
Abstract
Background
The NOR-SWITCH main and extension trials demonstrated that switching from originator to biosimilar infliximab (CT-P13) is efficacious and safe across six diseases. However, a subgroup analysis of Crohn’s disease (CD) in the main trial displayed a close to significant difference favouring originator infliximab, and more scientific data have therefore been requested.
Objective
The aim was to assess treatment efficacy, safety, and immunogenicity in an explorative subgroup analysis in CD and ulcerative colitis (UC) in the NOR-SWITCH trials.
Patients and Methods
The 52-week, randomised, non-inferiority, double-blind, multicentre, phase 4 NOR-SWITCH study was followed by a 26-week open extension trial where all patients received treatment with CT-P13. Treatment efficacy, safety, and immunogenicity in CD and UC were assessed throughout the 78-week study period.
Results
The main and extension trials included 155 and 93 patients with CD and 93 and 80 patients with UC, respectively. Demographic and baseline characteristics were comparable in both treatment arms within patient groups. There were no differences in the main and extension trials regarding changes in activity indices, C-reactive protein, faecal calprotectin, patient’s and physician’s global assessment of disease activity and patient-reported outcome measures in CD and UC. Moreover, comparable results were also demonstrated for trough serum levels, presence of anti-drug antibodies, and reported adverse events.
Conclusion
Efficacy, safety, and immunogenicity of both the originator and biosimilar infliximab were comparable in CD and UC in the NOR-SWITCH main and extension trials. These explorative subgroup analyses confirm that there are no significant concerns related to switching from originator infliximab to CT-P13 in CD and UC.
Trial Registration
ClinicalTrials.gov, number NCT02148640.
Similar content being viewed by others

The randomised, controlled, double-blind NOR-SWITCH study demonstrated that switching from originator infliximab to CT-P13 is efficacious and safe across six indications. However, a subgroup analysis of Crohn’s disease displayed a close to significant difference in terms of efficacy favouring originator infliximab. |
This explorative subgroup analysis of Crohn’s disease and ulcerative colitis in the NOR-SWITCH main and extension trials displays comparable results regarding clinical, biochemical, immunogenicity, and patient-reported outcome measures between originator infliximab and CT-P13. |
Long-term treatment with CT-P13 is efficacious and safe in inflammatory bowel disease. |
1 Introduction
Tumour necrosis factor alpha (TNFα) inhibitors and other biologic agents have had a substantial, positive impact on the treatment of several immune-mediated chronic inflammatory disorders, including inflammatory bowel disease (IBD) [1]. However, access to biologic treatment varies globally, with limited drug availability in many countries due to cost [2]. Biosimilars are less expensive reproductions of their originator counterparts and provide a potential opportunity to improve patient access.
The approval of the use of biosimilar infliximab CT-P13 (Remsima®, Inflectra®, Celltrion, Inchon, South Korea) in IBD is extrapolated from two pivotal studies conducted in patients with rheumatic diseases (PLANETAS and PLANETRA) [3, 4]. Although there are similarities between IBD and rheumatic diseases, monoclonal antibodies such as infliximab may interact in subtly different ways in different diseases; in IBD, blocking of both soluble and membrane bound TNFα is of importance [5]. Thus, it has been suggested that the clinically relevant mechanisms of action of infliximab might differ in different diseases [6] and there were some concerns in the gastroenterology community when biosimilar infliximab became available for use in clinical practice [7, 8].
So far, no real-life clinical studies have shown any unexpected effects when starting patients on CT-P13 or switching from infliximab originator (IFX) (Remicade®, Janssen, Beerse, Belgium) to CT-P13 [9,10,11,12,13,14,15,16,17,18]. The NOR-SWITCH study showed that switching from IFX to CT-P13 was not inferior to continued treatment with IFX regarding efficacy, safety, and immunogenicity across six indications [19]. However, sub-analyses of the primary endpoint in Crohn’s disease (CD) demonstrated a confidence interval (CI) for CT-P13 just within the prespecified non-inferiority margin (risk difference −14.3%, 95% CI −29.3 to 0.7) [19]. This finding might indicate a less favourable outcome after switching from reference product to biosimilars in CD, and more scientific data have been requested [20,21,22,23].
Here we present the complete results regarding efficacy, safety and immunogenicity from the subgroup analyses of CD and ulcerative colitis (UC) patients from both the main and extension part of the NOR-SWITCH study [19, 24].
