
Abstract
Fibrosis of the kidney glomerulus and interstitium are characteristic features of almost all chronic kidney diseases. Fibrosis is tightly associated with destruction of capillaries, inflammation, and epithelial injury which progresses to loss of nephrons, and replacement of kidney parenchyma with scar tissue. Understanding the origins and nature of the cells known as myofibroblasts that make scar tissue is central to development of new therapeutics for kidney disease. Whereas many cell lineages in the body have become defined by well-established markers, myofibroblasts have been much harder to identify with certainty. Recent insights from genetic fate mapping and the use of dynamic reporting of cells that make fibrillar collagen in mice have identified with greater clarity the major population of myofibroblasts and their precursors in the kidney. This review will explore the nature of these cells in health and disease of the kidney to understand their central role in the pathogenesis of kidney disease.
Similar content being viewed by others

Introduction
Chronic kidney disease (CKD), affecting approximately 26 million people—a total of more than 8 % of the population in the USA, can be initiated by many insults to the kidney including toxic, ischemic, infectious, paraneoplastic, congenital, genetic, endocrine and immunological diseases as well as responses to xenobiotics [1]. Regardless of the initial insults, CKD is characterized by fibrosis of the glomeruli (glomerulosclerosis), interstitial fibrosis, injury with flattening (atrophy), loss of tubule epithelium, inflammation due to leukocyte recruitment, and loss of peritubular capillaries [2, 3]. The pathological syndrome of CKD frequently does not recover even when the initiating insults are removed or treated but frequently becomes self-sustaining. CKD itself stimulates further kidney injury, resulting in disease progression. Despite the very high prevalence of CKD there are no specific therapies except inhibition of signaling the angiotensin receptor. This inhibition has not abated the progression of CKD to organ failure [4].
Scarring results when tissues are damaged and normal wound-healing responses persist or become dysregulated. This occurs frequently in response to iterative or sustained injury. In CKD, myofibroblasts accumulate in the interstitial space. Myofibroblasts are the primary cells, which synthesize and deposit pathological constituents of fibrillar matrix. Accumulation of fibrotic matrix progressively destroys the normal kidney tissue architecture by contraction and increased stiffness as well as by increasing the space between vascular and tubular structures, resulting in disrupted blood flow supply and nephron function [5]. Fibrosis, is not simply matrix, however, but also the cells that deposit matrix. Recent studies indicate myofibroblasts are themselves an important inflammatory cell, producing an array of chemokines and innate inflammatory cytokines as wells as toxic oxygen radicals. Therefore the myofibroblast is not only a cell that deposits matrix but also contributes directly to parenchymal and microvascular injury or destruction.
The origin of the myofibroblasts has become a major focus in fibrosis research over the past 20 years and remains somewhat controversial [6]. The controversies persist due to the lack of ideal markers identifying only cells of the fibroblast/myofibroblast phenotype, and inter-organ and inter-species differences expression of those markers in current use. For more than a decade, it was widely thought that injured epithelial cells were the primary progenitors of myofibroblasts through a process of transdifferentiation known as epithelial-to-mesenchymal transition (EMT) [7, 8]. Recent use of reporter mice that mark cells producing fibrillar collagen has helped to define myofibroblasts with greater certainty. State-of-the-art fate mapping studies of poorly-appreciated mesenchymal cells in the kidney which become pericytes and resident fibroblasts, in conjunction with fate mapping studies of epithelial cells of the nephron have provided strong evidence that epithelial cells are not a significant source myofibroblasts. Rather, pericytes and resident fibroblasts are the major, if not the only, source of myofibroblasts in animal models of CKD [9••, 10•, 11]. Independent fate mapping studies in skin, muscle, lung, liver, pancreas and central nervous system all have identified pericytes (also known as perivascular cells or pericyte-like cells) as the major source of myofibroblasts in these respective organs, suggesting a unified model of fibrogenesis across our organs whereby discrete mesenchymal cells attached to capillaries are the major precursors of myofibroblasts [12, 13].
In this mini-review, we will describe fate mapping studies that lead to the current state of knowledge; explore recent insights into cellular and molecular mechanisms by which pericytes transdifferentiate into myofibroblasts during kidney disease, as well as the consequence of pericyte detachment from capillaries for capillary integrity in fibrogenesis.
