
Abstract
Background
HLX04 is a proposed biosimilar of bevacizumab.
Objective
This phase III study aimed to evaluate the efficacy, safety, and immunogenicity of HLX04 compared with reference bevacizumab in combination with XELOX or mFOLFOX6 as first-line treatment for recurrent/metastatic colorectal cancer (CRC).
Methods
In this double-blind, parallel-group study, patients were randomized 1:1 to receive HLX04 or bevacizumab (7.5 mg/kg every 3 weeks when combined with XELOX; 5 mg/kg every 2 weeks when combined with mFOLFOX6). The primary endpoint was progression-free survival rate at week 36 (PFSR36w) per Response Evaluation Criteria in Solid Tumors (RECIST v1.1). Prespecified equivalence margins of PFSR36w were set as − 11 to 15% (rate difference) and 0.8 to 1.25 (rate ratio). Secondary endpoints included efficacy, safety, immunogenicity, and pharmacokinetics.
Results
A total of 677 patients were randomized (HLX04 n = 340; bevacizumab n = 337) between April 2018 and April 2020. PFSR36w was 46.4% (95% confidence interval [CI] 41.1–51.8) with HLX04 and 50.7% (95% CI 45.4–56.1) with bevacizumab. The rate difference (− 4.2%; 90% CI − 10.6 to 2.1) and rate ratio (0.92; 90% CI 0.80–1.05) both fell within the prespecified equivalence margins. No notable differences were observed between treatment groups in any efficacy endpoints or their subgroup analyses. Safety, immunogenicity, and pharmacokinetic profiles were comparable between the two treatment groups.
Conclusions
HLX04 demonstrated equivalent efficacy with similar safety and immunogenicity profiles to reference bevacizumab among patients with recurrent/metastatic CRC, thus offering an alternative treatment option to patients.
Trial registration
Chinadrugtrials.org.cn, CTR20171503 (18 March 2018); ClinicalTrials.gov, NCT03511963 (30 April 2018).
Plain Language Summary
Colorectal cancer (CRC) is the third most common cancer worldwide. Approximately 20% of patients with CRC have metastases at their first visit. Bevacizumab is a biologic antibody approved in many countries for the treatment of metastatic CRC. However, high treatment costs significantly limit patient access to bevacizumab. Therefore, HLX04, a potential bevacizumab biosimilar, which is almost identical to bevacizumab but less expensive and more accessible, has been developed. This randomized clinical trial was designed to evaluate the efficacy (ability of a drug to produce the desired treatment effects), safety, and immunogenicity (ability of a drug to induce immune response that would affect its efficacy and safety) of HLX04 compared with the reference bevacizumab in patients with recurrent/metastatic CRC. Efficacy of the tested drug was evaluated by comparing the proportion of patients without disease progression or death at week 36 (PFSR36w). Safety was monitored using adverse events and other clinical evaluations. Immunogenicity was assessed by the incidence of antidrug antibodies. Of the 677 patients enrolled in the study, 340 received HLX04 and 337 received bevacizumab. Statistical analyses showed that HLX04 was equivalent to bevacizumab in efficacy evaluations (the difference in PFSR36w between the two treatment groups fell within the prespecified “equivalence margins”). Moreover, the two treatments were similar with respect to safety and immunogenicity evaluations. In summary, patients responded equally well to HLX04 and bevacizumab, supporting the development of HLX04 as a proposed biosimilar to bevacizumab for patients with recurrent/metastatic CRC.
Similar content being viewed by others


This phase III equivalence study was designed to determine the clinical equivalence between HLX04, a potential bevacizumab biosimilar, and its reference bevacizumab in patients with recurrent or metastatic colorectal cancer. |
No significant differences were observed between HLX04 and bevacizumab in any study endpoints, including efficacy, safety, immunogenicity, and pharmacokinetics. |
HLX04 provides an alternative treatment option for patients with recurrent/metastatic colorectal cancer as a biosimilar candidate to bevacizumab. |
1 Introduction
Colorectal cancer (CRC), accounting for ~ 10% of cancer-related mortality worldwide, is the third most common cancer, with an estimated 1.8 million new cases globally in 2018 [1]. In China, there were 376,000 new cases of CRC and 191,000 deaths in 2018, ranking it as the third most common cancer and the fifth leading cause of cancer-related death [2]. Approximately 20% of patients with CRC will present with metastasis at initial diagnosis [3]. The current 5‐year survival rate for metastatic CRC (mCRC) is ~ 10% [3,4,5]. Novel biologic therapies targeting either vascular endothelial growth factor (VEGF) or epidermal growth factor receptor have improved clinical outcomes, doubling the overall survival (OS) to ~ 30 months in 20 years [6,7,8].
Bevacizumab (Avastin®; Roche Pharma [Schweiz] Ltd.) is a recombinant humanized monoclonal antibody against VEGF-A, preventing binding of all isoforms of VEGF-A to its receptor, VEGFR, on the surface of endothelial cells, thereby inhibiting VEGFR-mediated endothelial cell proliferation and angiogenesis [9, 10]. Bevacizumab in combination with backbone chemotherapy has been approved in Europe, the USA, and China for the first-line treatment of mCRC [11,12,13,14] and has been recommended by the European Society for Medical Oncology, National Comprehensive Cancer Network, and National Health Commission of the People’s Republic of China as the standard of care in the first-line treatment of mCRC [2, 8, 15].
