
Abstract
The subcutaneous route of administration has provided convenient and non-inferior delivery of therapeutic proteins compared to intravenous infusion, but there is potential for enhanced immunogenicity toward subcutaneously administered proteins in a subset of patients. Unwanted anti-drug antibody response toward proteins or monoclonal antibodies upon repeated administration is shown to impact the pharmacokinetics and efficacy of multiple biologics. Unique immunogenicity challenges of the subcutaneous route have been realized through various preclinical and clinical examples, although subcutaneous delivery has often demonstrated comparable immunogenicity to intravenous administration. Beyond route of administration as a treatment-related factor of immunogenicity, certain product-related risk factors are particularly relevant to subcutaneously administered proteins. This review attempts to provide an overview of the mechanism of immune response toward proteins administered subcutaneously (subcutaneous proteins) and comments on product-related risk factors related to protein structure and stability, dosage form, and aggregation. A two-wave mechanism of antigen presentation in the immune response toward subcutaneous proteins is described, and interaction with dynamic antigen-presenting cells possessing high antigen processing efficiency and migratory activity may drive immunogenicity. Mitigation strategies for immunogenicity are discussed, including those in general use clinically and those currently in development. Mechanistic insights along with consideration of risk factors involved inspire theoretical strategies to provide antigen-specific, long-lasting effects for maintaining the safety and efficacy of therapeutic proteins.
Similar content being viewed by others

Immune response toward subcutaneously administered proteins likely entails two waves of antigen presentation by both migratory skin-resident and lymph node-resident dendritic cells, which likely drive immunogenicity. |
Subcutaneous route of administration as a factor of immunogenicity is intertwined with product-related risk factors including impurities, biophysical characteristics, aggregation, and subvisible particle concentration. |
Some promising immunogenicity mitigation strategies in the investigative research stage are tolerance induction, T cell engineering, protein de-immunization and tolerization, use of chaperone molecules, and combination approaches. |
1 Introduction
1.1 Introduction to Immunogenicity of Therapeutic Proteins
Immunogenicity is the propensity of a therapeutic protein to induce unwanted immune response toward itself or endogenous proteins [1]. An anti-drug antibody (ADA) response can develop after a single dose and upon repeated administration of a therapeutic protein. ADA with neutralizing or binding capabilities directly or indirectly affect therapeutic protein efficacy, respectively [2]. Neutralizing antibodies targeting active site(s) on the protein can cause direct loss of efficacy. Several important examples underscore the impact of ADA against a therapeutic protein. Hemostatic efficacy of factor VIII (FVIII) is compromised by development of anti-FVIII antibodies with neutralizing activity (termed ‘inhibitors’) in approximately 30% of severe hemophilia A (HA) patients [3, 4]. Neutralizing antibody development in mild to moderate HA patients led to spontaneous bleeding episodes due to cross-reaction with endogenous FVIII [5]. Clinical response to Pompe disease treatment is negatively impacted by sustained antibody development toward recombinant human acid-alpha glucosidase (rhGAA), which is more common in infantile-onset patients with negative status for cross-reactive immunological material [6]. Binding ADA can impact pharmacokinetics and pharmacodynamics (PK/PD) of therapeutic proteins by increasing clearance, and anti-adalimumab antibody response is associated with decreased adalimumab serum concentrations and diminished therapeutic response in rheumatoid arthritis patients [7, 8]. Anti-infliximab antibodies increase infliximab clearance, leading to treatment failure and acute hypersensitivity reactions [9].
Although less frequent, immunologically based adverse events have been associated with ADA development during replacement therapy, such as recombinant erythropoietin (EPO), thrombopoietin, interferon (IFN)-β, and factor IX [10,11,12,13,14,15,16]. Increased relapse rate during recombinant IFNβ therapy has been observed for multiple sclerosis patients that develop neutralizing anti-IFNβ ADA, and multiple studies have found neutralizing ADA against recombinant IFNβ1a and IFNβ1b are cross-reactive and neutralize endogenous IFNβ [12, 17,18,19,20]. Other well-known examples include pure red-cell aplasia and thrombocytopenia development in patients receiving recombinant EPO or thrombopoietin, respectively, associated with detection of neutralizing ADA that cross-react with endogenous protein [13, 14, 21].
Food and Drug Administration (FDA) Guidance for Industry published in 2014 presents a risk-based approach for evaluation and mitigation of immune responses to therapeutic proteins that limit efficacy and negatively impact safety profiles [1]. Efforts to assess risk of immunogenicity have considered the currently known influential factors of immunogenicity, including a multitude of product-, treatment-, and patient-related factors. Examples of patient-related factors are age, immune status, genetic factors such as human leukocyte antigen (HLA) haplotype, and autoimmune condition [22]. Product-related factors include protein structure, stability, and dosage form, and intrinsic features of recombinant proteins can impact immunogenicity, such as sequence variation, post-translational modifications (PTM), immunodominant epitopes, and cellular expression system [23, 24]. Treatment-related factors include dose, duration and frequency of treatment, and route of administration [23]. Subcutaneous (SC) administration has unique immunogenicity challenges for some products compared to intravenous (IV) administration that are likely due to differences in immune system exposure and antigen presentation mechanisms [25, 26]. Vaccine development elucidated the capacity of antigens to induce a more efficient and effective host immune response following SC administration compared to IV infusion, likely a consequence of frequent encounter by dynamic skin antigen-presenting cells (APCs) [26,27,28,29]. Understanding how route of administration and product-related factors impact immunogenic risk will be critical for mitigating immunogenicity and designing safer biologics for SC delivery.
1.2 Anatomy of the Subcutaneous Space and Skin-Resident Immune Cells
1.2.1 The Epidermis and Langerhans Cells
Human skin is composed of three main layers: the epidermis, dermis, and hypodermis or SC fat. In the epidermis, keratinocytes form a layer of stratified epithelium with tight junctions to provide water-impermeable barrier protection, and cytokine secretion by keratinocytes promotes inflammation during infection or injury [27, 30, 31]. Other featured cells are melanocytes producing melanin pigment, Merkel cells communicating with neurons, memory T cells, and infiltrating innate immune cells [32]. Langerhans cells (LCs) are key APCs in the epidermis that spread dendritic processes to probe for and recognize invading antigens [33]. LCs develop from yolk sac-derived progenitors and acquire a dendritic phenotype and morphology immediately following birth, then in situ proliferation and keratinocyte-derived interleukin (IL)-34 help maintain their population [27, 34, 35]. Tight cellular connections render epidermal stromal and immune cells primarily fixed in place, until LC motility upon maturation is prompted by downregulation of E-cadherin interactions with keratinocytes [27, 30].
Adaptive immune responses initiated by LCs are diverse. Upon maturation, LCs migrate to regional lymph nodes to induce T helper 2 (TH2) polarization of naïve CD4+ T cells via thymic stromal lymphopoietin (TSLP) signaling, as well as T helper 1 (TH1) polarization to IFNγ-producing CD4+ T cells [36, 37]. LCs are also involved in T follicular helper (TfH) differentiation and germinal center (GC) B cell responses [38]. A major role for LCs in cellular immunity is differentiation of naïve CD8+ T cells into potent cytotoxic T lymphocytes (CTLs), but they have contrasting tolerogenic functions in the skin [37, 39]. LCs suppress contact hypersensitivity by interaction with cognate CD4+ T cells in the context of IL-10 [40]. They induce multiple types of regulatory T (Treg) cells during epicutaneous allergen immunotherapy in previously sensitized mice [41].
1.2.2 The Dermis and Dermal Dendritic Cells
The basement membrane regulates protein and cell movement between the epidermis and dermis [30, 42]. The major structural and functional protein components of the skin extracellular matrix (ECM) are produced by dermal fibroblasts [30, 43]. Intertwined collagen and elastin fibers provide structure and elasticity and facilitate migration of immune cells, such as dermal dendritic cells (DCs), along a ‘highway system’ to perform immunosurveillance [27, 30]. Compared to DCs, dermal macrophages have poor antigen presenting capacity and migratory activity but high phagocytic activity, thus they clean up debris to maintain homeostasis and facilitate wound repair/resolution [27]. Skin-resident macrophages arise from precursor pools established prenatally and from blood monocytes after birth, then reside in skin for long periods to provide early host defense [27, 44]. During immune response, dermal blood vessels facilitate recruitment and infiltration of circulating innate and effector immune cells into the skin. Endothelial cells regulate extravasation by production of cytokines, chemokines, and leukocyte adhesion molecules [30]. Macrophages also initiate infiltration of granulocytes into the skin, and perivascular macrophages are the main source of chemoattractants (CXCL1, CXCL2) in the dermis promoting neutrophil extravasation at post-capillary venules in response to bacterial infection [45]. Monocytes are recruited to the skin during homeostasis and in response to infection to differentiate into macrophages or myeloid DCs [30]. Effector cells recruited to the skin temporarily or that become skin-resident cells include CD8+ cytotoxic T cells, CD4+ TH cells, and CD4+ Treg cells [30].
The conventional DC (cDC) class is highly abundant in the healthy dermis, with major human and mouse subsets being CD1c+ and CD11b+ cDCs, respectively [27]. Under resting conditions, cDCs acquire self-antigens in the periphery and undergo homeostatic maturation followed by migration to lymph nodes licensed by morphological and phenotypical changes, including upregulation of major histocompatibility complex II (MHC II) [27]. By presentation of skin-derived self-antigens to T cells, cDCs can eliminate autoreactive T cells to maintain peripheral tolerance [46]. Maturation of cutaneous cDCs upon pathogen stimulation is unique from homeostatic maturation where co-stimulatory molecules are upregulated, and cDCs migrate to lymph nodes to promote differentiation and proliferation of naïve antigen-specific T cells [27]. Dermal CD1a+ DCs in the upper human dermis can induce TH2 polarization of naïve CD4+ T cells as well as differentiation of naïve CD8+ T cells into potent CTLs, although not as effective as LCs [37]. The CD14+ DC subset produces key anti-inflammatory cytokines, IL-10 and tumor growth factor-β (TGFβ), and a role for CD14+ DCs in B cell differentiation is suggested by their ability to induce CD4+ T cell production of TfH-associated chemokine CXCL13 [37].
1.2.3 The Hypodermis or Subcutaneous Fat
Underlying the dermis, the SC fat layer contains nerves, blood vessels, and lymphatic vessels, along with adipocytes that sequester potentially inflammatory lipids and produce proinflammatory cytokines upon stimulation [30]. Adipose tissue is separated into fat cell chambers by septa of connective tissue with heterogeneous structures in upper, middle, and lower layers of the hypodermis [47]. Connective tissue septa comprise the ECM and SC tissue architecture, which is composed of fibrous proteins and viscoelastic gel with the main components being collagen, elastin, glycosaminoglycans (GAGs), and proteoglycans [43, 48, 49]. Highly polar and negatively charged GAGs, including hyaluronic acid, are vastly abundant and contribute to the net negative charge of the ECM [50]. Along with high viscosity in the interstitium, collagen and hyaluronic acid constitute a major barrier to protein movement and dispersion in the SC ECM, and injection volume is limited [48, 51]. Binding of hyaluronic acid to water, creating a gel-like substance, and low hydraulic conductivity of the ECM consequently limit dispersion in the SC space [52, 53]. In the SC space, therapeutic proteins could encounter diverse cell populations including invading dermal DCs, LCs, or innate and effector immune cells recruited from circulation or lymph nodes.
1.2.4 Skin-Derived Immune Cell Migration
LCs, dermal CD1a+ DCs, and dermal CD14+CD1a− DCs are skin-derived migratory DC subsets in human axillary lymph nodes that mediate transport and presentation of skin-derived antigens [54]. Upon exit to draining lymph nodes (DLNs), dermal DCs are of a mature phenotype, and their functional specializations, including TH cell polarization and cross-presentation ability, remain unchanged by migration into lymph nodes [54, 55]. CCR7 signaling is required for DC migration under steady-state and inflammatory conditions. Through CCR7-mediated chemotaxis, migratory skin-derived DCs enter into lymphatic vessels in the skin in response to chemokine (CCL21) expression by lymphatic endothelial cells [56,57,58]. CCL17-deficient mice have demonstrated that CCL17 is strongly associated with LC migration to DLNs, and CCL17 also sensitized activated bone marrow-derived DCs in vitro for CCR7- and CXCR4-dependent migration [59]. Furthermore, TH2 differentiation of naïve CD4+ T cells by CD11bhigh migratory DCs required CCL17 expression, along with CCR7 upregulation, in response to TSLP signaling [60]. Mechanisms and stimuli for cell migration out of the skin are important elements of the immune response to subcutaneously administered proteins.
1.3 ‘First-Pass’ Interactions with Immune System Following Subcutaneous and Intravenous Delivery
Immunogenicity differences based on route of administration could arise from disparities in initial interactions between protein and the immune system as well as subsequent antigen processing and presentation mechanisms. First-pass interactions for SC proteins could occur in the injection site with immune cells, including skin-resident DCs, monocyte-derived DCs, and possibly innate or effector immune cells recruited into the skin during immune response [38, 61]. First-pass interactions could also occur later in the lymphatic system. Unlike IV administration, subcutaneously administered protein has to be absorbed from the injection site into the blood circulation [62]. Proteins or peptides less than 16 kDa in size can be transported from the SC injection site to systemic circulation by diffusing into blood capillaries; however, for larger molecular weight (MW) proteins, lymphatic uptake also plays a role in transport to systemic circulation [49, 63, 64]. A likely location for absorption is at initial lymphatics that start from ‘blind stumps’ and have leakier vessel walls than blood capillaries [64,65,66]. Under increased interstitial fluid pressure, stretching of connective tissue fibers creates tension on the anchoring filaments connecting endothelial cells to collagen, leading to opening of lymphatic lumen and intercellular channels [66, 67]. At this point, interstitial fluid containing water, macromolecules, and possibly therapeutic proteins, easily enters lymphatic capillaries with little protein exclusion [68].
Lymph drains into large lymphatic trunks then lymphatic collectors in the hypodermis that lead to the first DLN [49]. Lymph passes through at least one lymph node; thus, first-pass interactions between protein and immune cells could occur in DLNs, which constantly drain and monitor skin-derived antigens [65, 69]. Upon arrival in DLNs, lymph-borne protein antigen can encounter skin-derived lymph node-resident DCs located in close proximity to lymphatic vessel entry points, an ideal position for antigen uptake [69]. Thus, subcutaneously administered protein may encounter dynamic skin-derived APC populations that are highly specialized for antigen processing, presentation, and lymph node migration [70, 71].
