hepatitis C virus infection. Hepatology. 2018;68(4):1298–307.
Balistreri WF, Murray KF, Rosenthal P, Bansal S, Lin CH, Kersey K, et al. The safety and effectiveness of ledipasvir-sofosbuvir in adolescents 12–17 years old with hepatitis C virus genotype 1 infection. Hepatology. 2017;66(2):371–8.
Chuang WL, Chien RN, Peng CY, Chang TT, Lo GH, Sheen IS, et al. Ledipasvir/sofosbuvir fixed-dose combination tablet in Taiwanese patients with chronic genotype 1 hepatitis C virus. J Gastroenterol Hepatol. 2016;31(7):1323–9.
Murray KF, Balistreri WF, Bansal S, Whitworth S, Evans HM, Gonzalez-Peralta RP, et al. Safety and efficacy of ledipasvir-sofosbuvir with or without ribavirin for chronic hepatitis C in children ages 6–11. Hepatology. 2018;68(6):2158–66.
Walsh CE, Workowski K, Terrault NA, Sax PE, Cohen A, Bowlus CL, et al. Ledipasvir-sofosbuvir and sofosbuvir plus ribavirin in patients with chronic hepatitis C and bleeding disorders. Haemophilia. 2017;23(2):198–206.
Wei L, Xie Q, Hou JL, Tang H, Ning Q, Cheng J, et al. Ledipasvir/sofosbuvir for treatment-naive and treatment-experienced Chinese patients with genotype 1 HCV: an open-label, phase 3b study. Hepatol Int. 2018;12(2):126–32.
Isakov V, Gankina N, Morozov V, Kersey K, Lu S, Osinusi A, et al. Ledipasvir-sofosbuvir for 8 weeks in non-cirrhotic patients with previously untreated genotype 1 HCV infection ± HIV-1 co-infection. Clin Drug Investig. 2018;38(3):239–47.
Feld JJ, Ramji A, Shafran SD, Willems B, Marotta P, Huchet E, et al. Ledipasvir-sofosbuvir plus ribavirin in treatment-naive patients with hepatitis C virus genotype 3 infection: an open-label study. Clin Infect Dis. 2017;65(1):13–9.
Smolders EJ, de Kanter CT, van Hoek B, Arends JE, Drenth JP, Burger DM. Pharmacokinetics, efficacy, and safety of hepatitis C virus drugs in patients with liver and/or renal impairment. Drug Saf. 2016;39(7):589–611.
Gandhi Y, Eley T, Fura A, Li W, Bertz RJ, Garimella T. Daclatasvir: a review of preclinical and clinical pharmacokinetics. Clin Pharmacokinet. 2018;57(8):911–28.
German P, Mathias A, Brainard D, Kearney BP. Clinical pharmacokinetics and pharmacodynamics of ledipasvir/sofosbuvir, a fixed-dose combination tablet for the treatment of hepatitis C. Clin Pharmacokinet. 2016;55(11):1337–51.
Bourliere M, Bronowicki JP, de Ledinghen V, Hezode C, Zoulim F, Mathurin P, et al. Ledipasvir-sofosbuvir with or without ribavirin to treat patients with HCV genotype 1 infection and cirrhosis non-responsive to previous protease-inhibitor therapy: a randomised, double-blind, phase 2 trial (SIRIUS). Lancet Infect Dis. 2015;15(4):397–404.
Reddy KR, Bourliere M, Sulkowski M, Omata M, Zeuzem S, Feld JJ, et al. Ledipasvir and sofosbuvir in patients with genotype 1 hepatitis C virus infection and compensated cirrhosis: an integrated safety and efficacy analysis. Hepatology. 2015;62(1):79–86.
Sanai FM, Altraif IH, Alswat K, AlZanbagi A, Babatin MA, AlMousa A, et al. Real life efficacy of ledipasvir/sofosbuvir in hepatitis C genotype 4-infected patients with advanced liver fibrosis and decompensated cirrhosis. J Infect. 2018;76(6):536–42.
