
Abstract
Background
Selective toxicity antibacteribiotics is considered to be due to interactions with targets either being unique to bacteria or being characterized by a dichotomy between pro- and eukaryotic pathways with high affinities of agents to bacterial- rather than eukaryotic targets. However, the theory of selective toxicity oversimplifies the complex modes of action of antibiotics in pro- and eukaryotes.
Methods and objective
This review summarizes data describing multiple modes of action of antibiotics in eukaryotes.
Results
Aminoglycosides, macrolides, oxazolidinones, chloramphenicol, clindamycin, tetracyclines, glycylcyclines, fluoroquinolones, rifampicin, bedaquillin, ß-lactams inhibited mitochondrial translation either due to binding to mitosomes, inhibition of mitochondrial RNA-polymerase-, topoisomerase 2ß-, ATP-synthesis, transporter activities. Oxazolidinones, tetracyclines, vancomycin, ß-lactams, bacitracin, isoniazid, nitroxoline inhibited matrix-metalloproteinases (MMP) due to chelation with zinc and calcium, whereas fluoroquinols fluoroquinolones and chloramphenicol chelated with these cations, too, but increased MMP activities. MMP-inhibition supported clinical efficacies of ß-lactams and daptomycin in skin-infections, and of macrolides, tetracyclines in respiratory-diseases. Chelation may have contributed to neuroprotection by ß-lactams and fluoroquinolones. Aminoglycosides, macrolides, chloramphenicol, oxazolidins oxazolidinones, tetracyclines caused read-through of premature stop codons. Several additional targets for antibiotics in human cells have been identified like interaction of fluoroquinolones with DNA damage repair in eukaryotes, or inhibition of mucin overproduction by oxazolidinones.
Conclusion
The effects of antibiotics on eukaryotes are due to identical mechanisms as their antibacterial activities because of structural and functional homologies of pro- and eukaryotic targets, so that the effects of antibiotics on mammals are integral parts of their overall mechanisms of action.
Similar content being viewed by others

Introduction
The theory of selective toxicity was established by Paul Ehrlich. He defined antibacterial agents as “substances with an exclusive affinity for bacteria acting deleteriously or lethally on these alone, while at the same time, they possess no affinity for the normal constituents of the body…” [1,2,3]. The selective toxicity of ß-lactams, for example, is considered to be due to their affinity to penicillin binding proteins (PBPs) and inhibition of biosynthesis of bacterial cell walls, both being unique to prokaryotes [4, 5]. Sulfonamides are antimetabolites of para-aminobenzoic acid (PABA), so that they compete with folic acid synthesis in bacteria, whereas eukaryotes lack this system [6, 7]. Thus, antibacterial agents interact specifically with targets either being unique to bacteria or being characterized by a dichotomy between pro- and eukaryotic pathways with high affinities of agents to the bacterial- rather than eukaryotic target.
However, it is well documented that aminoglycosides [8, 9], fluoroquinolones [10], tetracyclines [11, 12], macrolides [13, 14], and even ß-lactams [15, 16], optimized for treatment of bacterial infections exert antiviral-, antifungal-, antiparasitic-, and/or antineoplastic effects. It is difficult to comprehend that a monocausal activity relationship allows enough flexibility for such polypharmacological effects. It has been demonstrated that e.g. aminoglycosides [17,18,19] and tetracyclines [20] bind to a variety of targets. Such alternative binding sites may help to explain the pleiotropic actions of antibacterial agents against pro- and/or eukaryotic organisms.
The interaction of antibacterial agents (antibiotics in the following) with mammalian targets is known since long. The discovery of hypoglycaemic sulfonamides can be attributed to the observation in 1942 that treatment of typhoid fever with the sulphonamide 2254RP caused hypoglycaemia in some patients [21]. A number of sulfonamides are used in the treatment of protozoal infections in animals and humans. In addition, immunomodulatory activities in particular of macrolides [22, 23], fluoroquinolones [24, 25], and almost every other drug class [26, 27] demonstrate that antibiotics affect eukaryotic cells, too, probably due to a physicochemical interaction with membranes thus triggering intracellular signaling cascades in pro- and eukaryotes [28], so that interspecies and interkingdom communication is affected [29,30,31,32]. These examples indicate the paradigm of selective toxicity is oversimplified. Instead, agents interfere with multiple targets in pro- as well as eukaryotic cells due to compound- and target promiscuity [33,34,35] and target homology as will be demonstrated below.
This review summarizes data describing multiple modes of action of antibiotics and is limited exclusively to a synopsis of studies describing interactions of antibiotics with pro- and eukaryoric targets although the clinical condition may be complex and multifactorial. Thus, clinical improvement may result from pleiotropic actions of an antibiotic which, however, should not be mentioned as this review is focussed on the targets. Only those agents are mentioned which are used clinically for treatment of bacterial infections; their structural derivatives optimized for non-antiinfective activities will not be discussed. Phenotypic descriptions of drug effects will not be discussed either. Comprehensive reviews have aggregated a plethora of data describing the phenotypes of action of polyfunctional antibiotics [36,37,38,39,40,41,42]. The reader is kindly referred to these summaries for further information. Adverse drug reactions, drug/drug interactions, and indirect effects mediated by oxidant scavenging activities, immunomodulation, and beneficial- or detrimental effects resulting from microbiome imbalance, etc. will not be discussed either. To emphasize a priori, most of the therapeutic opportunities mentioned below have not been reviewed by regulatory authorities and the preclinical and/or clinical evidence supporting these potential novel indications varies widely. The majority of preclinical studies were planned as hypothesis generating experiments and most clinical studies have been designed as retrospective observational rather than prospective, randomized clinical studies. But still these data suggest that therapeutic options of an antibiotic treatment may likely be more diverse than expected.
Non-antimicrobial activities of antibiotics beyond structural and mechanistic boundaries
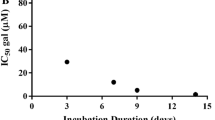
Detailed information about non-antimicrobial actions of antibiotics is provided in the supplemental material. Data compiled in Tables S1 and S2 confirm that antibiotics of almost every drug class interact with cellular- and in particular mitochondrial functions like autophagy and apoptosis/caspase mediated proteolysis as well as activities of matrix metalloproteinases (MMPs) thus exerting pleiotropic effects including anti-neoplastic activities. However, review and analysis of published data is difficult, since four variables had a significant impact on the data generated. First, the use of different methods results in divergent outcomes [43]. Second, antibiotics affected cellular functions also indirectly via interaction with cytokines and/or signaling cascades. Such systems are frequently redundant and pleomorph. Many cytokines are synthesized by more than one cell and different cells may secrete different cytokines with dissimilar activities and different stimuli may trigger diverse effects [24, 25]. Consequently, indirect effects of antibiotics on cellular functions via immunomodulation are not uniform but differ according to cell lines used and targets studied. Three examples should be mentioned pars pro toto: azithromycin and moxifloxacin decreased respiratory epithelial cell derived MMP1 and -3 concentrations, whereas both agents increased MMP secretion from MRC-5 fibroblasts [44]. Concentrations of doxycycline to inhibit MMP-9 synthesis to 50% (IC50) ranged in different cell lines from 1 to 608 μM [43, 45]. IC50-values of azithromycin for inhibition of proliferation and induction of apoptosis in HeLa-, cervical- and gastric cancer cells were 15.66, 26.05 and 91.00 mg/L [46]. Third, the parameter measured is not necessarily congruent to the mode of action of an antibiotic. Rifampicin and para-aminosalicylic acid, for example, inhibited secretion of MMPs secondary to an inhibition of prostaglandin synthesis [47, 48], so that these antibiotics targeted the endocrine system but not MMP synthesis. Fourth, antibiotic concentrations used in in vitro experiments scatter over a broad range and were frequently unphysiologically high. However, humanized doses were administered to experimental animals and standard doses were used in clinical studies on e.g. anti-neoplastic effects of antibiotics – even in monotherapy—or inhibition of MMP activities. For example, pancreatic cancer cell lines were exposed to 400 mg/L each of ciprofloxacin, moxifloxacin, and gatifloxacin; their maximal serum concentrations following oral standard doses are as low as 2.3-, 5.0-, and 3.8 mg/L [49, 50]. Patients with rosacea or periodontitis are treated with subantimicrobial doses of 40 and 20 mg b.i.d. doxycycline. This apparent discrepancy between pre-clinical and clinical study data and authority-approved indications remains unexplained. The more important it is to scrutinize the relevance of preclinical findings in proof of principle studies at the earliest possible time using alternative strategies [51, 52].
Antibiotics target mitochondrial functions and inhibit cancer cell growth
Mitochondrial metabolism is essential for e.g. energy conversion and regulation of membrane potential. Mammalian mitochondrial functions represent targets for antibiotics as documented already > 50 years ago [53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69]. Antibiotics interact not only with prokaryotic- and mitochondrial ribosomes but with eukaryotic 80S-ribosomes as well, so that human mitochondria and 80S-ribosomes represent a cancer target for antibiotics, too [70,71,72,73,74]. Data summarized in Table S1 demonstrate that cancer cell growth was affected by aminoglycosides, macrolides, tetracyclines, oxazolidinones, chloramphenicol, clindamycin, rifampicin targeting mitochondrial translation, quinolones targeting mitochondrial topoisomerases, and bedaquilin targeting mitochondrial ATP-synthesis [75]. However, the mitochondrial membrane may constitute a permeation barrier thus explaining that oxazolidinones, chloramphenicol and tetracyclines were significant inhibitors, while macrolides, clindamycin and aminoglycosides were poor inhibitors of mitochondrial protein synthesis [68]. Beta-lactams affected mitochondrial functions due to inhibition of carnitine/acylcarnitine transporter (Table S1). In addition, the agents also affected signaling cascades, micro-RNAs, and acted as immunomodulators or anti-inflammatory agents, so that antineoplastic activities of antibiotics may be due to interactions with multiple targets. Clinical trials indicated that aminoglycosides, macrolides, tetracyclines and fluoroquinolones, clarithromycin, and ciprofloxacin in particular, reduced mortalities in cancer patients, even as mono-therapeutics [76,77,78,79,80,81,82]. Synergistic effects were recorded in combination with anti-neoplastic agents. Thus, it may be plausible to prefer these agents to other antibiotics in cancer patients to benefit from their anti-neoplastic activities and their synergistic effects with other cancer treatment modalities. Antibiotics could be used not only for prophylaxis- or treatment of bacterial infections, but also for adjuvant therapy in cancer patients.
