
Abstract
Introduction
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive, fatal and under-recognized disease. This targeted literature review assessed the extent and consequences of diagnostic delay and misdiagnosis in ATTR-CM.
Methods
The Embase database was searched together with proceedings of eight cardiology conferences to identify publications or abstracts on ATTR-CM. Outcomes of interest were time from symptom onset to diagnosis, rates of delayed diagnosis and misdiagnosis, and costs, healthcare resource use or clinical outcomes whilst undiagnosed/misdiagnosed.
Results
Twenty-three articles were included. Weighted means of reported mean and median diagnostic delays were 6.1 and 3.4 years for wild-type (ATTRwt-CM) and 5.7 and 2.6 years for hereditary (ATTRv-CM). Misdiagnosis occurred in 34–57% of patients when reported. Evaluation and misdiagnosis by multiple healthcare providers before receiving an ATTR-CM diagnosis was common, and there was evidence that patients undergo unnecessary or inappropriate evaluations or treatments while misdiagnosed. Diagnostic “red flags” were reported to be underused. Data on the consequences of delay for patients and health systems were sparse, but given the progressive nature of ATTR-CM, delay is likely to have adverse consequences.
Conclusion
ATTR-CM patients commonly experience diagnostic delay and misdiagnosis. Efforts are required to provide timely diagnosis so that patients can benefit from earlier access to new disease-modifying therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a progressive and fatal disease that is often delayed in its recognition or misdiagnosed. |
This study reviewed the literature on diagnostic delay and its impact on patients and health systems. |
Mean and median diagnostic delays were 6.1 and 3.4 years for ATTRwt-CM and 5.7 and 2.6 years for ATTRv-CM (weighted means of reported delays). |
Patients reported evaluation by multiple healthcare providers before receiving a diagnosis of ATTR-CM, and incorrect or incomplete diagnoses were common. |
Efforts are required to provide timely diagnosis so that patients can benefit from earlier access to new disease-modifying therapies. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14333732.
Introduction
Transthyretin amyloid cardiomyopathy (ATTR-CM) is a fatal disease characterized by the deposition of misfolded transthyretin (TTR) amyloid fibrils in the heart muscle (myocardium) [1], resulting in progressive symptoms of heart failure and increased risk of arrhythmias and conduction abnormalities [1, 2]. Patients experience increasing functional disability and significantly impaired health-related quality of life (HRQoL) as the disease progresses [3]. Prognosis is poor, with life expectancy between 2 and 6 years from diagnosis [4]. There are two subtypes of ATTR-CM: wild-type (ATTRwt-CM), which appears to be associated with aging, and a hereditary form, also known as variant (ATTRv-CM), caused by inherited genetic mutations of TTR [5].
The prevalence of ATTR-CM is not well characterized, and global estimates could not be identified. A prevalence estimate for ATTRwt-CM of 3 per 10,000 has been reported in the European Union [6]; however, only a small proportion of these patients are currently diagnosed. Increasing evidence suggests that ATTR-CM may be found in patients with other conditions that are associated with aging, such as heart failure with preserved injection fraction (HFpEF) and aortic stenosis [7,8,9,10]. Hereditary ATTR-CM is a rare disease; its prevalence is unknown, and varies between regions due to geographical clustering of TTR mutations [7]. Little information is available on the incidence of ATTR-CM; a study from France suggested an incidence rate for both subtypes combined of 1.8/100,000 in 2017 [11].
ATTR-CM is often delayed in its recognition or misdiagnosed [7, 12]. Overlap of symptoms with other conditions and fragmented knowledge, limited chiefly to specialists, are believed to have contributed to low awareness and consequent underdiagnosis, even among heart failure specialists [7, 12]. In addition, the need for endomyocardial biopsy with histological testing, previously regarded as the gold standard for diagnosis [13], is thought to have discouraged or delayed investigation, as it is expensive, requires specialist expertise and carries a procedural risk [12,13,14]. A consensus paper on a noninvasive diagnostic algorithm involving nuclear bone scintigraphy imaging was published in 2016 [13], and recent multi-societal expert consensus recommendations for noninvasive diagnostic techniques [4, 5, 15] and diagnostic “red flags” [7] for ATTR-CM have contributed to greater awareness. Until the recent introduction of tafamidis for the treatment of ATTR-CM there were no Food and Drug Administration (FDA)- or European Medicines Agency (EMA)-approved pharmacologic therapies, which further reduced the incentive to appropriately diagnose these patients. Now that a proven disease-modifying therapy is available [16], delayed diagnosis represents a missed opportunity for timely treatment.
Although diagnostic delay and misdiagnosis are recognized problems in ATTR-CM, no review and synthesis of the literature on this topic has yet been published. The objective of this targeted literature review was to assess the extent of diagnostic delays and errors in ATTR-CM and their consequences for patients and healthcare systems.
Methods
Literature Search
This targeted review was undertaken with reference to the PRISMA-P (Preferred Reporting Items for Systematic Review and Meta-Analysis Protocols) guidance [17]. The Embase database was searched via Embase/Elsevier from inception to May 6, 2020. Search strategies used variations around terms such as “transthyretin cardiac amyloidosis”, “delayed diagnosis” and “misdiagnosis”. In addition, proceedings of eight major cardiology conferences from January 2015 to 15 May 2020 were searched, together with the reference lists of relevant studies, and a free-text internet search was undertaken. Searches were restricted to English language publications. Search strategies and conference proceedings searched are shown in supplementary material Tables S1 and S2, respectively.