Lim JK, Liapakis AM, Shiffman ML, Lok AS, Zeuzem S, Terrault NA, et al. Safety and effectiveness of ledipasvir and sofosbuvir, with or without ribavirin, in treatment-experienced patients with genotype 1 hepatitis C virus infection and cirrhosis. Clin Gastroenterol Hepatol. 2018;16(11):1811–1819.e4.
Abaalkhail F, Elsiesy H, Elbeshbeshy H, Shawkat M, Yousif S, Ullah W, et al. Treatment of patients with hepatitis C virus infection with ledipasvir-sofosbuvir in the liver transplant setting. Transplantation. 2017;101(11):2739–45.
Modi AA, Nazario HE, Gonzales GR, Gonzalez SA. Safety and efficacy of ledipasvir/sofosbuvir with or without ribavirin in hepatitis C genotype 1 patients including those with decompensated cirrhosis who failed prior treatment with simeprevir/sofosbuvir. Aliment Pharmacol Ther. 2018;47(10):1409–15.
Marshall WL, Feng HP, Wenning L, Garrett G, Huang X, Liu F, et al. Pharmacokinetics, safety, and tolerability of single-dose elbasvir in participants with hepatic impairment. Eur J Drug Metab Pharmacokinet. 2018;43(3):321–9.
Roth D, Nelson DR, Bruchfeld A, Liapakis A, Silva M, Monsour H Jr, et al. Grazoprevir plus elbasvir in treatment-naive and treatment-experienced patients with hepatitis C virus genotype 1 infection and stage 4–5 chronic kidney disease (the C-SURFER study): a combination phase 3 study. Lancet. 2015;386(10003):1537–45.
Zeuzem S, Ghalib R, Reddy KR, Pockros PJ, Ben Ari Z, Zhao Y, et al. Grazoprevir-elbasvir combination therapy for treatment-naive cirrhotic and noncirrhotic patients with chronic hepatitis C virus genotype 1, 4, or 6 infection: a randomized trial. Ann Intern Med. 2015;163(1):1–13.
Mogalian E, Mathias A, Brainard D, McNally J, Moorehead L, Hernandez M, et al. The pharmacokinetics of GS-5816, a pan-genotypic HCV NS5A inhibitor, in HCV-uninfected subjects with moderate and severe hepatic impairment. J Hepatol. 2014;60(1):S317.
Mogalian E, Brainard DM, Osinusi A, Moorehead L, Murray B, Ling KHJ, et al. Pharmacokinetics and safety of velpatasvir and sofosbuvir/velpatasvir in subjects with hepatic impairment. Clin Pharmacokinet. 2018;57(11):1449–57.
Kosloski MP, Wang H, Pugatch D, Mensa FJ, Gane E, Lawitz E, et al. Pharmacokinetics and safety of glecaprevir and pibrentasvir in HCV-negative subjects with hepatic impairment. Eur J Clin Pharmacol. 2019;75(2):217–26.
Chayama K, Suzuki F, Karino Y, Kawakami Y, Sato K, Atarashi T, et al. Efficacy and safety of glecaprevir/pibrentasvir in Japanese patients with chronic genotype 1 hepatitis C virus infection with and without cirrhosis. J Gastroenterol. 2018;53(4):557–65.
Bourliere M, Gordon SC, Schiff ER, Tran TT, Ravendhran N, Landis CS, et al. Deferred treatment with sofosbuvir-velpatasvir-voxilaprevir for patients with chronic hepatitis C virus who were previously treated with an NS5A inhibitor: an open-label substudy of POLARIS-1. Lancet Gastroenterol Hepatol. 2018;3(8):559–65.
Xia Y, Friedmann P, Yaffe H, Phair J, Gupta A, Kayler LK. Effect of HCV, HIV and coinfection in kidney transplant recipients: mate kidney analyses. Am J Transpl. 2014;14(9):2037–47.
Colombo M, Aghemo A, Liu H, Zhang J, Dvory-Sobol H, Hyland R, et al. Treatment with ledipasvir-sofosbuvir for 12 or 24 weeks in kidney transplant recipients with chronic hepatitis C virus genotype 1 or 4 infection: a randomized trial. Ann Intern Med. 2017;166(2):109–17.