Antibiotics inhibit metallo-matrix-proteinases
MMPs are Ca2+ containing endopeptidases with an essential Zn2+ bound to three histidine residues in the conserved catalytic region. MMPs are characterized by a broad spectrum of substrate specificities and are essential for migration, invasion and degeneration of cells. MMPs are involved in e.g. bone remodelling, angiogenesis, and wound healing, etc., but also in arthritic destruction, pulmonary fibrosis, and cancer development, etc. [83,84,85]. MMP activity is regulated amongst others by tissue inhibitors of metalloproteins (TIMPs), which chelate the catalytic zinc atom thus inactivating MMPs [86]. Therefore, antibiotics known to chelate with bi- and trivalent cations like tetracyclines [87,88,89] and fluoroquinolones [90] should theoretically inactivate MMPs. The oxazolidin ring of linezolid and its peptide bond may chelate with zinc/calcium. Furthermore, vancomycin [91], bacitracin [92, 93], isoniazid [89], macrolides [94,95,96], ß-lactams [97,98,99,100,101,102,103,104,105,106,107], nitroxoline [108, 109], and chloramphenicol [110] formed chelates or complexes with zinc and calcium, whereas streptomycin did not [89].
Data summarized in Table S2 show that expectedly all these agents inhibited MMPs except fluoroquinolones and chloramphenicol, which unexpectedly induced MMPs. It is difficult to distinguish whether the described effects are due to inhibition of enzyme activities or inhibition of MMP synthesis. Few investigators only have analysed enzyme activities or gene expression, whereas MMP concentrations in supernatants of cell cultures have been analysed in most of the studies. A decrease in MMP concentrations as a consequence of antibiotic treatment could be demonstrated in patients with orthopaedic infections treated with ß-lactams [111, 112] and daptomycin [113], as well as pulmonary diseases treated with clarithromycin and azithromycin [114, 115]; doxycycline reduced MMP concentrations in patients with CF, COPD, and tuberculosis [116,117,118,119], chronic wounds [120,121,122,123,124,125,126,127,128], abdominal aortic aneurysm [129,130,131,132], ophthalmological diseases [133, 134], and in patients with acute lung injury following cardiac bypass [135]. Marketing authorizations have been granted for treatment of rosacea and periodontal diseases with sub-antimicrobial doses of doxycycline due to MMP inhibition [136,137,138,139,140,141,142]. Interestingly, several disinfectants inhibiting MMPs via cation-chelation are used clinically in dentistry [143,144,145,146,147,148,149,150,151,152]. Aminoglycosides exhibited anti-neoplastic effects as well, which were due to drug-induced increases in read-through of premature stop codons and not, as expected, inhibition of MMPs due to a lack of chelation with zinc (Table S2).
Drug class specific non-antimicrobial activities of antibiotics
Sulfonamides
Although sulfonamides are antimetabolites of PABA [6, 7], they are multifunctional drugs as their -SO2NH- (or -OSO2NH-, -NHSO2NH-) moieties interact with metal ions, amino acid residues, as well as DNA- or RNA-moieties. The antibacterial sulfonamide 2254RP exerted hypoglycaemic activities [6, 7, 21]. Sulfanilamide (Prontosil®) inhibited human carbonic acid anhydrases playing crucial physiological roles including pH regulation, gluconeogenesis, respiration, etc. due to the interaction of its thiol group with the Zn2+ ion in the active centre of the enzyme. Other sulfonamide-derivatives exhibited anti-obesity-, diuretic-, antithyroid-, anti-tumor-, anti-neuropathic pain-, anti-inflammatory activities, and acted as serotonin antagonists and protease inhibitors, too [153, 154]. However, non-antibacterial sulfonamides are almost inactive against bacteria due to the fact that the best position of the aniline group and the acidic sulfonamide moiety to mimic PABA is the para-substitution about the benzene ring. This positioning is essential for antibacterial activities of sulfonamides [155].
ß-lactams
Beta-lactam antibiotics mimic the structure of the acyl-d-Ala-d-Ala C peptide chain terminus of the growing cell wall and react with PBPs to form an acyl-enzyme complex [4, 5]. The D-Ala-D-Ala building block as well as the structure of the bacterial cell wall is considered to be unique to prokaryotes. Consequently, PBPs and class A, C, and D ß-lactamases being acyl serine transferases are considered to be bacteria specific [156]. But structural homologies between bacterial- and human serine proteases are conceivable as eukaryotic acyl serine transferases like trypsin, thrombin, etc. transfer the electrophilic group of e.g. peptide carbonyl donors to an acceptor. The carbonyl donor is transpeptidated with the acceptor when the acceptor carries an aminogroup – as happens in the course of cell wall biosynthesis. Most of the approximately 1,000 bacterial serine proteases like the D-Ala-D-Ala carboxypeptidases are single domain proteases in prokaryotes with functional specialisation [157], while e.g. trypsin-, subtilisin-, and Lon-protease families account for multi-domain proteases with inter-kingdom distribution [157,158,159]. The structures of the catalytic domains, however, are highly conserved, so that ß-lactams may interact with mammalian serine proteases, too.
Cefoperazone prevented α1-antitrypsin inactivation [160] and ceftazidime inhibited neutrophil elastase activity [161]. Additional reports are scarce although non-antibacterial lactams interacted with many mammalian targets [15, 16, 162,163,164,165]. Whether this can be attributed to the marginal activities of commercially available ß-lactams, or if they have not been exploited systematically is uncertain. Vice versa, it has been shown that inhibitors of eukaryotic serine proteases increase activities of ß-lactams in susceptible and even resistant bacteria just because of structural homologies between PBPs and human serine proteases.
The PBP- and ß-lactamase core protein fold is highly conserved and is present in human enzymes, too [166,167,168]. These human metallo-ß-lactamases (hMBLs) catalyse pleiotropic reactions on which various catalytic, regulatory and structural activities are based [169,170,171,172,173]. One of the ≥ 18 known hMBL is the intra-mitochondrial membrane organizing serine ß-lactamase like protein (LACTB) which binds two metal ions and acts as a tumor suppressor that modulates lipid metabolism and cell state [174,175,176,177]. Other hMBLs inactivate not only anti-neoplastic agents but also penicillin G but are inhibited by sulbactam and clavulanic acid [178] as well as 7-aminocephalosporinic acid, cephalosporin C, cefotaxime, and ceftriaxone, while compounds with a penicillin-, carbapenem- or monobactam-core were inactive [179]. Although the cephalosporins and ß-lactamase inhibitors tested were active against purified hMBLs only but exhibited no activities in cell based assays these data could provide a basis for the development of selective hMBL-inhibitors thus inhibiting inactivation of anti-neoplasic agents ot interacting with tumorigenesis. Furthermore, ß-lactams inhibited tumor growth due to inhibition of mitochondrial carnitine/acylcarnitine transporter (Table S1). In addition, sequence homologies between PBPs and DNA-polymerase-α have been described, so that ß-lactams affect proliferating eukaryotic cells in the S-phase [180,181,182,183,184,185]. Cephalosporins inhibited growth of various cell lines by three- to 25-fold lower concentrations than penicillins, whereas clavulanate, sulbactam, and monobactams were inactive. If the anti-neoplastic effect of some ß-lactams summarized in Table S1 may or may not be linked to inhibition of DNA-polymerase-α and/or inhibition of mitochondrial functions remains an open question.
Beta-lactams, ceftriaxone in particular, act as neuroprotectants in various models of neurological and neurodegenerative diseases, such as Alzheimer's disease, Parkinson, epilepsy, strokes, etc. which have been linked to dysfunction of excitatory amino acid transporters (EAATs). EAATs exhibit structural homologies between eukaryotic, bacterial and archaeal glutamate transporters [186,187,188,189,190,191,192,193,194,195,196,197,198,199,200,201]. The activities of ß-lactams were due to an increased expression of glutamate transporter GLT1 (EAAT2) [202,203,204,205,206]. However, the mechanisms by which ß-lactams enhance gene expression uniquely of glutamate transporter EAAT2 and not other glutamate transporters remains unanswered. Hypothetically, ß-lactams either enhance EAAT2 gene expression through NF-κB mediated modulation of EAAT2 promotor activity, and/or act as chelators. Binding of NF-κB to one out of four NF-κB binding sites of the EAAT2 promotor was increased by ceftriaxone thus activating transcription. Ceftriaxone activated also other NF-κB signalling pathways being indirectly involved in ceftriaxone mediated EAAT2 promotor activation [207, 208]. Alternatively or in parallel ß-lactams may chelate with copper, an essential trace element. Ceftriaxone bound copper effectively, so that it could ameliorate neurodegenerative diseases [103, 106]. In addition, ß-lactams modulate cytokine activity and regulate gene expression in mammals through a covalent binding to cytokines or proteins [209,210,211,212,213,214]. Not only ß-lactams but minocycline, rapamycin inhibiting a serine/threonine protein kinase, and rifampicin inhibiting bacterial RNA polymerase acted as neuroprotectants [215, 216]. These findings indicate that the mechanism(s) of neuroprotective functions of such diverse classes of antibiotics are far from being understood.