Studies eligible for inclusion were those describing adult patients with ATTR-CM (ATTRwt-CM or ATTRv-CM) and reporting outcomes relating to the following: time from onset of symptoms to diagnosis; rate of misdiagnosis or delayed diagnosis; healthcare resource use (healthcare provider visits, hospitalization, medication) or indirect costs whilst undiagnosed or misdiagnosed; and disease progression, anxiety or mental health outcomes whilst undiagnosed or misdiagnosed.
Titles and abstracts of publications identified by the searches were screened for eligibility by a single reviewer. The full text of potentially eligible publications was reviewed against the eligibility criteria, and reasons for exclusion were documented. Data were extracted by a single reviewer into a bespoke extraction grid; checks were made by a second reviewer and discrepancies resolved by consensus. Data extracted from eligible publications included patient characteristics (including genotype, age at onset and age at diagnosis), time to diagnosis, rates of misdiagnosis, common misdiagnoses, and clinical consequences and resource use associated with delayed diagnosis or misdiagnosis.
Data Analysis
Data were summarized descriptively. For instances where diagnostic delay was not reported but age at symptom onset and age at diagnosis were available, delay durations were derived as the difference between the reported values. Delay durations were summarized across study-defined ATTRwt-CM and ATTRv-CM cohorts, weighting reported mean and median delays by the corresponding cohort size. This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Results
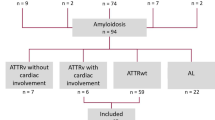
A total of 629 reports were identified excluding any duplicates. Fifty-nine articles were retrieved for full-text review, of which 23 met the inclusion criteria (Fig. 1). Eleven were full publications and 12 were conference abstracts. Electronic copies of the posters associated with the conference abstracts were obtained where available.

PRISMA flow diagram
Twelve studies collected data in North America [18,19,20,21,22,23,24,25,26,27,28,29], three in Asia [30,31,32], six in Europe [3, 33,34,35,36,37], and two were conducted internationally [38, 39]. Fourteen publications reported data for ATTRwt-CM [40], eleven for ATTRv-CM [3, 18, 21, 22, 24,25,26, 29, 30, 33, 39] and seven for mixed ATTR-CM populations (ATTRwt-CM and ATTRv-CM [18, 19, 23, 24, 27, 28, 37]). Study populations were predominantly male (69.6–98.0%) and elderly; mean age ranged from 60.9 to 78.2 years at symptom onset and 65.3 to 81.7 years at diagnosis. Details of the included studies are provided in supplementary table S3.
Diagnostic Delay (Time to Diagnosis)
Twenty-two studies [3, 18,19,20,21,22,23, 25,26,27,28,29,30,31,32,33,34,35,36,37,38,39] reported on diagnostic delay or had information on age at symptom onset and age at diagnosis from which time to diagnosis could be derived (Table 1). Symptom onset was patient-reported in some studies, while others reported delay from first documented presentation with cardiac or other specified symptoms. The nature of symptoms that were retrospectively considered as the first symptom was unspecified in most studies, or referred to simply as cardiac symptoms. Fatigue, shortness of breath and peripheral edema were the most common cardiac symptoms where a breakdown was reported. Several studies included carpal tunnel syndrome as a presenting symptom.
Patients faced substantial delays between onset of symptoms and a definitive ATTR-CM diagnosis. Mean time to diagnosis in whole-group ATTR-CM populations (i.e. not including values reported by subgroups other than ATTR type) ranged from 1.3 to 7.2 years for ATTRwt-CM [21, 22, 31, 32, 34, 35, 38] and 1.8–6.5 years for ATTRv-CM [21, 22] (Table 1). Median delays ranged from 0.8 to 3.9 years for ATTRwt-CM [3, 20, 22, 36, 38, 39] and 2.1–3.7 years for ATTRv-CM [3, 18, 22, 30, 39] populations.
Synthesis of the reported data suggests that overall, diagnostic delay is somewhat longer for ATTRwt-CM than ATTRv-CM. Weighted means of the reported mean and median diagnostic delays across the overall study populations were 6.1 and 3.4 years for ATTRwt-CM and 5.7 and 2.6 years for ATTRv-CM (Fig. 2). However, not all studies that reported on both groups reflected this trend; for example, Grogan et al. [22], reporting on a relatively large (n = 425) multinational sample from the Transthyretin Amyloidosis Outcomes Survey (THAOS), found that time to diagnosis was similar between the two types.

Overall study population diagnostic delays
The largest cohort of ATTRwt-CM patients identified in the literature is that described by Garcia-Pavia et al. [38], comprising 758 patients from THAOS and likely containing some of the same patients as the Grogan et al. [22] publication. The majority (69.3%) were from the USA and 86.3% were Caucasian. The median time from symptom onset to diagnosis (n = 651) was 3.9 years, with a 90th percentile time of 17.8 years illustrating the extremely long delay experienced by some ATTR-CM patients.
Another large cohort is the 1034 patients from the UK National Amyloidosis Centre, described by Lane et al. [3]. In the subset of 534 English patients with hospital usage data available, 42% of ATTRwt-CM patients waited > 4 years for a diagnosis after first presenting for consultation with cardiac symptoms; median (IQR) diagnostic delay was 39 (8–78) months. Median delay in ATTRv-CM diagnosis was shorter, at 25 (4–60) months. The proportion of patients waiting > 4 years for diagnosis was also reported by Lousada et al. 2018 [25], at 25% for ATTRwt-CM and 16% for ATTRv-CM; these data were taken from an online patient and caregiver survey, the Amyloidosis Research Consortium Cardiac Amyloidosis Survey, comprising 70 respondents with ATTRwt-CM and 114 with ATTRv-CM.