Definition of Kidney Myofibroblasts
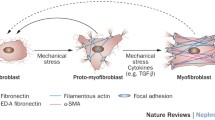
Identification of myofibroblasts in interstitial kidney disease has long been debated until recently. Myofibroblasts are contractile mesenchymal cells that deposit pathological fibrillar matrix in the virtual spaces between organ units. Myofibroblasts were originally defined by electron microscopy (EM) studies in the skin as contractile cells with characteristic cytoplasmic features including dense rough ER, moderate stress fibers of the actin cytoskeleton, but a paucity of lysosomes [14]. Myofibroblasts can be readily identified in CKD specimens by EM by similar criteria and distinguished from disease-associated fibroblasts by the extent of stress fibers, but EM has limited practical uses [15]. Instead, the intermediate filament α-smooth muscle actin (αSMA) has been used as a marker of myofibroblasts by many investigators. However, this is an imperfect marker because it is not specific (it also labels vascular smooth muscle and neonatal pericytes and stromal cells), and because in vitro many cells cultured on plastic express this protein. Therefore all cells in vitro can be considered myofibroblasts, but in vivo the situation has proven to be very different. Moreover, αSMA is at best an indirect marker of pathological matrix forming cells because it is not a matrix protein. In addition, antibodies used to identify αSMA protein are prone to artifact in mouse tissues since the antibodies are raised from mouse hybridomas, and therefore have high (non-specific) affinity for leukocytes [15]. These artifacts can be overcome by the use of directly conjugated antibodies and stringent blocking methods, but these are not widely employed [15]. Although detection of other intermediate filaments including desmin and vimentin have been used, these have proven to be even less specific. A more specific marker of myofibroblasts is the production of fibrillar collagen proteins, but such proteins are predominantly found extracellularly and inflammatory leukocytes frequently internalize collagen matrix, making such a protein marker imprecise. Our laboratories therefore generated a reporter mouse model of cells producing Collagen Iα1 (Col1a1) protein which express nuclear and cytoplasmic intracellular GFP which can be readily detected without amplification methods [10•, 16]. This mouse has been shown to faithfully report Collagen protein producing cells and mirrors the situation in human tissues [10•, 17, 18]. Col1a1-producing cells are present in normal adult kidney and have processes embedded in the basement membrane of peritubular capillaries. In adult kidney they lack αSMA expression, having downregulated its expression by 4 weeks of age [10•]. In models of kidney disease of greater than 4 days’ duration, all Col1a1 protein producing cells express αSMA, although not all αSMA+ cells produce Col1a1 protein [19] (Fig. 1). Therefore in the interstitium of chronically diseased kidney, αSMA does detect myofibroblasts and these are the cells which generate fibrillar collagen. When myofibroblasts were characterized using this reporter mouse they lacked epithelial, endothelial or leukocyte markers or S100A4 (also known as fibroblast specific protein-1) and were restricted to the interstitium [10•, 19]. However rigorous analysis of Col1a1-producing cells by flow cytometry of dissociated kidney cell preparations identified exceptionally rare leukocytes producing modest amounts of Col1a1 protein [10•]. Bone marrow chimera mice in which the Col1a1-GFP transgene (herein known as Coll-GFP Tg) was present only in bone marrow cells, confirmed that fewer than 1:1,000 Col1a1-producing cells were derived from bone marrow. In addition these leukocyte-derived Col1a1+ cells did not express αSMA, were not seen in the interstitium, rather they were restricted to perivenular areas. Finally, such leukocyte-derived Col1a1+ cells were not present in the kidney until 7 days after the commencement of injury [10•, 19]. Such studies provide strong evidence that in animal models of kidney disease, cells of leukocyte origin are not a significant source of fibrillar matrix forming cells or myofibroblasts [10•, 20, 21•, 22]. Importantly similar findings have been reported using stringent experiments in liver, lung, skin and muscle [23, 24].

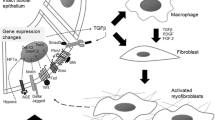
Model of myofibroblast appearance in the kidney interstitium and images of kidneys from mice in which the fate of progenitor cells was mapped in adult kidney disease. Schema showing the origin of kidney interstitial myofibroblasts as a population of mesenchymal cells (pericytes and resident fibroblasts) derived from FOXD1+ kidney progenitors. These cells become the major source of myofibroblasts in kidney diseases. Other cells including endothelial cells, epithelial cells, lymphocytes and macrophage populations can stimulate fibrogenesis by interacting with the resident mesenchymal cells. Left middle shows split panel image of mesenchymal cells (mainly pericytes) in normal kidney cortico-medullary junction derived from FOXD1+ progenitors (red). These cells constitutively produce Collagen Iα1 protein (green). Right middle shows split panel image of the fate of those same mesenchymal cells at d7 of the ureteral obstruction model of kidney disease. The mapped cells (red) have expanded in number and completely overlap with αSMA+ marker of myofibroblasts (green). Lower left shows split panel image of Collagen Iα1 protein producing (green) cells in Ureteral obstruction (d7) completely overlap with αSMA+ marker of myofibroblasts (red). Right lower shows monocyte derived cells (due to expression of the myeloid lysosomal protein LysM) in the kidney at d7 of the ureteral obstruction (red). The mapped cells show no overlap with Collagen Iα1 protein producing myofibroblasts (green) (Color figure online)
Genetic Fate Mapping Indicates that Interstitial Myofibroblasts Derive from Resident Perivascular Cells and These Derive from FOXD1+ Nephrogenic Progenitors
In normal adult mouse kidney, αSMA expression is restricted to the vascular smooth muscle of arterioles. In adult human, rat and neonatal mouse kidney, αSMA is expressed at low levels and to a variable extent by discrete perivascular cells [10•, 19]. By contrast, an extensive network of discrete Collagen producing cells in perivascular locations are readily identified by fluorescence microscopy in normal kidney of the Col1a1 reporter mouse [19] (Fig. 1). These Col1a1-producing cells express a number of typical pericyte or mesenchymal stem cell markers including PDGFRβ, PDGFRα, CD73, CD44 and variable levels of CD105 and CD90 as well as a number of other mesenchymal markers including CD248 [25, 26]. In neonatal kidney many express NG2 and P75NGR, but expression is downregulated in adult kidney [27]. Normal kidney Col1a1-producing cells do not express leucocyte, endothelial or epithelial markers [10•] and are therefore mesenchymal cells. The majority of these cells are attached to peritubular capillaries and have processes in capillary basement membrane. They are therefore called perivascular cells or pericytes [10•, 28]. When the cells are not attached to capillaries they may be referred to as resident fibroblasts. Surprisingly these discrete cells are extensive, particularly in the medulla of the kidney and numerically represent approximately 5 % of all adult kidney cells.