Aminoglycosides
Aminoglycosides inhibit bacterial protein synthesis by binding to the A-site on the 16S ribosomal RNA of the 30S ribosome thus inducing codon misreading resulting in mistranslation. In addition, aminoglycosides interact with a great variety of RNAs. They bind to hammerhead ribozyme, tRNA-Phe, the Rev response element transcriptional activation region in human immunodeficiency virus, the ribozyme from hepatitis delta virus, group I self-splicing introns, etc. because of their polycationic nature [17,18,19]. Contrary to the previous assumption, aminoglycosides interact with eukaryotic ribosomes, too. Analysis of crystal structures of 80S ribosomes in complex with aminoglycosides revealed that aminoglycosides bind to multiple sites on both subunits, so that multiple modes of action on the translation mechanism are probable [217,218,219]. Translation, elongation and termination are altered in a manner that induced read-through of premature stop codons (PTCs) (220,221,222, and Table S1). These findings have led to their consideration as potent drugs to treat human diseases caused by PTCs [221]. Although the modes of action of aminoglycosides on eukaryotic ribosomes and their potential clinical efficacy in treatment of non-infectious diseases has been elucidated quite recently [220,221,222], their efficacies in treatment of genetic disorders has been described phenotypically already in 1985, so that many data could be accumulated. Comprehensive recent reviews demonstrate that aminoglycoside-induced mutation suppression has not only been confirmed in a variety of preclinical in vitro and in vivo models but translates also into the clinical arena [223,224,225,226] (Table 1). Gentamicin, the investigational agents geneticin (G-418) and G-418 derivatives were the most frequently studied aminoglycosides; tobramycin, amikacin, paromomycin, neomycin, sisomycin, kanamycin, lividomycin, and hygromycinB were evaluated as well. While the major gentamicin compnts lack read-through activity the minor compnts, B1, X1, and G-418 caused read-through of PTCs [227, 228]. Aminoglycosides were initially used clinically as read-through agents in particular in the treatment of cystic fibrosis patients but concerns over the safety profile of aminoglycosides led to a search for alternatives.
Several specific features possibly limit the use of aminoglycosides in the treatment of genetic disorders: First, the identity of the stop codon and second, the sequence context surrounding it have an impact on the aminoglycoside mediated readthrough [222, 229,230,231,232,233], so that aminoglycosides can not suppress PTCs equally well in all patients, thus probably necessitating individualized diagnostic procedures. Third, mitochondrial dysfunction may occur in patients with specific polymorphisms in their 12S rRNA [234]. Fourth, aminoglycosides impair cell respiration leading to a superoxide overproduction. Furthermore, oxidative damage of mitochondrial aconitase leads to an accumulation of free ferrous iron in mitochondria, so that ultimately cells undergo apoptosis via the Fenton reaction (235).
Macrolides
Macrolides inhibit protein synthesis by binding to the bacterial ribosome in the region of the nascent peptide exit tunnel. Binding of a macrolide in the exit tunnel obstructs the passage of the nascent peptides, so that overall protein synthesis rapidly declines and peptidyl-tRNAs are accumulated. Therefore, it was previously suggested that macrolides inhibit the production of cellular polypeptides completely by preventing the exit of nascent peptide chains. Recent findings revealed that macrolides, instead of directly binding to the peptidyl transferase centre and disrupting its structure, bind in the exit tunnel one or more nanometres from the peptidyl transferase centre, so that the diameter of the exit tunnel is decreased. This results in steric clashes with nascent peptides leading to a stalling of ribosome function. Despite the narrowing of the exit tunnel sufficient room is still left, so that some proteins may bypass the narrowed exit tunnel and become partially or fully synthesized, while other proteins cannot bypass the obstruction. Inhibition of protein synthesis by macrolides depends on the properties of the polypeptide being synthesized by the drug-bound ribosome. The ability to bypass is not dependent on the entire sequence of the synthesized protein, but rather on its N-terminal sequence. In addition/or other mechanisms may cause differential impacts of macrolides on protein synthesis. Perturbed conformation of rRNAs in the P-site, which can lead to a decrease in translation rate, or reorientation of rRNAs might affect the stereochemistry of the A-site thereby preventing peptide bond formation [236,237,238,239,240,241]. Macrolides stall not only the ribosome within specific sequence contexts, but also induce translation errors [238, 242]. This effect is probably due to an inhibition of peptide bond formation and/or peptide release. This event could in turn alter kinetics of translation resulting in an increased chance of read-through. All the 14-membered and 16-membered lactone ring macrolides caused an extensive stop codon read-through of PTCs in the in vitro system studied [242] (Table 1). In addition, macrolides can also stimulate ribosomal frameshifting, which, however, is linked to resistance mechanisms [233, 237] and thus not relevant in the context of this review.
Read-through activities of macrolides were recorded in eukaryotes although only bacterial- or mitochondrial ribosomes are susceptible to macrolides, whereas ribosomes isolated from archaea or eukaryotic cytoplasmic ribosomes are resistant [243,244,245,246]. If read-through in eukaryotes is due to an interference with mitochondrial functions only has not been addressed. Nevertheless, a clinical study investigating the efficacy of erythromycin treatment for read-through of APC gene stop codon mutations in familial adenomatous polyposis has been initiated [247] and the impact of macrolides on stop codon read-through has been proven in pro- and eukaryotic species as well as patients (Table 1) [242, 248,249,250,251,252,253].
Furthermore, macrolides are prokinetic agents due to their motilin receptor stimulating activities. Fourteen- and 15- but not 16 membered macrolides act as motilin receptor agonists. Erythromycin affects two different pathways; at a low dose (40 mg) it activates an intrinsic cholinergic pathway, whereas higher doses (200–350 mg) act on motilin receptors on enteric nerves and smooth muscle [254,255,256,257,258]. Erythromycin itself exerts almost no prokinetic activity but its antibacterially inactive degradation intermediate 8,9-anhydro-6,9-hemiketal serves as a motilin receptor agonist and is then further metabolized into erythromycin-6,9;9,12-spiroketal [259]. The disadvantages of the use of macrolides as prokinetic agents are that motilin-induced contractions induce hunger feelings through a cholinergic pathway [256] and that macrolide-resistance might develop.
Another target for macrolides in eukaryotes has been described recently. Roxithromycin inhibited the cellular differentiation of the rice blast fungus Magnaporthe oryzae [260]. The gene mocdc27 (M. oryzae Cell Division Cycle 27) encoding appressorium formation is involved in growth inhibition of the fungus by roxithromycin. However, mocdc27 knock down mutants formed appressoria identical to those of the wild-type fungi. Therefore, it may also be likely that a complex of roxithromycin-MoCDC27 affects another molecule involved in appressorium formation. mocdc27 encodes a protein that is highly homologous to the anaphase promoting complex/cyclosome(APC/C) subunit (CDC27/APC3) of other eukaryotes. CDC27 homologs have been detected in yeasts, plants, and mammals. CDC27 plays a key role in colorectal cancer as CDC27 expression is significantly correlated with tumor progression and poor patient survival [261]. These findings and the interaction of macrolides with MoCDC27 may help to explain why macrolides exert anti-neoplastic effects (Table S1).
Chloramphenicol
Chloramphenicol inhibits translation by binding with its nitrobenzyl ring to several nucleotides of the 23S rRNA at the A site of the peptidyl transferase centre. The aromatic ring of the ribosome-bound chloramphenicol overlaps with the A-site, so that the aminoacyl moiety of incoming aminoacyl-tRNA cannot properly attach to peptidyl transferase centre. Originally, inhibition of translation was thought to be universal. However, the inhibitory activity of chloramphenicol is context specific. Inhibition depends on the nature of specific amino acids in the nascent chain and the identity of the residue entering the A site [262,263,264,265]. Chloramphenicol readily binds to mitochondrial- but not to mammalian cytoplasmic ribosomes [266,267,268,269,270]. Data summarized in Table 1 demonstrate that chloramphenicol affected read-through in eukaryotes, too [242, 271,272,273,274]. The impact of chloramphenicol on gene expression in eukaryotes is possibly due to alternative mechanisms or downstream effects to inhibition of mitochondrial translation resulting in decreases in cell surface transferrin expression, de novo ferritin synthesis, thus affecting iron dependent respiratory chain activity resulting in an reduced ATP synthesis and eventually in reduced tumor cell growth (Table S1). It has been shown recently that chloramphenicol inhibited appressorium formation in Magnaporthe oryzae because of an inhibition of MoDullard, a serine/threonine phosphatase [275]. Alignment and comparison of amino acid sequences of fungal and human origin revealed that five MoDullard orthologs could be identified from the human genome but only caboxy-terminal domain RNA polymerase II polypeptide A small phosphatase 1 (CTDSP1) complemented the MoDullard function and could be inhibited by chloramphenicol. Small serine phosphatases can exhibit multiple functions in a variety of cellular and biological processes. Human CTDSP1 interacts with a variety of proteins like cell division cycle associated protein 3, myelin basic protein, RE1-silencing transcription factor, some of which are involved in cell differentiation [275]. If CTDSP1 may represent a novel target for chloramphenicol in humans has to be verified or falsified. It also has to be examined if interaction of chloramphenicol with CTDSP1 may have an impact on CTDSP1-networks with a variety of proteins being involved in cell differentiation and carcinogenesis. But anyway, an unexpected chloramphenicol target has been identified in humans.