Cipriani et al. [34] reported delay from clinical suspicion of cardiac amyloidosis (on the evidence of typical clinical, electrocardiogram [ECG] and echocardiographic features) to definitive diagnosis in the 18 patients diagnosed with ATTRwt-CM at their Italian university hospital between 2015 and 2019. Mean diagnostic delay was 2.3 ± 1 years, leading the authors to conclude that long diagnostic delays continue to occur despite increasing clinical awareness and improved diagnostic modalities.
Similarly, Ladefoged et al. [36] reported delay from first cardiologic examination in 50 consecutive patients diagnosed with ATTRwt-CM at a Danish university hospital between 2017 and 2019. Median delay was 13 (2–47) months. What they termed as “red flag” examinations (correct diagnosis of ECG low voltage, utilization of left ventricular global longitudinal strain [LV-GLS] and measurement of the troponin T (TnT) and N-terminal pro B-type natriuretic peptide (NT-proBNP) biomarkers) were performed significantly less often in patients with longer diagnostic delay (> 3 months vs. ≤ 3 months, p < 0.001). They concluded that established red flags for ATTR-CM are poorly utilized in the diagnostic process.
Factors Associated with Diagnostic Delay
Multivariate analyses conducted by Bishop et al. [18] identified carpal tunnel syndrome (relative risk [RR] 4.57, 2.58–8.09), age < 70 at symptom onset (RR 3.67, 2.05–6.56) and TTR variant status (RR 2.23, 1.44–3.43 [no vs. yes]) as significant predictors of longer times to diagnosis, relative to the absence of these factors. The study was a retrospective chart review of 82 patients diagnosed with cardiac amyloidosis at a US center.
Several studies contained information on delay broken down by specific demographic or symptom groups, as shown in Supplementary table S4. Factors associated with longer delay are summarized in Table 2. However, information on each group was limited.
Age
Grogan et al. [22] found that median time to diagnosis in ATTRwt-CM was similar irrespective of age, whereas for ATTRv-CM it was longer in patients diagnosed at ≥ 70 years than in those diagnosed at a younger age.
Sex
Gonzalez-Lopez et al. [35] found that time to diagnosis of ATTRwt-CM was similar between the sexes, although females were significantly older than males at symptom onset (82.3 vs. 76.1 years, p = 0.002).
Phenotype
Bishop et al. [18] found that ATTRv-CM patients with a predominantly cardiomyopathy phenotype had a significantly longer diagnostic delay than those with a mixed phenotype (median [IQR] delay 53.1 [29.3–93.7] vs. 26.2 [9.4–59.0] months, p = 0.04). A similar trend was seen in Cilberti et al. [33], where the mean (SD) delay was 4 (3.2) years in cardiac phenotype patients and 3 (2.0) years in mixed phenotype. Bishop et al. [18] also found that median diagnostic delay was significantly longer in individuals without a family history of amyloid than in those with such a history (54.5 [26.1–94.5] vs. 24.5 [7.4–49.7] months; p = 0.0057).
Symptoms and Functional Classifications
There was little evidence on whether clinical outcome measures or functional classifications at presentation (for example, New York Heart Association [NYHA] class) are associated with length of diagnostic delay. The only study providing information on this was Gonzalez-Lopez et al. [35], where no relationship was identified. Nonspecific symptoms that present early in the disease course could be expected to be associated with the longest diagnostic delay. Only one study, Papoutsidakis et al. [28], examined diagnostic delay from first presenting symptom; it analyzed a mixed hereditary and wild-type group of 34 ATTR-CM patients in the Yale Cardiomyopathy Program in the USA. The symptom associated with the longest delay was carpal tunnel syndrome, with a median delay of 4.4 years. Erectile dysfunction, ocular problems and peripheral neuropathy were the only other symptoms associated with a median delay of greater than 2 years in this study.
Our review did not comprehensively examine the literature on presenting symptoms in ATTR-CM, as the included studies were required to contain information on diagnostic delay. However, several included studies stress the importance of carpal tunnel syndrome in raising suspicion of ATTR-CM. Ladefoged et al. [36] found that carpal tunnel syndrome was present in 45% (15/33) of ATTRwt-CM patients who had a diagnostic delay > 3 months, compared with 18% (3/17) of those with a shorter delay (p = 0.07). Lousada et al. [25] noted that 64% of ATTRwt-CM and 57% of ATTRv-CM patients had received a diagnosis of carpal tunnel syndrome before diagnosis with ATTR-CM.
Diagnostic Method
Information on the influence of diagnostic method on diagnostic delay was sparse. Gonzalez-Lopez et al. [35], reporting on 108 consecutive ATTRwt-CM patients at two Spanish centers over a period of up to 16 years, found a shorter median (IQR) time to diagnosis in patients diagnosed by noninvasive means (7 [1–45] months, vs. 12 [5–35] months for invasive diagnosis), but the difference was not statistically significant.