To define whether Col1a1-producing perivascular cells are the precursors of scar forming myofibroblasts, Humphreys et al. [9••] developed Cre/Lox reporter mice targeting the embryonic progenitors that give rise kidney stroma (mesenchymal cells) in response to activation of a critical transcription factor FOXD1. By using Cre/Lox reporter mice [29] all cells of the FOXD1 lineage were permanently labeled in mouse kidneys confirming that the stromal cells derived from FOXD1+ progenitors become vascular smooth muscle cells (VSMCs), pericytes, perivascular fibroblasts and mesangial cells of the kidney only. A comparison of Col1a1-producing cells with FOXD1-derived cells in adult normal kidney indicates nearly 100 % overlap of these two populations (Fig. 1), indicating that most FOXD1-derived cells are perivascular cells or pericytes. Triggering of interstitial kidney diseases in these lineage reporting mice resulted in an abundance of myofibroblasts (Col1a1 + αSMA + cells) in the interstitial space. All cells in the interstitium that expressed αSMA were derived from FOXD1 mesenchymal progenitors (Fig. 1) [9••]. Since FOXD1 is only expressed by nephrogenic progenitors during embryonic development, and not reactivated in this lineage in adult kidney or kidney disease these studies represent a true fate map and suggest strongly that the vast majority of Col1a1 protein producing αSMA+ cells in animal models of kidney disease arise from these progenitor cells.
Humphreys and colleagues used a similar strategy to genetically label all the daughter cells of the progenitors that give rise to kidney epithelium. To do this they targeted Cre expression to the transcription factor SIX2 which is restricted to the progenitors of epithelial cells in kidney development and is not reactivated in kidney diseases. They were able to map the fate of all epithelium by this strategy. In the setting of interstitial kidney diseases myofibroblasts were abundant in the interstitial space but no cells of epithelial lineage could be identified in the interstitium and none of the injured epithelial cells expressed αSMA [9••]. At least three independent investigations using similar fate mapping approaches have reported the same findings [30, 31].
Independent studies mapped mesenchymal cells even earlier in development and identified a population of progenitors expressing a myelin protein, P-zero, that completed migration from the neural crest region to the kidney metanephric mesenchyme by E13.5 [32]. These cells populated the zone of metanephric mesenchyme directly overlying ‘cap mesenchyme’ and known as cortical stroma, which is identical to the FOXD1+ mesenchymal area, before migrating into the kidney stroma, suggesting P-zero mapped cells are the precursors of FOXD1+ kidney progenitors. The P-zero progenitor derived cells became PDGFRβ+, CD73+ mesenchymal cells of the adult kidney (exactly the same as FOXD1+ progenitor derived cells). The group performed injury studies and showed that P-zero lineage cells, not epithelial or endothelial cells, were the source of kidney myofibroblasts using similar techniques to Humphreys and colleagues. Although the authors named them as resident fibroblasts of the adult kidney, rather than pericytes, they showed they are attached to capillaries and can generate erythropoietin. It is most likely therefore that P-zero lineage cells are overlapping or even the same as FOXD1+ lineage cells [9••, 32], and these studies provide strong supportive evidence that only mesenchymal cells become scar forming myofibroblasts. They also provide tantalizing evidence that mesenchymal cells may arise very early in development as neural crest progenitors.
Finally, although bone marrow chimera studies indicate that myeloid cells do not become myofibroblasts [10•, 19] we have evaluated whether myeloid lineage become myofibroblasts by mapping the fate of monocytes in the diseased kidney by Cre/Lox methods. Monocytes and neutrophils express lysosomal M protein and this protein is not expressed by other lineages [33]. Expression of Cre recombinase at the LysM locus enables permanent activation of a ubiquitous reporter gene (tdTomato fluorophore) only in cells of myeloid lineage (Fig. 1). These mice were bred with mice expressing the dynamic GFP reporter of Col1a1 protein production. In normal kidneys no monocyte derived cells expressed Col1a1 protein. We induced kidney disease in these mice (Fig. 1) and none of the Col1a1 protein expressing myofibroblasts expressed the monocyte lineage marker (Fig. 1).
Structure and Function of Pericytes
The major population of FOXD1 progenitor-derived cells in the kidney are attached to greater or lesser extent to peritubular capillaries and may therefore be considered to be perivascular cells or pericytes. Pericytes are contractile cells of mesenchymal origin that wrap around and support the microvasculature. In kidney, pericytes are attached to peritubular capillaries [9••, 34]. Pericytes and endothelial cells are embedded within the same basement membrane that they contribute to form by synthesizing laminins and collagens [35, 36]. Pericytes have intimate communications with endothelial cells by direct physical contact in a number of cytoplasmic regions including specialized invaginations of cytoplasm called “peg and socket” where paracrine signaling is believed to occur [37, 38]. Pericytes can be distinguished from perivascular fibroblasts (also known as fibrocytes or adventitial cells), which surround arterioles and have no connection with endothelium [39]. They can be distinguished from VSMCs, which surround larger blood vessels, by location, since VSMC are separated from endothelial cells by an internal elastic lamina and do not completely share molecular marker expression [3, 39]. Several recent reviews provide a list of validated markers to identify pericytes including those reported here [3, 39].