Oxazolidinones
Most of the studies on the mode of action of oxazolidinones have been performed with linezolid. Linezolid and earlier oxazolidinone derivatives were found to interfere with the early phase of protein synthesis, i.e. the formation of a functional 70S initiation complex with the two ribosomal subunits due to binding of linezolid to both subunits [276,277,278,279]. However, biochemical- and target binding studies suggested that oxazolidinones not only interfere with the formation of the initiation complex but interfere with almost each and every step of protein synthesis like elongation factor G dependent translocation during elongation and frameshifting, termination of translation, and nonsense suppression [280]. Also, resistance mutations mapping provided conflicting data. Docking studies and crystallography revealed that linezolid bound to the 50S ribosomal subunit in the A site pocket at the peptidyltransferase centre [280, 281]. Binding to the A site impeded the proper placement of incoming aminoacyl-tRNAs and also binding of other protein synthesis inhibitors like chloramphenicol [280,281,282]. Linezolid inhibits peptide-bond formation not globally, but rather blocks translation at specific locations within the mRNA in a context-specific manner [283]. Linezolid binds exclusively to mitochondrial ribosomes and leaves cytoplasmic ribosomes unaffected [284]. Linezolid and to a higher degree the investigational oxazolidinone R χ -01, increased frameshifting and stop codon read-through with nonsense suppression [271, 285]. Consequently, linezolid and tedizolid caused mitochondrial dysfunction thus suppressing cancer cell growth and increasing apoptosis [286,287,288,289]. Linezolid also inhibited overexpression of mucin MUC5AC in human airway epithelial cells at concentrations of 5 and 20 mg/L due to inhibition of phosphorylation of ERK1/2, which caused the activation of a MAPK pathway member resulting in overexpression of MUC5AC. This effect is analogous to that seen with azithromycin, which reduced mucin production via the NF-κB pathway [290,291,292].
Tetracyclines
Tetracyclines inhibit bacterial protein synthesis by preventing the association of aminoacyl-tRNA with the bacterial ribosome. Tetracyclines bind to the 30S ribosomal subunit at high-occupancy tetracycline-binding site (Tet-1) and five other minor binding sites in 16S rRNA. Tetracycline most likely binds complexed with two Mg2+ ions at the Tet-1 site near the A-site. A comparison of tigecycline- and tetracycline binding sites to the 70S ribosome and tigecycline binding to the 30S ribosome, respectively, showed that tigecycline was bound only to the Tet-1 site, and secondary binding sites were not observed. Thus, the significance of the other five tetracycline-binding sites is unclear [293, 294]. In addition, tetracyclines bind to various synthetic double-stranded RNAs, suggesting that the double-stranded structures may play a more important role in binding of tetracyclines to RNA than binding to specific base pairs [20]. This also implies that tetracyclines bind to double-stranded RNAs of pro- as well as eukaryotic origin. Furthermore, doxycycline bound to human cytosolic 80S ribosomes, so that human 80S ribosomal translation was modified and the cellular integrated stress response was activated [295, 296]. These findings contribute to an explanation why tetracyclines induced read-through of PTCs [272, 273] and exerted anti-proliferative effects on human cancer cell lines and in patients (Table S1). Favourable clinical outcomes have also been observed with tetracyclines in indications like rheumatoid arthritis, osteoarthritis, Fragile X syndrome, etc. [129, 131, 297,298,299,300,301,302,303,304]. A comprehensive summary of pleiotropic non-antibacterial actions and on-going clinical trials was published in 2010 [11] listing 54 and 88 clinical trials for minocycline and doxycycline, respectively, in indications like e.g. asthma, autism, Parkinson’s disease, amyothrophic lateral sclerosis.
Tetracyclines also exhibit an anti-parasitic effect and affect growth of e.g.
Plasmodium falciparum, Entamoeba histolytica, Giardia lamblia, Leishmania major, Trichomonas vaginalis, and Toxoplasma gondii [20]. The antiparasitic activity is explained by protein synthesis inhibition in mitochondria. However, T. vaginalis, G. lamblia, and E. histolytica are devoid of mitochondria but are nevertheless susceptible to tetracyclines. Thus, tetracyclines should affect structures in eukaryotes other than just mitochondria. Five not necessarily mutually exclusive theories are discussed: First, tetracyclines may inhibit translation in bacterial endosymbionts, like Wolbachia in helminths, being essential for parasite survival and reproduction [20, 305, 306]. Second, eukaryotes without mitochondria may contain mitochondria derived organelles like mitosomes and hydrogenosomes [307], so that such organisms contain genes of mitochondrial ancestry. Tetracycline targeted the hydrogenosome of T. vaginalis thus causing cell death [308]. P. falciparum harbours a plastid (apicoplast) that originated from an eukaryotic algal lineage. This organelle, too, is inhibited by tetracycline [309]. Third, binding of the tetracyclines to double stranded RNA may represent an alternative to the ribosomal target site thus allowing tetracyclines to affect organisms that lack the 16S ribosomal RNA (such as RNA-viruses and some protozoa) [20, 310]. Fourth, tetracyclines are strong chelators of divalent cations other than zinc [311,312,313,314], so that tetracyclines interact not only with metallo matrixproteinases (see paragraph 2.2. and Tables S1, S2), but affected also the interaction with double stranded RNA and thus their own uptake into Gram-negative bacteria, and their affinities to pro- or eukaryotic targets [20, 294]. Fifth, tetracyclines acted as pro-apoptotics in various cell-lines [11, 20, 315,316,317,318,319]. Furthermore, tetracyclines are used as valuable tools in biomedical research. The tetracycline-controlled Tet-Off and Tet-On gene expression systems are used to regulate the activity of genes in mammalian cells under various experimental conditions [320,321,322,323,324,325,326].
These data imply that tetracyclines could in theory be clinically effective in treatment of a broad variety of non-infectious diseases. However, the question of expanding the indications has not been addressed systematically, so that these findings represent interesting observations but do not yet translate into the clinical arena. Exceptions are marketing authorizations for treatment of rosacea and periodontal diseases with sub-antimicrobial doses of doxycycline.
Fluoroquinolones
In bacteria fluoroquinolones target both type II topoisomerases, i.e. DNA gyrase and topoisomerase IV [327, 328]. A critical role in the Mg2+ dependent interaction between topoisomerases and fluoroquinolones play two key residues in the GyrA and ParC/GrlA subunits of the heterotetrameric structure of topoisomerases. An invariant Ser, or sometimes a Thr, and Asp/Glu in the heterotetramer anchor water-metal ion bridges. Absence of these residues leads to quinolone-resistance. Affinities of fluoroquinolones to topoisomerases are surprisingly selective although some sequence similarities between human- and bacterial type II topoisomerases exist. Selectivity is due to the facts that first the A and B subunits of human topoisomerases are fused, so that they function as homodimers. This structure is different from bacterial heterotetramers. Second, human type II topoisomerases lack the key residues anchoring the metal-ion bridges [328,329,330,331,332]. The highly selective mode of action of fluoroquinolones suggests that these agents may likely not affect mammals at clinically relevant concentrations, and if so, fluoroquinolones should interact with targets other than topoisomerases. However, high fluoroquinolone-concentrations ranging from 80 mg/L to 5.9 g/L inhibited eukaryotic type II topoisomerases, depleted mitochondrial DNA and inhibited mitochondrial functions [333,334,335,336,337,338,339,340,341,342].
DNA damage repair mechanisms are essential in pro- and eukaryotes in response to noxial events like UV-exposure. While bacteria repair DNA damages preferentially via the SOS response system [343, 344], the micro-RNA biogenesis system plays a role in DNA damage repair in eukaryotes apart from their essential impact on posttranscriptional regulation of gene expression modulating the RNA interference pathway [345,346,347,348]. The RNA interference pathway related endoribonucleases DICER and DROSHA promote DNA damage response activation by generating small non-coding RNAs with the sequence of the DNA flanking the double strand breaks. Micro-RNAs play also a role in various human diseases including carcinogenesis [348, 349]. From the quinolones tested ciprofloxacin, ofloxacin, norfloxacin, pipemidic acid, oxolininc acid, dilfloxacin, enoxacin [350], and also moxifloxacin [351] only norfloxacin [350], enoxacin [350, 352,353,354,355], pefloxacin [356] affected micro-RNA-, DICER- or DROSHA-activities thus exerting anti-neoplastic effects [357,358,359,360] (Table S1). Furthermore, downregulation of micro-RNA concentrations is associated with multiple forms of amyotrophic lateral sclerosis and also depression. The effect of enoxacin on neuromuscular function was examined in two mouse models in which micro-RNA biogenesis was enhanced and neuromuscular function was improved [361, 362]. Enoxacin reverted in vitro in human pluripotent stem cells the general micro-RNA downregulation related to ALS disease [363]. Enoxacin enhanced micro-RNA concentrations in vitro and in rat frontal cortex in animals with learned helpless behaviour as compared to animals with normal adaptive responses [364]. Thus, enoxacin and possibly other fluoroquinolones as well may ameliorate depressive behaviour.
Helicases share amino acid sequence- and enzyme activity homologies in bacteria, viruses, yeasts, and humans [365,366,367]. of the helicases contributing significantly to the genomic integrity in bacteria and humans is RecQ. Humans possess five RecQ helicases, i.e. RecQL1, − 4, − 5, WRN, and BLM. RecQL4 interacts with several replisome factors, amongst others with MCM2-7 [368]. Humans with RecQ mutations are likely to develop cancer and age prematurely [369]. Ciprofloxacin inhibited MCM2-7 [370]. RNA helicases have been implicated in proofreading processes. of these helicases, i.e. DHX9 has a key function in the regulation of biological processes, including tumorigenesis. Enoxacin inhibited DHX9 which is found in patients with lung- and prostate cancers [371, 372].
Ciprofloxacin, norfloxacin and enrofloxacin inhibited in HEK293 cells three α-ketoglutarate-dependent dioxygenases that require iron as a co-factor, which chelates with fluoroquinols [373]. Inhibition of α-ketoglutarate-dependent dioxygenases may on the hand explain fluoroquinol-induced nephrotoxicity and tendinopathy. On the other hand, fluoroquinol mediated dioxygenase inhibition lead unexpectedly to a reduction of hypoxia inducible transcription factor- (HIF)-1α and -2α concentrations due to inhibition of HIF mRNA transcription. Tumor hypoxia induces the up-regulation of genes associated with angiogenesis, glycolysis, adaptation to pH, and apoptosis via HIF-1α and HIF-2α, so that disruption of this pathway may be relevant in cancer therapy.