Following IV administration, first-pass interactions between blood-borne protein and immune cells would occur more diffusely within systemic circulation and secondary lymphoid organs. IV administered albumin in mice had rapid distribution throughout the body, with accumulation in the liver area observed within minutes [72]. First-pass encounters of blood-borne protein could be with soluble factors, such as preexisting ADAs or binding proteins [73]. Upon ADA binding, immune complex (IC) formation may initiate additional distribution pathways or accelerated clearance [74]. Blood-borne protein will likely encounter cells of the mononuclear phagocyte system (MPS), comprising circulating blood monocytes, DCs, and tissue macrophages that make intimate connections with endothelial and epithelial cells [75]. Following IV administration, biodistribution of aggregated fluorescently labeled mouse serum albumin revealed fluorescence hotspots in the liver, lungs, and spleen, suggesting entrapment in organs with the MPS [72]. The liver may be a key site for first-pass interactions with tissue macrophages, called Kupffer cells, that clear soluble proteins and aggregates from circulation and internalize antigen-antibody complexes using Fc receptor (FcR) and complement receptor (CR) recognition [76]. Beyond their role in phagocytosis and sequestration of antigen, thought to support hyporesponsiveness, Kupffer cells may be able to promote antigen-specific immunity [77]. Thus, circulating proteins, aggregates, or ICs are likely to be captured by Kupffer cells, but it is not entirely clear whether induction of immunity and/or tolerance responses would occur.
Noteworthy first-pass interactions could also occur in the spleen, a secondary lymphoid organ with lymph node-like structures (white pulp [WP]) and functions [78]. The spleen WP contains distinct lymphoid sheaths based on chemokine signaling: B cell populations reside in B cell follicles, while CCL19 and CCL21 attract CCR7+ T cells and DCs to the periarticular lymphoid sheath (PALS) or T cell zone surrounding the central arteriole [78,79,80,81]. Protein antigen is delivered into the spleen at terminal arterioles ending in sinuses in the marginal zone (MZ) (or perifollicular zone) surrounding the WP [82]. There, protein could encounter MZ-resident APCs, such as macrophages, DCs, and B cells, located in ideal positions to screen and respond to blood-borne antigens via T-independent and T-dependent mechanisms [80, 83]. MZ macrophages and B cells recognize antigen by CRs or pattern recognition receptors (PRRs), and FcR or CR interaction on spleen macrophages mediates clearance of protein antigen arriving as ICs bound to erythrocytes [82, 84]. Protein antigen may accumulate in splenic MZ following IV infusion; for example, FVIII accumulated in splenic MZ of HA mice co-localized with metallophilic macrophages and CD11c+ DCs [85]. When accumulated in the MZ or spleen parenchyma, known as red pulp (RP), blood-borne protein may be phagocytosed and processed by sinusoidal macrophages, pericapillary macrophages, or RP macrophages [75, 83]. RP macrophages commonly clear large particles (> 200 nm) that are retained longer in the RP, while smaller particles are primarily internalized by MZ macrophages [86].
The spleen and MZ macrophages play central roles in immune response toward therapeutic proteins delivered intravenously, demonstrated by splenectomy or depletion of splenic macrophages and CD11c+CD8α− DC populations, which significantly reduced anti-FVIII immune response in FVIII-deficient mice [85]. Adaptive immune response requires antigen entry into the WP, and protein antigen greater than 60 kDa must be transported from the MZ across cellular borders by specialized DCs and MZ B cells [78]. CD8α− cDCs that capture blood-borne protein antigen in the MZ or RP can initiate CD4+ T cell responses in the WP, otherwise endocytosed antigen can be transferred to CD8α+ cDCs in PALS for T cell activation [82, 83]. In conjunction with antigen uptake, activation of immature splenic DCs by innate immune stimuli can license DCs to mature, alter chemokine receptor expression, and migrate toward WP [87]. Thus, splenic DCs mirror migration patterns of tissue-resident DCs moving from peripheral tissue (i.e., spleen parenchyma) toward lymphoid tissue (i.e., splenic WP), and in both cases CCR7 upregulation directs migration into T cell areas [58, 87]. When compared to the splenic immune response following IV administration, more frequent interactions of protein with dynamic skin APC subsets that have highly efficient antigen processing and presentation ability, as well as migratory activity, could drive the immunogenicity of SC administration.
2 Immunogenicity of the Subcutaneous Route of Administration
2.1 Mechanistic Insights into Immune Response Toward Subcutaneously Administered Proteins
A two-wave mechanism of antigen presentation following SC injection of therapeutic proteins has been proposed previously, in which both migratory skin-resident DCs and lymph node-resident DCs drive immunogenicity (Fig. 1) [25]. Location of protein entry and subpopulations of skin DCs involved in first encounters could influence the nature of subsequent immune response following different routes of administration [30]. For example, the nonthreatening nature of entry of epidermal antigen is more likely to initiate a tolerogenic response by LCs. However, direct antigen encounter with dermal DCs may instigate a proinflammatory response because these cells are positioned to encounter pathogens, such as viruses, that would enter the dermis systemically or through skin disruption. Local inflammation generates host cellular components such as lipids, metabolites, or nucleic acids that are damage-associated molecular patterns (DAMPs) [88]. DAMPs activate intracellular and/or cell surface PRRs on DCs and provide dangerous context to protein antigen uptake that warrants proinflammatory response [89]. Thus, they are danger signals that license skin-derived DCs for maturation, which upregulates antigen processing, presentation in the context of MHC II, co-stimulatory molecule expression, proinflammatory cytokine secretion, and migration [90]. Local skin inflammation can also generate small fragments or oligosaccharides of hyaluronic acid, which activate Toll-like receptor (TLR) 4 on DCs [91]. Furthermore, product-related attributes such as altered-self molecular patterns, impurities, host cell proteins, or aggregates have potential to serve as danger signals [24].

Schematic representation of the proposed two-wave mechanism of antigen presentation following subcutaneous injection of protein. a Lymph-borne antigen is delivered to skin-derived LN-resident DCs in DLNs, which begin antigen processing and presentation to antigen-specific naïve CD4+ T cells within T cell areas. b Skin-derived migratory DCs and LCs are recruited into the injection site to acquire antigen. Upregulation of chemokine receptors, CCR7 and CXCR4, on dermal DCs and LCs drives cell migration to initial lymphatics and DLNs. Antigen-loaded migratory DCs and LCs arrive in DLNs for the second wave of presentation to antigen-specific naïve CD4+ T cells, and migratory DCs also transfer antigen to LN-resident DCs [25]. DC dendritic cell, DLN draining lymph node, LC Langerhans cell, LN lymph node
The first wave of antigen presentation following SC injection begins when skin-derived lymph node-resident DCs in DLNs are delivered lymph-borne protein antigen early post-injection [69]. The first wave continues for hours by lymph node-resident DCs containing intermediate levels of intact protein acquired in the lymph node [92]. Initial recognition of peptide antigen in the context of MHC II by naïve antigen-specific CD4+ T cells occurs within T cell areas of DLNs. These DCs display low levels of peptide:MHC II complexes and initiate CD4+ T cell responses toward protein antigen via T cell activation (CD69+ phenotype), IL-2 production, and clonal proliferation [69, 93]. Effector T cells are thus generated to mediate immune response in secondary lymphoid and non-lymphoid peripheral tissues [55]. Lymphoid-resident DCs also selectively retain antigen-specific lymphocytes in inflamed DLNs via MHC II expression and antigen presentation [93].
The second wave of antigen presentation occurs later, for example, 24 h post-injection, when skin-derived migratory DCs arrive in DLNs carrying large amounts of protein acquired at the injection site [69]. Cell migration to DLNs for the second wave is driven by receptor-ligand interaction of CCR7 and CXCR4 upregulated on mature dermal DCs with ligands expressed within lymphatic vessels [94, 95]. In addition to chemokine signaling, matrix metalloproteinase (MMP) enzymes are essential for movement of LCs and DCs through the skin. LC production of MMP-2 and MMP-9, in addition to CXCL12 signaling of CXCR4 on LCs, facilitates translocation of activated LCs through the basement membrane toward the dermis [90]. MMPs also degrade collagen, which could assist DC movement in the dermis toward initial lymphatics, and MMP9 induced by prostaglandin E2 during inflammation is essential for DC migration to DLNs [90, 96]. Proinflammatory cytokines, tumor necrosis factor (TNF)-α and IL-1β, enhance lymphatic trafficking of migratory LCs and dermal DCs by upregulating vascular endothelial growth factor-C (VEGF-C), to increase lymphatic vessels in the inflammatory site, and reducing expression of adhesion molecule E-cadherin on LCs [90, 95]. Upon SC injection, mechanical injury to the skin could enhance and prolong LC and dermal DC migration [57].
The second wave of antigen presentation to CD4+ T cells by migratory DCs, expressing high levels of peptide:MHC II complexes, is required to sustain T cell IL-2 receptor expression necessary for effector CD4+ T cell generation [69]. The CD4+ T cell activation program integrates signals from multiple encounters with antigen-bearing DCs before and after T cell commitment to cell division [97]. Subsequent contacts between recently activated CD4+ T cells and antigen-bearing DCs promotes T cell CD25 expression and IFNγ production. Increased opportunities for DC:T cell reencounter reinforces CD4+ T cell activation; therefore, the number of antigen-bearing DCs arriving in DLNs could be an important parameter for SC immunogenicity [97]. Lymph node-resident DCs may participate in the second wave of antigen presentation following antigen transfer by migratory DCs [98]. Migratory DCs and LCs also have an important role in TfH cell and GC responses within DLNs [38]. Physical removal of the injection site at 1 h post-intradermal antigen administration, thus preventing skin DC migration, significantly reduced GC B cells and antibody-secreting cells as well as limited expansion of TfH cells. Even though nanoparticulate antigen efficiently transited to DLNs within 1 h (unaffected by injection site removal), the first wave of antigen presentation mediated by lymph node-resident DCs alone was not able to efficiently promote TfH cell and GC B cell responses. Although mechanistic understanding is incomplete, it is clear that both resident DCs in the DLNs and, more significantly, migratory skin-derived DCs could drive immunogenicity of subcutaneously administered protein.
Receptor-mediated uptake of proteins and mAbs by APCs could impact subsequent antigen processing and presentation. For example, mannose-sensitive receptor-mediated uptake into APCs could impact immunogenicity of recombinant human proteins with exposed mannose moieties. Averting uptake of FVIII by mannose-sensitive receptors reduced the ability of DCs to activate a human CD4+ T cell clone specific for a non-glycosylated FVIII epitope, and mannose receptor (CD206)-expressing human splenic myeloid DCs were hypothesized to play a role in splenic anti-FVIII immune response [99]. Furthermore, SC immune response may involve mannose-sensitive uptake given that mannose receptor-expressing APCs have been identified in peripheral lymph nodes [100, 101]. Receptor-mediated uptake by neonatal FcR (FcRn) could also impact antigen presentation due to its expression by professional APCs and apparent association with MHC II-associated invariant chain in the endocytic pathway [102,103,104]. Monoclonal antibodies (mAbs) or Fc-fusion proteins that can be recycled by FcRn could be recycled out of APCs thus decreasing lysosomal processing and the probability of antigen presentation. FcRn binding can also direct the fate of monomeric and multimeric immunoglobulin G (IgG) ICs upon uptake; monomeric ICs are protected from degradation and recycled, whereas multimeric ICs are routed into degradative compartments where peptides can be loaded into MHC II [105, 106]. If IC formation between mAbs or between drug and ADA occurs prior to uptake by APCs, FcRn recognition of monomeric ICs could lead to recycling out of cells, while recognition of multimeric ICs could lead to lysosomal degradation and increased antigen processing and presentation. Fcγ receptor (FcγR) may initiate APC uptake of IgG ICs followed by hand off to FcRn in acidified compartments [105]. Furthermore, FcγRIII engagement is involved in the enhanced ability of ICs, compared to free antigen, to upregulate CCR7 expression and MMP-9 production by DCs in vitro, as well as boost skin-resident DC migration to DLNs following SC injection [107]. Complex interactions of proteins with lymph node-resident DCs and skin-resident migratory DCs could introduce immunogenicity challenges for SC delivery.
2.2 Evidence for Immunogenicity of the Subcutaneous Route
Some biologics formulated for SC delivery have demonstrated enhanced immunogenicity by this route of administration; however, this notion has been contrasted by a number of proteins that demonstrate comparable or higher immunogenicity by IV administration. Clinical evidence for immunogenicity is variable between products and individuals due to the multitude of product-, treatment-, and patient-related factors, but the SC route of administration is known to exhibit immunogenicity challenges. In order to compare therapeutic protein immunogenicity following SC and IV administration, available data should be examined where dosing by both routes was directly compared and ADA development was measured concurrently. However, there is not an extensive number of clinical trials that have directly compared safety and efficacy of SC and IV dosing regimens for therapeutic proteins or mAbs.
2.2.1 Preclinical Evidence
Investigation into the impact of route of administration on immunogenicity of FVIII demonstrated that the SC route was more immunogenic than the IV route only in terms of total anti-FVIII titer, with no significant effect on neutralizing ADA (inhibitor) development [108]. It was hypothesized that modified epitopes of FVIII created upon proteolytic degradation at the injection site, with corresponding loss of conformational epitopes at the active site (likely inhibitor targets), could explain increased total anti-FVIII titers without increased inhibitors. Binding ADA are not inconsequential seeing as they could impact systemic exposure or clinical response rates by altering protein PK and clearance [109]. Because IFNα is administered by multiple routes clinically and induces ADA response in a significant patient population, impact of route of administration on IFNα immunogenicity has been investigated [110]. In BALB/c mice administered equivalent doses of IFNα, the SC route was most immunogenic followed by intraperitoneal (IP), intramuscular (IM), and then IV route based on anti-IFNα titers. Changes in IFNα half-life following SC administration along with exposure of a higher frequency of APCs to IFNα for longer times at higher concentrations could explain high titers induced at earlier times following SC administration [110]. Administration by the above routes is shown to impact kinetics and organ distribution of aggregated and monomeric albumin in mice; thus ,administration by different routes could expose therapeutic protein to altered cell populations in lymphoid and non-lymphoid organs [72]. Furthermore, therapeutic proteins administered subcutaneously exhibit a relatively slower rate of absorption and prolonged terminal half-life compared to that observed following IV administration [64, 66].