Pericytes contribute critically to the stability and integrity of the microvasculature, and are required for basement membrane synthesis and content [34, 40]. They regulate vascular tone, blood flow [41, 42], and capillary diameter [43]. In the kidney, medullary pericytes have been studied in greatest detail [41, 44–46]. They line the descending vasa recta, and relatively spare the ascending vasa recta. Medullary pericytes have been shown to regulate the distribution of blood flow from cortex to medulla, which is critical for regulation of salt and water homeostasis. Signaling between the thin descending limb of the loop of Henle and medullary pericytes plays a role in this regulation and a number of short acting factors including release of physiological levels of NO and H2O2 have been implicated in this process. More recently, it has become appreciated that pericytes are sensors of oxygen tension generating constitutively high levels of HIF-2 transcripts, and are the likely source of erythropoietin production in the kidney [32].
Studies have shown that a failure of integration of pericytes into the vascular wall during angiogenesis leads to collapse, aneurysm or hemorrhage of the vessel [47]. Likewise in response to sustained tissue injury or inappropriate activation, pericytes detach from the vessel wall. This process may initially be beneficial, since it permits endothelial activation, angiogenesis and leukocyte migration [48], but also leads to increased capillary permeability and instability, which ultimately resulting in capillary destruction [21].
Model of Fibrogenic Mechanisms in the Kidney Interstitium
The genetic fate mapping and bone marrow chimera studies lead us to a new model of fibrogenesis, whereby discrete cells resident to the kidney can activate to become fibrillar matrix forming cells. These discrete cells perform important homeostatic and potentially reparative functions in the kidney but in response to sustained injury detach from their niche [10•, 21•, 49•] enter the interstitial space and become cells that deposit pathological matrix. In doing so they lose certain functions of their precursors. Overwhelming evidence from many studies indicate that the injured epithelium of the kidney is a potent stimulus for fibrogenesis as is injury or dysfunction to the microvasculature. In addition ablation studies from laboratories including our own indicate that macrophages may potently stimulate the fibrogenic process. However most evidence from carefully performed in vivo studies indicate that epithelium, endothelium and leukocytes contribute to the interstitial fibrogenic process predominantly by indirect means (Fig. 1). The consequence from such a model is the pressing need to understand the cell signaling mechanisms that regulate this process as well as the functional changes when pericytes become myofibroblasts.
Molecular Mechanisms of Perivascular Cell Transdifferentiation into Myofibroblasts
The studies presented lead to a powerful hypothesis that by identifying factors regulating: pericyte detachment from capillaries; transdifferentiation into myofibroblasts; and regulation of myofibroblast activation or survival we will identify novel therapeutic strategies to treat inflammation, fibrosis and parenchymal destruction in CKD. This area of research is in its infancy, but several important cell pathways have already been identified that may rapidly lead to identification and development of drug candidates.
Platelet Derived Growth Factor Signaling
Endothelial, epithelial cells and macrophages are important sources of PDGFs in kidney disease. PDGFRα and β are expressed by kidney pericytes and resident fibroblasts during homeostasis and in short-term models of adult kidney disease, these receptors remain restricted to FOXD1 lineage cells including myofibroblasts. Blockade of these receptors by antibodies or soluble receptors (acting as ligand traps) [18, 21•], has been reported to attenuate detachment pericytes and their differentiation into myofibroblasts, and to reverse the evolution of fibrotic disease. Moreover, these receptor-blocking strategies markedly attenuate immune cell recruitment and microvascular disease [21•], implicating myofibroblasts and their activation state in leukocyte recruitment. Strategies to attenuate activation at these receptors may be useful in human CKD but further studies are required. Several kinase inhibitors which inhibit phosphorylation of PDGFRβ are in clinical trials (Novartis and Boehringer-Ingelheim) to treat fibrosis in several organs, including lung, although the inhibit activation of other receptors including VEGFR2 and FGFRs [15].
TGFβ Signaling
TGFβ signaling is widely known to promote epithelial injury and fibrosis and therapies to block TGFβ or its receptor are in clinical trials for fibrosing diseases. As well as the PDGF pathway, most cells in the kidney generate TGFβ including endothelium, epithelium and macrophages. The receptors are widely expressed. TGFβRI and RII are expressed by pericytes and signaling from these receptors is sufficient to trigger myofibroblast transdifferentiation in vivo and in vitro [27]. Non-canonical signaling pathways via MAPKs may be a dominant signaling pathway in myofibroblasts and their precursors, and further studies are required to understand the significance of canonical signaling via βcatenin nuclear activity in these cells.
Regulation of Metalloproteinase and Integrin Activity
Studies from our and collaborator’s laboratories suggest that pericyte detachment from capillaries is early critical step in the fibrogenic process that may be amenable to therapeutic targeting [10•, 49•]. Understanding of detachment is in its infancy, but studies have implicated metalloproteinases in this process, including the ADAM and ADAMTS families. ADAMs are a family of metalloproteinases, which cleave specific proteins including capillary basement membrane proteins such as versican, but also regulate VEGF signaling, migration, and inhibit normal angiogenesis [40, 50, 51]. In kidney ADAMTS1 is highly upregulated in pericytes early after injury and its natural inhibitor, TIMP3 is highly expressed normally but is rapidly downregulated after injury [40]. Targeting specific ADAM or ADAMTS activity may be beneficial. Integrin activity and binding may also be important regulators of the attachment, investment process in pericytes and is an area requiring further study.