Various fluoroquinolones were found to exert anti-parasitic actions [10, 374,375,376,377,378,379,380,381,382,383,384,385,386] due to an interference with mitochondria derived organelles [269, 387,388,389]. Thus, patients with parasitic infections may benefit from fluoroquinolone treatment twofold: they are going to be treated not only of bacterial- but also of opportunistic parasitic infections, so that fluoroquinolone therapy should probably not be withdrawn upon elimination or exclusion of the bacterial pathogen.
Conclusions and open questions
A synopsis of non-antimicrobial activities of antibiotics is provided in Table 2. In general, antibiotics interacted with multiple targets and exerted pleiotropic effects in eukaryotes. Many of these targets are multi-functional, so that one specific effect cannot be attributed to one specific target and agent. In addition, different drug classes caused identical effects in eukaryotes. Thus, the anyhow complex modes of actions of antibiotics in eukaryotes may be even more multifaceted.
It is important to emphasize that the majority of studies reviewed above were planned as hypothesis generating preclinical experiments or retrospective observational clinical studies rather than prospective, randomized clinical trials, so that evidence supporting these potential novel indications varies widely. But still these data demonstrate that antibiotics interact with eukaryotic targets, so that therapeutic options of an antibiotic treatment may likely be more diverse than expected. Although none of the antibiotics should be used at the time being for monotherapy of non-infectious diseases, they could be used as adjunctive therapeutics in cancer patients or patients with parasitic diseases. Macrolides, tetracyclines, aminoglycosides, and fluoroquinolones have been shown to be clinically effective as anti-neoplastic agents, even in monotherapy, and were found to synergise with cancer drugs. Therefore, antibiotics could possibly be used not only for prophylaxis- or treatment of bacterial infections, but also for adjuvant therapy in cancer patients. Treatment modalities could hypothetically be individualized by selecting agents for prophylaxis or treatment of bacterial infections which are active against the specific cancer type and which synergize with the specific cancer drug administered to the patient. Likewise, tetracyclines and fluoroquinolones exerting anti-parasitic activities should probably not be withdrawn upon elimination or exclusion of the bacterial pathogen but should be continued in parallel to administration of the specific anti-parasitic agent. Their activities against RNA-viruses [10,11,12] could complement the spectrum of activities. Several additional targets for antibiotics in human cells have been identified (Table 2) apart from mitochondrial functions and MMPs. This may open up new opportunities for the focused evaluation of non-antimicrobial activities of antibiotics.
However, following effect dichotomies have to be considered: antibiotics could be used as adjunctive therapeutics but long-term use may possibly promote resistance development, tumorigenesis, and obesity. Antibiotics were used in some of the indications for several weeks and even months and treatment of genetic disorders may last for life. Toxicological implications of long term antibacterial treatment have not yet been assessed with the majority of antibiotics although it is documented that prolonged use of linezolid and chloramphenicol was associated with adverse events. The impact on the microbiome and resistance development has also not been analysed in these studies. Systematic reviews have revealed that increased consumption of antibiotics was associated with resistance development at the individual patient level and also in the community. Moreover, antibiotic-resistance was not always, but usually, associated with a significant economic burden resulting from (re-)admission to hospital, need for i.v.-administration, or even use of a less well tolerated antibiotic [390,391,392,393,394,395,396]. Antibiotics causing mitochondrial dysfunction may promote tumorigenesis [397,398,399,400], obesity [401], and psychiatric disorders [402, 403]. Furthermore, this review has demonstrated that antibiotics may have an unpredictable impact on cell culture metabolism, gene expression and signalling cascades thus supporting a previous study entitled “are cell culture data skewed?” [404]. These open questions should be carefully considered at the present time and be addressed henceforth.
The effects of antibiotics on eukaryotes are due to identical mechanisms as their antibacterial activities because of structural and functional homologies of pro- and eukaryotic targets, so that the effects of antibiotics on mammals are integral parts of their overall mechanisms of action. A purposeful use of antibiotics not only as antibacterial agents but also as agents targeting the human body and his functions should be considered.
Addendum search strategy
Publications addressing four topics were screened: first, impact of antibiotics on growth of eukaryotic/mammalian cells/cell cultures. Second, impact of antibiotics on mitochondrial functions, eukaryotic translation/transcription, and/or enzyme activities like metallo-matrix-proteinases, serine proteases, etc., read-through, premature stop codons. Third, interaction with mitosomes, 80S ribosome, RNA, DNA. Fourth, interaction of antibiotics with eukaryotic targets. Search strategy and selection criteria were based on the combination of key words sulphonamides, ß-lactams, aminoglycosides, macrolides, chloramphenicol, oxazolidinones, tetracyclines, fluoroquinolones, the corresponding single agents of these drug-classes, and antibiotics in general. Growth inhibition, tumor growth, inhibitory concentration IC50, homologous, orthologs, paralogs, chelation, complexation. Articles summarized in recent reviews were excluded from this synopsis and the reviews are quoted instead.
References
Kaufmann SHE. Paul Ehrlich: founder of chemotherapy. Nat Rev Drug Discov. 2008;7:373. https://doi.org/10.1038/nrd2582.
Bäumler E (1997) Paul Ehrlich, Forscher für das Leben. Edition Wötzel, Frankfurt am Main, dritte durchgesehene Auflage, dritter Teil: Der Weg zur Chemotherapie, 1997; pp 161–192. ISBN 3-925831-21-5
Gradmann C. Magic bullets and moving targets: antibiotic resistance and experimental chemotherapy, 1900–1940. Dynamis. 2011;31:305–21. https://doi.org/10.4321/s0211-95362011000200003.
Stewart GT. Toxicity of the penicillins. Postgrad Med J. 1964;40(Suppl):160–5. https://doi.org/10.1136/pgmj.40.Suppl.160.
Park JT, Strominger JI. Mode of action of penicillin. Biochemical basis for the mechanism of action of penicillin and for its selective toxicity. Science. 1957;125:99–101. https://doi.org/10.1126/science.125.3238.99.
Woolley DW. Water-soluble vitamins. Annu Rev Biochem. 1947;16:359–86. https://doi.org/10.1146/annurev.bi.16.070147.002043.
Woolley DW. A study of the basis of selectivity of action of antimetabolites with analogues of pimelic acid. J Biol Chem. 1950;183:495–505. https://doi.org/10.1111/j.1749-6632.1950.tb54027.x.
Dalhoff A (1987) Pleiotropic actions of aminoglycosides. In: Döring G, Holder IA, Botzenhart K (eds). Basic Research and Clinical Aspects of Pseudomonas aeruginosa. International Symposium on Pseudomonas aeruginosa, Tübingen, June 1986. Antibiot Chemother. Basel, Karger, vol 39, pp 182–204 https://doi.org/10.1159/000414345
Cohen JI. New activities of old aminoglycosides. Nat Microbiol. 2018;3:531–2. https://doi.org/10.1038/s41564-018-0152-4.
Dalhoff A. Antiviral, antifungal, and antiparasitic activities of fluoroquinols optimized for treatment of bacterial infections: a puzzling paradox or a logical consequence of their mode of action? Eur J Clin Microbiol Infect Dis. 2015;34:661–8. https://doi.org/10.1007/s10096-014-2296-3.
Griffin MO, Fricovsky E, Ceballos G, Villarreal F. Tetracyclines: a pleiotropic family of compounds with promising therapeutic properties. Review of the literature. Am J Physiol Cell Physiol. 2010a;299:C539–48. https://doi.org/10.1152/ajpcell.00047.2010.
Nagarakanti S, Bishburg E. Is minocycline an antiviral agent? A review of current literature. Basic Clin Pharmacol Toxicol. 2016;118:4–8. https://doi.org/10.1111/bcpt.12444.
Wong EHC, Porter JD, Edwards MR, Johnston SL. The role of macrolides in asthma: current evidence and future directions. Lancet Respir Med. 2014;2:657–70. https://doi.org/10.1016/S2213-2600(14)70107-9.
Min JY, Jang YJ. Macrolide therapy in respiratory viral infections. Mediators Inflamm. 2012. https://doi.org/10.1155/2012/649570.
Hamilton-Miller JMT. ß-lactams: variations on a chemical theme, with some surprising biological results. J Antimicrob Chemother. 1999;44:729–34. https://doi.org/10.1093/jac/44.6.729.
Kuhn D, Coates C, Daniel K, Chen D, Bhuiyan M, Kazi A, Turos E, Dou QP. Beta-lactams and their potential as novel anticancer chemotherapeutic drugs. Front Biosci. 2004;9:2605–17. https://doi.org/10.2741/1420.
Kotra LP, Haddad J, Mobashery S. Aminoglycosides: perspectives on mechanisms of action and resistance and strategies to counter resistance. Antimicrob Agents Chemother. 2000;44:3249–56. https://doi.org/10.1128/AAC.44.12.3249-3256.2000.
Schroeder R, Waldsich C, Wank H. Modulation of RNA function by aminoglycoside antibiotics. EMBO J. 2000;19:1–9. https://doi.org/10.1093/emboj/19.1.1.
Tekos A, Tsagla A, Stathopoulos C, Drainas D. Inhibition of eukaryotic ribonuclease P activity by aminoglycosides: kinetic studies. FEBS Lett. 2000;485:71–5. https://doi.org/10.1016/S0014-5793(00)02190-6.
Chuckwudi C. rRNA binding sites and the molecular mechanism of action of the tetracyclines. Antimicrob Agents Chemother. 2016;60:4433–41. https://doi.org/10.1128/AAC.00594-16.
Loubatieres-Mariani MM. The discovery of hypoglycemic sulphonamides. J Soc Biol. 2007;20:121–5. https://doi.org/10.1051/jbio:2007014.
Altenburg J, De Graaff CS, Van Der Werf TS, Boersma WG. Immunomodulatory effects of macrolide antibiotics–par 1: biological mechanisms. Respiration. 2011;81:67–74. https://doi.org/10.1159/000320319.
Labro MT, Abdelghaffar H. Immunomodulation by macrolide antibiotics. J Chemother. 2001;13:3–8. https://doi.org/10.1179/joc.2001.13.1.3.