Contrasting results for recombinant human IFNβ identified the IV route to be most immunogenic upon administration to immune-tolerant, transgenic mice, proposed to be a result of high aggregate content in some IFNβ products [111, 112]. Upon repeated IV administration, protein aggregates may have enhanced uptake and processing by splenic macrophages, compared to uptake of monomeric protein, with sustained activation of MZ B cells [111]. Similarly, murine growth hormone aggregates were immunogenic by IV administration, with higher IgG2c and IgG3 titers compared to SC delivery, suggesting involvement of T-independent type 2 response. However, IgG1 titers were high and comparable following SC and IV administration [113]. Aggregates can be considered an immunogenicity challenge for SC and IV administration, where mechanisms responsible likely differ.
2.2.2 Clinical Evidence
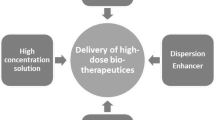
A mAb administered subcutaneously that has demonstrated considerable immunogenicity, where efficacy is impacted by ADA development, is adalimumab. In a long-term follow-up study for adalimumab in rheumatoid arthritis patients, 28% developed anti-adalimumab antibodies, 67% of which developed within the first 28 weeks of treatment [114]. Anti-adalimumab antibody development was associated with lower serum concentrations and lower likelihood of achieving minimal disease activity or clinical remission. However, without directly comparable clinical IV immunogenicity data, it is unclear whether the relatively high immunogenicity of adalimumab is due to the SC route or other intrinsic or extrinsic factors. Where available, comparative immunogenicity data, represented by incidence of total and neutralizing ADA response, within the same clinical trial have been collected, expanding on previous analysis by Hamuro et al. [73]. ADA incidence sourced from product labels or peer-reviewed publications are presented in Table 1 for ten currently approved biologics. Herceptin® (trastuzumab) formulated for SC administration has demonstrated enhanced ADA incidence following SC delivery. A higher incidence of anti-trastuzumab antibodies (16%) was observed following treatment with SC Herceptin Hylecta™ (formulated with recombinant human hyaluronidase [rHuPH20]) compared to IV trastuzumab (10%) (Table 1) [115]. Additionally, 21% of patients treated with Herceptin Hylecta™ developed anti-rHuPH20 antibodies—a common observation for products formulated with this permeation enhancer.
SC rituximab, which is much more concentrated than the IV formulation, is also formulated with rHuPH20 to facilitate larger injection volumes and improve antibody dispersion and absorption by temporarily hydrolyzing hyaluronic acid [52, 116]. Observed immunogenicity of rituximab in SC and IV formulations is low; treatment-induced/enhanced anti-rituximab antibody incidence in previously untreated patients with follicular lymphoma was 2.0% and 1.9% in the SC and IV groups, respectively [117]. SC rituximab treatment also induces or enhances levels of anti-rHuPH20 antibodies in 15% of patients. Pooled clinical trial results for SC trastuzumab, rituximab, insulin, and human IgG co-administered with rHuPH20 show an overall incidence of 1.7–18.1% for induced or boosted anti-rHuPH20 antibody development, plus a 3.3–12.1% incidence of pre-existing anti-rHuPH20 antibodies [118]. No neutralizing anti-rHuPH20 antibodies were observed, and adverse events were not associated with anti-rHuPH20 positivity regardless of boosting after rHuPH20 exposure. Antibody positivity to rHuPH20 has been found in 5.2% of a large cohort not previously exposed to rHuPH20, and rates were significantly higher in males compared to females and varied with age [119]. The reasons for baseline prevalence of anti-rHuPH20 antibodies are not clear, but then rHuPH20 immunogenicity seems modest with no observed effects on adverse events or efficacy.
Marginally higher incidence of immunogenicity following SC administration compared to IV is observed for peginesatide, mepolizumab, golimumab, and Phesgo™ (pertuzumab, trastuzumab, and rHuPH20), although ADA incidence was approximately 5% or less (Table 1) [120,121,122,123]. Overall low immunogenicity of the protein itself seems to confound significant comparison of immunogenic risk between routes of administration in some clinical trials. Low and comparable immunogenicity of SC and IV administration has been observed for daratumumab and vedolizumab (Table 1) [124, 125]. In some examples, including tezepelumab (human anti-TSLP IgG2λ) and inebilizumab (humanized, afucosylated anti-CD19 IgG1κ), no ADA incidence was detected for either route of administration [126, 127]. The direct impact of B cell-depleting agents, rituximab and inebilizumab, on humoral responses may explain their observed overall low immunogenicity.
A phase IIIb clinical trial for the fusion protein abatacept, human IgG Fc plus extracellular domain of cytotoxic T lymphocyte-associated protein 4 (CTLA-4), demonstrated similar total ADA rates (anti-abatacept or anti-CTLA-4-T antibodies) between SC (1.1%) and IV (2.3%) administration [128]. However, in the long-term extension period where patients received SC abatacept, 23.2% were positive for anti-abatacept antibodies [129]. No correlations between anti-abatacept seropositivity and adverse events, infusion reactions, or efficacy changes have been observed [130, 131]. Similarly, for tocilizumab comparable efficacy and immunogenicity profiles are observed for SC and IV formulations [132,133,134]. ADA positivity rates in patients administered tocilizumab subcutaneously or intravenously were estimated to be 1.5% and 1.2%, respectively, based on a meta-analysis of 14 studies, indicating overall low risk of tocilizumab immunogenicity [135]. Although more ADA-positive patients who received tocilizumab subcutaneously had neutralizing ADA (85.1%) compared to ADA-positive patients who received tocilizumab intravenously (78.3%), none of these patients in either treatment group experienced loss of efficacy. Tocilizumab’s low immunogenicity profile with limited ADA development may result from its suppression of IL-6-dependent B cell differentiation and TfH cell activity [136].
Comparative immunogenicity results for SC and IV administration are available for some mAbs currently undergoing clinical trials. In a phase I clinical trial for PF-06480605 (human anti-TNF-like ligand 1A [anti-TL1A] IgG1) conducted in healthy participants, high ADA incidence (81.8–83.3%) was observed for SC and IV administration in single-dose or multiple-dose groups [137]. Lower exposure of PF-06480605 was seen in participants positive for neutralizing ADA at lower doses, but not at higher doses, and ADA were not associated with adverse events. In a phase I trial for PF-06801591 (humanized anti-programmed cell death protein 1 [anti-PD-1] IgG4) conducted in patients with advanced solid tumors, ADA incidence was marginally higher in IV groups (9.5%) compared to the SC group (7.1%) [138]. No neutralizing ADA were detected. Clinical evidence for immunogenicity is variable between products and individuals, and although a range of products show comparable, low ADA incidence (< 5%), there are immunogenicity challenges for SC delivery of certain biologics.
2.3 Interplay Between Subcutaneous Immunogenicity and Product-Related Risk Factors
2.3.1 Biophysical Characteristics
Lymphatic transport of proteins, and thus absorption and distribution, are impacted by protein biophysical characteristics—molecular size and charge. Transit of proteins and peptides greater than 16 kDa through SC interstitium into lymphatic capillaries occurs by convection with limited diffusion [49]. Large MW proteins in interstitial fluid can enter highly permeable lymphatic vessels through intercellular channels opened under increased interstitial pressure [68]. Protein will traffic through lymphatic capillaries and pass into lymph vessels draining into local lymph nodes, eventually reaching systemic circulation by draining through efferent lymph vessels into larger collecting vessels and the thoracic duct [49, 68]. Studies in sheep models have demonstrated direct correlation between protein MW and proportion of dose absorbed by lymphatic uptake, and large proteins (30–40 kDa and above) have demonstrated almost complete dose recovery in peripheral lymph [63, 139]. Insulin bioavailability following SC administration in sheep significantly involves lymphatic absorption; however, species differences caution against generalizing preclinical results [28, 49, 140].
Studies in mice have further demonstrated that rate of loss of protein from the SC injection site correlates inversely with protein MW in the range of 23–149 kDa, with 149-kDa protein cleared the slowest [141]. Also, lymphatic exposure, or fraction of injected dose recovered, at the draining, axillary lymph nodes following front foot pad injection increased proportionally relative to protein MW. Lymph node distribution and PK studies demonstrate arrival of subcutaneously administered proteins in DLNs within hours [25]. Mean tmax value, or time to reach maximum protein levels, in draining, axillary lymph nodes obtained in mice was almost 3 h for 149-kDa protein following SC injection compared to shorter times just under or over 1 h for proteins 23, 44.3, and 66 kDa [141]. It is possible that slower rate of exit from the SC space and increased immunological exposure of larger MW proteins at the lymph nodes could translate into heightened immune response.
Very large protein and particulates could become trapped in the ECM due to convection limitations and steric hindrance. Furthermore, positively charged proteins sized 20–78 kDa appear in lymph at delayed times compared to negatively charged, size-matched proteins [142]. Biologics with slight positive charge at local physiological pH, due to an isoelectric point of 7–9, could interact with negatively charged GAGs that are highly abundant in the ECM [28, 49]. Protein molecular charges can be heterogeneous from deamination, isomerization, and PTM, and surface charge may change from surrounding pH variations in the SC space during transition and dispersion of the protein from the stable formulation state [28, 143]. An additional consideration is whether protein chemical stability pathways, for example, oxidation, could generate modified epitopes and impact immunogenicity [144,145,146,147]. Overall, increased retention time due to charge interactions, or steric hindrance, could slow absorption and prolong exposure of therapeutic protein to invading LCs and dermal DCs (Fig. 2). The presumed mechanism of vaccine adjuvants demonstrates how SC immune response can be enhanced via facilitation of phagocytosis and slowing antigen exit from the injection site to promote uptake and trafficking of antigen by migratory DCs [57, 148].

Product-related risk factors for immunogenicity of subcutaneously administered therapeutic proteins. Structural or conformational modifications related to instability pathways or proteolytic degradation could generate new/modified epitopes. Protein aggregates or precipitates present in the formulation or formed post-injection can have longer SC retention time. Charge interactions between slight positive charge on mAbs at local physiological pH and negative charge density in ECM may increase SC retention time. Enhanced retention time of protein could confer immunogenic risk by increasing opportunities for encounter with invading dermal DCs and LCs post-injection. Innate immune stimulation by adjuvant-like drug product impurities (e.g., host cell proteins, leachates, and endotoxins) at the injection site can trigger maturation and migration of dermal DCs and LCs. Ag antigen, DC dendritic cell, ECM extracellular matrix, LC Langerhans cell, LN lymph node, mAb monoclonal antibody, SC subcutaneous
2.3.2 Protein Aggregation
Protein molecules acquire a strong propensity to aggregate when externally applied stresses induce changes in protein conformation or partial loss of native structure that increase surface exposure of hydrophobic domains [149, 150]. Bioprocessing stresses include high concentration, high temperature, changes in pH or ionic strength, shear stresses, and air–liquid or liquid–surface interfaces [24]. Even though classification systems are in place to determine aggregate features that confer immunogenic potential, there is an overall lack of understanding of the type and size of therapeutic protein aggregates universally implicated in immunogenicity [151,152,153]. Filipe et al. endeavored to correlate type and amount of stress-induced IgG aggregates with immunogenic potential, and not all aggregates had the same propensity to induce an immune response [152]. FDA Guidance for Industry recognized subvisible aggregates or particulates (0.1–10 μm) to have a strong potential to be immunogenic, but preclinical studies present contrasting results [1, 154].
Submicron-sized mAb aggregates (100–1000 nm) were demonstrated to be most immunogenic upon SC administration compared to soluble oligomers (< 100 nm) or micron-sized aggregates (1–100 μm) [155]. Conversely, native-like soluble oligomers (< 100 nm) induced higher antibody response in mice following SC administration compared to native mAb monomer or micron-sized non-native aggregates [153]. Subvisible aggregates of single-chain variable fragment (scFv) and ovalbumin induced significantly higher IgG2a titers compared to monomeric protein by SC injection in BALB/c mice, although total IgG and IgG1 titers were comparable. Skewing towards TH1-type immune response by aggregates was also suggested by cytokine profiles in DC co-culture experiments [156, 157]. Additionally, TH1-type immune response was observed for bevacizumab heat-triggered aggregates in a human artificial lymph node (HuALN) model, where delayed immune reactions can be monitored by long-term exposure of the system up to 28 days [158].
Human IgG aggregates induced by stirring and micron-sized particles coated with IgG induce B cell-mediated immune response in an immunologically tolerant murine model [159]. Thus, IgG-coated particles with multivalency were able to transiently break immunological tolerance upon SC immunization. The particulate nature of aggregates may be responsible; via presentation of repetitive surface antigens, multivalent protein aggregates may be uniquely capable of cross-linking B cell receptors, leading to antibody production without T cell help [160]. Also in human IgG transgenic mice, human IgG oligomers with chemical amino acid modifications from light stress were able to break tolerance and induce ADA recognizing native IgG, the mechanism of which depended on T cell help and presumably involved generation of ‘neo-epitopes’ [161]. Notably, IgG oligomers generated under process- or pH-related stress conditions were immunogenic in wild-type mice but did not break tolerance in the immune tolerant mouse model. Breaking of tolerance was also observed in mice following SC administration of recombinant murine growth hormone (rmGH) aggregates with a presumed TH1-type antibody response [113]. IV administration of the same aggregates, however, showed a mixed TH1/TH2-type response with possible T-independent type 2 response. Encounter of IV-injected aggregates with splenic MZ B cells may make T-independent type 2 response more likely following IV administration.
Upon SC administration, submicron- to micron-sized aggregates can experience longer retention in the SC space, enrichment in DLNs, and slower clearance than monomeric protein [72, 162, 163]. Removal of albumin aggregates from the SC injection site in mice is slower than monomeric protein, and complete clearance was not observed after 48 h [72]. Retention of protein aggregates in the SC space or enrichment in DLNs could prolong interactions with skin-derived DCs and heighten ADA response (Fig. 2). Furthermore, the complex, unpredictable nature of protein aggregates alludes to potential for changes in composition and size distribution once introduced to the body. Upon dilution in human serum, subvisible IgG aggregates formed by pH-related stress immediately became smaller and more numerous; then, incubation at 37 °C led to formation of large, subvisible aggregates, with evidence suggesting association of serum components [164]. The type or composition of aggregates, host immune tolerance status, and immunogenicity of the monomer itself could impact immunogenic risk [165]. Because a minute amount of aggregates could enhance immune response and current analytical techniques could overlook these particles, it will be important to prevent or moderate aggregate formation [166, 167].