Connective Tissue Growth Factor (CTGF) Signaling
Connective tissue growth factor (CTGF), a member of the CCN family of small, cysteine-rich secreted modular proteins that can integrate signals of integrins and growth factors or morphogens. Expression of CTGF has been recognized as a fibrogenic factor in kidney and other diseases [52]. It is highly produced by pericytes and podocytes and plays important roles in angiogenesis and maintenance of the vasculature in development and homeostasis. Stimulation of pericytes by CTGF also directly triggers a migratory and myofibroblast phenotype, which may be independent of TGFβ signaling. This signaling is also is dependent on MAPK signaling in kidney pericytes [49•]. It appears therefore that CTGF can also signal pathologically to stimulate myofibroblast appearance and activation, but that the nature of this signaling is distinct from its effects on kidney epithelial cells [49•]. Currently antibodies that bind to CTGF are in clinical trials as anti-fibrotic therapies [15].
WNT Signaling
Activated pericytes and myofibroblasts show marked upregulation of the WNT/β-catenin pathway. The WNT/β-catenin pathway can be blocked by exogenous administration into the circulation of an endogenous regulator of the pathway, known as Dickkopf related protein-1 (DKK-1) which binds to the WNT co-receptor known as LRP-6 at the cell surface. Systemically delivery of DKK-1 remarkably attenuates inflammation, fibrosis capillary disease and epithelial injury. A major target for DKK-1 FOXD1-lineage of cells including activated pericytes and mature myofibroblasts [49•]. Intriguingly the WNT receptor LRP-6 appears necessary for PDGFR, CTGF and TGFβR signaling in pericytes and myofibroblasts that results in MAPK pathway activation in myofibroblasts, so that DKK-1 is able to attenuate the deleterious effects of these signaling pathways in fibrogenes. Studies suggest that in the FOXD1 lineage of cells in the kidney the WNT pathway and these other fibrogenic signaling pathways converge physically at the plasma-membrane to effect pathological signaling [49•, 53]. DKK-1 and extracellular blockade of the WNT signaling complex appears to be a novel therapeutic target in CKD but further studies are required.
Vascular Endothelial Growth Factor Receptor Signaling
VEGF receptor 2 (VEGFR2) is restricted to endothelial cells in the kidney. Genetic deletion of VEGF in podocytes leads to unstable glomerular capillaries and microthrombi formation [54, 55], but attenuated VEGFR2 signaling in interstitial kidney disease is beneficial [21•]. This paradox is complex to understand, but may hinge around the fact that in disease states splice variants of VEGFA are generated in the kidney that may lead to aberrant signaling in pertitubular capillaries at VEGFR2 and its co-receptors neuropilins. Excessive VEGFR2 signaling activates endothelial cells, which may stimulate pericyte activation, recruit leukocytes and stimulate fibrogenesis. Although VEGFR2 signaling is also an attractive therapeutic target in CKD, further understanding of the distinction between glomerular and peritubular capillary signaling is required in order that signaling in the tubulointerstitium can be targeted without detrimentally affecting glomerular functions. Small molecule inhibitors such as tyrosine kinase receptors BIBF1120 that include inhibition of VEGFR2 are in currently in advanced clinical trials in fibrotic disease in lung and the efficacy and side effect profile will be carefully studied during those trials.
Other Developmental Cell Signaling Pathways
The Eph:ephrinB signaling pathway is known to play roles in axon guidance, but these signaling receptors are expressed more widely. In skin and in cancer, signaling at these receptors has been reported to play important roles in maintenance of normal pericyte endothelial interactions during patterning of the vasculature. This physiological signaling has been reported to be disrupted in fibrogenic and pathological microvascular disease [56, 57]. Bidirectional signaling of Eph:ephrinB pathway is active in kidney microvascular endothelium and in pericytes [58]. When disrupted, it affects endothelial behavior and the ability of pericytes to maintain microvascular stability [58]. Disrupted signaling by these receptors provokes fibrogenesis and microvascular destruction. Similarly, in the glomerulus Eph:ephrinB signaling by podocytes has been shown to be important in glomerular development and regeneration after injury. This receptor signaling pathway may be an important homeostatic mechanisms that could be targeted to stimulate normal repair processes. Recent studies have also identified the Hedgehog signaling pathway in the fibrogenic process. In development, this signaling pathway plays important roles in vasculogenesis and in bidirectional signaling from endothelium to pericytes. In adult kidney disease the tubules are an important source of Hedgehog ligands but Hedgehog pathway receptors Gli-1 and Gli-2 are restricted to pericytes and perivascular fibroblasts [59]. Although this pathway is reactivated in disease, blocking studies have produced variable results leading to current uncertainties about the importance of this signaling pathway in pericyte activation in response to tissue injury indicating further studies are merited [59, 60].