However, access to bevacizumab is suboptimal, with treatment costs acting as the main barrier [6, 16]. Biosimilars, which are highly similar to the licensed reference biologics in efficacy and safety, have the potential to increase patients’ accessibility. HLX04 (Shanghai Henlius Biotech, Inc.), a proposed bevacizumab biosimilar, has demonstrated comparable pharmacokinetics, safety, and immunogenicity to bevacizumab in a phase I study in Chinese healthy male volunteers [17].
Here, we report the results of a phase III equivalence study comparing the efficacy, safety, and immunogenicity of HLX04 with those of reference bevacizumab in patients with mCRC. The primary objective of this trial was to demonstrate the equivalence in efficacy of HLX04 and reference bevacizumab in combination with chemotherapy (XELOX [capecitabine plus oxaliplatin] or modified FOLFOX6 [mFOLFOX6: 5-fluorouracil, leucovorin, and oxaliplatin) as first-line treatment for recurrent CRC/mCRC. Secondary objectives were to compare the safety, immunogenicity, and pharmacokinetics of HLX04 with reference bevacizumab.
2 Methods
2.1 Study Design and Treatment
This randomized, double-blind, multicenter, parallel-group, phase III clinical trial was conducted from 13 April 2018 to 15 April 2020 at 63 centers in China (Table S1 in the electronic supplementary material [ESM]) [18]. Eligible patients were randomized 1:1 to receive HLX04 or bevacizumab (China-sourced and European Union-sourced) in combination with mFOLFOX6 or XELOX.
HLX04 or bevacizumab was administered intravenously before oxaliplatin (7.5 mg/kg 3-week cycle when given with XELOX or 5 mg/kg 2-week cycle when given with mFOLFOX6). XELOX consisted of an intravenous infusion of oxaliplatin 130 mg/m2 on day 1 followed by oral capecitabine 1000 mg/m2 twice daily on days 1 through 14 of a 3-week cycle [9]. mFOLFOX6 consisted of an intravenous infusion of oxaliplatin 85 mg/m2 followed by leucovorin 400 mg/m2, and an intravenous bolus of 5-fluorouracil 400 mg/m2 on day 1 with subsequent 2400 mg/m2 × 46 h continuous intravenous infusion [19]. HLX04 or bevacizumab dose adjustments due to toxicity were not permitted; if necessary, treatment could be withheld for up to 6 weeks or discontinued. Treatment continued until disease progression (PD), unacceptable toxicity, or the patient becoming eligible for curative resection or radiotherapy, whichever occurred first.
The study was conducted in accordance with the International Council for Harmonisation guidelines on Good Clinical Practice and all applicable regulatory requirements, including the Declaration of Helsinki, and was approved by the ethics committees at each participating center. Each patient provided written, informed consent prior to enrollment in the study.
2.2 Patients
Patients aged 18‒75 years were eligible for inclusion in the study if they had histologically or cytologically confirmed colorectal adenocarcinoma with recurrent/metastatic lesions that could not be cured by surgery, with an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 or 1, a life expectancy of longer than 6 months, and at least one measurable lesion according to Response Evaluation Criteria in Solid Tumors version 1.1 (RECIST v1.1) within 4 weeks before randomization. Key exclusion criteria included prior adjuvant therapy containing targeted drugs for mCRC; cerebral and/or leptomeningeal metastasis; bleeding tendency, at high risk of bleeding, or coagulation dysfunction; and poorly controlled hypertension. Additional details of participant eligibility criteria are provided in Table S2 in the ESM.
2.3 Randomization and Masking
Stratified dynamic randomization was performed using an interactive web response system. Randomization was stratified with respect to three planned stratification factors: ECOG PS score (0 vs. 1), chemotherapy regimen (mFOLFOX6 vs. XELOX), and primary tumor site (left vs. right colon). Investigators, patients, the sponsor, and the contract research organization were blinded to the randomized treatment assignment throughout the study. An independent data monitoring committee regularly reviewed safety data and the interim analysis.
2.4 Study Endpoints and Clinical Assessments
The primary efficacy endpoint was progression-free survival rate at week 36 (PFSR36w), defined as the proportion of patients without PD (per RECIST v1.1) or death within 36 weeks from the date of randomization. Tumor assessments (computed tomography, magnetic resonance imaging) were conducted within 28 days before the first dose and repeated thereafter every 6 weeks until treatment discontinued. Review of these radiographic images was performed by a central, independent, blinded Central Images Reading (CIR) group or investigators. Secondary efficacy endpoints included 48-week best objective response rate (BORR); objective response rate (ORR) at weeks 6, 12, 18, 24, 30, 36, 42, and 48; 12-month overall survival rate (OSR); progression-free survival (PFS); time to response (TTR; time from randomization to overall response of complete response [CR] or partial response [PR]); and duration of response (DOR; time between first documentation of CR or PR and the date of PD or death, whichever came first). Analyses of efficacy endpoints were further stratified by chemotherapy, primary tumor site, ECOG PS, age, sex, and KRAS/BRAF mutation status. Efficacy was evaluated for up to 1 year after inclusion of the last patient.