2.3.3 Dosage Form Considerations
Presence of product impurities or contaminants can exacerbate immune response following SC administration by introducing danger signals (Fig. 2). Host cell proteins, leachates, and endotoxins are adjuvant-like contaminants that could enhance DC migration and antigen presentation, demonstrated experimentally by lipopolysaccharide (LPS) [24, 168, 169]. Even a small amount of residual impurities present in the drug product may activate the innate immune system, for example, via TLR stimulation, to promote immunogenicity [170, 171]. Immune response could also be exacerbated by pre-existing inflammatory/autoimmune conditions that increase sensitized APCs or if patient skin-resident DC populations are activated by DAMPs from tissue inflammation/injury [73, 88]. An additional dosage form consideration for SC immunogenicity is the presence of proteinaceous or non-proteinaceous particulates, possibly enhanced by use-related stress. Adalimumab samples shaken in siliconized syringes contained a significant concentration of silicone oil droplets and soluble to subvisible particles; these samples were more immunogenic upon SC administration to BALB/c mice compared to unstressed or non-siliconized syringe stressed samples [172]. Besides agitation, freeze–thaw cycles or continuous low temperature storage (96 h) increased subvisible particle concentration in multiple TNFα inhibitors [173]. The potential immunogenic risk of subvisible particle concentration in therapeutic protein preparations requires further investigation.
Solubility challenges are a key dosage form consideration for SC administration due to requirement for high protein concentration in small injection volumes [28]. Protein crowding and aggregation are concerns for high concentration formulations, and excipients and stabilizers are added to maintain conformational and colloidal stability [28, 174, 175]. Low solubility within skin ECM is also an issue, and precipitation at the injection site is a possible consequence. mAbs with poor solubility at neutral pH and formulated at high concentrations could spontaneously precipitate due to pH change after SC dosing [176]. Following SC administration of such a mAb, precipitated antibody was retained at the injection site, but cellular immune response within lymph nodes and ADA development were not enhanced. Precipitation in this case may be reversible upon dilution in vivo due to concentration-dependent solubility as the driving factor. Also, precipitated antibody could be cleared by phagocytic cells without inducing a strong immune response; co-localization of the mAb with CD68+ cells (likely macrophages or monocytes) in the skin was observed along with no increase in systemic cytokine response [176]. No correlation between immunogenic risk and protein precipitation after SC delivery was established. To avoid solubility challenges, depot formulations with hyaluronidase (rHuPH20) and protein stabilizers can facilitate administration of increased injection volumes [177]. The use of hyaluronidase could address slow and incomplete absorption of proteins to limit immunological exposure, and pre-existing or induced anti-rHuPH20 antibodies have not impacted efficacy or safety in tested products [73, 118].
3 Existing and Future Strategies to Minimize Subcutaneous Immunogenicity
3.1 Immune Suppression and Lymphocyte Manipulation
Conventional strategies to mitigate immunogenicity of biologics, whether dosed subcutaneously and/or intravenously, have varying degrees of success clinically and rely on immune suppression using small molecule drugs, such as methotrexate, rapamycin, bortezomib, and cyclophosphamide (Fig. 3) [7]. The immunomodulatory drugs azathioprine and methotrexate have been used in combination with TNFα blockers infliximab and adalimumab [178]. Kishnani and colleagues have combined rituximab with methotrexate and IV gamma globulin to successfully prevent and reverse anti-rhGAA antibody response in infantile Pompe disease patients [179, 180]. Anti-rhGAA titer development has been prevented by anti-CD3 antibody treatment in preclinical models, which also provided modest reduction of pre-existing titers [181]. This non-FcR-binding anti-CD3ε F(ab’)2 fragment protects HA mice from total and inhibitory anti-FVIII antibody formation, the mechanism of which involves increased CD25 expression on peripheral effector CD4+CD25− cells [181, 182]. The importance of antigen-specific Treg cells (CD4+CD25+) inspired a strategy to transduce FVIII-specific CD4+ T cells with forkhead box P3 (FoxP3), thus imparting a Treg-like phenotype [183]. In addition to preventing inhibitor formation upon adoptive transfer, combination with anti-murine CD20 (anti-mCD20) antibody provided modest reversal of pre-existing inhibitors. However, inhibitors rise upon discontinuation of anti-mCD20 therapy. Note that important CD20− cell populations survive anti-CD20 therapy, particularly long-lived plasma cells [184]. To achieve FVIII-specific B cell depletion, cytotoxic CD8+ T cells have been engineered with B cell antibody-targeting receptor (BAR) expressing immunodominant FVIII A2 and C2 domains [185]. Lymphocyte manipulation strategies should ideally deliver a long-lasting, antigen-specific protective effect.

Selection of existing and theoretical strategies to minimize immunogenicity of subcutaneously administered therapeutic proteins. a Conventional strategies rely on nonspecific immune suppression using small molecule drugs, such as methotrexate, rapamycin, bortezomib, and cyclophosphamide. A combination approach uses the lymphocyte depletion agent rituximab (anti-CD20) with methotrexate and intravenous Ig. b Example lymphocyte modulation strategies are anti-CD3 antibody, engineered antigen-specific FoxP3+ Treg cells, and cytotoxic BAR CD8+ T cells. c Reduction of product-related factors by ex vivo human cell-based assay screening, removal of non-native IgG aggregate precursors by specific absorption to AF.2A1 magnetic beads, and chaperone molecules to improve protein stability. d Inhibition of CCL19/CCL21 directed DC migration to lymph nodes by the small molecule CCR7 inhibitor cosalane (anti-HIV agent). e mAb humanization by incorporation of fully human content apart from complementarity-determining regions. In silico prediction of T or B cell epitopes on proteins to perform de-immunization or incorporation of Treg cell epitopes (Tregitopes). f Peripheral tolerance induction by co-administration of OPLS with therapeutic protein subcutaneously to induce tolerogenic DCs and antigen-specific Treg responses [7, 178, 179, 183, 185, 186, 191, 192, 197, 209]. BAR B cell antibody-targeting receptor, CDR complementarity determining region, DC dendritic cell, DLN draining lymph node, FoxP3 forkhead box P3, HIV human immunodeficiency virus, IDO indoleamine 2,3-dioxygenase, Ig immunoglobulin, IL interleukin, mAb monoclonal antibody, MHC major histocompatibility complex, OPLS O-phospho-L-serine, RA retinoic acid, TCR T cell receptor, TGFβ tumor growth factor-β, Treg regulatory T cell
3.2 Inhibition of Skin-Derived Immune Cell Migration
Theoretical mitigation strategies for SC immunogenicity could co-administer inhibitors or function-blocking antibodies directed against chemokine receptors CCR7 and CXCR4 to interrupt cutaneous DC migration to DLNs during the second wave of antigen presentation [25]. The small molecule drug cosalane, identified by high-throughput screening, inhibits CCR7-mediated mouse and human cell chemotaxis in vitro in response to CCL19/CCL21 stimulation [186]. CD11c+ DC maturation markers and CCL21-directed migration in vitro are also impaired by etanercept (TNF inhibitor) [187]. Collagen-induced arthritic mice treated with etanercept had significantly reduced myeloid and plasmacytoid DCs in lymph nodes compared to controls while peripheral blood DC numbers remained unchanged, and CXCR4 downregulation partially explains reduced DC migration [187]. IL-4 also modulates DC migratory capacity. DCs matured by TNFα in the presence of IL-4 have reduced CCR7 expression and impaired CCL19/CCL21-directed migratory activity [188]. Interrupting DC migration from the injection site will not impact antigen presentation by lymph node-resident DCs, thus ADA development could still occur.
3.3 Reduction of Product-Related Risk Factors
Reduction of protein aggregates and impurities, such as residual host cell proteins or endotoxins, could alleviate potential product-related immunogenic risk factors [168, 169]. Protein samples can be screened in human ex vivo cell-based assays for impurities and contaminants [189]. Prediction of aggregation propensity and susceptible sites for PTMs would help mitigate immunogenic and hypersensitivity risks of aggregates and non-human glycosylation [189]. Sequence-based prediction tools can be employed to identify potential aggregation-prone sequence and structural motifs on proteins [190]. Prevention of aggregation during long-term storage is ideal, but small aggregate precursors are not removed by 0.22-μm filtration during manufacturing. A specific approach for IgG formulations removes non-native IgG molecules and small aggregate precursors by adsorption to magnetic beads conjugated with AF.2A1 protein [191]. Addition of chaperone molecules to protein formulations could prevent aggregation; a chaperone molecule (miglustat) for rhGAA that improves PK and protein stability in circulation is under investigation as a replacement therapy for Pompe disease [192]. FVIII is also prone to form aggregates with high immunogenic risk; however, onset of aggregation is delayed by the stabilizing interaction of O-phospho-L-serine (OPLS) with the aggregation-prone, immunogenic region on FVIII [193, 194]. Moreover, FVIII-OPLS complex was significantly less immunogenic than free protein upon SC administration.
3.4 Protein Modification and Re-engineering
Less immunogenic therapeutic proteins could be designed through de-immunization or tolerization. Protein modification is especially relevant for immunogenicity driven by non-self recognition, for example, replacement therapies in patients that lack central tolerance to endogenous protein or proteins with non-human sequences [195]. Humanization incorporates fully human sequences into mAbs without changing the complementarity-determining regions, but inadequacy of humanization is revealed by unfortunate ADA rates for fully human adalimumab [114, 196, 197]. Zurdo et al. reviewed quality by design methodologies and early risk assessment for immunogenic potential of proteins in development [189]. De-immunization or T cell epitope removal should limit high-affinity, long-lived ADA development by abrogating T cell responses [189]. De Groot and Martin developed web-accessible tools to perform in silico protein re-engineering and screen then rank candidate protein sequences for immunogenic potential [198]. Less immunogenic, but efficacious, versions of FVIII and immunotoxin LMB-2 (anti-CD25) were engineered by de-immunization of immunodominant T cell epitopes [199, 200]. Additionally, B cell epitope modification is expected to interfere with binding of pre-existing ADA or memory B cells [201]. Beyond de-immunization, sequences known to be regulatory T cell epitopes, i.e., Tregitopes, can be introduced into the protein to promote tolerogenic responses [197].
3.5 Tolerance Induction
Tolerance induction to therapeutic proteins would avoid severe issues associated with immune suppression, such as susceptibility to secondary infections [7]. Strategies can take advantage of natural peripheral tolerance mechanisms involving antigen presentation by migratory DCs to antigen-specific CD4+ T cells in the context of MHC II but with low co-stimulatory signals [202,203,204,205,206]. Regulatory mediators produced by tolerogenic DCs, such as IL-10, TGFβ, IL-35, and indoleamine 2,3-dioxygenase (IDO), induce the generation of antigen-specific Treg cells [206]. Retinoic acid, another important mediator of Treg induction, is produced by skin CD103−CD11b+ DCs in mice [207]. Strategies for antigen-specific tolerance induction, including those employing SC administration, have been reviewed by Luo et al. [208]. Certain skin-resident migratory cell populations, including dermal CD103+ DCs and LCs, have been targeted in such strategies to induce or expand Treg cells [205]. A reverse vaccination strategy employs SC pre-exposure to low-dose protein in the presence of OPLS to induce tolerance, and mice were rendered hyporesponsive upon re-exposure to protein alone [209, 210]. OPLS co-administration generates DCs with a tolerogenic profile including high secretion of regulatory TGFβ and normal migratory capability (Fig. 4). SC co-administration of OPLS and rhGAA in a mixed formulation induced hyporesponsiveness to rhGAA in Pompe disease mice [211]. Furthermore, reverse vaccination by SC pre-administration of Lyso-phosphatidylserine (Lyso-PS)-containing nanoparticles loaded with FVIII significantly reduced anti-FVIII antibody response during re-exposure to FVIII intravenously, the mechanism of which involved a specific PS receptor, TIM-4 [212].

Immune tolerance induction using SC co-administration of OPLS and therapeutic protein to mitigate immunogenicity. Top: (1) Uptake and processing of protein by skin-derived immature DCs licenses DC maturation and migration to DLNs. (2) DCs present peptide:MHC II complexes to naïve CD4+ T cells in T cell areas and induce (3) differentiation and proliferation of effector CD4+ T cells. (4) B cell activation and differentiation in germinal centers generates (5) memory B cells and plasma cells producing ADA (e.g., IgG). Bottom: (1) FVIII and OPLS are mixed immediately before SC administration. Uptake and processing of FVIII in the presence of OPLS by skin-derived immature DCs induces tolerogenic DCs, with downregulation of proinflammatory cytokine production and co-stimulatory molecule expression, but similar MHC II expression compared to mature DCs. (2) Tolerogenic DCs retain migratory activity and reach DLNs to present peptide:MHC II complexes to naïve CD4+ T cells in T cell areas. (3) Antigen presentation in the context of regulatory mediators with low co-stimulatory signals induces Treg cells. (4) Treg cells can suppress effector T cell function via metabolic disruption, adenosine A2A receptor activation, cytolysis, and deprivation of crucial cytokines (e.g. IL-2). (5) Treg cells produce key regulatory cytokines. (6) Interaction of Treg CTLA-4 with DC co-stimulatory molecule B7 induces tolerogenic DCs that suppress effector T cells via regulatory mediators [202,203,204, 209]. Note that steps 4–6 (bottom) represent general pathways of tolerance induction by Treg cells; the mechanism of OPLS-mediated hyporesponsiveness beyond tolerogenic DCs requires further investigation. ADA anti-drug antibody, CD40L CD40 ligand, CTLA-4 cytotoxic T lymphocyte-associated protein 4, DC dendritic cell, DLN draining lymph node, FVIII factor VIII, IDO indoleamine 2,3-dioxygenase, Ig immunoglobulin, IL interleukin, MHC II major histocompatibility complex II, OPLS O-phospho-L-serine, RA retinoic acid, SC subcutaneous, TCR T cell receptor, TGFβ tumor growth factor-β, TNFα tumor necrosis factor-α, Treg regulatory T cell
4 Conclusion
The SC route of administration provides convenient and non-inferior delivery of therapeutic proteins compared to IV infusion, but unwanted ADA response can occur upon repeated administration. Based on available preclinical and clinical studies, there is evidence both supporting and refuting the notion that the SC route of administration increases risk of immunogenicity. Mechanistic insight into molecular and cellular contributors that may drive immunogenicity of subcutaneously administered therapeutic proteins is critical to rationally develop safe and efficacious protein-based therapies. A key contributor may be the large population of dynamic APCs in the skin with high antigen processing efficiency and migratory activity. Product-related factors of immunogenicity are also particularly relevant to SC formulations. Molecular characteristics, presence of protein aggregates, and impurities have the potential to increase SC retention time, upregulate immune cell migration, and/or enhance local inflammation. Current mitigation strategies for immunogenicity are lacking antigen-specific, long-lasting effects. Mechanistic insights and risk factors for SC immunogenicity inspire future approaches to prevent or reduce immunogenicity, which merit further investigation.