2 Methods
2.1 Study Design and Participants
The NOR-SWITCH main study was designed as a 52-week, randomised, double-blind, parallel-group, multicentre, non-inferiority, comparative, phase 4 study, in a hospital setting. Patient recruitment, inclusion, and exclusion criteria have previously been described in detail [19]. A nationwide project group including representatives from all relevant specialties together with patient representatives planned and conducted the study. Adult patients with a diagnosis of CD and UC on stable treatment with IFX for at least 6 months were eligible for participation. The patients who completed the main study period (week 0 to week 52) were recruited to the open, 26-week follow-up extension trial (week 52 to week 78). All patients received verbal and written information about the main and extension study and signed an informed consent. The study was conducted in compliance with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. The study protocol and consent documents were approved by an independent ethics committee (Regional Committees for Medical and Health Research Ethics South East; reference number 2014/848), by appropriate institutional review boards, and by the Norwegian Medicines Agency (reference number 14/07192-11).
2.2 Randomisation and Masking
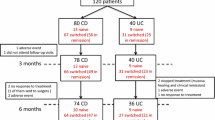
In the main study, eligible patients were randomised 1:1 to either continued treatment with IFX or a switch to CT-P13 treatment. The type of IFX treatment was blinded, and the dose and infusion intervals of patients’ treatment regimens were kept unchanged from those before randomisation. After completion of the main study, the 26-week extension study recruited consenting patients who were all given open-label CT-P13 treatment with an unchanged regimen in terms of dose and infusion intervals. The patients treated with CT-P13 in the main trial continued this treatment in the extension phase (CT-P13 maintenance group), whereas patients treated with IFX switched to CT-P13 in the extension phase (IFX/CT-P13 switch group). Consequently, the CT-P13 maintenance group received CTP13 for 18 months and the IFX/CT-P13 switch group received CT-P13 for 6 months (after 12 months with IFX in the main study) (Fig. 1) [24]. The blinded treatment allocation in the main study was not revealed until after completion of the extension study period.

Patient disposition in IBD in the NOR-SWITCH main and extension study. CD Crohn’s disease, IBD inflammatory bowel disease, IFX infliximab, PPS per-protocol set, UC ulcerative colitis. 1 Four CD patients withdrew consent, two CD and one UC patients had major change in immunosuppressive treatment, two CD patients were unblinded during the study, two CD patients developed adverse events, and one CD patient was excluded for other reasons. 2 Seven CD and four UC patients withdrew consent, two CD and five UC patients had major change in immunosuppressive treatment, two CD patients were unblinded during the study, one CD and two UC patients developed adverse events, three CD patients and one UC patient were excluded for other reasons
2.3 Study Assessments
Data were collected at every infusion visit, including clinical assessments by a trained study nurse and/or physician using an electronic case report form (Viedoc®). Blood samples for protocol-specified laboratory tests were obtained before infusions, including measurements of drug concentrations and anti-drug antibodies (ADAbs), and for storage in a biobank. IFX and CT-P13 trough serum levels and ADAbs were analyzed using in-house assays automated on the AutoDELFIA® (PerkinElmer, Waltham, MA) immunoassay platform [19]. ADAbs were not analyzed in samples with IFX/CT-P13 levels above 5 mg/L, since high drug concentrations cause interference in our assays. The patients were asked to deliver a faecal sample for calprotectin measurements after each visit (CalproLab, Calpro AS, Oslo, Norway; Buhlmann Laboratories AG, Basel, Switzerland). The number of study visits differed according to treatment regimen. Full details about the collected variables can be found in the main study report [19].
2.4 Outcomes
The primary endpoint was disease worsening during the 52-week main study and the subsequent 26-week extension study [19, 24]. Disease worsening was defined by worsening in Harvey-Bradshaw Index (HBI) for CD (a change from baseline of 4 points or more and a total score of 7 points or greater) and partial Mayo Score (PMS) for UC (a change from baseline of more than 3 points and a total score of 5 or greater) or a consensus on disease worsening between investigator and patient leading to major change in treatment. Secondary endpoints included clinical remission status (HBI ≤ 4 points, PMS ≤ 2 points), levels of C-reactive protein (CRP) and faecal calprotectin, changes in disease variables and investigator and patient global assessments (follow-up minus baseline), patient-reported outcome measures (PROMs), drug concentrations (trough measurements), and ADAb assessments. Measures of safety included clinical and laboratory adverse events (AEs). The coding of AEs was performed according to the Medical Dictionary for Regulatory Activities (MedDRA, v.13.0).