Dalhoff A, Shalit I. Immunomodulatory effects of quinols. Lancet Infect Dis. 2003;3:359–71. https://doi.org/10.1016/s1473-3099(03)00658-3.
Dalhoff A. Immunomodulatory activities of fluoroquinols. Infection. 2005;33:55–70. https://doi.org/10.1007/s15010-005-8209-8.
Labro T. Interference of antibacterial agents with phagocyte functions: immunomodulation or “immuno-fairy tales”? Clin Microbiol Rev. 2000;13:615–50. https://doi.org/10.1128/CMR.13.4.615.
Kwiatkowska B, Maslinska M, Przygodzka M, Dmowska-Chalaba J, Dabrowska J, Sikorska-Siudek K. Immune system as a new therapeutic target for antibiotics. Adv Biosci Biotechnol. 2013;4:91–101. https://doi.org/10.4236/abb.2013.44A013.
Dalhoff A (2018) Membrane interactions of antibacterial agents. Trend Clin Microbiol 1: 04–48. https://www.gratisoa.org/journals/index.php/TCMY/article/view/1244/1173
Petra AI, Panagiotidou S, Hatziagelaki E, Stewart M, Conti P, Theoharides TC. Gut-microbiota-brain axis and its effect on neuropsychiatric disorders with suspected immune dysregulation. Clin Ther. 2015;37:984–95. https://doi.org/10.1016/j.clinthera.2015.04.002.
Gillings MR. Evolutionary consequences of antibiotic use for the resistome, mobilome and microbial pangenome. Front Microbiol. 2013;4:4. https://doi.org/10.3389/fmicb.2013.00004.
Taga ME, Bassler BL. Chemical communication among bacteria. Proc Nat Acad Sci. 2003;100(2):14549–54. https://doi.org/10.1073/pnas.1934514100.
Wecke T, Mascher T. Antibiotic research in the age of omics: from expression profiles to interspecies communication. J Antimicrob Chemother. 2011;66:2689–704. https://doi.org/10.1093/jac/dkr373.
Hu Y, Gupta-Ostermann D, Bajorath J. Exploring compound promiscuity patterns and multi-target activity spaces. Computat Structural Biotechnol J. 2014;9:e201401003. https://doi.org/10.5936/csbj.201401003.
Hopkins AL, Mason JS, Overington JP. Can we rationally design promiscuous drugs? Curr Opin Struct Biol. 2006;16:127–36. https://doi.org/10.1016/j.sbi.2006.01.013.
Hopkins AL. Network pharmacology: the next paradigm in drug discovery. Nat Chem Biol. 2008;4:682–90. https://doi.org/10.1038/nbt1007-1110.
Brown D. Antibiotic resistance breakers: can repurposed drugs fill the antibiotic discovery void? Nature Rev Drug Discovery. 2015;14:821–32. https://doi.org/10.1038/nrd4675.
Smith CJ, Heal C, Vail A, Jeans AR, Westendorp WF, Nederkoorn P, van de Beek D, Kalra L, Montaner J, Woodhead M, Meisel A. Antibiotic class and outcome in post-stroke infections: an individual participant data pooled analysis of VISTA-Acute. Front Neurol. 2019;10:504. https://doi.org/10.3389/fneur.2019.00504.
Ratcliff WC, Denison RF. Alternative actions for antibiotics. Science. 2011;332:547–8. https://doi.org/10.1126/science.1205970.
Pasquale TR, Tan JS. Nonantimicrobial effects of antibacterial agents. Clin Infect Dis. 2005;40:127–35. https://doi.org/10.1086/426545.
Sadarangani P, Estes LL, Steckelberg JM. Non–anti-infective effects of antimicrobials and their clinical applications: a review. Mayo Clin Proc. 2015;90:109–27. https://doi.org/10.1016/j.mayocp.2014.09.006.
Kalayci J. Antimicrobial properties of various non-antimicrobial drugs against microorganisms. Bioanal Biomed. 2016;8:4. https://doi.org/10.4172/1948-593X.1000e142.
Kruszewska H, Zareba T, Tyski S. Search of antimicrobial activity of selected non-antibiotic drugs. Acta Pol Pharm. 2002;59:436–9 ((PMID: 12669766)).
Hanemaaijer R, van Lent N, Sorsa T, Salo T, Yrj Ö, Konttinen T, Lindemann J. Inhibition of matrix metalloproteinases (MMPs) by tetracyclines. In: Nelson M, Hillen W, Greenwald RA, editors. Tetracyclines in Biology, Chemistry and Medicine. Basel: Birkhäuser; 2001. https://doi.org/10.1007/978-3-0348-8306-1_11 ((Print ISBN 978-3-0348-9511-8)).
Singh S, Kubler A, Singh UK, Singh A, Gardiner H, Prasad R, Elkington PT, Friedland JS. Antimycobacterial drugs modulate immunopathogenic matrix metalloproteinases in a cellular model of pulmonary tuberculosis. Antimicrob Agents Chemother. 2014;58:4657–65. https://doi.org/10.1128/AAC.02141-13.
Modheji M, Olapour S, Khodayar MJ, Jalili A, Yaghooti H. Minocycline is more potent than tetracycline and doxycycline in Inhibiting MMP-9 in vitro. Jundishapur J Nat Pharm Prod. 2016;11(2):e27377. https://doi.org/10.17795/jjnpp-27377.
Zhou X, Zhang Y, Li Y, Hao X, Liu X, Wang Y. Azithromycin synergistically enhances anti-proliferative activity of vincristine in cervical and gastric cancer cells. Cancers. 2012;4:1318–32. https://doi.org/10.3390/cancers4041318.
Yuhas Y, Azoulay-Alfaguter I, Berent E, Ashkenazi S. Rifampin inhibits prostaglandin E2 production and arachidonic acid release in human alveolar epithelial cells. Antimicrob Agents Chemother. 2007;51:4225–30. https://doi.org/10.1128/AAC.00985-07.
Rand L, Green JA, Saraiva L, Friedland JS, Elkington PT. Matrixmetalloproteinase-1 is regulated in tuberculosis by a p38 MAPK-dependent, p-aminosalicylic acid-sensitive signaling cascade. J Immunol. 2009;182:5865–72. https://doi.org/10.4049/jimmunol.0801935.
Yadav V, Varshney P, Sultana S, Yadav J, Saini N. Moxifloxacin and ciprofloxacin induces S-phase arrest and augments apoptotic effects of cisplatin in human pancreatic cancer cells via ERK activation. BMC Cancer. 2015;15:581. https://doi.org/10.1186/s12885-015-1560-y.
Yadav V, Sultana S, Yadav J, Saini N. Gatifloxacin induces S and G2-phase cell cycle arrest in pancreatic cancer cells via p21/p27/p53. PLoS One. 2012. doi:10.1371/journal.pone.0047796.
Dalhoff A, Weintraub A, Nord CE. Alternative strategies for proof-of-principle studies of antibacterial agents. Antimicrob Agents Chemother. 2014;58:4257–63. https://doi.org/10.1128/AAC.02473-14.
Vente A, Bentley C, Lückermann M, Tambyah P, Dalhoff A. Early clinical assessment of the antimicrobial activity of finafloxacin compared to ciprofloxacin in subsets of microbiologically characterized isolates. Antimicrob Agents Chemother. 2018;62:e02325-e2417. https://doi.org/10.1128/AAC.02325-17.
Kinsky SC, Gronau GR, Weber MM. Interaction of polyene antibiotics with subcellular membrane systems I. Mitochondria Mol Pharmacol. 1965;1:190–201 ((PMID: 5294438)).
Asahi T, Majima R. Effect of antibiotics on biogenesis of mitochondria during aging of sliced sweet potato root tissue. Plant Cell Physiol. 1969;10:317–23. https://doi.org/10.1093/oxfordjournals.pcp.a074410.
Mitani M, Otake N. Studies on the ionophorous antibiotics. XVI J Antibiot. 1978;31:888–93. https://doi.org/10.7164/antibiotics.31.888.
Wallace DC, Pollack Y, Bunn CL, Eisenstadt JM. Cytoplasmic inheritance in mammalian tissue culture cells. Vitro. 1976;12:758–76. https://doi.org/10.1007/BF02835451.
Wallace DC. Why do we have a maternally inherited mitochondrial DNA? Insights from evolutionary medicine. Annu Rev Biochem. 2008;76:781–821.
Wallace DC, Chalkia D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb Perspect Biol. 2013;5:a021220. https://doi.org/10.1101/cshperspect.a021220.
Doersen CJ, Stanbridge EJ. Cytoplasmic inheritance of erythromycin resistance in human cells. Proc Natl Acad Sci. 1979;76:4549–53. https://doi.org/10.1073/pnas.76.9.4549.
Kroon AM, Van den Bogert C. Antibacterial drugs and their interference with the biogenesis of mitochondria in animal and human cells. Pharm Weekbl Sci. 1983;5:81–7. https://doi.org/10.1007/bf01960982.
Kalghatgi S, Spina C, Costello JC, Liesa M. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in mammalian cells. Sci Transl Med. 2013;5:19285. https://doi.org/10.1126/scitranslmed.3006055.
Duewelhenke N, Krut O, Eysel P. Influence on mitochondria and cytotoxicity of different antibiotics administered in high concentrations on primary human osteoblasts and cell lines. Antimicrob Agents Chemother. 2007;51:54–63. https://doi.org/10.1128/AAC.00729-05.
Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, Scotiga F, Lisanti MP. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 2015;6:4569–84. https://doi.org/10.18632/oncotarget.3174.
Parrasiaa S, Mattareib A, Furlan A, Zorattia M, Biasutto L. Small-molecule modulators of mitochondrial channels as chemotherapeutic agents. Cell Physiol Biochem. 2019;53:11–43. https://doi.org/10.33594/000000192.