Safety was assessed throughout the study by means of vital signs, physical examinations, laboratory tests, electrocardiograms, ECOG PS, and drug exposure, along with incidence and severity of adverse events (AEs), serious AEs (SAEs), and predefined AEs of special interest (AESIs). AEs were graded according to National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03 (NCI CTCAE v4.03). AESIs for HLX04 and bevacizumab included gastrointestinal perforation, fistula involving internal organ, bleedings, thromboembolic events, wound healing complications, proteinuria, nephrotic syndrome, hypertension, and infusion-related adverse reactions.
Serum samples for assessment of immunogenicity were collected prior to drug administration on day 1 of dosing cycle 1, cycle 2, weeks 6, 18, 36, 48, and 4 weeks after the last dose of study drug. Antidrug antibodies (ADAs) were measured by a validated, semi-quantitative bridging electrochemiluminescence immunoassay (ECLIA) on Meso Scale Discovery (MSD) platform (Meso Scale Diagnostics, LLC.; Rockville, MD, USA). Only samples with positive ADA results were further assessed for neutralizing antibodies (NAbs) by a validated, semi-quantitative bridging competitive ligand binding ECLIA on MSD platform.
For pharmacokinetic evaluations, serum drug concentrations were measured in samples collected before dosing on day 1 of cycle 1, cycle 2, weeks 18, 36, 48, and 4 weeks after the last dose, as well as after dosing on day 1 of cycle 1 and week 18. Concentrations were determined by a validated, quantitative enzyme-linked immunosorbent assay. Pharmacokinetic analysis was performed using Phoenix WinNonlin®, version 7.0 (Pharsight Corporation; CA, USA).
Immunogenicity and pharmacokinetic analyses were carried out at a central lab (WuXi AppTec Co., Ltd.; Shanghai, China).
2.5 Statistical Analysis
Based on a previous phase III study of bevacizumab in mCRC [9], a sample size of 638 was estimated to provide 80% power by two one-sided tests (α = 0.05) to demonstrate therapeutic equivalence between HLX04 and bevacizumab on PFSR36w, assuming PFSR36w of 55% with rate ratio equivalence margins of 0.8–1.25 and a 10% dropout rate. Therapeutic equivalence was demonstrated by comparing the two-sided 90% confidence interval (CI) of the PFSR36w rate ratio and rate difference between HLX04 and bevacizumab with the prespecified equivalence margins (rate ratio 0.8–1.25; rate difference − 11 to 15%) via a Cochran–Mantel–Haenszel (CMH) test with three planned stratification factors. A sensitivity analysis for the primary endpoint was conducted by comparing the rate ratio and rate difference of PFS rate (PFSR) at 36, 42, and 48 weeks and 9, 10, 11, and 12 months, excluding patients eligible for curative resection or radiotherapy.
All efficacy analyses were performed in both the full analysis set (FAS; all randomized patients receiving at least one dose of HLX04 or bevacizumab, following the intention-to-treat principle) and the per protocol set (PPS; a subset of FAS that consisted of all randomized patients who received at least two doses of study drug, had at least one valid tumor assessment result, and did not experience a protocol deviation that affected their evaluation of the primary efficacy endpoint). Safety and immunogenicity were assessed in the safety set (SS), which comprised all randomized patients who were treated with the study drug and received a safety assessment. The pharmacokinetic set (PKS) included all randomized subjects who were treated with the study drug and had at least one pharmacokinetic parameter assessment and did not experience a major protocol violation that affected the evaluation of pharmacokinetics.
Wald asymptotic confidence limits were used to calculate ORR 95% CI. A CMH test stratified by three planned factors was used to detect the ORR difference (two-sided α = 0.05). For OSR, PFS, TTR, and DOR, a Cox proportional-hazards model stratified by three planned factors was used to estimate the hazard ratio (HR) with 95% CI. Curves and medians of OSR, PFS, TTR, and DOR were estimated using the Kaplan–Meier method, and comparisons of curves between the treatment groups were performed using the stratified log-rank test (p < 0.05 was considered statistically significant). Baseline demographic and clinical data were summarized using descriptive statistics. Number of observations, mean, median, standard deviation (SD), minimum, and maximum were calculated for continuous variables; number and percentage of patients in each category were listed for categorical variables.
All statistical analyses were calculated using SAS® software, version 9.4 (SAS Institute; Cary, NC, USA).
3 Results
3.1 Patients and Exposure
In total, 873 patients were screened, of whom 677 were randomized to the HLX04 group (n = 340) or the bevacizumab group (n = 337). A patient not receiving HLX04 after randomization was excluded from the FAS, PKS, and SS in the HLX04 group. Another patient diagnosed with primary liver cancer was excluded from the FAS in the HLX04 group. Thus, 676 patients (HLX04, n = 339; bevacizumab, n = 337) were included in the PKS and SS; 675 patients (HLX04, n = 338; bevacizumab, n = 337) were included in the FAS (Fig. 1). In total, 650 patients (HLX04, n = 322; bevacizumab, n = 328) were included in the PPS. Primary reasons for exclusion from the PPS were lack of valid tumor assessment and receiving fewer than two doses of study drug.