2.5 Statistical Analyses
The statistical analyses have previously been described in detail [19]. A power calculation to demonstrate non-inferiority of 15% was performed for the entire study population including the six subgroups. The per-protocol set (PPS) consisted of subjects without major protocol deviations throughout the main and extension study, while the full analysis set (FAS) comprised all subjects entering the main and extension study, respectively. We analysed the primary outcome and secondary dichotomous endpoints using logistic regression, with treatment as a fixed effect, adjusted for diagnosis and the treatment duration of IFX at baseline (week 0), providing estimates (by the delta method) of adjusted risk difference and adjusted relative risk for the treatment difference. Continuous outcomes were analyzed using linear mixed effect models, with treating time as a categorical variable and including a time by treatment group interaction. Missing data for remission and disease worsening in the FAS were imputed with last observation carried forward.
2.6 Role of the Funding Source
The funder of the study (the Norwegian Ministry of Health and Care Services) had no role in study design, data collection, data analysis, data interpretation, writing of this article, or decision to submit. All authors reviewed and approved the final manuscript. The corresponding author (KKJ) together with the statistician (JS) had full access to all the data in the study and had final responsibility for the decision to submit for publication.
3 Results
3.1 Patients
Between October 2014 and July 2016, 248 IBD patients (155 CD, 93 UC) were enrolled and randomised into the NOR-SWITCH main trial at 19 gastroenterology study centres. Based on randomisation, 123 patients switched treatment from IFX to CT-P13 and 125 patients continued treatment with IFX (Fig. 1). Of the 230 patients who completed the 52-week main trial, 207 (90%) (127 CD, 80 UC) continued into the 26-week open extension study (Fig. 1). Two of the 19 study centres did not recruit patients to the extension phase.
3.1.1 Main Study
In the FAS, the mean age at inclusion was 38.8 years (SD 13.8) in CD patients and 45.2 years (SD 14.4) in UC patients. The mean disease duration was 13.6 (SD 8.8) and 11.3 years (SD 8.3) and the mean duration of ongoing IFX treatment at randomisation was 5.5 (SD 3.4) and 4.3 years (SD 2.3) in CD and UC patients, respectively. The demographics and baseline characteristics of the randomised patients with IBD are listed in Table 1. The majority of patients were biologic treatment–naïve prior to initiating ongoing IFX treatment, and almost half of the patients used concomitant immunosuppressive treatment (Table 1). Both the general and the disease-specific measures suggested low disease activity at baseline, with no differences between groups (Table 1).
3.1.2 Extension Study
In the FAS, there were 65 (61%) and 62 (62%) CD patients and 42 (39%) and 38 (38%) UC patients in the CT-P13 maintenance and IFX/CT-P13 switch groups, respectively (Fig. 1). The demographics and baseline characteristics (week 52) were comparable to the cohort of the main study (see the Electronic Supplementary Material, Table S1). As in the main study, disease activity at baseline (week 52) in both CD and UC patients was generally low with no differences between groups (Table S1).
3.2 Primary Endpoint
3.2.1 Main Study
In CD, disease worsening occurred in 23 out of 63 patients (36.5%) in the CT-P13 group and in 14 out of 66 patients (21.2%) in the IFX group (PPS). The 95% CI of the adjusted risk difference of −14.3% was −29.3 to 0.7 [19]. A total of 46 disease worsening events were recorded (in some patients, more than one event). Of these, 18 were associated with a change in study drug dosage, and one with study drug discontinuation. In UC, disease worsening occurred in five out of 42 patients (11.9%) and three out of 33 patients (9.1%) in the CT-P13 and IFX group, respectively (adjusted risk difference −2.6%, 95% CI −15.2 to 10.0) [19]. Of these, two were associated with a change in study drug dosage; no study drug discontinuations occurred.
3.2.2 Extension Study
In CD, disease worsening occurred in 13 out of 63 patients (20.6%) in the CT-P13 maintenance group and in eight out of 61 patients (13.1%) in the IFX/CT-P13 switch group (PPS). The 95% CI of the adjusted risk difference of 7.9% was −5.2 to 21 [24]. In 11 patients, disease worsening was recorded for the first time during the study period; six in the maintenance group and five in the IFX/CT-P13 switch group. A total of 28 disease worsening events were recorded (in some patients, more than one event). Seven events were associated with a change in study drug dosage. In UC, disease worsening occurred in six out of 39 patients (15.4%) in the CT-P13 maintenance group and in one out of 35 patients (2.9%) in the IFX/CT-P13 switch group (adjusted risk difference 12.4%, 95% CI −0.1 to 25) [24]. In six patients, disease worsening was recorded for the first time during the study period; five in the maintenance group and one in the IFX/CT-P13 switch group. A total of nine disease worsening events were recorded (in some patients, more than one event); four of these were associated with a change in study drug dosage. No study drug discontinuations were associated with disease worsening in the CD and UC patients in the study groups.