References
U.S. Food and Drug Administration. Guidance for Industry: Immunogenicity Assessment for Therapeutic Protein Products. Silver Spring, MD: FDA; 2014. https://www.fda.gov/downloads/drugs/guidances/ucm338856.pdf Accessed 12 Jun 2020.
De Groot AS, Scott DW. Immunogenicity of protein therapeutics. Trends Immunol. 2007;28(11):482–90. https://doi.org/10.1016/j.it.2007.07.011.
Lee CA, Kessler CM, Varon D, Martinowitz U, Heim M, Jacquemin MG, et al. Factor VIII immunogenicity. Haemophilia. 1998;4(4):552–7. https://doi.org/10.1046/j.1365-2516.1998.440552.x.
Tieu P, Chan A, Matino D. Molecular mechanisms of inhibitor development in hemophilia. Mediterr J Hematol Infect Dis. 2020;12(1):e2020001. https://doi.org/10.4084/mjhid.2020.001.
Lee CA, Kessler CM, Varon D, Martinowitz U, Heim M, Hay CRM. Factor VIII inhibitors in mild and moderate-severity haemophilia A. Haemophilia. 1998;4(4):558–63. https://doi.org/10.1046/j.1365-2516.1998.440558.x.
Banugaria SG, Prater SN, Ng YK, Kobori JA, Finkel RS, Ladda RL, et al. The impact of antibodies on clinical outcomes in diseases treated with therapeutic protein: lessons learned from infantile Pompe disease. Genet Med. 2011;13(8):729–36. https://doi.org/10.1097/GIM.0b013e3182174703.
Dingman R, Balu-Iyer SV. Immunogenicity of Protein Pharmaceuticals. J Pharm Sci. 2019;108(5):1637–54. https://doi.org/10.1016/j.xphs.2018.12.014.
Bartelds GM, Wijbrandts CA, Nurmohamed MT, Stapel S, Lems WF, Aarden L, et al. Clinical response to adalimumab: relationship to anti-adalimumab antibodies and serum adalimumab concentrations in rheumatoid arthritis. Ann Rheum Dis. 2007;66(7):921–6. https://doi.org/10.1136/ard.2006.065615.
Edlund H, Steenholdt C, Ainsworth MA, Goebgen E, Brynskov J, Thomsen OO, et al. Magnitude of increased infliximab clearance imposed by anti-infliximab antibodies in crohn’s disease is determined by their concentration. AAPS J. 2017;19(1):223–33. https://doi.org/10.1208/s12248-016-9989-8.
Krishna M, Nadler SG. Immunogenicity to biotherapeutics – the role of anti-drug immune complexes. Front Immunol. 2016;7(21). https://doi.org/10.3389/fimmu.2016.00021.
Ross C, Clemmesen KM, Svenson M, Soelberg Sørensen P, Koch-Henriksen N, Lange Skovgaard G, et al. Immunogenicity of interferon-β in multiple sclerosis patients: influence of preparation, dosage, dose frequency, and route of administration. Ann Neurol. 2000;48(5):706–12. https://doi.org/10.1002/1531-8249(200011)48:5%3c706::AID-ANA3%3e3.0.CO;2-V.
Sorensen PS, Ross C, Clemmesen KM, Bendtzen K, Frederiksen JL, Jensen K, et al. Clinical importance of neutralising antibodies against interferon beta in patients with relapsing-remitting multiple sclerosis. The Lancet. 2003;362(9391):1184–91. https://doi.org/10.1016/S0140-6736(03)14541-2.
Bennett CL, Luminari S, Nissenson AR, Tallman MS, Klinge SA, McWilliams N, et al. Pure red-cell aplasia and epoetin therapy. N Engl J Med. 2004;351(14):1403–8. https://doi.org/10.1056/NEJMoa040528.
Li J, Yang C, Xia Y, Bertino A, Glaspy J, Roberts M, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood. 2001;98(12):3241–8. https://doi.org/10.1182/blood.V98.12.3241.
Warrier I, Ewenstein BM, Koerper MA, Shapiro A, Key N, DiMichele D, et al. Factor IX inhibitors and anaphylaxis in hemophilia B. J Pediatr Hematol Oncol. 1997;19(1):23–7. https://doi.org/10.1097/00043426-199701000-00003.
Bon A, Morfini M, Dini A, Mori F, Barni S, Gianluca S, et al. Desensitization and immune tolerance induction in children with severe factor IX deficiency; inhibitors and adverse reactions to replacement therapy: a case-report and literature review. Ital J Pediatrics. 2015;41(1):12. https://doi.org/10.1186/s13052-015-0116-8.
Bertolotto A, Malucchi S, Milano E, Castello A, Capobianco M, Mutani R. Interferon β neutralizing antibodies in multiple sclerosis: neutralizing activity and cross-reactivity with three different preparations. Immunopharmacology. 2000;48(2):95–100. https://doi.org/10.1016/S0162-3109(00)00182-X.
Khan OA, Dhib-Jalbut SS. Neutralizing antibodies to interferon β-1a and interferon β-1b in MS patients are cross-reactive. Neurology. 1998;51(6):1698. https://doi.org/10.1212/WNL.51.6.1698.
Sethu S, Govindappa K, Quinn P, Wadhwa M, Stebbings R, Boggild M, et al. Immunoglobulin G1 and immunoglobulin G4 antibodies in multiple sclerosis patients treated with IFNβ interact with the endogenous cytokine and activate complement. Clinical Immunology. 2013;148(2):177–85. https://doi.org/10.1016/j.clim.2013.05.008.
Sominanda A, Lundkvist M, Fogdell-Hahn A, Hemmer B, Hartung HP, Hillert J, et al. Inhibition of endogenous interferon beta by neutralizing antibodies against recombinant interferon beta. Arch Neurol. 2010;67(9):1095–101. https://doi.org/10.1001/archneurol.2010.218.
Casadevall N, Nataf J, Viron B, Kolta A, Kiladjian J-J, Martin-Dupont P, et al. Pure red-cell aplasia and antierythropoietin antibodies in patients treated with recombinant erythropoietin. N Engl J Med. 2002;346(7):469–75. https://doi.org/10.1056/NEJMoa011931.
Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS. T-cell dependent immunogenicity of protein therapeutics: Preclinical assessment and mitigation. Clinical Immunology. 2013;149(3):534–55. https://doi.org/10.1016/j.clim.2013.09.006.
Schellekens H. Factors influencing the immunogenicity of therapeutic proteins. Nephrol Dial Transpl. 2005;20(suppl_6):iv3–9. https://doi.org/10.1093/ndt/gfh1092.
Singh SK. Impact of Product-Related Factors on Immunogenicity of Biotherapeutics. J Pharm Sci. 2011;100(2):354–87. https://doi.org/10.1002/jps.22276.
Fathallah AM, Bankert RB, Balu-Iyer SV. Immunogenicity of subcutaneously administered therapeutic proteins–a mechanistic perspective. AAPS J. 2013;15(4):897–900. https://doi.org/10.1208/s12248-013-9510-6.
Schunk MK, Macallum GE. Applications and optimization of immunization procedures. Ilar j. 2005;46(3):241–57. https://doi.org/10.1093/ilar.46.3.241.
Malissen B, Tamoutounour S, Henri S. The origins and functions of dendritic cells and macrophages in the skin. Nat Rev Immunol. 2014;14(6):417–28. https://doi.org/10.1038/nri3683.
Turner MR, Balu-Iyer SV. Challenges and opportunities for the subcutaneous delivery of therapeutic proteins. J Pharm Sci. 2018;107(5):1247–60. https://doi.org/10.1016/j.xphs.2018.01.007.
Cook IF. Evidence based route of administration of vaccines. Hum Vaccin. 2008;4(1):67–73. https://doi.org/10.4161/hv.4.1.4747.
Richmond JM, Harris JE. Immunology and skin in health and disease. Cold Spring Harb Perspect Med. 2014;4(12):a015339-a. https://doi.org/10.1101/cshperspect.a015339.
Pasparakis M, Haase I, Nestle FO. Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol. 2014;14(5):289–301. https://doi.org/10.1038/nri3646.
Woo S-H, Lumpkin EA, Patapoutian A. Merkel cells and neurons keep in touch. Trends Cell Biol. 2015;25(2):74–81. https://doi.org/10.1016/j.tcb.2014.10.003.
Kashem SW, Haniffa M, Kaplan DH. Antigen-Presenting Cells in the Skin. Annu Rev Immunol. 2017;35(1):469–99. https://doi.org/10.1146/annurev-immunol-051116-052215.
Collin M, Milne P. Langerhans cell origin and regulation. Curr Opin Hematol. 2016;23(1):28–35. https://doi.org/10.1097/MOH.0000000000000202.
Wang Y, Szretter KJ, Vermi W, Gilfillan S, Rossini C, Cella M, et al. IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat Immunol. 2012;13(8):753–60. https://doi.org/10.1038/ni.2360.
Nakajima SMD, Igyártó BZP, Honda TMDP, Egawa GMDP, Otsuka AMDP, Hara-Chikuma MP, et al. Langerhans cells are critical in epicutaneous sensitization with protein antigen via thymic stromal lymphopoietin receptor signaling. J Allergy Clin Immunol. 2012;129(4):1048-55.e6. https://doi.org/10.1016/j.jaci.2012.01.063.
Klechevsky E, Morita R, Liu M, Cao Y, Coquery S, Thompson-Snipes L, et al. Functional specializations of human epidermal Langerhans cells and CD14+ dermal dendritic cells. Immunity. 2008;29(3):497–510. https://doi.org/10.1016/j.immuni.2008.07.013.
Levin C, Bonduelle O, Nuttens C, Primard C, Verrier B, Boissonnas A, et al. Critical role for skin-derived migratory DCs and Langerhans cells in TFH and GC responses after intradermal immunization. J Investig Dermatol. 2017;137(9):1905–13. https://doi.org/10.1016/j.jid.2017.04.016.
Celluzzi CM, Falo LD Jr. Epidermal dendritic cells induce potent antigen-specific CTL-mediated immunity. J Invest Dermatol. 1997;108(5):716–20. https://doi.org/10.1111/1523-1747.ep12292095.
Kaplan DH, Igyártó BZ, Gaspari AA. Early immune events in the induction of allergic contact dermatitis. Nat Rev Immunol. 2012;12(2):114–24. https://doi.org/10.1038/nri3150.
Dioszeghy V, Mondoulet L, Laoubi L, Dhelft V, Plaquet C, Bouzereau A, et al. Antigen uptake by langerhans cells is required for the induction of regulatory T cells and the acquisition of tolerance during epicutaneous immunotherapy in OVA-sensitized mice. Front Immunol. 2018;9:1951. https://doi.org/10.3389/fimmu.2018.01951.
LeBleu VS, Macdonald B, Kalluri R. Structure and function of basement membranes. Exp Biol Med (Maywood). 2007;232(9):1121–9. https://doi.org/10.3181/0703-mr-72.
Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2010;123(Pt 24):4195–200. https://doi.org/10.1242/jcs.023820.
Franken L, Schiwon M, Kurts C. Macrophages: sentinels and regulators of the immune system. Cell Microbiol. 2016;18(4):475–87. https://doi.org/10.1111/cmi.12580.
Abtin A, Jain R, Mitchell AJ, Roediger B, Brzoska AJ, Tikoo S, et al. Perivascular macrophages mediate neutrophil recruitment during bacterial skin infection. Nat Immunol. 2014;15(1):45–53. https://doi.org/10.1038/ni.2769.
Waithman J, Allan RS, Kosaka H, Azukizawa H, Shortman K, Lutz MB, et al. Skin-derived dendritic cells can mediate deletional tolerance of class I-restricted self-reactive T cells. J Immunol. 2007;179(7):4535–41. https://doi.org/10.4049/jimmunol.179.7.4535.
Mirrashed F, Sharp JC, Krause V, Morgan J, Tomanek B. Pilot study of dermal and subcutaneous fat structures by MRI in individuals who differ in gender, BMI, and cellulite grading. Skin Res Technol. 2004;10(3):161–8. https://doi.org/10.1111/j.1600-0846.2004.00072.x.
Jackisch C, Müller V, Maintz C, Hell S, Ataseven B. Subcutaneous administration of monoclonal antibodies in oncology. Geburtshilfe Frauenheilkd. 2014;74(4):343–9. https://doi.org/10.1055/s-0034-1368173.
Richter WF, Bhansali SG, Morris ME. Mechanistic determinants of biotherapeutics absorption following SC administration. AAPS J. 2012;14(3):559–70. https://doi.org/10.1208/s12248-012-9367-0.
Schaefer L, Schaefer RM. Proteoglycans: from structural compounds to signaling molecules. Cell Tissue Res. 2010;339(1):237–46. https://doi.org/10.1007/s00441-009-0821-y.
Zijlstra E, Jahnke J, Fischer A, Kapitza C, Forst T. Impact of injection speed, volume, and site on pain sensation. J Diabetes Sci Technol. 2018;12(1):163–8. https://doi.org/10.1177/1932296817735121.
Frost GI. Recombinant human hyaluronidase (rHuPH20): an enabling platform for subcutaneous drug and fluid administration. Expert Opin Drug Deliv. 2007;4(4):427–40. https://doi.org/10.1517/17425247.4.4.427.
Locke KW, Maneval DC, LaBarre MJ. ENHANZE(®) drug delivery technology: a novel approach to subcutaneous administration using recombinant human hyaluronidase PH20. Drug Deliv. 2019;26(1):98–106. https://doi.org/10.1080/10717544.2018.1551442.
Segura E, Valladeau-Guilemond J, Donnadieu MH, Sastre-Garau X, Soumelis V, Amigorena S. Characterization of resident and migratory dendritic cells in human lymph nodes. J Exp Med. 2012;209(4):653–60. https://doi.org/10.1084/jem.20111457.
Gerner MY, Mescher MF. Antigen processing and MHC-II presentation by dermal and tumor-infiltrating dendritic cells. J Immunol. 2009;182(5):2726–37. https://doi.org/10.4049/jimmunol.0803479.
Ohl L, Mohaupt M, Czeloth N, Hintzen G, Kiafard Z, Zwirner J, et al. CCR7 governs skin dendritic cell migration under inflammatory and steady-state conditions. Immunity. 2004;21(2):279–88. https://doi.org/10.1016/j.immuni.2004.06.014.