Participant disposition. A patient who had not received HLX04 was excluded from the full analysis set, the pharmacokinetic set, and the safety set of the HLX04 group after randomization. Another HLX04-treated patient was excluded from the full analysis set because of diagnosis of primary liver cancer. Patients were excluded from the per protocol set because of a lack of valid tumor assessment (HLX04, n = 11; bevacizumab, n = 5), receiving fewer than two doses of study drug (HLX04, n = 7; bevacizumab, n = 4), noncompliance (HLX04, n = 2; bevacizumab, n = 4), and major protocol violation (HLX04, n = 4; bevacizumab, n = 1). FAS full analysis set, PKS pharmacokinetic set, PPS per protocol set, SS safety set
Demographic and baseline characteristics were well-balanced between the two treatment groups in the FAS (Table 1). Of the 675 patients, 404 (59.9%) were male, with the proportion of males in the HLX04 group slightly lower than that in the bevacizumab group (56.2 vs. 63.5%). The mean age was 57.0 years (HLX04, 56.7 years; bevacizumab, 57.4 years). The majority of the patients were Han Chinese (HLX04, 94.4%; bevacizumab, 95.8%). The primary reason for treatment discontinuation was PD in 185 (54.4%) patients receiving HLX04 and in 171 (50.7%) patients receiving bevacizumab.
Exposure to HLX04 or bevacizumab in the two treatment groups was comparable. The mean ± SD number of doses was 12.3 ± 8.0 and 12.6 ± 7.5 for HLX04 and bevacizumab, respectively. The mean duration of treatment was similar (230.4 vs. 236.2 days), with comparable mean infusion time (51.2 vs. 50.4 minutes) between the two groups. Exposure to chemotherapy regimens (XELOX or mFOLFOX6) was also similar.
3.2 Primary Endpoint
The study met its primary endpoint. In the FAS, the CIR-assessed PFSR36w was 46.4% (95% CI 41.1–51.8) in the HLX04 group and 50.7% (95% CI 45.4–56.1) in the bevacizumab group. The rate difference was − 4.2% (90% CI − 10.6 to 2.1), and the rate ratio was 0.92 (90% CI 0.80–1.05); both fell within the prespecified equivalence margins (Fig. 2a). Eight (2.4%) patients in the HLX04 group and 16 (4.7%) patients in the bevacizumab group were eligible for curative resection or radiotherapy through 36 weeks. PD or death within 36 weeks was reported in 69 (20.4%) and 71 (21.1%) patients in the HLX04 and bevacizumab groups, respectively.

PFSR36w by CIR in a FAS and b PPS. CI confidence interval, CIR Central Images Reading Group, FAS full analysis set, PFSR36w progression-free survival rate at week 36, PPS per protocol set
The results produced from the PPS were consistent with those from the FAS. In the PPS, CIR-assessed PFSR36w was 48.1% (95% CI 42.7–53.6) in the HLX04 group and 52.1% (95% CI 46.7–57.5) in the bevacizumab group. Both rate difference (− 4.1%; 90% CI − 10.5 to 2.4) and rate ratio (0.92; 90% CI 0.81–1.05) fell within the prespecified equivalence margins (Fig. 2b). Investigator-assessed PFSR36w also displayed equivalence in both the FAS and the PPS (Tables S3 and S4, respectively, in the ESM).
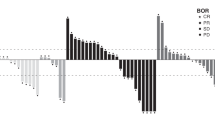
Equivalence of the primary endpoint was observed across all subgroup analyses stratified by chemotherapy, primary tumor site, ECOG PS, age, sex, and KRAS/BRAF mutation status (Fig. S1 in the ESM). Equivalence was also observed in the sensitivity analysis when excluding patients who were eligible for curative resection or radiotherapy, with PFSR rate difference and rate ratio staying within the prespecified equivalence margins (Table S5 in the ESM).
3.3 Secondary Endpoints
In the FAS, a similar number of patients in the two treatment groups achieved a CR (HLX04, n = 1; bevacizumab, n = 0) or PR (HLX04, n = 203; bevacizumab, n = 210) by week 48 (Table 2). No significant between-group differences were observed in BORR at week 48 or in ORR at weeks 6, 12, 18, 24, 30, 36, 42, and 48 (Table 2). No significant differences were observed between the HLX04 and bevacizumab groups in 12-month OSR (HR 0.92; 95% CI 0.66–1.29; p = 0.62) (Fig. 3a), PFS (HR 1.07; 95% CI 0.83–1.37; p = 0.62) (Fig. 3b), TTR (HR 1.11; 95% CI 0.91–1.34; p = 0.30) (Fig. 3c), or DOR (HR 1.14; 95% CI 0.80–1.61; p = 0.48) (Fig. 3d).


a Overall survival rate within 12 months, b progression-free survival by CIR, c time to response by CIR, and d duration of response by CIR (full analysis set). CI confidence interval, CIR Central Images Reading Group, CR complete response, DOR duration of response, HR hazard ratio, NE not estimable, NR not reached, OS overall survival, PD disease progression, PFS progression-free survival, PR partial response, TTR time to response
The results from the PPS (data not shown) were consistent with those from the FAS.