No predictive factors for disease worsening in CD and UC in the main and extension part of the study were identified (Table S3a and S3b).
3.3 Secondary Endpoints
At the end of the main study (week 52), clinical remission was present in CD in 41 out of 63 patients (65%) and 46 out of 66 patients (70%) in the CT-P13 and IFX groups, respectively (adjusted risk difference 5.6%, 95% CI −11 to 22.2) (PPS) [19]. In UC, 39 out of 42 patients (93%) and 29 out of 33 patients (88%) were in clinical remission in the CT-P13 and IFX groups, respectively (adjusted rate difference −5.9%, 95% CI −21.7 to 9.9) [19]. At the end of the extension study (week 78), clinical remission was present in CD in 41 out of 63 patients (65%) and 46 out of 61 patients (75%) in the CT-P13 maintenance and IFX/CT-P13 switch groups, respectively (adjusted risk difference −11.3%, 95% CI −27.2 to 4.5) [24]. In UC, 32 out of 39 patients (82%) and 30 out of 35 patients (86%) were in clinical remission in the CT-P13 maintenance group and IFX/CT-P13 switch group, respectively (adjusted risk difference −3.3%, 95% CI −20 to 13.4). [24].
Of the patients entering the extension study, 90 CD and 75 UC patients were in clinical remission (last visit in main study), compared to 88 and 68 patients, respectively, at the last visit in the extension study.
Disease state at baseline and changes in the disease variables from baseline (week 0) to the end of the extension study (week 78) demonstrated no significant differences between the two groups in CD and UC (PPS) (see Tables 2a and 2b, and Tables S2a and S2b in the Electronic Supplementary Material). Likewise, the changes in PROMs were generally similar in the two groups, in both CD and UC (Tables 2a and 2b, Tables S2a and S2b).
Figures 2 and 3 present the disease-specific composite measures, CRP, faecal calprotectin, serum infliximab, and patient’s and physician’s global assessment of disease worsening in CD (Fig. 2) and UC (Fig. 3) throughout the main and extension study (week 0–78). No significant differences for any of these parameters were found between treatment arms in the two groups.

Secondary efficacy endpoints in Crohn’s disease from randomisation in main study (baseline/week 0) to study end (week 78), per-protocol set. IFX infliximab

Secondary efficacy endpoints in ulcerative colitis from randomisation in main study (baseline/week 0) to study end (week 78), per-protocol set. IFX infliximab
Throughout the main study, 13 CD patients discontinued the study: six in the CT-P13 group and seven in the IFX group (FAS). In UC, six patients discontinued the study: one in the CT-P13 group and five in the IFX group (FAS). Two CD patients (one due to low serum levels, one due to “other reasons”) and three UC patients (two due to AEs, one due to low serum levels) discontinued the study drug.
Throughout the extension study, nine patients discontinued the study: three CD patients and six UC patients (FAS) (Fig. 1). One patient with CD discontinued the study drug (due to “other reasons”).
4 Adverse Events
Similar numbers of patients had at least one treatment emergent AE in the two treatment groups in CD and UC in the main and extension study (FAS) (Table 3). Likewise, the number of patients with a treatment-emergent serious AE did not differ between the two groups in the two diseases (Table 3). The most frequent treatment-emergent AEs were related to infections in both diseases (Table 3). No deaths occurred.
4.1 Anti-drug Antibodies
The incidence of ADAbs detected at any time during the main study (excluding patients with detectable ADAbs at baseline) in CD was four out of 77 (5%) in the CT-P13 group and three out of 78 (4%) in the IFX group (FAS). In UC, eight out of 46 patients (17%) in the CT-P13 group and five out of 47 patients (11%) in the IFX group developed new ADAbs. In the extension study, none of the CD patients in the CT-P13 maintenance group and one out of 62 (1.6%) in the IFX/CT-P13 switch group developed ADAbs. In UC, one out of 42 (2.4%) in the CT-P13 maintenance group and one out of 38 (2.6%) in the IFX/CT-P13 switch group developed ADAbs. Trough drug concentrations were similar in the two groups for both diseases (Figs. 2 and 3).
5 Discussion
The NOR-SWITCH study is the first randomised study demonstrating that switching from IFX to CT-P13 is safe [19], but due to the large difference in efficacy favouring IFX in CD patients this conclusion has been questioned for this subgroup [20,21,22]. In the exploratory sub-analyses of IBD patients involving both the main and extension part of the NOR-SWITCH trial, we demonstrate comparable results for IFX and CT-P13 across treatment arms in terms of efficacy, safety, and immunogenicity. Hence, the suspicion of inferiority of CT-P13 compared to IFX in CD is refuted.