Tomura M, Hata A, Matsuoka S, Shand FH, Nakanishi Y, Ikebuchi R, et al. Tracking and quantification of dendritic cell migration and antigen trafficking between the skin and lymph nodes. Sci Rep. 2014;4:6030. https://doi.org/10.1038/srep06030.
Comerford I, Harata-Lee Y, Bunting MD, Gregor C, Kara EE, McColl SR. A myriad of functions and complex regulation of the CCR7/CCL19/CCL21 chemokine axis in the adaptive immune system. Cytokine Growth Factor Rev. 2013;24(3):269–83. https://doi.org/10.1016/j.cytogfr.2013.03.001.
Stutte S, Quast T, Gerbitzki N, Savinko T, Novak N, Reifenberger J, et al. Requirement of CCL17 for CCR7- and CXCR4-dependent migration of cutaneous dendritic cells. Proc Natl Acad Sci USA. 2010;107(19):8736–41. https://doi.org/10.1073/pnas.0906126107.
Kitajima M, Ziegler SF. Cutting edge: identification of the thymic stromal lymphopoietin-responsive dendritic cell subset critical for initiation of type 2 contact hypersensitivity. J Immunol (Baltimore, Md: 1950). 2013;191(10):4903–7. https://doi.org/10.4049/jimmunol.1302175.
Chakarov S, Fazilleau N. Monocyte-derived dendritic cells promote T follicular helper cell differentiation. EMBO Mol Med. 2014;6(5):590–603. https://doi.org/10.1002/emmm.201403841.
Wang W, Wang EQ, Balthasar JP. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 2008;84(5):548–58. https://doi.org/10.1038/clpt.2008.170.
McLennan DN, Porter CJ, Charman SA. Subcutaneous drug delivery and the role of the lymphatics. Drug Discov Today Technol. 2005;2(1):89–96. https://doi.org/10.1016/j.ddtec.2005.05.006.
Porter CJ, Charman SA. Lymphatic transport of proteins after subcutaneous administration. J Pharm Sci. 2000;89(3):297–310. https://doi.org/10.1002/(SICI)1520-6017(200003)89:3%3c297::AID-JPS2%3e3.0.CO;2-P.
Moore JE Jr, Bertram CD. Lymphatic system flows. Annu Rev Fluid Mech. 2018;50:459–82. https://doi.org/10.1146/annurev-fluid-122316-045259.
Richter WF, Jacobsen B. Subcutaneous absorption of biotherapeutics: knowns and unknowns. Drug Metab Dispos. 2014;42(11):1881. https://doi.org/10.1124/dmd.114.059238.
Skobe M, Detmar M. Structure, function, and molecular control of the skin lymphatic system. J Investig Dermatol Symp Proc. 2000;5(1):14–9. https://doi.org/10.1046/j.1087-0024.2000.00001.x.
Swartz MA. The physiology of the lymphatic system. Adv Drug Deliv Rev. 2001;50(1–2):3–20. https://doi.org/10.1016/s0169-409x(01)00150-8.
Itano AA, McSorley SJ, Reinhardt RL, Ehst BD, Ingulli E, Rudensky AY, et al. Distinct dendritic cell populations sequentially present antigen to CD4 T cells and stimulate different aspects of cell-mediated immunity. Immunity. 2003;19(1):47–57. https://doi.org/10.1016/s1074-7613(03)00175-4.
Matucci A, Vultaggio A, Danesi R. The use of intravenous versus subcutaneous monoclonal antibodies in the treatment of severe asthma: a review. Respir Res. 2018;19(1):154. https://doi.org/10.1186/s12931-018-0859-z.
Haniffa M, Gunawan M, Jardine L. Human skin dendritic cells in health and disease. J Dermatol Sci. 2015;77(2):85–92. https://doi.org/10.1016/j.jdermsci.2014.08.012.
Kijanka G, Prokopowicz M, Schellekens H, Brinks V. Influence of aggregation and route of injection on the biodistribution of mouse serum albumin. PLoS ONE. 2014;9(1):e85281. https://doi.org/10.1371/journal.pone.0085281.
Hamuro L, Kijanka G, Kinderman F, Kropshofer H, Bu DX, Zepeda M, et al. Perspectives on subcutaneous route of administration as an immunogenicity risk factor for therapeutic proteins. J Pharm Sci. 2017;106(10):2946–54. https://doi.org/10.1016/j.xphs.2017.05.030.
Vugmeyster Y, Xu X, Theil F-P, Khawli LA, Leach MW. Pharmacokinetics and toxicology of therapeutic proteins: Advances and challenges. World J Biol Chem. 2012;3(4):73–92. https://doi.org/10.4331/wjbc.v3.i4.73.
Hume DA. Differentiation and heterogeneity in the mononuclear phagocyte system. Mucosal Immunol. 2008;1(6):432–41. https://doi.org/10.1038/mi.2008.36.
Finbloom DS, Abeles D, Rifai A, Plotz PH. The specificity of uptake of model immune complexes and other protein aggregates by the murine reticuloendothelial system. J Immunol. 1980;125(3):1060–5.
Richman LK, Klingenstein RJ, Richman JA, Strober W, Berzofsky JA. The Murine Kupffer Cell. J Immunol. 1979;123(6):2602.
Lewis SM, Williams A, Eisenbarth SC. Structure and function of the immune system in the spleen. Sci Immunol. 2019;4(33):eaau6085. https://doi.org/10.1126/sciimmunol.aau6085.
Reif K, Ekland EH, Ohl L, Nakano H, Lipp M, Förster R, et al. Balanced responsiveness to chemoattractants from adjacent zones determines B-cell position. Nature. 2002;416(6876):94–9. https://doi.org/10.1038/416094a.
Velásquez-Lopera MM, Correa LA, García LF. Human spleen contains different subsets of dendritic cells and regulatory T lymphocytes. Clin Exp Immunol. 2008;154(1):107–14. https://doi.org/10.1111/j.1365-2249.2008.03734.x.
Gunn MD, Kyuwa S, Tam C, Kakiuchi T, Matsuzawa A, Williams LT, et al. Mice lacking expression of secondary lymphoid organ chemokine have defects in lymphocyte homing and dendritic cell localization. J Exp Med. 1999;189(3):451–60. https://doi.org/10.1084/jem.189.3.451.
Bronte V, Pittet MJ. The spleen in local and systemic regulation of immunity. Immunity. 2013;39(5):806–18. https://doi.org/10.1016/j.immuni.2013.10.010.
Hey YY, O’Neill HC. Murine spleen contains a diversity of myeloid and dendritic cells distinct in antigen presenting function. J Cell Mol Med. 2012;16(11):2611–9. https://doi.org/10.1111/j.1582-4934.2012.01608.x.
Rojko JL, Evans MG, Price SA, Han B, Waine G, DeWitte M, et al. Formation, clearance, deposition, pathogenicity, and identification of biopharmaceutical-related immune complexes: review and case studies. Toxicol Pathol. 2014;42(4):725–64. https://doi.org/10.1177/0192623314526475.
Navarrete A, Dasgupta S, Delignat S, Caligiuri G, Christophe OD, Bayry J, et al. Splenic marginal zone antigen-presenting cells are critical for the primary allo-immune response to therapeutic factor VIII in hemophilia A. J Thromb Haemost. 2009;7(11):1816–23. https://doi.org/10.1111/j.1538-7836.2009.03571.x.
Cataldi M, Vigliotti C, Mosca T, Cammarota M, Capone D. Emerging role of the spleen in the pharmacokinetics of monoclonal antibodies, nanoparticles and exosomes. Int J Mol Sci. 2017;18(6):1249. https://doi.org/10.3390/ijms18061249.
Calabro S, Liu D, Gallman A, Nascimento MS, Yu Z, Zhang TT, et al. Differential Intrasplenic Migration of Dendritic Cell Subsets Tailors Adaptive Immunity. Cell Rep. 2016;16(9):2472–85. https://doi.org/10.1016/j.celrep.2016.07.076.
Jounai N, Kobiyama K, Takeshita F, Ishii KJ. Recognition of damage-associated molecular patterns related to nucleic acids during inflammation and vaccination. Front Cell Infect Microbiol. 2013;2:168. https://doi.org/10.3389/fcimb.2012.00168.
Nace G, Evankovich J, Eid R, Tsung A. Dendritic cells and damage-associated molecular patterns: endogenous danger signals linking innate and adaptive immunity. J Innate Immun. 2012;4(1):6–15. https://doi.org/10.1159/000334245.
Förster R, Braun A, Worbs T. Lymph node homing of T cells and dendritic cells via afferent lymphatics. Trends Immunol. 2012;33(6):271–80. https://doi.org/10.1016/j.it.2012.02.007.
Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, et al. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195(1):99–111. https://doi.org/10.1084/jem.20001858.
Iezzi G, Frohlich A, Ernst B, Ampenberger F, Saeland S, Glaichenhaus N, et al. Lymph node resident rather than skin-derived dendritic cells initiate specific T cell responses after Leishmania major infection. J Immunol. 2006;177(2):1250–6. https://doi.org/10.4049/jimmunol.177.2.1250.
Allenspach EJ, Lemos MP, Porrett PM, Turka LA, Laufer TM. Migratory and lymphoid-resident dendritic cells cooperate to efficiently prime naive CD4 T cells. Immunity. 2008;29(5):795–806. https://doi.org/10.1016/j.immuni.2008.08.013.
Kabashima K, Shiraishi N, Sugita K, Mori T, Onoue A, Kobayashi M, et al. CXCL12-CXCR4 engagement is required for migration of cutaneous dendritic cells. Am J Pathol. 2007;171(4):1249–57. https://doi.org/10.2353/ajpath.2007.070225.
Hwang ST. Homeward bound: how do skin dendritic cells find their way into the lymph system? J Invest Dermatol. 2012;132(4):1070–3. https://doi.org/10.1038/jid.2012.39.
Yen JH, Khayrullina T, Ganea D. PGE2-induced metalloproteinase-9 is essential for dendritic cell migration. Blood. 2008;111(1):260–70. https://doi.org/10.1182/blood-2007-05-090613.
Celli S, Garcia Z, Bousso P. CD4 T cells integrate signals delivered during successive DC encounters in vivo. J Exp Med. 2005;202(9):1271–8. https://doi.org/10.1084/jem.20051018.
Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006;25(1):153–62. https://doi.org/10.1016/j.immuni.2006.04.017.
Dasgupta S, Navarrete A-M, Bayry J, Delignat S, Wootla B, André S, et al. A role for exposed mannosylations in presentation of human therapeutic self-proteins to CD4+ T lymphocytes. Proc Natl Acad Sci USA. 2007;104(21):8965–70. https://doi.org/10.1073/pnas.0702120104.
Linehan SA. The mannose receptor is expressed by subsets of APC in non-lymphoid organs. BMC Immunol. 2005;6:4. https://doi.org/10.1186/1471-2172-6-4.
McKenzie EJ, Taylor PR, Stillion RJ, Lucas AD, Harris J, Gordon S, et al. Mannose receptor expression and function define a new population of murine dendritic cells. J Immunol. 2007;178(8):4975. https://doi.org/10.4049/jimmunol.178.8.4975.
Ye L, Liu X, Rout SN, Li Z, Yan Y, Lu L, et al. The MHC class II-associated invariant chain interacts with the neonatal Fc gamma receptor and modulates its trafficking to endosomal/lysosomal compartments. J Immunol. 2008;181(4):2572–85. https://doi.org/10.4049/jimmunol.181.4.2572.
Ward ES, Ober RJ. Chapter 4: multitasking by exploitation of intracellular transport functions the many faces of FcRn. Adv Immunol. 2009;103:77–115. https://doi.org/10.1016/s0065-2776(09)03004-1.
Akilesh S, Christianson GJ, Roopenian DC, Shaw AS. Neonatal FcR expression in bone marrow-derived cells functions to protect serum IgG from catabolism. J Immunol. 2007;179(7):4580–8. https://doi.org/10.4049/jimmunol.179.7.4580.
Pyzik M, Rath T, Lencer WI, Baker K, Blumberg RS. FcRn: the architect behind the immune and nonimmune functions of IgG and albumin. J Immunol. 2015;194(10):4595. https://doi.org/10.4049/jimmunol.1403014.
Qiao SW, Kobayashi K, Johansen FE, Sollid LM, Andersen JT, Milford E, et al. Dependence of antibody-mediated presentation of antigen on FcRn. Proc Natl Acad Sci USA. 2008;105(27):9337–42. https://doi.org/10.1073/pnas.0801717105.
Clatworthy MR, Aronin CEP, Mathews RJ, Morgan NY, Smith KGC, Germain RN. Immune complexes stimulate CCR7-dependent dendritic cell migration to lymph nodes. Nat Med. 2014;20(12):1458–63. https://doi.org/10.1038/nm.3709.
Peng A, Gaitonde P, Kosloski MP, Miclea RD, Varma P, Balu-Iyer SV. Effect of route of administration of human recombinant factor VIII on its immunogenicity in Hemophilia A mice. J Pharm Sci. 2009;98(12):4480–4. https://doi.org/10.1002/jps.21765.
Chirmule N, Jawa V, Meibohm B. Immunogenicity to therapeutic proteins: impact on PK/PD and efficacy. AAPS J. 2012;14(2):296–302. https://doi.org/10.1208/s12248-012-9340-y.
Braun A, Kwee L, Labow MA, Alsenz J. Protein aggregates seem to play a key role among the parameters influencing the antigenicity of interferon alpha (IFN-alpha) in normal and transgenic mice. Pharm Res. 1997;14(10):1472–8. https://doi.org/10.1023/a:1012193326789.
Kijanka G, Jiskoot W, Schellekens H, Brinks V. Effect of treatment regimen on the immunogenicity of human interferon Beta in immune tolerant mice. Pharm Res. 2013;30(6):1553–60. https://doi.org/10.1007/s11095-013-0992-9.
van Beers MM, Jiskoot W, Schellekens H. On the role of aggregates in the immunogenicity of recombinant human interferon beta in patients with multiple sclerosis. J Interferon Cytokine Res. 2010;30(10):767–75. https://doi.org/10.1089/jir.2010.0086.
Christie M, Torres RM, Kedl RM, Randolph TW, Carpenter JF. Recombinant murine growth hormone particles are more immunogenic with intravenous than subcutaneous administration. J Pharm Sci. 2014;103(1):128–39. https://doi.org/10.1002/jps.23794.
Bartelds GM, Krieckaert CL, Nurmohamed MT, van Schouwenburg PA, Lems WF, Twisk JW, et al. Development of antidrug antibodies against adalimumab and association with disease activity and treatment failure during long-term follow-up. JAMA. 2011;305(14):1460–8. https://doi.org/10.1001/jama.2011.406.