3.4 Safety and Tolerability
In the SS, 336 (99.1%) patients in the HLX04 group and 334 (99.1%) patients in the bevacizumab group experienced at least one treatment-emergent AE (TEAE) (Table 3). Grade 3 or higher TEAEs were reported by 222 (65.5%) patients in the HLX04 group and 238 (70.6%) in the bevacizumab group. In both groups, the most frequently reported grade 3 or higher TEAEs were decreased neutrophil count (20.6 vs. 20.2%), decreased platelet count (10.3 vs. 10.1%), and hypertension (7.4 vs. 12.5%) (Table S6 in the ESM). The incidence of hypertension in the HLX04 group was lower than that in the bevacizumab group, and this was the only grade 3 or higher TEAE or adverse drug reaction (ADR) with a >3% difference in incidence between the two groups (Tables S6 and S7 in the ESM). SAEs were experienced by 104 (30.7%) patients in the HLX04 group and 91 (27.0 %) in the bevacizumab group. The most frequently reported SAEs were decreased platelet count (4.1 vs. 2.4%), intestinal obstruction (3.5 vs. 3.3%), and incomplete intestinal obstruction (2.4 vs. 1.5%). No clinically meaningful differences in the incidence of any AESI were observed (61.7 vs. 65.6%) (Table 3). The most frequently reported AESIs were proteinuria (26.5 vs. 27.9%) and hypertension (17.7 vs. 22.0%).
A total of 20 patients experienced grade 5 TEAEs, including 11 (3.2%) in the HLX04 group and 9 (2.7%) in the bevacizumab group. ADRs leading to death were reported in six patients (1.8%) in the HLX04 group (one sepsis, one intracranial hemorrhage, one gastrointestinal infection and bone marrow failure accompanied by multiple organ dysfunction syndrome, one abdominal infection and intestinal perforation, one acute kidney injury, and one large intestine perforation) and three patients (0.9%) in the bevacizumab group (one gastrointestinal bleeding, one hypovolemic shock and upper gastrointestinal bleeding, one bone marrow failure and respiratory failure).
3.5 Immunogenicity
The observed rate of immunogenicity was low, with ADAs and NAbs detected in comparable percentages of patients in the two groups. In the SS, only one patient in the HLX04 group receiving mFOLFOX6 tested ADA positive, giving a positive rate of 0.29% (1/339). No patient in the bevacizumab group tested ADA positive, and no patient in either treatment arm tested positive for NAb.
3.6 Pharmacokinetics
Serum concentrations of HLX04 and bevacizumab were comparable at all time points of pharmacokinetic sample collection, indicating that exposures to HLX04 and bevacizumab were similar regardless of the combined chemotherapy regimen. No notable differences were observed in samples collected before drug administration at weeks 18, 36, and 48 in both groups, suggesting that serum concentrations of HLX04 and bevacizumab had reached steady state at week 18 (Fig. 4).

Mean serum concentrations of HLX04 and bevacizumab (pharmacokinetic set). C cycle, mFOLFOX6 5-fluorouracil, leucovorin, and oxaliplatin, W week, XELOX capecitabine plus oxaliplatin
4 Discussion
PFSR36w was chosen as the primary endpoint for this study following a comprehensive evaluation of available bevacizumab clinical data [20, 21]. The study demonstrated similarity between HLX04 and reference bevacizumab based on the prespecified equivalence margins. Results of sensitivity analysis and subgroup analyses stratified by chemotherapy, primary tumor site, ECOG PS, age, sex, and KRAS/BRAF mutation status further validated the therapeutic equivalence. Although subgroup analyses of PFSR36w were in favor of bevacizumab in patients with right-sided (primary tumor site) colon cancer or BRAF mutations, and those aged ≥ 65 years according to Fig. S1 in the ESM, the differences were not statistically significant. In addition, no clinically meaningful differences were observed between the two groups in secondary efficacy endpoints, safety, immunogenicity, or pharmacokinetics.
No new safety signals were identified in the HLX04 group compared with the established safety profile of reference bevacizumab. The safety profile of HLX04 was comparable to that of reference bevacizumab, including incidences of TEAEs, SAEs, ADRs, and AESIs [11,12,13]. A high incidence of grade 3 or higher TEAEs, including neutropenia, decreased platelet count, hypertension, arterial thromboembolic events, and gastrointestinal perforations reported were in line with those previously reported [11,12,13, 22, 23]. The difference in incidence of hypertension between the two groups was ~ 5% regardless of TEAE, ADR, or AESI; this could be related to factors that influence blood pressure during measurement, such as position of the patient’s arm, blood pressure differences between left and right arms, and so on [24]. Although the incidence of hypertension differed between the two groups, it was consistent with those reported in previous clinical studies [13].
Several grade 5 TEAEs observed in the present study were reported in previous bevacizumab trials, including gastrointestinal perforation (in up to 2.7% of patients with mCRC), bleeding events (grade 3–5 in 0.4–6.9% of patients), and infections (grade 3–5, in up to 24% of patients with cervical cancer) [11,12,13].
Immunogenicity was similar between HLX04 and reference bevacizumab, with a 0.29% ADA-positive rate in the HLX04 group, which is similar to the 0.6% reported by previous clinical studies of bevacizumab in the treatment of solid tumors [12]. Pharmacokinetic characteristics were also similar between HLX04 and reference bevacizumab in the current trial.