Disease worsening based on HBI and PMS as the primary endpoint has clear weaknesses since these activity indices mainly consist of subjective assessments of symptoms by the patient. However, the secondary endpoints of the study, i.e. pharmacokinetics and the inflammatory markers (CRP and faecal calprotectin), provide more objective information that clearly supports biosimilarity. Endoscopic assessment is important to evaluate disease activity in IBD patients. Unfortunately, this was not included in our study due to practical reasons and possible negative impact on patient inclusion. However, previous studies have demonstrated a clear correlation between endoscopic findings and faecal calprotectin [25]. Throughout the study fewer UC than CD patients experienced disease worsening. Due to the low number of UC patients reaching the primary endpoint, no conclusions can be drawn regarding the risk difference between treatment arms in the extension part of the study.
There were no new safety signals during the main study and the extension study period, and AEs were evenly distributed between study arms in both CD and UC patients. As expected, the most frequent AEs reported were infections. Immunogenicity with the formation of ADAbs is of great importance in relation to both AEs and loss of response to treatment. Drug levels and ADAbs were therefore measured at every visit throughout the entire study period, and no differences were demonstrated between the two treatment arms in UC and CD. Thus, the results support that there is no increased risk of immunogenicity after switching to CT-P13 in IBD patients.
The NOR-SWITCH trial has both strengths and weaknesses. The randomised multicentre design and the involvement of patient representatives in the planning and conduct of the study are obvious strengths. Moreover, the study was financed by the Norwegian Government and monitored within the healthcare system, completely independent of pharmaceutical companies. One limitation of the subgroup analyses of IBD is that the power calculation to demonstrate non-inferiority applies for the entire study population and not each diagnostic group. However, the majority of patients included in the study had IBD (51%), with CD as the most common diagnosis (n = 155; 32%). In our opinion, these facts together with the controversy that was raised around the potential non-inferiority of CT-P13 when compared to IFX for CD justifies a separate and more comprehensive presentation of the IBD subgroup analyses. We consider the results to contribute to our knowledge in the field of biosimilars where randomised data are sparse, especially in IBD. Furthermore, valuable information is added to the discussion regarding the possible loss of response in CD under CT-P13 treatment.
To our knowledge, the only randomised controlled IFX switch trial besides the NOR-SWITCH study is described in a paper by Ye et al. [26]. They included anti-TNF–naïve CD patients with moderate to severe disease, who received either treatment with CT-P13 or IFX. At week 30, half of the patients in each group switched to IFX and CT-P13, respectively, and comparable results regarding efficacy and safety were seen in all four groups at week 54 [26]. Similar to our study, which lacked the power to separately evaluate the non-inferiority of switching in the IBD subpopulation, this study was not powered to assess the non-inferiority of switching beyond week 30. The patient cohort, encompassing CD patients with short treatment duration, is not directly comparable to our study cohort with stable CD patients with long-term treatment of IFX before randomisation.
The nocebo effect is an important issue when switching from originator to biosimilar medicines, because of a possible impact on treatment success [27, 28]. Studies in IBD have demonstrated a nocebo effect after switching from originator to biosimilar infliximab; Boone et al. found an overall nocebo response of 13% 6 months after switch in a pragmatic switch study involving mainly CD and UC patients [28]. No significant changes in objective effectiveness and safety were proven. On the contrary, Razanskaite et al. demonstrated no deterioration in clinical and patient-reported outcomes after an open prospective switch in 143 IBD patients [29]. Importantly, in the NOR-SWITCH main study, we avoided the nocebo effect on a group level due to the double-blind design. However, when looking at the overall patient’s and physician’s global assessment of disease activity throughout this study, an upward drift can be observed in the patient’s assessment in both study arms, indicating a nocebo effect [30]. Our IBD substudy does not demonstrate a similar drift as previously described, not in CD, nor in UC. In the extension part of our study, one might expect a nocebo effect due to the open study design. However, both our patients and the healthcare workers remained blinded to what treatment the patients had been given during the main study period. Perhaps for these reasons, we do not see a clearly delineated nocebo effect in the CD and UC patients in the extension part of the trial.
6 Conclusions
In summary, we conclude that there were no significant differences regarding efficacy, safety and immunogenicity between patients treated with IFX or CTP-13 within the CD and UC patient groups when evaluating the NOR-SWITCH trials together. Therefore, the subgroup analyses support that a switch from originator to biosimilar infliximab can be recommended in IBD patients.