Misdiagnosis
Misdiagnosis (i.e. a diagnosis other than ATTR-CM to explain the patient’s cardiac symptoms) was common where reported, occurring in 34–57% of patients (five studies [24,25,26, 35, 37]; Table 2 and supplementary table S5). Gonzales-Lopez et al. [35] described 108 consecutive ATTRwt-CM patients diagnosed at two Spanish hospitals over a period of up to 16 years. Approximately one-third had previously been misdiagnosed with other cardiac diseases (including being given diagnoses that overlapped with ATTR-CM but did not recognize ATTR-CM as the underlying cause of heart failure). The most common were hypertensive heart disease (35.3% of those misdiagnosed), hypertrophic cardiomyopathy (23.5%) and ischemic heart disease (11.8%). These findings were similar whether patients had been diagnosed with ATTR-CM by noninvasive or invasive techniques. Similarly, in a cohort of 116 consecutive ATTR-CM (ATTRwt-CM or ATTRv-CM) patients diagnosed in a Spanish hospital over 10 years, reported by López-Sainz et al. [37], 45% had previously received an alternative diagnosis. HFpEF (10% of all patients) and hypertensive disease (9.4%) were most common.
Three publications by Lousada et al. [24,25,26] reported on previous misdiagnosis using data from the Amyloidosis Research Consortium online survey of patients and caregivers. Sample sizes ranged from 144 to 214, and there appeared to be some overlap between the populations. Across the publications, 39% of ATTRwt-CM and 44–57% of ATTRv-CM respondents reported receiving an alternative diagnosis. In ATTRwt-CM, the most common was unspecified heart failure (32% of 214 respondents [26]); this information was not available for ATTRv-CM patients. Incorrect or incomplete diagnoses were given by cardiologists as well as by generalists and specialists in other disciplines. In the largest sample [26], just under a third of ATTRwt-CM who reported a misdiagnosis said it came from a cardiologist, as did a slightly smaller proportion of ATTRv-CM patients.
Clinical Consequences of Delay or Misdiagnosis
Data on the clinical consequences of diagnostic delay or misdiagnosis were sparse, but the information available indicates that long delays are detrimental to patients, and many authors alluded to this in their discussions (summarized in Table 2). Ladefoged et al. [36] found that in ATTRwt-CM, a delay of ≥ 3 months was associated with significantly higher NYHA classification at diagnosis, indicating more advanced disease (Fig. 3). However, there was no significant difference between early and late diagnosis groups in the distribution of disease stages based on estimated glomerular filtration rate (eGFR) and NT-proBNP.

NYHA class distribution at diagnosis by delay duration. Data source: Ladefoged et al. [36]
Bishop et al. [18] found that delay in diagnosis ≥ 1 year was significantly associated with a number of adverse cardiac markers at time of diagnosis, after adjusting for amyloid type and amount of deposition: these were higher NT-proBNP (4451 vs. 2559 pg/mL, p = 0.016), prolonged PR interval (225 vs. 162 ms, p < 0.001), and a higher prevalence of intraventricular conduction delay (65 vs. 13%, p = 0.001) and atrial fibrillation (41 vs. 11%, p = 0.038) at the time of diagnosis. There was no association between delay and ejection fraction. Although this analysis population contained 30 patients with light-chain (AL) amyloidosis in addition to 52 with ATTRv-CM, the authors noted that adjustments for amyloid type and amount exclude potential differences in pathogenesis as confounders to the consequences of delay. They concluded that delayed diagnosis of either amyloidosis type has a detrimental effect on cardiac function.
Lane et al. [3] found that ATTR-CM patients had poor HRQoL at diagnosis, as measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ); the relationship between diagnostic delay and HRQoL was not explicitly explored, but it was noted that lengthy diagnostic delays in the face of a progressive disease likely contributed to poor HRQoL at the time of diagnosis.
Use of inappropriate treatment or diagnostic tests is another potential consequence of diagnostic delay or error, with implications for both patients and resource use. Lousada et al. [24] reported that approximately 75% of patients who had received a different diagnosis prior to ATTR-CM were treated for the misdiagnosed condition. For both ATTR types combined, 43% of misdiagnosed patients had received treatment with beta blockers and 26% with angiotensin-converting enzyme (ACE) inhibitors. The authors noted that these treatments may be inappropriate or poorly tolerated in ATTR-CM. These data provide an indication of the potential extent of inappropriate treatment but are subject to the limitations of the study format (online patient/caregiver questionnaire dependent on recall-based reporting).
Ladefoged et al. [36] noted that before their ATTRwt-CM diagnosis was established, 60% of patients were suspected of at least two other cardiac or noncardiac diseases and underwent various noninvasive and invasive diagnostic tests. Although misdiagnosis rates and consequent treatment were not specifically quantified, they noted that treatments were often applied until the suspected non-ATTR diagnosis was rejected. They further commented that these tests are associated with patient discomfort and procedural risk, and an increased economic burden.
Our review identified no data on mortality or effects on life expectancy associated with diagnostic delay.
Consequences of Delay/Misdiagnosis for Resource Use
Only four studies [3, 23, 24, 26] provided information on healthcare resource associated with diagnostic delay or misdiagnosis, all reporting on healthcare provider visits prior to ATTR-CM diagnosis (Table 3). Evaluation by multiple healthcare professionals before a definitive ATTR-CM diagnosis was common. Lousada et al. reported information from two samples from a patient and caregiver survey. In the larger sample [26], 16% of ATTRwt-CM patients reported seeing at least three different physicians before diagnosis, and 11% saw five or more. For ATTRv-CM, the proportions were 13% and 23%, respectively. Seventy-one percent of ATTRwt-CM patients and 59% of ATTRv-CM patients reported seeing two or more different cardiologists before diagnosis.
Kessler et al. [23] characterized resource use for 68 American and Canadian ATTR-CM patients enrolled in an online survey. Patients commonly reported seeing three or more healthcare professionals before diagnosis (69%), and a third (32%) had seen six or more. Most patients had visited primary care providers (76.5%) and cardiologists (75%), making a median of three visits to primary care providers and two visits to cardiologists prior to diagnosis with ATTR-CM.