Peiris-Pagès M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti P. Cancer stem cell metabolism. Breast Cancer Res. 2016;18:1–10. https://doi.org/10.1186/s13058-016-0712-6.
Shin MK, Cheong JH. Mitochondria-centric bioenergetic characteristics in cancer stem-like cells. Arch Pharm Res. 2019;42:113–27. https://doi.org/10.1007/s12272-019-01127-y.
Zhang L, Ging NC, Komoda T, Hanada T, Suzuki T, Watanabe K. Antibiotic susceptibility of mammalian mitochondrial translation. FEBS Lett. 2005;579:6423–7. https://doi.org/10.1016/j.febslet.2005.09.103.
McKee EE, Ferguson M, Bentley AT, Marks TA. Inhibition of mammalian mitochondrial protein synthesis by oxazolidins. Antimicrob Agents Chemother. 2006;50:2042–9. https://doi.org/10.1128/AAC.01411-05.
Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, Jovaisaite V, Frochaux MV, Quiros PM, Deplancke B, Houtkooper RH, Auwerx J. Tetracyclines disturb mitochondrial function across eukaryotic models: a call for caution in biomedical research. Cell Rep. 2015;10:1681–91. https://doi.org/10.1016/j.celrep.2015.02.034.
Myasnikov AG, Natchiar SK, Nebout M, Hazemann I, Imbert V, Khatter H, Peyron JF, Klaholz BP. Structure–function insights reveal the human ribosome as a cancer target for antibiotics. Nature Commun. 2016;7:12856. https://doi.org/10.1002/9783527808465.EMC2016.6610.
Yusupova G, Yusupov M. Crystal structure of eukaryotic ribosome and its complexes with inhibitors. Phil Trans R Soc B. 2017;372:20160184. https://doi.org/10.1098/rstb.2016.0184.
Gadaleta MN, Greco M, Sacc C. The effect of rifampicin on mitochondrial RNA polymerase from rat liver. FEBS Lett. 1970;10:54–6. https://doi.org/10.1016/0014-5793(70)80414-8.
Awad D, Prattes M, Kofler L, Rössler I, Loibl M, Pertl M, Zisser G, Wolinski H, Pertschy P, Bergler H. Inhibiting eukaryotic ribosome biogenesis. BMC Biol. 2019;17:46. https://doi.org/10.1186/s12915-019-0664-2.
Sulima SO, Kampen KR, De Keersmaecker K. Cancer biogenesis in ribosomopathies. Cells. 2019;8:229. https://doi.org/10.3390/cells8030229.
Luo M, Zhou W, Patel H, Srivastava AP, Symerdky J, Bonar MM, Feraldo-Gomez JD, Liao M, Mueller DM. Bedaquiline inhibits the yeast and human mitochondrial ATP synthases. Commun Biol. 2020;3:452. https://doi.org/10.1038/s42003-020-01173-z.
Van Nuffel AM, Sukhatme V, Pantziarka P, Meheus L, Sukhatme VP, Bouche G. Repurposing Drugs in Oncology (ReDO)—clarithromycin as an anti-cancer agent. Ecancermedicalscience. 2015. https://doi.org/10.3332/ecancer.2015.513.
Chukhlovin AB. Drug repurposing in leukemia treatment and hematopoietic stem cell transplantation. Cell Ther Transplant. 2019;8:12–9. https://doi.org/10.18620/ctt-1866-8836-2019-8-1-12-19.
Dong Z, Abbas MN, Kausar S, Yang J, Li L, Tan L, Cui H. Biological functions and molecular mechanisms of antibiotic tigecycline in the treatment of cancers. Int J Mol Sci. 2019;20:3577. https://doi.org/10.3390/ijms20143577.
Xu Z, Yan Y, Li Z, Qian L, Gong Z. The antibiotic drug tigecycline: a focus on its promising anticancer properties. Front Pharmacol. 2016;7:473. https://doi.org/10.3389/fphar.2016.00473.
Gafter-Gvili A, Fraser A, Paul M, Leibovici L. Meta-analysis: antibiotic prophylaxis reduces mortality in neutropenic patients. Ann Intern Med. 2005;142:979–95. https://doi.org/10.7326/0003-4819-142-12_Part_1-200506210-00008.
Paul M, Gafter-Gvili A, Fraser A, Leibovici L. The anti-cancer effects of quinol antibiotics. Eur J Clin Microbiol Infect Dis. 2007;26:825–31. https://doi.org/10.1007/s10096-007-0375-4.
Koltai T. 2016. Is ciprofloxacin an anti-cancer drug? A minireview. https://www.researchgate.net/publication/305319162. https://doi.org/10.13140/RG.2.1.3255.1920. Accessed May 29, 2020.
Mandal M, Mandal A, Das S, Chakraborti T, Chakraborti S. Clinical implications of matrix metalloproteinases. Mol Cell Biochem. 2003;252:305–29. https://doi.org/10.1023/A:1025526424637.
Löffek S, Schilling O, Franzke CW. Biological role of matrix metalloproteinases: a critical balance. Eur Respir J. 2011;38:191–208. https://doi.org/10.1183/09031936.00146510.
Rodríguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39–54. https://doi.org/10.1016/j.bbamcr.2009.09.015.
Nagase H, Visse R, Murphy G. Structure and function of matrix metalloproteinases and TIMPs. Cardiovasc Res. 2006;69:562–73. https://doi.org/10.1016/j.cardiores.2005.12.002.
Stephens CR, Murai K, Brunings KJ, Woodward RB. Acidity constants of the tetracycline antibiotics. J Am Chem Soc. 1956;78:4155–8. https://doi.org/10.1021/ja01597a081.
Jin L, Amaya-Mazo X, Apel ME, Sankisa SS, Johnson E, Zbyszynska MA, Han A. Ca2+ and Mg2+ bind tetracycline with distinct stoichiometries and linked deprotonation. Biophys Chem. 2007;128:185–96. https://doi.org/10.1016/j.bpc.2007.04.005.
Weinberg ED. The mutual effects of antimicrobial compounds and metallic cations. Bacteriol Rev. 1957;21:46–68 ((PMID: 13412621)).
Uivarosi V. Metal complexes of quinol antibiotics and their applications: an update. Molecules. 2013;18:11153–97. https://doi.org/10.3390/molecules180911153.
Zarkan A, Macklyne HR, Truman AW, Hesketh AR, Hong J. The frontline antibiotic vancomycin induces a zinc starvation response in bacteria by binding to Zn (II). Sci Rep. 2016;6:19602. https://doi.org/10.1038/srep19602.
St KJ, Strominger JL. Mechanism of action of bacitracin: complexation with metal ion and C55-isoprenyl pyrophosphate. Proc Nat Acad Sci. 1971;68:3223–7. https://doi.org/10.1073/pnas.68.12.3223.
Craig LC, Phillips WF, Bacitracin BM, A, . Isolation by counter double-current distribution and characterization. Biochemistry. 1969;8:2348–56. https://doi.org/10.1021/bi00834a015.
Sultana, Aryne MS, Sabri R. Erythromycin synergism with essential and trace elements. Pak J Pharm Sci 2005; 18: 35–39. https://www.researchgate.net/publication/215597765
Arayne S, Sultana N, Shamin S, Naz A. Synthesis, characterization and antimicrobial activities of azithromycin metal complexes. Mod Chem Appl. 2014;2:3. https://doi.org/10.4172/2329-6798.1000133.
Hamdan II. Comparative in vitro investigations of the interaction between some macrolides and Cu (II), Zn (II) and Fe (II). Pharmazie. 2003;58:223–4 ((PMID: 12685822)).
Arayne MS, Sultana N, Khanum F, Ali MA. Antibacterial studies of cefixime copper, zinc and cadmium complexes. Pak J Pharm Sci. 2002;15:1–8 ((PMID: 16414863)).
Auda SH, Knütter I, Bretschneider B, Brandsch M, Mrestani Y, Große C, Neubert RH. Effect of different metal ions on the biological properties of cefadroxil. Pharmaceuticals. 2009;2:184–93. https://doi.org/10.3390/ph2030184.
Zaman R, Rehman W, Hassan M, Mumtaz M, Khan MM, Anjum Z, Asad S, Shah H, Abbas SR. Synthesis, characterization and biological activities of cephalosporin metals complexes. Int J Biosci. 2016;9:163–72. https://doi.org/10.12692/ijb/9.5.163-172.
Siddiqi KS, Mohd A, Khan AAP, Bano S. Interaction of CFP with metal ions: complex formation of CFP with metal ion by absorption and fluorescence spectrophotometery. J Korean Chem Soc. 2009;53:152–8. https://doi.org/10.5012/jkcs.2009.53.2.152.
Iqbal MS, Ahmad AR, Sabir M, Asad SM. Preparation, characterization and biological evaluation of copper (II) and zinc (II) complexes with cephalexin. J Pharm Pharmacol. 1999;51:371–5. https://doi.org/10.1211/0022357991772556.
Alekseev VG. Metal complexes of penicillins and cephalosporins. Pharm Chem J. 2012;45:679–97. https://doi.org/10.1007/s11094-012-0703-6.
Ji HF, Shen L, Zhang HY. β-Lactam antibiotics are multipotent agents to combat neurological diseases. Biochem Biophys Res Commun. 2005;333:661–3. https://doi.org/10.1016/j.bbrc.2005.05.014.
Auda SH, Mrestani Y, Fetouh MI, Neubert RHH. Characterization and activity of cephalosporin metal complexes. Pharmazie. 2008;63:555–61. https://doi.org/10.1691/ph.2008.08.8532.
Anacona JR, Acosta F. Synthesis and antibacterial activity of cephradine metal complexes. J Coord Chem. 2006;59:621–7. https://doi.org/10.1080/00958970500393208.
Anacona JR, Rodriguez A. Synthesis and antibacterial activity of ceftriax metal complexes. Transition Met Chem. 2005;30:897–901. https://doi.org/10.1007/s11243-005-6219-0.