References
Kuek A, Hazleman BL, Ostor AJ. Immune-mediated inflammatory diseases (IMIDs) and biologic therapy: a medical revolution. Postgrad Med J. 2007;83(978):251–60.
Putrik P, Ramiro S, Kvien TK, Sokka T, Pavlova M, Uhlig T, et al. Inequities in access to biologic and synthetic DMARDs across 46 European countries. Ann Rheum Dis. 2014;73(1):198–206.
Park W, Hrycaj P, Jeka S, Kovalenko V, Lysenko G, Miranda P, et al. A randomised, double-blind, multicentre, parallel-group, prospective study comparing the pharmacokinetics, safety, and efficacy of CT-P13 and innovator infliximab in patients with ankylosing spondylitis: the PLANETAS study. Ann Rheum Dis. 2013;72(10):1605–12.
Yoo DH, Hrycaj P, Miranda P, Ramiterre E, Piotrowski M, Shevchuk S, et al. A randomised, double-blind, parallel-group study to demonstrate equivalence in efficacy and safety of CT-P13 compared with innovator infliximab when coadministered with methotrexate in patients with active rheumatoid arthritis: the PLANETRA study. Ann Rheum Dis. 2013;72(10):1613–20.
Billmeier U, Dieterich W, Neurath MF, Atreya R. Molecular mechanism of action of anti-tumor necrosis factor antibodies in inflammatory bowel diseases. World J Gastroenterol. 2016;22(42):9300–13.
Feagan BG, Choquette D, Ghosh S, Gladman DD, Ho V, Meibohm B, et al. The challenge of indication extrapolation for infliximab biosimilars. Biologicals. 2014;42(4):177–83.
Danese S, Gomollon F, Governing B, Operational Board of E. ECCO position statement: the use of biosimilar medicines in the treatment of inflammatory bowel disease (IBD). J Crohns Colitis. 2013;7(7):586-9.
Ben-Horin S, Vande Casteele N, Schreiber S, Lakatos PL. Biosimilars in inflammatory bowel disease: facts and fears of extrapolation. Clin Gastroenterol Hepatol. 2016;14(12):1685–96.
Gecse KB, Lovasz BD, Farkas K, Banai J, Bene L, Gasztonyi B, et al. Efficacy and safety of the biosimilar infliximab CT-P13 treatment in inflammatory bowel diseases: a prospective, multicentre, nationwide cohort. J Crohns Colitis. 2016;10(2):133–40.
Armuzzi A, Fiorino G, Variola A, Manetti N, Fries W, Orlando A, et al. The PROSIT cohort of infliximab biosimilar in IBD: a prolonged follow-up on the effectiveness and safety across Italy. Inflamm Bowel Dis. 2019;25(3):568–79.
Glintborg B, Sorensen IJ, Loft AG, Lindegaard H, Linauskas A, Hendricks O, et al. A nationwide non-medical switch from originator infliximab to biosimilar CT-P13 in 802 patients with inflammatory arthritis: 1-year clinical outcomes from the DANBIO registry. Ann Rheum Dis. 2017;76(8):1426–31.
Komaki Y, Yamada A, Komaki F, Micic D, Ido A, Sakuraba A. Systematic review with meta-analysis: the efficacy and safety of CT-P13, a biosimilar of anti-tumour necrosis factor-alpha agent (infliximab), in inflammatory bowel diseases. Aliment Pharmacol Ther. 2017;45(8):1043–57.
Meyer A, Rudant J, Drouin J, Weill A, Carbonnel F, Coste J. Effectiveness and safety of reference infliximab and biosimilar in crohn disease: a French equivalence study. Ann Intern Med. 2019;170(2):99–107.
Uhlig T, Goll GL. Reviewing the evidence for biosimilars: key insights, lessons learned and future horizons. Rheumatology (Oxford). 2017;56(suppl_4):iv49-iv62.
Jahnsen J, Detlie TE, Vatn S, Ricanek P. Biosimilar infliximab (CT-P13) in the treatment of inflammatory bowel disease: a Norwegian observational study. Expert Rev Gastroenterol Hepatol. 2015;9(Suppl 1):45–52.
Jahnsen J, Kaasen Jorgensen K. Experience with Biosimilar Infliximab (Remsima(R)) in Norway. Dig Dis. 2017;35(1–2):83–90.
Park W, Yoo DH, Miranda P, Brzosko M, Wiland P, Gutierrez-Urena S, et al. Efficacy and safety of switching from reference infliximab to CT-P13 compared with maintenance of CT-P13 in ankylosing spondylitis: 102-week data from the PLANETAS extension study. Ann Rheum Dis. 2017;76(2):346–54.