Lane et al. [3] noted that during the 3 years before diagnosis, ATTR-CM patients were hospitalized a median of 17 times, including 3 inpatient hospital admissions, suggesting missed opportunities for diagnosis.
Discussion
This review confirms that diagnostic delay and misdiagnosis of ATTR-CM are common. Synthesis of the data on diagnostic delay, weighted by study sample size, showed a mean time to diagnosis of 6.1 years (median 3.4) for ATTRwt-CM and 5.7 (median 2.6) years for ATTRv-CM. However, there was a paucity of data on rates of misdiagnosis and on the consequences of delay or misdiagnosis for patients and health systems.
The reasons for diagnostic delay are multifactorial and include symptom overlap with other conditions, low disease awareness, the historical need for invasive diagnosis, and until recently the lack of a disease-modifying treatment. Advances in cardiac imaging and increased availability of genetic testing have aided in diagnosing ATTR-CM: a recent simplified, noninvasive diagnostic algorithm and consensus recommendations have the potential to increase disease awareness and reduce the diagnostic delay [4, 13]. However, there is evidence that uptake of new methods for the diagnosis of cardiac amyloidosis is slow outside of specialist centers. Lousada et al. [25] noted that technetium-99m diphosphonate (Tc-DPD) or pyrophosphate (Tc-PYP) scintigraphy scans are noninvasive, inexpensive and reliable but are currently underused, and other authors have reported continuing long delays despite both increased awareness and improved diagnostic modalities [34]. An analysis from the THAOS survey published after the search date for this review found that despite the rising use of scintigraphy in diagnosis, half of ATTRwt-CM patients experienced diagnostic delay of > 4.7 years [40].
Diagnosing ATTR-CM can be difficult, as cardiac symptoms are consistent with more common types of heart failure (HFpEF or hypertrophic cardiomyopathy [4]) and extra-cardiac manifestations are heterogeneous and nonspecific [7]. Autopsy studies suggest that ATTRwt-CM may in fact be a common but undiagnosed cause of HFpEF [9, 10]. The principal differential diagnoses include heart failure with preserved or reduced ejection fraction, aortic stenosis and hypertrophic cardiomyopathy [41]; Fabry disease and lone atrial fibrillation are also possibilities. It is also vital to distinguish between ATTR-CM and AL amyloidosis, as the two conditions have different biologies and treatments [4, 42]. Hypertension can mask hypertrophy, and hypertensive heart disease was a common misdiagnosis (Table 2).
A set of “diagnostic clues” to ATTR-CM were proposed in the expert consensus recommendations by Maurer et al. [4]. In addition, Witteles et al. recently proposed a set of “red flag” diagnostic markers designed to raise the index of suspicion for ATTR-CM among patients with heart failure; these include bilateral carpal tunnel syndrome, symptoms of polyneuropathy and/or dysautonomia, repeated mild increases in troponin levels, and a range of characteristic findings in relation to longitudinal strain, left ventricular thickness, atrioventricular block, echocardiographic features and extracellular volume expansion [7, 43]. Lumbar spinal stenosis and biceps tendon rupture have also been identified as clinical manifestations of amyloid deposition in ATTRwt [7, 43]. However, all of these conditions are common in older people, and there are limitations in attributing secondary symptoms to amyloidosis. The red flags are therefore designed to be applied within the context of increased left ventricular wall thickness [7]. Once a diagnosis of ATTR amyloidosis is determined, genetic testing should be performed to differentiate ATTRv-CM from ATTRwt-CM [4].
Several authors highlighted the need for improved understanding of early presenting symptoms of ATTR-CM, particularly carpal tunnel syndrome [18, 25, 36], which is a frequent early sign of amyloidosis and can precede clinical heart failure by several years [7, 20, 28, 44]. Ladefoged et al. noted that the proposed red flags remain underused, and were less likely to have been used in patients with the longest diagnostic delay [36]. Wider use of red flags in the relevant clinical scenarios would in turn lead to more timely use of ATTR-specific investigations and reduce diagnostic delay.
The only study to examine diagnostic delay by presenting symptom, Papoutsidakis et al. [28] confirmed that carpal tunnel syndrome appeared earlier than any other manifestation. Although many of the studies identified did not specify the nature of “first symptoms” of ATTR-CM, variations in symptom definition may account for some of the differences in reported delays.
Delay appears to be slightly longer for wild-type than hereditary ATTR-CM. This is not unexpected, as earlier diagnosis of ATTRv-CM might be anticipated due to the familial component [26]. However, Lousada et al. found that a high percentage of ATTRv-CM patients went undiagnosed after symptom onset, and called for better awareness of ATTRv-CM among both healthcare providers and affected patient communities to ensure that early symptoms are recognized. Disparities in access to healthcare may also contribute to delayed diagnosis of ATTRv-CM [45]. In the USA, ATTRv-CM is found predominantly in African Americans [46], caused by the V122I variant which is present in 3.43% of the African American population [47]. African Americans are more likely to have difficulties in access to healthcare than the general US population [48], and there is a risk that communities who are underserved by healthcare provision will not benefit from efforts designed to promote early diagnosis of ATTR-CM [45]. V122I ATTRv-CM is also an under-recognized cause of heart failure in people of Afro-Caribbean descent in the UK [49]. In both countries, individuals of African descent have a higher prevalence of hypertensive heart disease, leading to a readiness to ascribe cardiomyopathy to hypertension or, when hypertensive heart disease is present as a concomitant condition, reduced likelihood of further investigation [46, 49].