Morales AL, Liriano-Ward L, Tierney A, Sang M, Lalos A, Hassan M, et al. Ledipasvir/sofosbuvir is effective and well tolerated in postkidney transplant patients with chronic hepatitis C virus. Clin Transpl. 2017. https://doi.org/10.1111/ctr.12941.
Kamar N, Marion O, Rostaing L, Cointault O, Ribes D, Lavayssiere L, et al. Efficacy and safety of sofosbuvir-based antiviral therapy to treat hepatitis C virus infection after kidney transplantation. Am J Transpl. 2016;16(5):1474–9.
Saxena V, Khungar V, Verna EC, Levitsky J, Brown RS Jr, Hassan MA, et al. Safety and efficacy of current direct-acting antiviral regimens in kidney and liver transplant recipients with hepatitis C: results from the HCV-TARGET study. Hepatology. 2017;66(4):1090–101.
Fernandez I, Munoz-Gomez R, Pascasio JM, Baliellas C, Polanco N, Esforzado N, et al. Efficacy and tolerability of interferon-free antiviral therapy in kidney transplant recipients with chronic hepatitis C. J Hepatol. 2017;66(4):718–23.
Lionetti R, Calvaruso V, Piccolo P, Mancusi RL, Mazzarelli C, Fagiuoli S, et al. Sofosbuvir plus daclatasvir with or without ribavirin is safe and effective for post-transplant hepatitis C recurrence and severe fibrosis and cirrhosis: a prospective study. Clin Transpl. 2018;32(2):e13165.
Atsukawa M, Tsubota A, Toyoda H, Takaguchi K, Kondo C, Okubo T, et al. Efficacy and safety of elbasvir/grazoprevir for Japanese patients with genotype 1b chronic hepatitis C complicated by chronic kidney disease, including those undergoing hemodialysis: a post hoc analysis of a multicenter study. J Gastroenterol Hepatol. 2019;34(2):364–9.
Suda G, Kurosaki M, Itakura J, Izumi N, Uchida Y, Mochida S, et al. Safety and efficacy of elbasvir and grazoprevir in Japanese hemodialysis patients with genotype 1b hepatitis C virus infection. J Gastroenterol. 2019;54(1):78–86.
Alric L, Ollivier-Hourmand I, Berard E, Hillaire S, Guillaume M, Vallet-Pichard A, et al. Grazoprevir plus elbasvir in HCV genotype-1 or -4 infected patients with stage 4/5 severe chronic kidney disease is safe and effective. Kidney Int. 2018;94(1):206–13.
Kosloski M, Dutta S, Zhao W, Pugatch D, Mensa FJ, Kort J, et al. Pharmacokinetics, safety, and tolerability of next generation direct acting antivirals ABT-493 and ABT-530 in subjects with renal impairment. The International Liver Congress—EASL; 13–17 Apr 2016; Barcelona. J Hepatol 64(2):S405–6. https://www.journal-of-hepatology.eu/article/S0168-8278(16)00643-7/abstract.
Lawitz E, Landis C, Maliakkal B, Bonacini M, Ortiz-Lasanta G, Zhang J, et al. Safety and efficacy of treatment with once-daily ledipasvir/sofosbuvir (90/400 mg) for 12 weeks in genotype 1 HCV-infected patients with severe renal impairment [abstract no. 1587]. AASLD: The Liver Meeting; 20–24 Oct 2017. Washington, DC. http://www.natap.org/2017/AASLD/AASLD_09.htm. Accessed 9 May 2019.
Gane E, Robson RA, Bonacini M, Maliakkal B, Kirby BJ, Liu LJ, et al. Safety, antiviral efficacy, and pharmacokinetics of sofosbuvir in patients with severe renal impairment [abstract no. 966]. In: 65th Annual Meeting of the American Association for the Study of Liver Diseases; 7 Nov 2014. Boston. http://www.natap.org/2014/AASLD/AASLD_85.htm. Accessed 9 May 2019.
Martin P, Gane EJ, Ortiz-Lasanta G, Liu L, Sajwani K, Kirby B, et al. Safety and efficacy of treatment with daily sofosbuvir 400 mg + ribavirin 200 mg for 24 weeks in genotype 1 and 3 HCV-infected patients with severe renal impairment [abstract no. 1128]. In: 66th Annual Meeting of the American Association for the Study of Liver Diseases; 13–17 Nov 2015. Boston. http://www.natap.org/2015/AASLD/AASLD_134.htm. Accessed 9 May 2019.