Understanding Myofibroblasts in Glomerular Disease
Mesangial cells are a glomerular pericyte and also derive from FOXD1+ embryonic progenitor cells, similar to pericytes of kidney peritubular capillaries. These cells have been thought to be a major cellular source of glomerular fibrosis [61]. Nodules of fibrotic tissue are frequently seen in mesangial areas of the glomerulus and the mesangial extracellular matrix is frequently expanded in diabetic kidney disease. But the role of the podocyte in loss of the glomerular filtration barrier and glomerular scarring has recently come to the forefront in diabetic and other glomerular diseases. A reduction in podocyte coverage of glomerular capillary loops by progressive loss of the population of podocytes in each glomerulus is not only a hallmark of glomerular disease but also believed to be functionally critical in disease [62, 63]. New lines of evidence indicate that although the podocyte has epithelial characteristics, expresses epithelial genes and transcription factors during nephrogenesis [64], it subsequently acquires pericyte characteristics and functions after nephrogenesis [55, 65]. Like the pericyte, podocytes synthesize capillary basement membrane, stabilize endothelial cells, maintain fenestrations, and regulate capillary permeability. Indeed emerging evidence suggests that mature podocytes have undergone an EMT type process compared to their embryonic counterparts. It is interesting therefore that in the normal glomerulus, podocytes, but not mesangial cells produce Col1a1 protein [10•, 66]. One explanation for this is that non-fibrillar Col1a1 protein is an important minor constituent of glomerular basement membrane. Another explanation is that Collagen I protein is generated but rapidly degraded (turnover). It is possible therefore that in disease states such as CKD, the diseased podocyte is a significant or even major source of pathological fibrillar matrix production in the glomerulus. In recent studies our laboratory showed that in a mouse model of antibody mediated crescentic glomerulonephritis with focal and segmental glomerulosclerosis (FSGS), podocytes were the only cell type to generate significant levels of Collagen I protein [19]. These findings implicate the diseased podocyte (rather than podocyte loss), directly in the fibrogenic process in glomerular tufts. Obviously, new studies are required to determine the extent to which diseased podocytes are a type of myofibroblast in chronic glomerular diseases, and the extent to which pericyte/myofibroblast gene-targets will benefit podocyte functions.
Conclusions
Resident mesenchymal cells derived from FOXD1+ nephrogenic progenitor cells are the major precursors of scar-forming myofibroblasts in the kidney interstitium. During injury, differentiation of perivascular cells (pericytes) into myofibroblasts not only contributes to deposition of pathological extracellular matrix, but also plays a role in capillary rarefaction and inflammation, which leads to CKD. Myofibroblasts play a central role during kidney fibrosis; however, epithelial, endothelial cells and leukocytes have also important roles during fibrogenesis by paracrine signaling mechanisms. Here, we have illustrated some of these mechanisms, but further studies are urgently needed to elucidate these paracrine signaling mechanisms.
References
Recently published papers of particular interest have been highlighted as: • Of important •• Of major importance
Snyder JJ, Foley RN, Collins AJ (2009) Prevalence of CKD in the United States: a sensitivity analysis using the National Health and Nutrition Examination Survey (NHANES) 1999–2004. Am J Kidney Dis 53:218–228. doi:10.1053/j.ajkd.2008.07.034
Grgic I, Duffield JS, Humphreys BD (2012) The origin of interstitial myofibroblasts in chronic kidney disease. Pediatr Nephrol 27:183–193. doi:10.1007/s00467-011-1772-6
Schrimpf C, Duffield JS (2011) Mechanisms of fibrosis: the role of the pericyte. Curr Opin Nephrol Hypertens 20:297–305. doi:10.1097/MNH.0b013e328344c3d4
National Kidney Foundation (2012) KDOQI clinical practice guideline for diabetes and CKD: 2012 update. Am J Kidney Dis 60:850–886. doi:10.1053/j.ajkd.2012.07.005
Wynn TA (2007) Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 117:524–529. doi:10.1172/JCI31487
Zeisberg M, Duffield JS (2010) Resolved: EMT produces fibroblasts in the kidney. J Am Soc Nephrol 21:1247–1253. doi:10.1681/ASN.2010060616
Kalluri R, Neilson EG (2003) Epithelial–mesenchymal transition and its implications for fibrosis. J Clin Invest 112:1776–1784. doi:10.1172/JCI20530
Zeisberg M, Kalluri R (2008) Fibroblasts emerge via epithelial–mesenchymal transition in chronic kidney fibrosis. Front Biosci 13:6991–6998
•• Humphreys BD, Lin S-L, Kobayashi A et al (2010) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176:85–97. doi:10.2353/ajpath.2010.090517. This study reveals that pericytes, not epithelial cells, are the major source of scar-forming myofibroblasts in animal models of interstitial kidney disease using fate-mapping approaches
• Lin S-L, Kisseleva T, Brenner DA, Duffield JS (2008) Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173:1617–1627. doi:10.2353/ajpath.2008.080433. This study shows that pericytes and the closely related perivascular fibroblasts are the major source of scar-forming myofibroblasts in animal models of interstitial kidney disease