Relatively few studies reported quantitatively on misdiagnosis. Where this information was provided, 34–57% of patients reported or were documented as receiving a prior cardiac diagnosis other than ATTR-CM, most commonly hypertensive or hypertrophic cardiomyopathy, HFpEF, or an unspecified heart failure diagnosis. Although the patient may indeed have these conditions, the diagnosis is incomplete, as the underlying cause (ATTR-CM) has not been recognized. Of note, the samples in the included studies are by definition patients who eventually received an ATTR-CM diagnosis. It is likely that many more are never diagnosed with ATTR-CM and remain under treatment for nonspecific heart failure or other cardiac conditions.
Data on the consequences of diagnostic delay or misdiagnosis are sparse. However, ATTR-CM is a progressive disease characterized by an inexorable decline in cardiac function [3]. Ladefoged et al. [36] found that longer delay was associated with higher NYHA class at diagnosis, and Bishop et al. [18] found a statistically significant association between diagnostic delay ≥ 1 year and several adverse cardiac markers. Lane et al. [3] stated that long diagnostic delay was likely to have contributed to the low HRQoL observed in their study cohort at diagnosis.
Patients may also receive potentially inappropriate tests or treatment whilst undiagnosed or misdiagnosed: Lousada et al. [24] found frequent use of ACE inhibitors or beta blockers, which may be poorly tolerated in patients with ATTR-CM [1]. However, use of these agents may be appropriate in some ATTR-CM patients, depending on the individual’s morbidities and tolerance. Similarly, Ladefoged et al. [36] found that patients underwent a range of tests and treatments for incorrect or incomplete diagnoses. Inappropriate treatment or testing has potential consequences for the patient, including inconvenience and the risk of adverse effects, and also for healthcare systems in the form of unnecessary resource use. Studies that reported on healthcare visits during patients’ diagnostic journey found that they often undergo evaluation by multiple healthcare providers before a diagnosis of ATTR-CM is confirmed [3, 23, 24]. A cohort of 534 ATTRwt-CM patients in the UK had a median of three inpatient hospitalizations in the 3 years before their ATTR-CM diagnosis, suggesting heavy use of healthcare resources [3]. The long diagnostic journey is likely to be distressing for patients and their families, although no data on the consequences of delayed diagnosis for emotional well-being were found.
Until recently there were no disease-modifying pharmacotherapies for ATTR-CM, and patients were managed by symptomatic treatment of heart failure [50]. However, the first disease-modifying treatment, tafamidis, has recently been approved. In the ATTR-ACT study, tafamidis was shown to prolong survival, reduce cardiovascular hospitalization and slow the decline in cardiac function and HRQoL [50]. The availability of a disease-modifying treatment means that diagnostic delay now represents a missed opportunity for intervention to slow the disease process.
In this study we present a comprehensive synthesis of the literature on diagnostic delay in ATTR-CM. However, the study has several limitations, primarily associated with the limitations inherent in the included publications. As some authors noted [18], it is not certain that symptoms deemed to be the first evidence of amyloidosis were truly an indicator of amyloidosis, as many of the symptoms noted are nonspecific. Secondly, although some studies used documented symptom onset dates, others depended on patient recall. Thirdly, some studies dated diagnostic delay from date of first clinical suspicion or first cardiologic examination, rather than symptom onset. This means that the true delay (i.e. from symptom onset) may be underestimated. Characteristics of the health systems in different countries may influence delay, so results from one region may not be generalizable to others. Finally, some of the publications were conference abstracts and provided limited information, and restricting the search to English-language publications may have resulted in relevant studies being missed.
Conclusion
Patients with ATTR-CM commonly experience diagnostic delay and misdiagnosis. Efforts are required to provide timely diagnosis so that patients can benefit from earlier access to disease-modifying therapies, and to avoid unnecessary resource use. The recognition of early red flags, the introduction of noninvasive diagnostic techniques and the availability of disease-modifying therapies are expected to increase disease awareness and facilitate early diagnosis.
References
Ruberg FL, Grogan M, Hanna M, et al. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73(22):2872–91.
Rapezzi C, Quarta CC, Riva L, et al. Transthyretin-related amyloidoses and the heart: a clinical overview. Nat Rev Cardiol. 2010;7(7):398–408.
Lane T, Fontana M, Martinez-Naharro A, et al. Natural history, quality of life, and outcome in cardiac transthyretin amyloidosis. Circulation. 2019;140(1):16–26.
Maurer MS, Bokhari S, Damy T, et al. Expert consensus recommendations for the suspicion and diagnosis of transthyretin cardiac amyloidosis. Circ Heart Fail. 2019;12(9):e006075.
Dorbala S, Ando Y, Bokhari S, et al. ASNC/AHA/ASE/EANM/HFSA/ISA/SCMR/SNMMI expert consensus recommendations for multimodality imaging in cardiac amyloidosis: part 2 of 2-diagnostic criteria and appropriate utilization. J Nucl Cardiol. 2020;27(2):659–73.
European Medicines Agency. Public summary of opinion on orphan designation: Tafamidis for treatment of senile systemic amyloidosis. 2012 [cited 2020 Sept 29]. https://www.ema.europa.eu/en/documents/orphan-designation/eu/3/12/1066-public-summary-opinion-orphan-designation-tafamidis-treatment-senile-systemic-amyloidosis_en.pdf.