Surendra M, Raju SB, Sridhar N, Vijay Kiran B, Rajesh G, Anvesh G, et al. Ledipasvir and Sofosbuvir for untreated HCV genotype 1 infection in end stage renal disease patients: a prospective observational study. Hemodial Int. 2018;22(2):217–21.
Borgia S, Dearden J, Lurie Y, Shafran SD, Brown A, Hyland R, et al. Sofosbuvir/velpatasvir for 12 weeks is safe and effective in patients undergoing dialysis. AASLD: The Liver Meeting; 9–13 Nov 2018. San Francisco.
Rockstroh JK, Nelson M, Katlama C, Lalezari J, Mallolas J, Bloch M, et al. Efficacy and safety of grazoprevir (MK-5172) and elbasvir (MK-8742) in patients with hepatitis C virus and HIV co-infection (C-EDGE CO-INFECTION): a non-randomised, open-label trial. Lancet HIV. 2015;2(8):e319–27.
Rosenthal E, Fougerou-Leurent C, Renault A, Carrieri MP, Marcellin F, Garraffo R, et al. Efficacy, safety and patient-reported outcomes of ledipasvir/sofosbuvir in NS3/4A protease inhibitor-experienced individuals with hepatitis C virus genotype 1 and HIV coinfection with and without cirrhosis (ANRS HC31 SOFTRIH study). HIV Med. 2018;19(3):227–37.
Lacombe K, Fontaine H, Dhiver C, Metivier S, Rosenthal E, Antonini T, et al. Real-world efficacy of daclatasvir and sofosbuvir, with and without ribavirin, in HIV/HCV coinfected patients with advanced liver disease in a French early access cohort. J Acquir Immune Defic Syndr. 2017;75(1):97–107.
Wang C, Ji D, Chen J, Shao Q, Li B, Liu J, et al. Hepatitis due to reactivation of hepatitis B virus in endemic areas among patients with hepatitis C treated with direct-acting antiviral agents. Clin Gastroenterol Hepatol. 2017;15(1):132–6.
Terrault NA, Lok ASF, McMahon BJ, Chang KM, Hwang JP, Jonas MM, et al. Update on prevention, diagnosis, and treatment of chronic hepatitis B: AASLD 2018 hepatitis B guidance. Hepatology. 2018;67(4):1560–99.
Stedman CAM, Hyland RH, Ding X, Pang PS, McHutchison JG, Gane EJ. Once daily ledipasvir/sofosbuvir fixed-dose combination with ribavirin in patients with inherited bleeding disorders and hepatitis C genotype 1 infection. Haemophilia. 2016;22(2):214–7.
Mehta R, Kabrawala M, Nandwani S, Desai P, Bhayani V, Patel S, et al. Safety and efficacy of sofosbuvir and daclatasvir for hepatitis C virus infection in patients with beta-thalassemia major. J Clin Exp Hepatol. 2018;8(1):3–6.
Nagao A, Hanabusa H. Brief report: the impact of ledipasvir/sofosbuvir on HIV-positive and HIV-negative Japanese hemophilia patients with 1, 4, and mixed-genotype HCV. J Acquir Immune Defic Syndr. 2017;74(4):418–22.
Mangia A, Sarli R, Gamberini R, Piga A, Cenderello G, Piazzolla V, et al. Randomised clinical trial: sofosbuvir and ledipasvir in patients with transfusion-dependent thalassaemia and HCV genotype 1 or 4 infection. Aliment Pharmacol Ther. 2017;46(4):424–31.
Moon J, Hyland RH, Zhang F, Brainard DM, Lanzkron S, McHutchison JG, et al. Efficacy and safety of ledipasvir/sofosbuvir for the treatment of chronic hepatitis C in persons with sickle cell disease. Clin Infect Dis. 2017;65(5):864–6.
American Association for the Study of Liver Diseases (AASLD), Infectious Diseases Society of America (IDSA). HCV guidance: recommendations for testing, managing, and treating hepatitis C. 2018. https://www.hcvguidelines.org/sites/default/files/full-guidance-pdf/HCVGuidance_May_24_2018b.pdf. Accessed 19 Mar 2019.