Biosimilars are expected to play an important role in improving patient access to biologics and in helping to address increasing healthcare expenditure, especially in the treatment of cancer [25,26,27]. In a recent survey of oncologists from the USA, Europe, Brazil, Mexico, and Turkey, mCRC topped the list of tumor types in which a bevacizumab biosimilar could have the greatest impact on patient outcomes [28]. However, approvals of bevacizumab-awwb (Mvasi™) and bevacizumab-bvzr (Zirabev™) in the USA and Europe since 2017 [29, 30], as well as approvals of QL1101 [31] and IBI305 [32] by the China National Medical Products Administration since 2019, were all based on pivotal studies in non-small-cell lung cancer. Although indication extrapolation avoids unnecessary clinical trials in all patient populations, some physicians are concerned by the lack of clinical data available for the extrapolated indications, potentially limiting therapeutic uptake [6]. The therapeutic equivalence demonstrated in patients with mCRC in the present study thus lays a solid foundation for the application of HLX04, as well as other bevacizumab biosimilars, in mCRC treatment, which has the potential to increase patient access and reduce the economic burden of this disease [2].
Although cross-trial comparisons should be made with caution, the median PFS (11.4 months; 95% CI 9.8–12.7) and median OS (21.3 months; 95% CI 17.6–not estimable) in patients treated with HLX04 plus XELOX or mFOLFOX6 were comparable to those observed in patients with mCRC treated with bevacizumab plus XELOX or FOLFOX-4 in the reference study (median PFS 9.4 months; median OS 21.3 months) [9]. In a systematic review of FOLFOXIRI plus bevacizumab in patients with unresectable mCRC, the pooled median PFS of 12.4 months (95% CI 10.0–14.3) was similar to the median PFS in the current study [33]. Moreover, the 48-week BORR (60.4%; 95% CI 55.1–65.6) of the HLX04 group was close to the pooled ORR (69%; 95% CI 65–72) in this systemic review [33]. These data have shown that the efficacy of HLX04 in combination with chemotherapy is comparable with bevacizumab tested in other clinical trials for the treatment of mCRC, which further support the use of HLX04 in mCRC management.
One potential limitation of the present study is that the generalizability may be compromised as only a limited patient population was enrolled compared with real-world clinical practice. Another potential limitation is that the duration of the current study was set as 1 year. Additionally, equivalence between HLX04 and bevacizumab in combination with oxaliplatin-based chemotherapy was satisfactorily demonstrated in this study, but the similarity between HLX04 and bevacizumab in combination with other chemotherapy will need further investigation. Therefore, long-term observations in real-world settings to further validate the equivalence between HLX04 and reference bevacizumab are necessary.
5 Conclusion
This phase III study demonstrated the efficacy equivalence between HLX04 and reference bevacizumab in patients with recurrent CRC/mCRC. Safety, immunogenicity, and pharmacokinetic profiles were also comparable between the two groups. HLX04 offers an alternative treatment option for patients with recurrent CRC/mCRC as a biosimilar candidate to bevacizumab.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. https://doi.org/10.3322/caac.21492.
National Health Commission of the People’s Republic of China. Chinese protocol of diagnosis and treatment of colorectal cancer (2020 edition). Zhonghua Wai Ke Za Zhi. 2020;58(8):561–85. https://doi.org/10.3760/cma.j.cn112139-20200518-00390.
Mody K, Baldeo C, Bekaii-Saab T. Antiangiogenic therapy in colorectal cancer. Cancer J. 2018;24(4):165–70. https://doi.org/10.1097/ppo.0000000000000328.
Riihimäki M, Hemminki A, Sundquist J, Hemminki K. Patterns of metastasis in colon and rectal cancer. Sci Rep. 2016;6:29765. https://doi.org/10.1038/srep29765.
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70(1):7–30. https://doi.org/10.3322/caac.21590.
Rosen LS, Jacobs IA, Burkes RL. Bevacizumab in colorectal cancer: current role in treatment and the potential of biosimilars. Target Oncol. 2017;12(5):599–610. https://doi.org/10.1007/s11523-017-0518-1.
Aggarwal H, Sheffield KM, Li L, Lenis D, Sorg R, Barzi A, et al. Primary tumor location and survival in colorectal cancer: a retrospective cohort study. World J Gastrointest Oncol. 2020;12(4):405–23. https://doi.org/10.4251/wjgo.v12.i4.405.
Van Cutsem E, Cervantes A, Adam R, Sobrero A, Van Krieken JH, Aderka D, et al. ESMO consensus guidelines for the management of patients with metastatic colorectal cancer. Ann Oncol. 2016;27(8):1386–422. https://doi.org/10.1093/annonc/mdw235.
Saltz LB, Clarke S, Díaz-Rubio E, Scheithauer W, Figer A, Wong R, et al. Bevacizumab in combination with oxaliplatin-based chemotherapy as first-line therapy in metastatic colorectal cancer: a randomized phase III study. J Clin Oncol. 2008;26(12):2013–9. https://doi.org/10.1200/jco.2007.14.9930.
Kim KJ, Li B, Houck K, Winer J, Ferrara N. The vascular endothelial growth factor proteins: identification of biologically relevant regions by neutralizing monoclonal antibodies. Growth Factors. 1992;7(1):53–64. https://doi.org/10.3109/08977199209023937.
Roche Pharma (Schweiz) Ltd. Avastin®Chinese prescribing information. 2020. https://www.roche.com.cn/content/dam/rochexx/roche-com-cn/roche_china/zh_CN/pdf-instructions/03_BeiFaZhuDankangZhusheye_20201009.pdf. Accessed 1 Nov 2020.