Witteles RM, Bokhari S, Damy T, et al. Screening for transthyretin amyloid cardiomyopathy in everyday practice. JACC Heart Fail. 2019;7(8):709–16.
Castaño A, Narotsky DL, Hamid N, et al. Unveiling transthyretin cardiac amyloidosis and its predictors among elderly patients with severe aortic stenosis undergoing transcatheter aortic valve replacement. Eur Heart J. 2017;38(38):2879–87.
González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585–94.
Mohammed SF, Mirzoyev SA, Edwards WD, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC Heart Fail. 2014;2(2):113–22.
Damy T, Bourel G, Slama M, et al. PCV67 epidemiology of transthyretin amyloid cardiomyopathy (ATTR-CM) in France: EPACT, a study based on the French Nationwide Claims Database Snds. Value Health. 2020;23:S498–9.
Rapezzi C, Lorenzini M, Longhi S, et al. Cardiac amyloidosis: the great pretender. Heart Fail Rev. 2015;20(2):117–24.
Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–12.
Maurer MS. Noninvasive identification of ATTRwt cardiac amyloid: the re-emergence of nuclear cardiology. Am J Med. 2015;128(12):1275–80.
American Society of Nuclear Cardiology. ASNC practice points: 99mTechnetium-pyrophosphate imaging for transthyretin cardiac amyloidosis. 2019 [cited 2020 Aug 23]. https://www.asnc.org/content.asp?contentid=290.
US FDA. FDA approves new treatments for heart disease caused by a serious rare disease, transthyretin mediated amyloidosis. 2019 [cited 2020 Aug 23]. https://www.fda.gov/news-events/press-announcements/fda-approves-new-treatments-heart-disease-caused-serious-rare-disease-transthyretin-mediated.
Shamseer L, Moher D, Clarke M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015: elaboration and explanation. BMJ. 2015;349:g7647.
Bishop E, Brown EE, Fajardo J, et al. Seven factors predict a delayed diagnosis of cardiac amyloidosis. Amyloid. 2018;25(3):174–9.
Chaudhary A, Alaarmi S, Leipsic J, et al. Comparison of AL and TTR cardiac amyloidosis: a single center Canadian experience [conference abstract]. Can J Cardiol. 2013;29(10):S114–5.
Connors LH, Sam F, Skinner M, et al. Heart failure resulting from age-related cardiac amyloid disease associated with wild-type transthyretin: a prospective, observational cohort study. Circulation. 2016;133(3):282–90.
Falk RH, Geller HI, Mirto T, et al. Val122Ile familial transthyretin (TTR) amyloid cardiomyopathy in African-American patients has a worse prognosis than wild-type TTR amyloid cardiomyopathy [conference abstract]. Eur Heart J. 2016;37:761.
Grogan M, Dispenzieri A, Carlsson M, et al. A survival analysis of subjects with transthyretin amyloid cardiomyopathy from the transthyretin amyloidosis outcomes survey. Poster presented at: 21st annual meeting of the Heart Failure Society of America (HFSA); 2017 September 16–19, Grapevine, TX, USA.
Kessler AS, Yarlas A, Pollock M, et al. Characterizing the journey to diagnosis for patients with transthyretin amyloidosis and accompanying congestive heart failure [conference abstract]. J Card Fail. 2019;25(8):S60.
Lousada I, Maurer M, Warner M, et al. Amyloidosis research consortium cardiac amyloidosis survey: Results from patients with ATTR amyloidosis and their caregivers. Poster session presented at: first European meeting for ATTR amyloidosis for doctors and patients; 2017 November 2–3, Paris, France.
Lousada I, Maurer M, Warner M, et al. Amyloidosis research consortium cardiac amyloidosis survey: results from patients with al and attr amyloidosis and their caregivers. In: Poster session presented at: XVIth international symposium on amyloidosis, 2018 March 26–29, Kumamoto, Japan; 2018.
Lousada I, Maurer MS, Warner MT, et al. Amyloidosis research consortium cardiac amyloidosis survey: results from patients with AL and ATTR amyloidosis and their caregivers poster session presented at: 23rd annual Heart Failure Society of America, 2019 September 13–16, Philadelphia, PA.
Papoutsidakis N, Jacoby D, Rodonski A, et al. How soon is now? Delay in the utilization of technetium-99m pyrophosphate scintigraphy for the diagnosis of cardiac transthyretin amyloidosis in patients with symptoms [conference abstract]. J Nucl Cardiol. 2017;24(4):1485.
Papoutsidakis N, Miller EJ, Rodonski A, et al. Time course of common clinical manifestations in patients with transthyretin cardiac amyloidosis: delay from symptom onset to diagnosis. J Card Fail. 2018;24(2):131–3.
Swiecicki PL, Zhen DB, Mauermann ML, et al. Hereditary ATTR amyloidosis: a single-institution experience with 266 patients. Amyloid. 2015;22(2):123–31.
He S, Tian Z, Guan H, et al. Clinical characteristics and prognosis of Chinese patients with hereditary transthyretin amyloid cardiomyopathy. Orphanet J Rare Dis. 2019;14(1):251.
Nakagawa M, Sekijima Y, Yazaki M, et al. Carpal tunnel syndrome: a common initial symptom of systemic wild-type ATTR (ATTRwt) amyloidosis. Amyloid. 2016;23(1):58–63.
Ochi Y, Kubo T, Nakashima Y, et al. Integrated diagnostic approach to wild-type transthyretin cardiac amyloidosis with the use of high-sensitivity cardiac troponin T measurement and 99mTc-pyrophosphate scintigraphy. J Cardiol. 2020;75(1):12–9.