Squires JE, Balistreri WF. Hepatitis C virus infection in children and adolescents. Hepatol Commun. 2017;1(2):87–98.
Bernstein HB, Dunkelberg JC, Leslie KK. Hepatitis C in pregnancy in the era of direct-acting antiviral treatment: potential benefits of universal screening and antepartum therapy. Clin Obstet Gynecol. 2018;61(1):146–56.
El-Karaksy H, Mogahed EA, Abdullatif H, Ghobrial C, El-Raziky MS, El-Koofy N, et al. Sustained viral response in genotype 4 chronic hepatitis C virus-infected children and adolescents treated with sofosbuvir/ledipasvir. J Pediatr Gastroenterol Nutr. 2018;67(5):626–30.
Yakoot M, El-Shabrawi MH, AbdElgawad MM, Mahfouz AA, Helmy S, Abdo AM, et al. Dual sofosbuvir/daclatasvir therapy in adolescent patients with chronic hepatitis C infection. J Pediatr Gastroenterol Nutr. 2018;67(1):86–9.
Abdel Ghaffar TY, El Naghi S, Abdel Gawad M, Helmy S, Abdel Ghaffar A, Yousef M, et al. Safety and efficacy of combined sofosbuvir/daclatasvir treatment of children and adolescents with chronic hepatitis C genotype 4. J Viral Hepat. 2019;26(2):263–70.
Gane EJ, Stedman CA, Hyland RH, Ding X, Svarovskaia E, Subramanian GM, et al. Efficacy of nucleotide polymerase inhibitor sofosbuvir plus the NS5A inhibitor ledipasvir or the NS5B non-nucleoside inhibitor GS-9669 against HCV genotype 1 infection. Gastroenterology. 2014;146(3):736–743.e1.
Grebely J, Mauss S, Brown A, Bronowicki JP, Puoti M, Wyles D, et al. Efficacy and safety of ledipasvir/sofosbuvir with and without ribavirin in patients with chronic HCV genotype 1 infection receiving opioid substitution therapy: analysis of phase 3 ION trials. Clin Infect Dis. 2016;63(11):1405–11.
Johnson SW, Ammirati SR, Hartis CE, Weber SF, Morgan MR, Darnell TA, et al. Effectiveness of ledipasvir/sofosbuvir in real-world patients with chronic hepatitis C: a collaborative treatment approach. Int J Antimicrob Agents. 2017;49(6):778–81.
Lawitz E, Poordad FF, Pang PS, Hyland RH, Ding X, Mo H, et al. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet. 2014;383(9916):515–23.
Suda G, Ogawa K, Yamamoto Y, Katagiri M, Furuya K, Kumagai K, et al. Retreatment with sofosbuvir, ledipasvir, and add-on ribavirin for patients who failed daclatasvir and asunaprevir combination therapy. J Gastroenterol. 2017;52(10):1122–9.
Wyles D, Pockros P, Morelli G, Younes Z, Svarovskaia E, Yang JC, et al. Ledipasvir-sofosbuvir plus ribavirin for patients with genotype 1 hepatitis C virus previously treated in clinical trials of sofosbuvir regimens. Hepatology. 2015;61(6):1793–7.
Kowdley KV, Gordon SC, Reddy KR, Rossaro L, Bernstein DE, Lawitz E, et al. Ledipasvir and sofosbuvir for 8 or 12 weeks for chronic HCV without cirrhosis. N Engl J Med. 2014;370(20):1879–88.
Zeng QL, Xu GH, Zhang JY, Li W, Zhang DW, Li ZQ, et al. Generic ledipasvir-sofosbuvir for patients with chronic hepatitis C: a real-life observational study. J Hepatol. 2017;66(6):1123–9.
Gane EJ, Hyland RH, Yang Y, Svarovskaia E, Stamm LM, Brainard DM, et al. Efficacy of ledipasvir plus sofosbuvir for 8 or 12 weeks in patients with hepatitis C virus genotype 2 infection. Gastroenterology. 2017;152(6):1366–71.