Duffield JS, Humphreys BD (2011) Origin of new cells in the adult kidney: results from genetic labeling techniques. Kidney Int 79:494–501. doi:10.1038/ki.2010.338
Kisseleva T, Cong M, Paik Y et al (2012) Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci 109:9448–9453. doi:10.1073/pnas.1201840109
Bhattacharyya S, Wei J, Varga J (2011) Understanding fibrosis in systemic sclerosis: shifting paradigms, emerging opportunities. Nat Rev Rheumatol 8:42–54. doi:10.1038/nrrheum.2011.149
Gabbiani G, Gabbiani G, Majno G, Majno G (1972) Dupuytren’s contracture: fibroblast contraction? An ultrastructural study. Am J Pathol 66:131–146
Duffield JS, Lupher M, Thannickal VJ, Wynn TA (2012) Host responses in tissue repair and fibrosis. Annu Rev Pathol. doi:10.1146/annurev-pathol-020712-163930
Krempen K, Grotkopp D, Hall K et al (1999) Far upstream regulatory elements enhance position-independent and uterus-specific expression of the murine alpha1(I) collagen promoter in transgenic mice. Gene Expr 8:151–163
Fligny C, Duffield JS (2013) Activation of pericytes: recent insights into kidney fibrosis and microvascular rarefaction. Curr Opin Rheumatol 25:78–86. doi:10.1097/BOR.0b013e32835b656b
Chen Y-T, Chang F-C, Wu C-F et al (2011) Platelet-derived growth factor receptor signaling activates pericyte–myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int 80:1170–1181. doi:10.1038/ki.2011.208
Campanholle G, Ligresti G, Gharib SA, Duffield JS (2013) Cellular mechanisms of tissue fibrosis. 3. Novel mechanisms of kidney fibrosis. AJP Cell Physiol 304:C591–C603. doi:10.1152/ajpcell.00414.2012
Castano AP, Lin SL, Surowy T et al (2009) Serum amyloid P inhibits fibrosis through Fc gamma R-dependent monocyte-macrophage regulation in vivo. Sci Transl Med 1:5ra13–5ra13. doi:10.1126/scitranslmed.3000111
• Lin SL, Chang FC, Schrimpf C et al (2011) Targeting endothelium–pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. AJPA 178:911–923. doi:10.1016/j.ajpath.2010.10.012. This study shows links between endothelium and pericytes through VEGF during kidney fibrosis and that attenuated VEGFR2 signaling is beneficial in interstitial kidney disease
Lin SL, Castano AP, Nowlin BT et al (2009) Bone marrow Ly6Chigh monocytes are selectively recruited to injured kidney and differentiate into functionally distinct populations. J Immunol 183:6733–6743. doi:10.4049/jimmunol.0901473
Scholten D, Reichart D, Paik YH et al (2011) Migration of fibrocytes in fibrogenic liver injury. Am J Pathol 179:189–198. doi:10.1016/j.ajpath.2011.03.049
Rock JR, Barkauskas CE, Cronce MJ et al (2011) Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc Natl Acad Sci 108:E1475–E1483. doi:10.1073/pnas.1117988108
Smith SW, Eardley KS, Croft AP et al (2011) CD248+ stromal cells are associated with progressive chronic kidney disease. Kidney Int 80:199–207. doi:10.1038/ki.2011.103
Smith SW, Schrimpf C, Parekh DJ et al (2012) Kidney pericytes: a novel therapeutic target in interstitial fibrosis. Histol Histopathol 27:1503–1514
Wu C-F, Chiang W-C, Lai C-F et al (2012) Transforming growth factor β-1 stimulates profibrotic epithelial signaling to activate pericyte–myofibroblast transition in obstructive kidney fibrosis. Am J Pathol. doi:10.1016/j.ajpath.2012.09.009
Allt G, Lawrenson JG (2001) Pericytes: cell biology and pathology. Cells Tissues Organs 169:1–11
Nagy A (2000) Cre recombinase: the universal reagent for genome tailoring. Genesis 26:99–109
Bielesz B, Sirin Y, Si H et al (2010) Epithelial Notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest 120:4040–4054. doi:10.1172/JCI43025
Koesters R, Kaissling B, Lehir M et al (2010) Tubular overexpression of transforming growth factor-beta1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol 177:632–643. doi:10.2353/ajpath.2010.091012
Asada N, Takase M, Nakamura J et al (2011) Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J Clin Invest 121:3981–3990. doi:10.1172/JCI57301
Clausen BE, Burkhardt C, Reith W et al (1999) Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res 8:265–277
Kida Y, Duffield JS (2011) Pivotal role of pericytes in kidney fibrosis. Clin Exp Pharmacol Physiol 38:467–473. doi:10.1111/j.1440-1681.2011.05531.x
Stratman AN, Malotte KM, Mahan RD et al (2009) Pericyte recruitment during vasculogenic tube assembly stimulates endothelial basement membrane matrix formation. Blood 114:5091–5101. doi:10.1182/blood-2009-05-222364
Stratman AN, Schwindt AE, Malotte KM, Davis GE (2010) Endothelial-derived PDGF-BB and HB-EGF coordinately regulate pericyte recruitment during vasculogenic tube assembly and stabilization. Blood 116:4720–4730. doi:10.1182/blood-2010-05-286872
Armulik A, Genové G, Betsholtz C (2011) Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21:193–215. doi:10.1016/j.devcel.2011.07.001
Bergers G, Song S (2005) The role of pericytes in blood-vessel formation and maintenance. Neurooncology 7:452–464. doi:10.1215/S1152851705000232
Rojas A, Chang F-C, Lin S-L, Duffield JS (2012) The role played by perivascular cells in kidney interstitial injury. Clin Nephrol 77:400–408
Schrimpf CC, Xin CC, Campanholle GG et al (2012) Pericyte TIMP3 and ADAMTS1 modulate vascular stability after kidney injury. J Am Soc Nephrol 23:868–883. doi:10.1681/ASN.2011080851
Pallone TL, Silldorff EP (2001) Pericyte regulation of renal medullary blood flow. Exp Nephrol 9:165–170