Genentech IRG. Avastin®prescribing information. 2020. https://www.gene.com/download/pdf/avastin_prescribing.pdf. Accessed 1 Nov 2020.
Genentech IRG. Avastin®Summary of product characteristics. 2017. https://www.ema.europa.eu/en/documents/product-information/avastin-epar-product-information_en.pdf. Accessed 1 Nov 2020.
China NMPA. Clinical trial guidelines for bevacizumab biosimilar. 2020. https://www.nmpa.gov.cn/directory/web/nmpa/images/1596531401417069296.pdf. Accessed 2 Nov 2020.
Benson AB, Venook AP, Al-Hawary MM, Cederquist L, Chen YJ, Ciombor KK, et al. NCCN guidelines insights: colon cancer, Version 2. 2018. J Natl Compr Canc Netw. 2018;16(4):359–69. https://doi.org/10.6004/jnccn.2018.0021.
Melosky B, Reardon DA, Nixon AB, Subramanian J, Bair AH, Jacobs I. Bevacizumab biosimilars: scientific justification for extrapolation of indications. Future Oncol. 2018;14(24):2507–20. https://doi.org/10.2217/fon-2018-0051.
Ding Y, Yu T, Sun J, Wu M, Chen Q, Qian H, et al. 105P: a China-manufactured bevacizumab biosimilar, HLX04, matches bevacizumab sourced from China, USA and the European Union. Ann Oncol. 2018;29:ix33–40. https://doi.org/10.1093/annonc/mdy431.019.
Qin S, Li J, Bai Y, Shu Y, Li W, Yin X, et al. 104P-Safety and efficacy of HLX04 versus reference bevacizumab in combination with XELOX or mFOLFOX6 as first-line treatment for metastatic colorectal cancer: a randomised, double-blind phase III study. Ann Oncol. 2020;6:S1273–86. https://doi.org/10.1016/annonc/annonc355.
Chinese Society of Clinical Oncology (CSCO) diagnosis and treatment guidelines for colorectal cancer working group. Chinese Society of Clinical Oncology (CSCO) diagnosis and treatment guidelines for colorectal cancer 2018 (English version). Chin J Cancer Res. 2019;31(1):117–34. https://doi.org/10.21147/j.issn.1000-9604.2019.01.07.
Hagman H, Frödin JE, Berglund Å, Sundberg J, Vestermark LW, Albertsson M, et al. A randomized study of KRAS-guided maintenance therapy with bevacizumab, erlotinib or metronomic capecitabine after first-line induction treatment of metastatic colorectal cancer: the Nordic ACT2 trial. Ann Oncol. 2016;27(1):140–7. https://doi.org/10.1093/annonc/mdv490.
Castellano D, Capdevila J, Sastre J, Alonso V, Llanos M, García-Carbonero R, et al. Sorafenib and bevacizumab combination targeted therapy in advanced neuroendocrine tumour: a phase II study of Spanish Neuroendocrine Tumour Group (GETNE0801). Eur J Cancer. 2013;49(18):3780–7. https://doi.org/10.1016/j.ejca.2013.06.042.
Wagner AD, Arnold D, Grothey AA, Haerting J, Unverzagt S. Anti-angiogenic therapies for metastatic colorectal cancer. Cochrane Database Syst Rev. 2009. https://doi.org/10.1002/14651858.CD005392.pub3.
Hapani S, Chu D, Wu S. Risk of gastrointestinal perforation in patients with cancer treated with bevacizumab: a meta-analysis. Lancet Oncol. 2009;10(6):559–68. https://doi.org/10.1016/s1470-2045(09)70112-3.
Wang W, Zhang W, Sun N, Lin J, Chen L, Wu K, et al. China blood pressure measurement guide. Chin J Hypertens. 2011;19(12):1101–15.
Zelenetz AD, Ahmed I, Braud EL, Cross JD, Davenport-Ennis N, Dickinson BD, et al. NCCN biosimilars white paper: regulatory, scientific, and patient safety perspectives. J Natl Compr Canc Netw. 2011;9(Suppl 4):S1-22. https://doi.org/10.6004/jnccn.2011.0136.
Tabernero J, Vyas M, Giuliani R, Arnold D, Cardoso F, Casali PG, et al. Biosimilars: a position paper of the European Society for Medical Oncology, with particular reference to oncology prescribers. ESMO Open. 2016;1(6):e000142. https://doi.org/10.1136/esmoopen-2016-000142.
Cherny N, Sullivan R, Torode J, Saar M, Eniu A. ESMO European Consortium Study on the availability, out-of-pocket costs and accessibility of antineoplastic medicines in Europe. Ann Oncol. 2016;27(8):1423–43. https://doi.org/10.1093/annonc/mdw213.
Monk BJ, Lammers PE, Cartwright T, Jacobs I. Barriers to the access of bevacizumab in patients with solid tumors and the potential impact of biosimilars: a Physician Survey. Pharmaceuticals (Basel). 2017. https://doi.org/10.3390/ph10010019.
Casak SJ, Lemery SJ, Chung J, Fuchs C, Schrieber SJ, Chow ECY, et al. FDA’s approval of the first biosimilar to bevacizumab. Clin Cancer Res. 2018;24(18):4365–70. https://doi.org/10.1158/1078-0432.Ccr-18-0566.