Brown A, Hezode C, Zuckerman E, Foster GR, Zekry A, Roberts SK, et al. Efficacy and safety of 12 weeks of elbasvir ± grazoprevir ± ribavirin in participants with hepatitis C virus genotype 2, 4, 5 or 6 infection: the C-SCAPE study. J Viral Hepat. 2018;25(5):457–64.
Ahmed OA, Safwat E, Khalifa MO, Elshafie AI, Fouad MHA, Salama MM, et al. Sofosbuvir plus daclatasvir in treatment of chronic hepatitis C genotype 4 infection in a cohort of Egyptian patients: an experiment the size of Egyptian village. Int J Hepatol. 2018;2018:9616234.
Ahmed OA, Kaisar HH, Badawi R, Hawash N, Samir H, Shabana SS, et al. Efficacy and safety of sofosbuvir-ledipasvir for treatment of a cohort of Egyptian patients with chronic hepatitis C genotype 4 infection. Infect Drug Resist. 2018;11:295–8.
Shiha G, Esmat G, Hassany M, Soliman R, Elbasiony M, Fouad R, et al. Ledipasvir/sofosbuvir with or without ribavirin for 8 or 12 weeks for the treatment of HCV genotype 4 infection: results from a randomised phase III study in Egypt. Gut. 2019;68(4):721–8. https://doi.org/10.1136/gutjnl-2017-315906.
Elbaz T, Abdo M, Omar H, Hassan EA, Zaghloul AM, Abdel-Samiee M, et al. Efficacy and safety of sofosbuvir and daclatasvir with or without ribavirin in elderly patients with chronic hepatitis C virus infection. J Med Virol. 2019;91(2):272–7.
Grebely J, Dalgard O, Conway B, Cunningham EB, Bruggmann P, Hajarizadeh B, et al. Sofosbuvir and velpatasvir for hepatitis C virus infection in people with recent injection drug use (SIMPLIFY): an open-label, single-arm, phase 4, multicentre trial. Lancet Gastroenterol Hepatol. 2018;3(3):153–61.
Poordad F, Pol S, Asatryan A, Buti M, Shaw D, Hezode C, et al. Glecaprevir/pibrentasvir in patients with hepatitis C virus genotype 1 or 4 and past direct-acting antiviral treatment failure. Hepatology. 2018;67(4):1253–60.
Pott-Junior H, Bricks G, Grandi G, Figueiredo Senise J, Castelo Filho A. Sofosbuvir in combination with daclatasvir or simeprevir for 12 weeks in noncirrhotic subjects chronically infected with hepatitis C virus genotype 1: a randomized clinical trial. Clin Microbiol Infect. 2019;25(3):365–71.
Chu X, Liao M, Shen H, Yoshida K, Zur AA, Arya V, et al. Clinical probes and endogenous biomarkers as substrates for transporter drug–drug interaction evaluation: perspectives from the international transporter consortium. Clin Pharmacol Ther. 2018;104(5):836–64.
Acknowledgments
We thank Jolien Freriksen and Minou van Seyen, both Ph.D. candidates at the Radboud University Medical Center, for providing information about HCV treatment in pregnant women.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
EJS declares having received travel grants from Abbvie and Gilead. AMEJ and PGJTH declare no conflicts of interest that are directly relevant to the content of this review. DMB is on the advisory boards for Gilead and Merck and has received sponsorship/research grants from BMS, Gilead, Janssen, Merck, and ViiV. DJB is on advisory boards for Abbvie, Gilead, and Merck and has received research support/educational grants from Abbvie, Gilead, Janssen, and Merck. JKR has received honoraria for consulting or speaking at educational events from Abbvie, Abivax, Gilead, Janssen, Siemens, Merck, and ViiV. These conflicts of interest did not influence the preparation of this review.
Funding
No funding was used in the preparation of this review.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Smolders, E.J., Jansen, A.M.E., ter Horst, P.G.J. et al. Viral Hepatitis C Therapy: Pharmacokinetic and Pharmacodynamic Considerations: A 2019 Update. Clin Pharmacokinet 58, 1237–1263 (2019). https://doi.org/10.1007/s40262-019-00774-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00774-0