Ciliberti P, Quarta CC, Riva L, et al. Hereditary transthyretin-related amyloidosis with exclusive cardiac phenotype [conference abstract]. Eur Heart J. 2009;30:539.
Cipriani A, Campagnolo M, Civera S, et al. Diagnostic approach to wild-type transthyretin cardiac amyloidosis, A single-centre experience [conference abstract]. G Ital Cardiol. 2019;20(12):213S.
González-López E, Gagliardi C, Dominguez F, et al. Clinical characteristics of wild-type transthyretin cardiac amyloidosis: disproving myths. Eur Heart J. 2017;38(24):1895–904.
Ladefoged B, Dybro A, Povlsen JA, et al. Diagnostic delay in wild type transthyretin cardiac amyloidosis—a clinical challenge. Int J Cardiol. 2020;304:138–43.
López-Sainz Á, Hernandez-Hernandez A, Gonzalez-Lopez E, et al. Clinical profile and outcome of cardiac amyloidosis in a Spanish referral center. Revista Española de Cardiología (English Edition). 2021;74(2):149–58.
Garcia-Pavia P, Grogan M, Dispenzieri A, et al., editors. A descriptive analysis of patients with wild-type ATTR cardiomyopathy from the transthyretin amyloidosis outcomes survey. In: Poster session presented at: 2019 European Society of Cardiology annual meeting, August 31–September 04, Paris, France; 2019.
Khella S, Drachman B, Damy T, et al. Prevalence and severity of neuropathy in THAOS subjects with wild-type and hereditary transthyretin cardiac amyloidosis. In: Poster session presented at: Peripheral Nerve Society annual meeting (PNS), 2018 July 22–25, Baltimore, Maryland, USA; 2018.
Nativi-Nicolau J, Siu A, Dispenzeri A, et al. Temporal trends of wild-Type ATTR amyloidosis in the transthyretin amyloidosis outcomes survey. In: Poster session presented at: Heart Failure Society of America (HFSA) virtual annual scientific meeting, 2020 September 30–October 6.
Hafeez AS, Bavry AA. Diagnosis of transthyretin amyloid cardiomyopathy. Cardiol Ther. 2020;9(1):85–95.
Maurer MS, Elliott P, Comenzo R, et al. Addressing common questions encountered in the diagnosis and management of cardiac amyloidosis. Circulation. 2017;135(14):1357–77.
Nativi-Nicolau JN, Karam C, Khella S, et al. Screening for ATTR amyloidosis in the clinic: overlapping disorders, misdiagnosis, and multiorgan awareness. Heart Fail Rev. 2021. https://doi.org/10.1007/s10741-021-10080-2.
Pinney JH, Whelan CJ, Petrie A, et al. Senile systemic amyloidosis: clinical features at presentation and outcome. J Am Heart Assoc. 2013;2(2):e000098.
Hauptman PJ, Wall JS, Maurer MS. Disparities, uncertainties, and societal cost: precision medicine and transthyretin amyloidosis. Am J Med. 2020;133(8):892–4.
Shah KB, Mankad AK, Castano A, et al. Transthyretin cardiac amyloidosis in black Americans. Circ Heart Fail. 2016;9(6):e002558.
Jacobson DR, Alexander AA, Tagoe C, et al. Prevalence of the amyloidogenic transthyretin (TTR) V122I allele in 14 333 African-Americans. Amyloid. 2015;22(3):171–4.
Centers for Disease Control and Prevention. African American Health 2017 [cited 2020 Sept 28]. https://www.cdc.gov/vitalsigns/aahealth/index.html.
Dungu JN, Papadopoulou SA, Wykes K, et al. Afro-Caribbean heart failure in the United Kingdom: cause, outcomes, and ATTR V122I cardiac amyloidosis. Circ Heart Fail. 2016;9(9):e003352.
Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–16.
Acknowledgements
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Medical Writing and Editorial Assistance
Medical writing support was provided by Sukhvir Rai of Health Economics and Outcomes Research Ltd, funded by Pfizer.
Compliance with Ethics and Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
All data generated or analyzed during this study are included in this published article/as supplementary information files.
Funding
This work was supported by Pfizer, who also funded the journal’s Rapid Service Fee.
Disclosures
Mark H. Rozenbaum, Samuel Large, Rahul Bhambri and Michelle Stewart are employees of Pfizer and own stock and/or stock options. Jo Whelan and Alexander van Doornewaard are employees of Health Economics and Outcomes Research Ltd. Health Economics and Outcomes Research Ltd. received fees from Pfizer in relation to this study and development of the manuscript. Jose Nativi-Nicolau’s institution received funding for clinical trials for Pfizer, Akcea and Eidos and educational grants from Pfizer. Noel Dasgupta has been a consultant for Pfizer, Ionis, Akcea, and Alnylam. Jose Nativi-Nicolau has been a consultant for Pfizer, Eidos, Akcea and Alnylam. Ahmad Masri’s institution received research grants from Pfizer, Akcea, and Ultromics. Ahmad Masri has been a consultant for Eidos, Ionis and Cytokinetics.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Rozenbaum, M.H., Large, S., Bhambri, R. et al. Impact of Delayed Diagnosis and Misdiagnosis for Patients with Transthyretin Amyloid Cardiomyopathy (ATTR-CM): A Targeted Literature Review. Cardiol Ther 10, 141–159 (2021). https://doi.org/10.1007/s40119-021-00219-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-021-00219-5