Herceptin Hylecta [package insert]. South San Francisco, CA: Genentech, Inc.; 2019 (revised). https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761106s000lbl.pdf. Accessed 7 Oct 2020.
Davies A, Merli F, Mihaljević B, Mercadal S, Siritanaratkul N, Solal-Céligny P, et al. Efficacy and safety of subcutaneous rituximab versus intravenous rituximab for first-line treatment of follicular lymphoma (SABRINA): a randomised, open-label, phase 3 trial. Lancet Haematol. 2017;4(6):e272–82. https://doi.org/10.1016/s2352-3026(17)30078-9.
Rituxan Hycela [package insert]. South San Francisco, CA: Genentech, Inc.; 2020 (revised). https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761064s008s010lbl.pdf. Accessed 7 Oct 2020.
Rosengren S, Dychter SS, Printz MA, Huang L, Schiff RI, Schwarz H-P, et al. Clinical immunogenicity of rHuPH20, a hyaluronidase enabling subcutaneous drug administration. AAPS J. 2015;17(5):1144–56. https://doi.org/10.1208/s12248-015-9782-0.
Rosengren S, Souratha J, Conway D, Muchmore DB, Sugarman BJ. Recombinant human PH20: baseline analysis of the reactive antibody prevalence in the general population using healthy subjects. BioDrugs. 2018;32(1):83–9. https://doi.org/10.1007/s40259-018-0260-y.
Omontys [package insert]. Deerfield, IL: Takeda Pharmaceuticals America, Inc.; 2012 (revised). https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/202799s001lbl.pdf Accessed 10 Oct 2020.
Ortega HG, Liu MC, Pavord ID, Brusselle GG, FitzGerald JM, Chetta A, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371(13):1198–207. https://doi.org/10.1056/NEJMoa1403290.
Zhuang Y, Xu Z, Frederick B, de Vries DE, Ford JA, Keen M, et al. Golimumab pharmacokinetics after repeated subcutaneous and intravenous administrations in patients with rheumatoid arthritis and the effect of concomitant methotrexate: an open-label, randomized study. Clin Ther. 2012;34(1):77–90. https://doi.org/10.1016/j.clinthera.2011.11.015.
Phesgo [package insert]. South San Francisco, CA: Genentech, Inc.; 2020 (revised). https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761170s000lbl.pdf. Accessed 7 Oct 2020.
Mateos MV, Nahi H, Legiec W, Grosicki S, Vorobyev V, Spicka I, et al. Subcutaneous versus intravenous daratumumab in patients with relapsed or refractory multiple myeloma (COLUMBA): a multicentre, open-label, non-inferiority, randomised, phase 3 trial. Lancet Haematol. 2020;7(5):e370–80. https://doi.org/10.1016/s2352-3026(20)30070-3.
Sandborn WJ, Baert F, Danese S, Krznaric Z, Kobayashi T, Yao X, et al. Efficacy and safety of vedolizumab subcutaneous formulation in a randomized trial of patients with ulcerative colitis. Gastroenterology. 2020;158(3):562-72e12. https://doi.org/10.1053/j.gastro.2019.08.027.
Agius MA, Klodowska-Duda G, Maciejowski M, Potemkowski A, Li J, Patra K, et al. Safety and tolerability of inebilizumab (MEDI-551), an anti-CD19 monoclonal antibody, in patients with relapsing forms of multiple sclerosis: Results from a phase 1 randomised, placebo-controlled, escalating intravenous and subcutaneous dose study. Mult Scler. 2019;25(2):235–45. https://doi.org/10.1177/1352458517740641.
Parnes JR, Sullivan JT, Chen L, Dias C. Pharmacokinetics, safety, and tolerability of tezepelumab (AMG 157) in healthy and atopic dermatitis adult subjects. Clin Pharmacol Ther. 2019;106(2):441–9. https://doi.org/10.1002/cpt.1401.
Genovese MC, Covarrubias A, Leon G, Mysler E, Keiserman M, Valente R, et al. Subcutaneous abatacept versus intravenous abatacept: a phase IIIb noninferiority study in patients with an inadequate response to methotrexate. Arthritis Rheum. 2011;63(10):2854–64. https://doi.org/10.1002/art.30463.
Genovese MC, Pacheco-Tena C, Covarrubias A, Leon G, Mysler E, Keiserman M, et al. Longterm safety and efficacy of subcutaneous abatacept in patients with rheumatoid arthritis: 5-year results from a phase IIIb trial. J Rheumatol. 2018;45(8):1085–92. https://doi.org/10.3899/jrheum.170344.
Schiff M. Subcutaneous abatacept for the treatment of rheumatoid arthritis. Rheumatology (Oxford). 2013;52(6):986–97. https://doi.org/10.1093/rheumatology/ket018.
Ruperto N, Lovell DJ, Quartier P, Paz E, Rubio-Pérez N, Silva CA, et al. Long-term safety and efficacy of abatacept in children with juvenile idiopathic arthritis. Arthritis Rheum. 2010;62(6):1792–802. https://doi.org/10.1002/art.27431.
Ogata A, Tanimura K, Sugimoto T, Inoue H, Urata Y, Matsubara T, et al. Phase III study of the efficacy and safety of subcutaneous versus intravenous tocilizumab monotherapy in patients with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2014;66(3):344–54. https://doi.org/10.1002/acr.22110.
Burmester GR, Rubbert-Roth A, Cantagrel A, Hall S, Leszczynski P, Feldman D, et al. A randomised, double-blind, parallel-group study of the safety and efficacy of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional disease-modifying antirheumatic drugs in patients with moderate to severe rheumatoid arthritis (SUMMACTA study). Ann Rheum Dis. 2014;73(1):69–74. https://doi.org/10.1136/annrheumdis-2013-203523.
Burmester GR, Rubbert-Roth A, Cantagrel A, Hall S, Leszczynski P, Feldman D, et al. Efficacy and safety of subcutaneous tocilizumab versus intravenous tocilizumab in combination with traditional DMARDs in patients with RA at week 97 (SUMMACTA). Ann Rheum Dis. 2016;75(1):68–74. https://doi.org/10.1136/annrheumdis-2015-207281.
Burmester GR, Choy E, Kivitz A, Ogata A, Bao M, Nomura A, et al. Low immunogenicity of tocilizumab in patients with rheumatoid arthritis. Ann Rheum Dis. 2017;76(6):1078–85. https://doi.org/10.1136/annrheumdis-2016-210297.
Sigaux J, Hamze M, Daien C, Morel J, Krzysiek R, Pallardy M, et al. Immunogenicity of tocilizumab in patients with rheumatoid arthritis. Jt Bone Spine. 2017;84(1):39–45. https://doi.org/10.1016/j.jbspin.2016.04.013.
Banfield C, Rudin D, Bhattacharya I, Goteti K, Li G, Hassan-Zahraee M, et al. First-in-human, randomized dose-escalation study of the safety, tolerability, pharmacokinetics, pharmacodynamics and immunogenicity of PF-06480605 in healthy subjects. Br J Clin Pharmacol. 2020;86(4):812–24. https://doi.org/10.1111/bcp.14187.
Johnson ML, Braiteh F, Grilley-Olson JE, Chou J, Davda J, Forgie A, et al. Assessment of subcutaneous vs intravenous administration of anti-PD-1 antibody PF-06801591 in patients with advanced solid tumors: a phase 1 dose-escalation trial. JAMA Oncol. 2019;5(7):999–1007. https://doi.org/10.1001/jamaoncol.2019.0836.
Supersaxo A, Hein WR, Steffen H. Effect of molecular weight on the lymphatic absorption of water-soluble compounds following subcutaneous administration. Pharm Res. 1990;7(2):167–9. https://doi.org/10.1023/a:1015880819328.
Charman SA, McLennan DN, Edwards GA, Porter CJ. Lymphatic absorption is a significant contributor to the subcutaneous bioavailability of insulin in a sheep model. Pharm Res. 2001;18(11):1620–6. https://doi.org/10.1023/a:1013046918190.
Wu F, Bhansali SG, Law WC, Bergey EJ, Prasad PN, Morris ME. Fluorescence imaging of the lymph node uptake of proteins in mice after subcutaneous injection: molecular weight dependence. Pharm Res. 2012;29(7):1843–53. https://doi.org/10.1007/s11095-012-0708-6.
Xie D, Hale VG. Factors affecting the lymphatic absorption of macromolecules following extravascular administration. Pharm Res. 1996;13(9 Suppl.):S-396.
Kinnunen HM, Mrsny RJ. Improving the outcomes of biopharmaceutical delivery via the subcutaneous route by understanding the chemical, physical and physiological properties of the subcutaneous injection site. J Control Release. 2014;182:22–32. https://doi.org/10.1016/j.jconrel.2014.03.011.
Peyron I, Dimitrov JD, Delignat S, Gangadharan B, Srivastava A, Kaveri SV, et al. Oxidation of factor VIII increases its immunogenicity in mice with severe hemophilia A. Cell Immunol. 2018;325:64–8. https://doi.org/10.1016/j.cellimm.2018.01.008.
Alderman CJJ, Shah S, Foreman JC, Chain BM, Katz DR. The role of advanced oxidation protein products in regulation of dendritic cell function. Free Radic Biol Med. 2002;32(5):377–85. https://doi.org/10.1016/S0891-5849(01)00735-3.
Kuriakose A, Chirmule N, Nair P. Immunogenicity of biotherapeutics: causes and association with posttranslational modifications. J Immunol Res. 2016. https://doi.org/10.1155/2016/1298473.
Hermeling S, Schellekens H, Maas C, Gebbink MFBG, Crommelin DJA, Jiskoot W. Antibody response to aggregated human interferon alpha2b in wild-type and transgenic immune tolerant mice depends on type and level of aggregation. J Pharm Sci. 2006;95(5):1084–96. https://doi.org/10.1002/jps.20599.
Hogenesch H. Mechanism of immunopotentiation and safety of aluminum adjuvants. Front Immunol. 2013;3:406. https://doi.org/10.3389/fimmu.2012.00406.
Ratanji KD, Derrick JP, Dearman RJ, Kimber I. Immunogenicity of therapeutic proteins: influence of aggregation. J Immunotoxicol. 2014;11(2):99–109. https://doi.org/10.3109/1547691x.2013.821564.
Poole, RA, Hawe A, Jiskoot W, Braeckmans K (2012) Fluorescence spectroscopy to characterize protein aggregates and particles. In: Mahler H-C, Jiskoot W (eds) Analysis of aggregates and particles in protein pharmaceuticals. https://doi.org/10.1002/9781118150573.ch9
Narhi LO, Schmit J, Bechtold-Peters K, Sharma D. Classification of protein aggregates. J Pharm Sci. 2012;101(2):493–8. https://doi.org/10.1002/jps.22790.
Filipe V, Jiskoot W, Basmeleh AH, Halim A, Schellekens H, Brinks V. Immunogenicity of different stressed IgG monoclonal antibody formulations in immune tolerant transgenic mice. mAbs. 2012;4(6):740–52. https://doi.org/10.4161/mabs.22066.
Fathallah AM, Chiang M, Mishra A, Kumar S, Xue L, Middaugh R, et al. The effect of small oligomeric protein aggregates on the immunogenicity of intravenous and subcutaneous administered antibodies. J Pharm Sci. 2015;104(11):3691–702. https://doi.org/10.1002/jps.24592.
Moussa EM, Panchal JP, Moorthy BS, Blum JS, Joubert MK, Narhi LO, et al. Immunogenicity of therapeutic protein aggregates. J Pharm Sci. 2016;105(2):417–30. https://doi.org/10.1016/j.xphs.2015.11.002.
Kijanka G, Bee JS, Korman SA, Wu Y, Roskos LK, Schenerman MA, et al. Submicron size particles of a murine monoclonal antibody are more immunogenic than soluble oligomers or micron size particles upon subcutaneous administration in mice. J Pharm Sci. 2018;107(11):2847–59. https://doi.org/10.1016/j.xphs.2018.06.029.
Ratanji KD, Dearman RJ, Kimber I, Thorpe R, Wadhwa M, Derrick JP. Editor’s highlight: subvisible aggregates of immunogenic proteins promote a Th1-type response. Toxicol Sci. 2016;153(2):258–70. https://doi.org/10.1093/toxsci/kfw121.
Eyes TJ, Austerberry JI, Dearman RJ, Johannissen LO, Kimber I, Smith N, et al. Identification of B cell epitopes enhanced by protein unfolding and aggregation. Mol Immunol. 2019;105:181–9. https://doi.org/10.1016/j.molimm.2018.11.020.
Kraus T, Lubitz A, Schließer U, Giese C, Reuschel J, Brecht R, et al. Evaluation of a 3D human artificial lymph node as test model for the assessment of immunogenicity of protein aggregates. J Pharm Sci. 2019;108(7):2358–66. https://doi.org/10.1016/j.xphs.2019.02.011.
Bi V, Jawa V, Joubert MK, Kaliyaperumal A, Eakin C, Richmond K, et al. Development of a human antibody tolerant mouse model to assess the immunogenicity risk due to aggregated biotherapeutics. J Pharm Sci. 2013;102(10):3545–55. https://doi.org/10.1002/jps.23663.
Rosenberg AS. Effects of protein aggregates: an immunologic perspective. AAPS J. 2006;8(3):E501–7. https://doi.org/10.1208/aapsj080359.
Bessa J, Boeckle S, Beck H, Buckel T, Schlicht S, Ebeling M, et al. The immunogenicity of antibody aggregates in a novel transgenic mouse model. Pharm Res. 2015;32(7):2344–59. https://doi.org/10.1007/s11095-015-1627-0.
Filipe V, Que I, Carpenter JF, Löwik C, Jiskoot W. In vivo fluorescence imaging of IgG1 aggregates after subcutaneous and intravenous injection in mice. Pharm Res. 2014;31(1):216–27. https://doi.org/10.1007/s11095-013-1154-9.
Kijanka G, Bee JS, Bishop SM, Que I, Lowik C, Jiskoot W. Fate of multimeric oligomers, submicron, and micron size aggregates of monoclonal antibodies upon subcutaneous injection in mice. J Pharm Sci. 2016;105(5):1693–704. https://doi.org/10.1016/j.xphs.2016.02.034.
Filipe V, Poole R, Oladunjoye O, Braeckmans K, Jiskoot W. Detection and characterization of subvisible aggregates of monoclonal IgG in serum. Pharm Res. 2012;29(8):2202–12. https://doi.org/10.1007/s11095-012-0749-x.