Yoo DH, Prodanovic N, Jaworski J, Miranda P, Ramiterre E, Lanzon A, et al. Efficacy and safety of CT-P13 (biosimilar infliximab) in patients with rheumatoid arthritis: comparison between switching from reference infliximab to CT-P13 and continuing CT-P13 in the PLANETRA extension study. Ann Rheum Dis. 2017;76(2):355–63.
Jorgensen KK, Olsen IC, Goll GL, Lorentzen M, Bolstad N, Haavardsholm EA, et al. Switching from originator infliximab to biosimilar CT-P13 compared with maintained treatment with originator infliximab (NOR-SWITCH): a 52-week, randomised, double-blind, non-inferiority trial. Lancet. 2017;389(10086):2304–16.
Michetti P. Efficacy of infliximab biosimilars in patients with Crohn’s disease. Lancet. 2017;390(10111):2435.
Ribaldone DG, Saracco GM, Astegiano M, Pellicano R. Efficacy of infliximab biosimilars in patients with Crohn’s disease. Lancet. 2017;390(10111):2435–6.
Jorgensen KK, Lundin KE, Olsen IC, Kvien TK, Jahnsen J. Efficacy of infliximab biosimilars in patients with Crohn’s disease - Authors’ reply. Lancet. 2017;390(10111):2436.
McKinnon RA, Cook M, Liauw W, Marabani M, Marschner IC, Packer NH, et al. Biosimilarity and Interchangeability: principles and evidence: a systematic review. BioDrugs. 2018;32(1):27–52.
Goll GL, Jorgensen KK, Sexton J, Olsen IC, Bolstad N, Haavardsholm EA, et al. Long-term efficacy and safety of biosimilar infliximab (CT-P13) after switching from originator infliximab: open-label extension of the NOR-SWITCH trial. J Intern Med. 2019;285(6):653–69.
D’Haens G, Ferrante M, Vermeire S, Baert F, Noman M, Moortgat L, et al. Fecal calprotectin is a surrogate marker for endoscopic lesions in inflammatory bowel disease. Inflamm Bowel Dis. 2012;18(12):2218–24.
Ye BD, Pesegova M, Alexeeva O, Osipenko M, Lahat A, Dorofeyev A, et al. Efficacy and safety of biosimilar CT-P13 compared with originator infliximab in patients with active Crohn’s disease: an international, randomised, double-blind, phase 3 non-inferiority study. Lancet. 2019;393(10182):1699–707.
Planes S, Villier C, Mallaret M. The nocebo effect of drugs. Pharmacol Res Perspect. 2016;4(2):e00208.
Boone NW, Liu L, Romberg-Camps MJ, Duijsens L, Houwen C, van der Kuy PHM, et al. The nocebo effect challenges the non-medical infliximab switch in practice. Eur J Clin Pharmacol. 2018;74(5):655–61.
Razanskaite V, Bettey M, Downey L, Wright J, Callaghan J, Rush M, et al. Biosimilar infliximab in inflammatory bowel disease: outcomes of a managed switching programme. J Crohns Colitis. 2017;11(6):690–6.
Kristensen LE, Alten R, Puig L, Philipp S, Kvien TK, Mangues MA, et al. Non-pharmacological effects in switching medication: the nocebo effect in switching from originator to biosimilar agent. BioDrugs. 2018;32(5):397–404.
Acknowledgements
This trial was supported by a direct grant from the Norwegian government, by the Ministry of Health and Care Services. The views and opinions expressed in this paper are those of the authors and do not necessarily reflect those of the Ministry. Many people contributed to this study, in particular study nurses at each study centre. The authors thank these individuals for the invaluable part they played in helping to identify and recruit participants, and to collect data. We thank Karoline J. Henanger at the Department of Medical Biochemistry at Oslo University Hospital for analysis and biobanking of serum samples. We sincerely thank all patients who participated in this study. The authors thank Martha Colban, Nina Flatner, Trond Smedsrud, Bjørn Solvang, Inger Hilde Zahl, Cecilie Moe, Sigrun Sæther, and the Norwegian Clinical Research Infrastructure Network (NorCRIN) for clinical research support and data monitoring during the study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding provided by Akershus University Hospital (AHUS). This work was supported by the Norwegian Ministry of Health and Care Services.