Herman IM, D apos Amore PA (1985) Microvascular pericytes contain muscle and nonmuscle actins. J Cell Biol 101:43–52
Peppiatt CM, Howarth C, Mobbs P, Attwell D (2006) Bidirectional control of CNS capillary diameter by pericytes. Nature 443:700–704. doi:10.1038/nature05193
Zhang Z, Lin H, Cao C et al (2010) Voltage-gated divalent currents in descending vasa recta pericytes. Am J Physiol Renal Physiol 299:F862–F871. doi:10.1152/ajprenal.00321.2010
Zhang W, Pibulsonggram T, Edwards A (2004) Determinants of basal nitric oxide concentration in the renal medullary microcirculation. Am J Physiol Renal Physiol 287:F1189–F1203. doi:10.1152/ajprenal.00125.2004
O apos Connor PM, Cowley AW (2012) Medullary thick ascending limb buffer vasoconstriction of renal outer-medullary vasa recta in salt-resistant but not salt-sensitive rats. Hypertension 60:965–972. doi:10.1161/HYPERTENSIONAHA.112.195214
Smith SW, Chand S, Savage COS (2012) Biology of the renal pericyte. Nephrol Dial Transplant 27:2149–2155. doi:10.1093/ndt/gfs134
Proebstl D, Voisin M-B, Woodfin A et al (2012) Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med 209:1219–1234. doi:10.1084/jem.20111622
• Ren S, Johnson BG, Kida Y et al (2013) LRP-6 is a coreceptor for multiple fibrogenic signaling pathways in pericytes and myofibroblasts that are inhibited by DKK-1. Proc Natl Acad Sci USA 110:1440–1445. doi:10.1073/pnas.1211179110. This study shows the detailed signaling pathways including the dominance of non-canonical WNT pathway signaling involved in pericyte proliferation and activation
Basile DP, Fredrich K, Chelladurai B et al (2008) Renal ischemia reperfusion inhibits VEGF expression and induces ADAMTS-1, a novel VEGF inhibitor. Am J Physiol Renal Physiol 294:F928–F936. doi:10.1152/ajprenal.00596.2007
Jönsson-Rylander A-C, Nilsson T, Fritsche-Danielson R et al (2005) Role of ADAMTS-1 in atherosclerosis: remodeling of carotid artery, immunohistochemistry, and proteolysis of versican. Arterioscler Thromb Vasc Biol 25:180–185. doi:10.1161/01.ATV.0000150045.27127.37
Yokoi H, Mukoyama M, Nagae T et al (2004) Reduction in connective tissue growth factor by antisense treatment ameliorates renal tubulointerstitial fibrosis. J Am Soc Nephrol 15:1430–1440
He W, Dai C, Li Y et al (2009) Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J Am Soc Nephrol 20:765–776. doi:10.1681/ASN.2008060566
Machado FG, Kuriki PS, Fujihara CK et al (2012) Chronic VEGF blockade worsens glomerular injury in the remnant kidney model. PLoS One 7:e39580. doi:10.1371/journal.pone.0039580
Eremina V, Jefferson JA, Kowalewska J et al (2008) VEGF inhibition and renal thrombotic microangiopathy. N Engl J Med 358:1129–1136. doi:10.1056/NEJMoa0707330
Salvucci O, Maric D, Economopoulou M et al (2009) EphrinB reverse signaling contributes to endothelial and mural cell assembly into vascular structures. Blood 114:1707–1716. doi:10.1182/blood-2008-12-192294
Foo SS, Turner CJ, Adams S et al (2006) Ephrin-B2 controls cell motility and adhesion during blood–vessel–wall assembly. Cell 124:161–173. doi:10.1016/j.cell.2005.10.034
Kida Y, Leronimakis N, Schrimpf C et al (2013) Defective ephrinB2 reverse signaling promotes capillary rarefaction and fibrosis after kidney injury. J Am Soc Nephrol 24(4):559–572
Fabian SL, Penchev RR, St-Jacques B et al (2012) Hedgehog-Gli pathway activation during kidney fibrosis. Am J Pathol 180:1441–1453. doi:10.1016/j.ajpath.2011.12.039
Ding H, Zhou D, Hao S et al (2012) Sonic hedgehog signaling mediates epithelial–mesenchymal communication and promotes renal fibrosis. J Am Soc Nephrol 23:801–813. doi:10.1681/ASN.2011060614
Johnson RJ, Floege J, Yoshimura A et al (1992) The activated mesangial cell: a glomerular “myofibroblast”? J Am Soc Nephrol 2:S190–S197
Reiser J, Sever S (2012) Podocyte biology and pathogenesis of kidney disease. Annu Rev Med. doi:10.1146/annurev-med-050311-163340
Jefferson JA, Alpers CE, Shankland SJ (2011) Podocyte biology for the bedside. Am J Kidney Dis 58:835–845. doi:10.1053/j.ajkd.2011.03.033
Kobayashi A, Valerius MT, Mugford JW et al (2008) Six2 defines and regulates a multipotent self-renewing nephron progenitor population throughout mammalian kidney development. Cell Stem Cell 3:169–181. doi:10.1016/j.stem.2008.05.020
Brunskill EW, Georgas K, Rumballe B et al (2011) Defining the molecular character of the developing and adult kidney podocyte. PLoS One 6:e24640. doi:10.1371/journal.pone.0024640
Hutchison N, Fligny C, Duffield JS (2012) Resident mesenchymal cells and fibrosis. Biochim Biophys Acta. doi:10.1016/j.bbadis.2012.11.015
Acknowledgments
The Duffield Lab is funded by NIH Grants DK93493, DK94768, and DK87389. Naoki Nakagawa is funded by Asahikawa Medical University. Thanks to Ivan Gomez for assistance with images.
Compliance with Ethics Guidelines
Conflict of Interest
Naoki Nakagawa declares that he has no conflict of interest. Jeremy S. Duffield is on scientific advisory boards of Promedior and Regulus Therapeutics; is a cofounder of Muregen; and has served as a consultant to Takeda, Boehringer Ingelheim, Bristol-Myers Squibb, GlaxoSmithKline, Biogen Idec, and Gilead Sciences.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nakagawa, N., Duffield, J.S. Myofibroblasts in Fibrotic Kidneys. Curr Pathobiol Rep 1, 189–198 (2013). https://doi.org/10.1007/s40139-013-0025-8
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40139-013-0025-8