Joubert MK, Hokom M, Eakin C, Zhou L, Deshpande M, Baker MP, et al. Highly aggregated antibody therapeutics can enhance the in vitro innate and late-stage T-cell immune responses. J Biol Chem. 2012;287(30):25266–79. https://doi.org/10.1074/jbc.M111.330902.
Ahmadi M, Bryson CJ, Cloake EA, Welch K, Filipe V, Romeijn S, et al. Small amounts of sub-visible aggregates enhance the immunogenic potential of monoclonal antibody therapeutics. Pharm Res. 2015;32(4):1383–94. https://doi.org/10.1007/s11095-014-1541-x.
Carpenter JF, Randolph TW, Jiskoot W, Crommelin DJ, Middaugh CR, Winter G, et al. Overlooking subvisible particles in therapeutic protein products: gaps that may compromise product quality. J Pharm Sci. 2009;98(4):1201–5. https://doi.org/10.1002/jps.21530.
Reis e Sousa C, Germain RN. Analysis of adjuvant function by direct visualization of antigen presentation in vivo: endotoxin promotes accumulation of antigen-bearing dendritic cells in the T cell areas of lymphoid tissue. J Immunol (Baltimore, Md: 1950). 1999;162(11):6552–61.
Zhong G, Reis e Sousa C, Germain RN. Antigen-unspecific B cells and lymphoid dendritic cells both show extensive surface expression of processed antigen-major histocompatibility complex class II complexes after soluble protein exposure in vivo or in vitro. J Exp Med. 1997;186(5):673–82. https://doi.org/10.1084/jem.186.5.673.
Verthelyi D, Wang V. Trace levels of innate immune response modulating impurities (IIRMIs) synergize to break tolerance to therapeutic proteins. PLoS ONE. 2010;5(12):e15252-e. https://doi.org/10.1371/journal.pone.0015252.
Haile LA, Puig M, Kelley-Baker L, Verthelyi D. Detection of innate immune response modulating impurities in therapeutic proteins. PLoS ONE. 2015;10(4):e15252-e. https://doi.org/10.1371/journal.pone.0125078.
Uchino T, Miyazaki Y, Yamazaki T, Kagawa Y. Immunogenicity of protein aggregates of a monoclonal antibody generated by forced shaking stress with siliconized and nonsiliconized syringes in BALB/c mice. J Pharm Pharmacol. 2017;69(10):1341–51. https://doi.org/10.1111/jphp.12765.
Vlieland ND, Nejadnik MR, Gardarsdottir H, Romeijn S, Sediq AS, Bouvy ML, et al. The impact of inadequate temperature storage conditions on aggregate and particle formation in drugs containing tumor necrosis factor-alpha inhibitors. Pharm Res. 2018;35(2):42. https://doi.org/10.1007/s11095-017-2341-x.
Rao G, Iyer V, Kosloski MP, Pisal DS, Shin E, Middaugh CR, et al. Use of a folding model and in situ spectroscopic techniques for rational formulation development and stability testing of monoclonal antibody therapeutics. J Pharm Sci. 2010;99(4):1697–706. https://doi.org/10.1002/jps.21938.
Shire SJ, Shahrokh Z, Liu J. Challenges in the development of high protein concentration formulations. J Pharm Sci. 2004;93(6):1390–402. https://doi.org/10.1002/jps.20079.
Kinderman F, Yerby B, Jawa V, Joubert MK, Joh NH, Malella J, et al. Impact of precipitation of antibody therapeutics after subcutaneous injection on pharmacokinetics and immunogenicity. J Pharm Sci. 2019;108(6):1953–63. https://doi.org/10.1016/j.xphs.2019.01.015.
Smith A, Manoli H, Jaw S, Frutoz K, Epstein AL, Khawli LA, et al. Unraveling the effect of immunogenicity on the PK/PD, efficacy, and safety of therapeutic proteins. J Immunol Res. 2016;2016:2342187. https://doi.org/10.1155/2016/2342187.
Strik AS, van den Brink GR, Ponsioen C, Mathot R, Löwenberg M, D’Haens GR. Suppression of anti-drug antibodies to infliximab or adalimumab with the addition of an immunomodulator in patients with inflammatory bowel disease. Aliment Pharmacol Ther. 2017;45(8):1128–34. https://doi.org/10.1111/apt.13994.
Messinger YH, Mendelsohn NJ, Rhead W, Dimmock D, Hershkovitz E, Champion M, et al. Successful immune tolerance induction to enzyme replacement therapy in CRIM-negative infantile Pompe disease. Genet Med. 2012;14:135. https://doi.org/10.1038/gim.2011.4.
Mendelsohn NJ, Messinger YH, Rosenberg AS, Kishnani PS. Elimination of antibodies to recombinant enzyme in Pompe’s disease. N Eng J Med. 2009;360(2):194–5. https://doi.org/10.1056/NEJMc0806809.
Ohashi T, Iizuka S, Shimada Y, Higuchi T, Eto Y, Ida H, et al. Administration of anti-CD3 antibodies modulates the immune response to an infusion of α-glucosidase in mice. Mol Ther. 2012;20(10):1924–31. https://doi.org/10.1038/mt.2012.133.
Waters B, Qadura M, Burnett E, Chegeni R, Labelle A, Thompson P, et al. Anti-CD3 prevents factor VIII inhibitor development in hemophilia A mice by a regulatory CD4+ CD25+-dependent mechanism and by shifting cytokine production to favor a Th1 response. Blood. 2009;113(1):193–203.
Herzog RW, Kuteyeva V, Saboungi R, Terhorst C, Biswas M. Reprogrammed CD4(+) T cells that express FoxP3(+) control inhibitory antibody formation in hemophilia A mice. Front Immunol. 2019;10:274. https://doi.org/10.3389/fimmu.2019.00274.
Hofmann K, Clauder A-K, Manz RA. Targeting B cells and plasma cells in autoimmune diseases. Front Immunol. 2018. https://doi.org/10.3389/fimmu.2018.00835.
Parvathaneni K, Scott DW. Engineered FVIII-expressing cytotoxic T cells target and kill FVIII-specific B cells in vitro and in vivo. Blood Adv. 2018;2(18):2332–40. https://doi.org/10.1182/bloodadvances.2018018556.
Hull-Ryde EA, Porter MA, Fowler KA, Kireev D, Li K, Simpson CD, et al. Identification of cosalane as an inhibitor of human and murine CC-chemokine receptor 7 signaling via a high-throughput screen. SLAS Discov. 2018;23(10):1083–91. https://doi.org/10.1177/2472555218780917.
Huang X, He Y, Han J, Zhuang J, He J, Sun E. Not only anti-inflammation, etanercept abrogates collagen-induced arthritis by inhibiting dendritic cell migration and maturation. Cent Eur J Immunol. 2019;44(3):237–45. https://doi.org/10.5114/ceji.2019.89595.
Chabot V, Martin L, Meley D, Sensebé L, Baron C, Lebranchu Y, et al. Unexpected impairment of TNF-α-induced maturation of human dendritic cells in vitro by IL-4. J Transl Med. 2016;14(1):93. https://doi.org/10.1186/s12967-016-0848-2.
Zurdo J, Arnell A, Obrezanova O, Smith N, de la Cuesta GR, Gallagher TR, et al. Early implementation of QbD in biopharmaceutical development: a practical example. Biomed Res Int. 2015. https://doi.org/10.1155/2015/605427.
Wang X, Das TK, Singh SK, Kumar S. Potential aggregation prone regions in biotherapeutics: A survey of commercial monoclonal antibodies. mAbs. 2009;1(3):254–67. https://doi.org/10.4161/mabs.1.3.8035.
Senga Y, Honda S. Suppression of aggregation of therapeutic monoclonal antibodies during storage by removal of aggregation precursors using a specific adsorbent of non-native IgG conformers. Bioconjug Chem. 2018;29(10):3250–61.
Xu S, Lun Y, Frascella M, Garcia A, Soska R, Nair A, et al. Improved efficacy of a next-generation ERT in murine Pompe disease. JCI Insight. 2019;4(5):e125358. https://doi.org/10.1172/jci.insight.125358.
Pisal DS, Kosloski MP, Middaugh CR, Bankert RB, Balu-Iyer SV. Native-like aggregates of factor VIII are immunogenic in von Willebrand factor deficient and hemophilia a mice. J Pharm Sci. 2012;101(6):2055–65. https://doi.org/10.1002/jps.23091.
Miclea RD, Purohit VS, Balu-Iyer SV. O-phospho-L-serine, multi-functional excipient for B domain deleted recombinant factor VIII. AAPS J. 2007;9(2):E251–9. https://doi.org/10.1208/aapsj0902028.
Salazar-Fontana LI, Desai DD, Khan TA, Pillutla RC, Prior S, Ramakrishnan R, et al. Approaches to mitigate the unwanted immunogenicity of therapeutic proteins during drug development. AAPS J. 2017;19(2):377–85. https://doi.org/10.1208/s12248-016-0030-z.
Adair JR, Athwal DS, Bodmer MW, Bright SM, Collins AM, Pulito VL, et al. Humanization of the murine anti-human CD3 monoclonal antibody OKT3. Hum Antibodies Hybridomas. 1994;5(1–2):41–7.
De Groot AS, Terry F, Cousens L, Martin W. Beyond humanization and de-immunization: tolerization as a method for reducing the immunogenicity of biologics. Expert Rev Clin Pharmacol. 2013;6(6):651–62. https://doi.org/10.1586/17512433.2013.835698.
De Groot AS, Martin W. Reducing risk, improving outcomes: Bioengineering less immunogenic protein therapeutics. Clin Immunol. 2009;131(2):189–201. https://doi.org/10.1016/j.clim.2009.01.009.
Ettinger RA, Liberman JA, Gunasekera D, Puranik K, James EA, Thompson AR, et al. FVIII proteins with a modified immunodominant T-cell epitope exhibit reduced immunogenicity and normal FVIII activity. Blood Adv. 2018;2(4):309–22. https://doi.org/10.1182/bloodadvances.2017013482.
Mazor R, Kaplan G, Park D, Jang Y, Lee F, Kreitman R, et al. Rational design of low immunogenic anti CD25 recombinant immunotoxin for T cell malignancies by elimination of T cell epitopes in PE38. Cell Immunol. 2017;313:59–66. https://doi.org/10.1016/j.cellimm.2017.01.003.
Pratt KP. Engineering less immunogenic and antigenic FVIII proteins. Cell Immunol. 2016;301:12–7. https://doi.org/10.1016/j.cellimm.2015.10.008.
Liu J, Cao X. Regulatory dendritic cells in autoimmunity: A comprehensive review. J Autoimmun. 2015;63:1–12. https://doi.org/10.1016/j.jaut.2015.07.011.
Adalid-Peralta L, Fragoso G, Fleury A, Sciutto E. Mechanisms underlying the induction of regulatory T cells and its relevance in the adaptive immune response in parasitic infections. Int J Biol Sci. 2011;7(9):1412–26. https://doi.org/10.7150/ijbs.7.1412.
Caridade M, Graca L, Ribeiro RM. Mechanisms underlying CD4+ Treg immune regulation in the adult: from experiments to models. Front Immunol. 2013;4:378. https://doi.org/10.3389/fimmu.2013.00378.
Idoyaga J, Fiorese C, Zbytnuik L, Lubkin A, Miller J, Malissen B, et al. Specialized role of migratory dendritic cells in peripheral tolerance induction. J Clin Invest. 2013;123(2):844–54. https://doi.org/10.1172/jci65260.
Hasegawa H, Matsumoto T. Mechanisms of tolerance induction by dendritic cells in vivo. Front Immunol. 2018;9:350. https://doi.org/10.3389/fimmu.2018.00350.
Guilliams M, Crozat K, Henri S, Tamoutounour S, Grenot P, Devilard E, et al. Skin-draining lymph nodes contain dermis-derived CD103(-) dendritic cells that constitutively produce retinoic acid and induce Foxp3(+) regulatory T cells. Blood. 2010;115(10):1958–68. https://doi.org/10.1182/blood-2009-09-245274.
Luo X, Miller SD, Shea LD. Immune tolerance for autoimmune disease and cell transplantation. Annu Rev Biomed Eng. 2016;18:181–205. https://doi.org/10.1146/annurev-bioeng-110315-020137.
Fathallah AM, Ramakrishnan R, Balu-Iyer SV. O-Phospho-l-Serine mediates hyporesponsiveness toward FVIII in hemophilia a-murine model by inducing tolerogenic properties in dendritic cells. J Pharm Sci. 2014;103(11):3457–63. https://doi.org/10.1002/jps.24173.
Purohit VS, Ramani K, Sarkar R, Kazazian HH Jr, Balasubramanian SV. Lower inhibitor development in hemophilia A mice following administration of recombinant factor VIII-O-phospho-L-serine complex. J Biol Chem. 2005;280(18):17593–600. https://doi.org/10.1074/jbc.M500163200.
Schneider JL, Balu-Iyer SV. Phosphatidylserine converts immunogenic recombinant human acid alpha-glucosidase to a tolerogenic form in a mouse model of Pompe disease. J Pharm Sci. 2016;105(10):3097–104. https://doi.org/10.1016/j.xphs.2016.06.018.
Glassman FY, Balu-Iyer SV. Subcutaneous administration of Lyso-phosphatidylserine nanoparticles induces immunological tolerance towards Factor VIII in a Hemophilia A mouse model. Int J Pharm. 2018;548(1):642–8. https://doi.org/10.1016/j.ijpharm.2018.07.018.
Drugs@FDA. Silver Spring, MD: U.S. Food and Drug Administration; 2020. https://www.accessdata.fda.gov/scripts/cder/daf/ Accessed 7 Oct 2020.
Acknowledgements
This work was financially supported by National Institutes of Health R01 grant HL-70227 to Dr. Sathy V. Balu-Iyer. The opinions expressed in this review are those of the authors and do not represent the opinion or position of SUNY University at Buffalo, the FDA, the National Institutes of Health, or any other corporate entity.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Funding was provided by National Institutes of Health R01 grant HL-70227 to Dr. Sathy V. Balu-Iyer.
Conflicts of interest/Competing interests
The authors declare that they have no conflicts of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and material
Not applicable.
Authors’ contributions
NLJ performed the literature search and drafted the manuscript. All authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Rights and permissions
About this article

Cite this article
Jarvi, N.L., Balu-Iyer, S.V. Immunogenicity Challenges Associated with Subcutaneous Delivery of Therapeutic Proteins. BioDrugs 35, 125–146 (2021). https://doi.org/10.1007/s40259-020-00465-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-020-00465-4