Genetics and Biosimilars Initiative. Clinical review of biosimilars approved in oncology. 2020. http://gabionline.net/Biosimilars/Research/Clinical-review-of-biosimilars-approved-in-oncology. Accessed 1 Nov 2020.
Baohui H, Kai L, Tianqing C, Minghong B, Helong Z, Yan Y et al. Efficacy and safety of biosimilar QL1101 compared with avastin in patients with non-squamous non-small cell lung cancer. The 20th World Conference on Lung Cancer 2019; September 8; Barcelona, Spain; 2019. p. 380.
Yang Y, Wu B, Huang L, Shi M, Liu Y, Zhao Y, et al. Biosimilar candidate IBI305 plus paclitaxel/carboplatin for the treatment of non-squamous non-small cell lung cancer. Transl Lung Cancer Res. 2019;8(6):989–99. https://doi.org/10.21037/tlcr.2019.12.23.
Tomasello G, Petrelli F, Ghidini M, Russo A, Passalacqua R, Barni S. FOLFOXIRI plus bevacizumab as conversion therapy for patients with initially unresectable metastatic colorectal cancer: a systematic review and pooled analysis. JAMA Oncol. 2017;3(7):e170278. https://doi.org/10.1001/jamaoncol.2017.0278.
Acknowledgements
Writing and editorial support was provided by Simon Foulcer, Ph.D., and Xiaoguang Xu, Ph.D., of Parexel, and Shiqi Zhong, Ph.D., of Shanghai Henlius Biotech, Inc. and funded by Shanghai Henlius Biotech, Inc. The authors thank the patients who participated in the trial, their families, the principal investigators, clinicians, study coordinators, nurses, and the clinical study team (Clinical Operations: Lu Luo, Haoyu Yu; Medical R&D: Wei Li; Statistics: Boyao Shan, Jiancheng Cheng; Pharmacokinetics: Xiaodong Dong; Medical Publication: Chen Hu), Ling Li and Wenjie Zhang from Shanghai Henlius Biotech, Inc.
The members of HLX04-mCRC03 Investigators are: Yuxian Bai, Yongqian Shu, Wei Li, Xianli Yin, Ying Cheng, Guoping Sun, Yanhong Deng, Haijun Zhong, Jin Li, Yunfeng Li, Xiaoping Qian, Shukui Qin, Liangming Zhang, Jingdong Zhang, Kehe Chen, Yuping Sun, Yuan Lin, Tianshu Liu, Li Bai, Shirong Cai, Hong Zong, Helong Zhang, Wei Wang, Sanyuan Sun, Jianping Xiong, Jianfeng Zhou, Bangwei Cao, Hongming Pan, Suxia Luo, Yi Ba, Nong Xu, Jianwei Lu, Jiemin Zhao, Tao Zhang, Zhendong Chen, Jun Liang, Qiu Li, Peiguo Cao, Dong Wang, Wangjun Liao, Yueyin Pan, Longzhen Zhang, Yan Tan, Yunpeng Liu, Xi Chen, Jianwei Yang, Tao Ma, Xiaoyan Lin, Shan Zeng, Minghui Zhang, Xiuli Wang, Enxiao Li, Yiye Wan, Guohua Yu, Weijian Guo, Ying Yuan, Yuansong Bai, Guangyu An, Jianming Xu, Lei Yang, Houjie Liang, Jiang Liu, Wenling Wang.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Funding
This study was sponsored by Shanghai Henlius Biotech, Inc.
Conflict of Interest
Wenying Kang is an employee of Shanghai Henlius Biotech, Inc. Shukui Qin, Jin Li, Yuxian Bai, Yongqian Shu, Wei Li, Xianli Yin, Ying Cheng, Guoping Sun, Yanhong Deng, Haijun Zhong, Yunfeng Li, Xiaoping Qian, Liangming Zhang, Jingdong Zhang, and Kehe Chen have no conflicts of interest that are directly relevant to the content of this article.
Availability of data and materials
Data are available from the corresponding author on reasonable request.
Ethics approval
The study was conducted in accordance with the International Council for Harmonisation guidelines on Good Clinical Practice and all applicable regulatory requirements, including the Declaration of Helsinki, and was approved by the ethics committees at each participating center.
Consent to participate
All participants provided written informed consent prior to study enrollment.
Author contributions
Shukui Qin and Jin Li contributed to the study conception and design. Material preparation, data collection, and analysis were performed by Yuxian Bai, Yongqian Shu, Wei Li, Xianli Yin, Ying Cheng, Guoping Sun, Yanhong Deng, Haijun Zhong, Yunfeng Li, Xiaoping Qian, Liangming Zhang, Jingdong Zhang, Kehe Chen and Wenying Kang. All authors reviewed and provided comments on the manuscript. All authors read and approved the final manuscript.
Additional information
The members of HLX04-mCRC03 Investigators are listed in acknowledgements.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Qin, S., Li, J., Bai, Y. et al. Efficacy, Safety, and Immunogenicity of HLX04 Versus Reference Bevacizumab in Combination with XELOX or mFOLFOX6 as First-Line Treatment for Metastatic Colorectal Cancer: Results of a Randomized, Double-Blind Phase III Study. BioDrugs 35, 445–458 (2021). https://doi.org/10.1007/s40259-021-00484-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-021-00484-9