Conflict of interest
KKJ reports personal fees from Intercept, Norgine, and Celltrion. GLG reports personal fees from AbbVie, Biogen, Eli Lilly, MSD, Novartis, Pfizer, Roche, Sandoz, Orion Pharma, Celltrion, and Boehringer Ingelheim. NB reports personal fees received from Orion Pharma, Roche, Napp Pharmaceuticals, Pfizer, and Takeda. ICO reports grants from the Norwegian Ministry of Health and Care Services during the conduct of the study and personal fees from Pfizer. EAH reports grants from AbbVie, Pfizer, UCB, Roche, and MSD. IPB reports personal fees from AbbVie, MSD, Takeda, Hospira, and Ferring. KEAL reports grants from MSD and personal fees from Takeda, Orion, AbbVie, Pfizer, and MSD. JJ reports personal fees from MSD, AbbVie, Celltrion, Orion Pharma, Takeda, Napp Pharm, AstroPharma, Hikma, and Pfizer. TK reports grants from the Norwegian Ministry of Health and Care Services during the conduct of the study and personal fees from AbbVie, Biogen, BMS, Boehringer Ingelheim, Celltrion, Eli Lilly, Epirus, Janssen, Merck-Serono, MSD, Mundipharma, Novartis, Oktal, Orion Pharma, Hospira/Pfizer, Roche, Sandoz, and UCB Pharma. SOF reports personal fees from Takeda, Pharmacosmos, Tillotts, Janssen, Intercept, and Pfizer. The remaining authors have no conflicts of interest to declare.
Ethics approval
The study protocol and consent documents were approved by an independent ethics committee (Regional Committees for Medical and Health Research Ethics South East; reference number 2014/848), by appropriate institutional review boards, and by the Norwegian Medicines Agency (reference number 14/07192-11). The study was conducted in compliance with the Declaration of Helsinki and the International Conference on Harmonization Guidelines for Good Clinical Practice. Trial registration numbers: ClinicalTrials.gov NCT-02148640, EudraCT Number: 2014-002056-40.
Consent
All patients received verbal and written information about the study and signed an informed consent form.
Availability of data and material
The corresponding author (KKJ) together with the statistician (JS) had full access to all the data in the study and had final responsibility for the decision to submit for publication. The study data are available on request from the authors.
Code availability
Statistical analyses were done in Stata version 14.1 and R 3.4.4. Key commands used in the statistical analyses are available upon request.
Author contributions
KKJ contributed to study design, oversaw the implementation of the study at all centres, provided support to study personnel at each site during the study, contributed to data interpretation, and drafted and critically revised the manuscript. She took final responsibility for the decision to submit for publication. JJ contributed to study conception and design, interpreted data, and drafted and critically revised the manuscript. He has had full access to all the data in the study. TK was the principal investigator who conceived and designed the study, interpreted data, and drafted and critically revised the report. He has had full access to all the data in the study. GLG contributed to study design, contributed to data interpretation, and drafted and critically revised the manuscript. JS and ICO were the study statisticians. JS performed the analyses, contributed to data interpretation, and drafted and critically revised the manuscript. ICO contributed to study conception, helped design the study, led the development of the electronic CRF, and contributed support to all sites during the study. He contributed to data interpretation and drafted and critically revised the manuscript. NB helped design the study, facilitated laboratory analyses of infliximab and anti-drug antibodies, developed the assay for anti-drug antibodies, and planned and organised the study biobank. He helped interpret data and critically revised the manuscript. KEAL contributed to study conception and design, in particular, contributing to planning and interpretation of the immunogenicity analyses, and critically revised the manuscript. EAH contributed to study conception, helped design the study, helped interpret data, and critically revised the manuscript. DJW developed the assay for infliximab drug levels, provided reagents for anti-drug antibody analysis, was instrumental in setting up the infrastructure for these analyses, helped interpret data, and critically revised the report. RK performed the immunogenicity analyses, helped organise the study biobank, and critically revised the report. ØA, IPB, IMB, KD, JF, SOF, MH, GHH, BM, GN, UP, JHR, LS, KAS, RT, CV, and CMY were main investigators at each study site, who implemented the study at their site, collected data, and critically revised the report. JH was the user representative and contributed to the design of the trial, data collection from a service user perspective, was crucial in distributing study information to relevant patient populations, and critically revised the report. All authors made substantial contributions to the conception or design of the study, the acquisition, analysis or interpretation of the data; commented on drafts of this paper; and approved the final version. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Jørgensen, K.K., Goll, G.L., Sexton, J. et al. Efficacy and Safety of CT-P13 in Inflammatory Bowel Disease after Switching from Originator Infliximab: Exploratory Analyses from the NOR-SWITCH Main and Extension Trials. BioDrugs 34, 681–694 (2020). https://doi.org/10.1007/s40259-020-00438-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40259-020-00438-7