Anacona JR, Riodriguez H. Metalloantibiotics: Synthesis and antibacterial activity of cefepime metal complexes. J Coord Chem. 2009;62:2212–9. https://doi.org/10.1080/00958970902769815.
Gale EF. The assimilation of aminoacids by bacteria: trace metals on glutamic acid assimilation and their inactivation by 8-hydroxyquinoline. J Gen Microbiol. 1949;3:369–84. https://doi.org/10.1099/00221287-3-3-369.
Pelletier C, Prognon P, Bourlioux P. Roles of divalent cations and pH in mechanism of action of nitroxoline against Escherichia coli strains. Antimicrob Agents Chemother. 1995;39:707–13. https://doi.org/10.1128/aac.39.3.707.
El-Wahed MA, Refat MS, El-Megharbel SM. Spectroscopic studies on the complexation of some transition metals with chloramphenicol drug. J Mol Struc. 2008;892:402–13. https://doi.org/10.1016/j.molstruc.2008.06.005.
Santavirta S, Takagi M, Kontinen YT, Sorsa T, Suda A. Inhibitory effect of cephalothin on matrix metalloproteinase activity around loose hip prosthesis. Antimicrob Agents Chemother. 1996;40:244–6. https://doi.org/10.1128/aac.40.1.244.
Cifcibasi E, Kantarci A, Badur S, Issever H, Cintan S. Impact of metronidazole and amoxicillin combination on matrix metalloproteinases-1 and tissue inhibitors of matrix metalloproteinases balance in generalized aggressive periodontitis. Eur J Dent. 2015;9:53–9. https://doi.org/10.4103/1305-7456.149642.
Ambrosch A, Halevy D, Fwity B, Brin T, Lobmann R. Effect of daptomycin on local interleukin-6, matrix metalloproteinase-9, and metallopeptidase inhibitor 1 in patients with MRSA-infected diabetic foot. Int J Low Extrem Wounds. 2013;12:100–5. https://doi.org/10.1177/1534734613490506.
Simpson JL, Powell H, Boyle MJ, Scott RJ, Gibson PG. Clarithromycin targets neutrophilic airway inflammation in refractory asthma. Am J Respir Crit Care Med. 2008;177:148–55. https://doi.org/10.1164/rccm.200707-1134oc.
Fernandez-Robredo P, Recalde S, Moreno-Orduña M, García-García L, Zarranz-Ventura J, García-Layana A. Azithromycin reduces inflammation in a rat model of acute conjunctivitis. Mol Vis. 2013;19:153–65 ((PMID: 23378729)).
Singh B, Ghosh N, Saha D, Sarkar S, Bhattacharyya P, Chaudhury K. Effect of doxycyline in chronic obstructive pulmonary disease—an exploratory study. Pulm Pharmacol Ther. 2019;58:101831. https://doi.org/10.1016/j.pupt.2019.101831.
Sabir N, Hussain T, Mangi MH, Zhao D, Zhou X. Matrix metalloproteinases: expression, regulation and role in the immunopathology of tuberculosis. Cell Prol. 2019;52(4):e12649. https://doi.org/10.1111/cpr.12649.
Walker NF, Clark SO, Oni T, Andreu N, Tezera L, Singh S, Saraiva L, Pedersen B, Kelly DL, Tree JA, D’Armiento JM, Friedland JS, Elkington PT. Doxycycline and HIV infection suppress tuberculosis-induced matrix metalloproteinases. Am J Respir Crit Care Med. 2012;185:989–97. https://doi.org/10.1164/rccm.201110-1769OC.
Xu X, Abdalla T, Bratcher PE, Jackson PL, Sabbatini G, Wells JM, Lou XY, Quinn R, Blalock JE, Clancy JP, Gaggar A. Doxycycline improves clinical outcomes during cystic fibrosis exacerbations. Eur Respir J. 2017;49:1601102. https://doi.org/10.1183/13993003.01102-2016.
Xu DH, Zhu Z, Fang Y. The effect of a common antibiotics doxycycline on non-healing chronic wound. Curr Pharm Biotechnol. 2017;18:360–4. https://doi.org/10.2174/1389201018666170519095339.
Wilcox JR, Covington DS, Paez N. Doxycycline as a modulator of inflammation in chronic wounds. Wounds. 2012;24:339–49 ((PMID: 25876218)).
Stechmiller J, Cowan L, Schultz G. The role of doxycycline as a matrix metalloproteinase inhibitor for the treatment of chronic wounds. Biol Res Nurs. 2010;11:336–44. https://doi.org/10.1177/1099800409346333.
Menke MN, Menke NB, Boardman CH, Diegelmann RF. Biologic therapeutics and molecular profiling to optimize wound healing. Gynecol Oncol. 2008;111:S87–91. https://doi.org/10.1016/j.ygyno.2008.07.052.
Acharya MR, Venitz J, Figg WD, Sparreboom A. Chemically modified tetracyclines as inhibitors of matrix metalloproteinases. Drug Resist Updat. 2004;7:195–208. https://doi.org/10.1016/j.drup.2004.04.002.
Sabino F, auf dem Keller U. Matrix metalloproteinases in impaired wound healing. Metalloproteinases Med. 2015;2:1–8. https://doi.org/10.2147/MNM.S68420.
Sadler GM, Wallace HJ, Stacey MC. Oral doxycycline for the treatment of chronic leg ulceration. Arch Dermatol Res. 2012;304:487–93. https://doi.org/10.1007/s00403-012-1211-y.
Cariati A, Piromalli E, Cariati P. Effects of compression therapy and antibiotics on lymphatic flow and chronic venous leg ulceration. Arch Dermatol Res. 2012;304:497–8. https://doi.org/10.1007/s00403-012-1220-x.
Liu J, Xiong W, Baca-Regen L, Nagase H, Baxter BT. Mechanism of inhibition of matrix metalloproteinase-2 expression by doxycycline in human aortic smooth muscle cells. J Vasc Surg. 2003;38:1376–83. https://doi.org/10.1016/S0741-5214(03)01022-X.
Lindeman JH, Abdul-Hussien H, van Bockel JH, Wolterbeek R, Kleemann R. Clinical trial of doxycycline for matrix metalloproteinase-9 inhibition in patients with an abdominal aneurysm. Doxycycline selectively depletes aortic wall neutrophils and cytotoxic T cells. Circulation. 2009;119:2209–16. https://doi.org/10.1161/CIRCULATIONAHA.108.806505.
Mosorin M, Juvn J, Biancari F, Satta J, Surcel HM, Leinn M, Saikku P, Juvn T. Use of doxycycline to decrease the growth rate of abdominal aortic aneurysms: a randomized, double-blind, placebo-controlled pilot study. J Vasc Surg. 2001;34:606–10. https://doi.org/10.1067/mva.2001.117891.
Hackmann AE, Rubin BG, Sanchez LA, Geraghty PA, Thompson RW, Curci JA. A randomized, placebo-controlled trial of doxycycline after endoluminal aneurysm repair. J Vasc Surg. 2008;48:519–26. https://doi.org/10.1016/j.jvs.2008.03.064.
Curci JA, Mao D, Bohner DG, Allen BT, Rubin BG, Reilly JM, Sicard GA, Thompson RW. Preoperative treatment with doxycycline reduces aortic wall expression and activation of matrix metalloproteinases in patients with abdominal aortic aneurysms. J Vasc Surg. 2000;31:325–42. https://doi.org/10.1016/S0741-5214(00)90163-0.
Wladis EJ, Bradley EA, Bilyk JR, Yen MT, Mawn LA. Oral antibiotics for meibomian gland-related ocular surface disease: a report by the American Academy of Ophthalmology. Ophthalmology. 2016;123:492–6. https://doi.org/10.1016/j.ophtha.2015.10.062.
Federici TJ. The non-antibiotic properties of tetracyclines: clinical potential in ophthalmic disease. Pharm Res. 2011;64:614–23. https://doi.org/10.1016/j.phrs.2011.06.013.
Wang CT, Zhang L, Wu HW, Wei L, Xu B, Li DM. Doxycycline attenuates acute lung injury following cardiopulmonary bypass: involvement of matrix metalloproteinases. Int J Clin Exp Pathol. 2014;7:7460 ((PMID: 25550781)).
Gu Y, Walker C, Ryan ME, Payne JB, Golub LM. Non-antibacterial tetracycline formulations: clinical applications in dentistry and medicine. J Oral Microbiol. 2012;4:19227. https://doi.org/10.3402/jom.v4i0.19227.
Walker SG, Golub LM. Host modulation therapy for periodontal disease: Subantimicrobial-dose doxycycline, medical as well as dental benefits. Oral Sci 2012; 11: 8. https://www.oralhealthgroup.com/features/host-modulation-therapy-for-periodontal-disease-subantimicrobial-dose-doxycycline-medical-as-well-as/ Accessed May 29, 2020
Golub LM, Lee HM. Periodontal therapeutics: current host-modulation agents and future directions. Periodontology. 2000;82:186–204. https://doi.org/10.1111/prd.12315.
Golub LM, Wolff M, Roberts S, Lee HM, Leung M, Payonk GS. Treating periodontal diseases by blocking tissue destructive enzymes. J Am Dent Assoc. 1994;125:163–9. https://doi.org/10.14219/jada.archive.1994.0261.
Golub LM, Lee HM, Ryan ME, Giannobile WV, Payne JB, Sorsa T. Tetracyclines inhibit connective tissue breakdown by multiple non-antimicrobial mechanisms. Adv Dent Res. 1998;12:12–26. https://doi.org/10.1177/08959374980120010501.
Valentín S, Morales A, Sánchez JL, Rivera A. Safety and efficacy of doxycycline in the treatment of rosacea. Clin Cosmet Investig Dermatol. 2009;2:129–40. https://doi.org/10.2147/ccid.s4296.
Schaller M, Schöfer H, Homey B, Gieler U, Lehmann P, Luger TA, Ruzicka T, Steinhoff M. State of the art: systemic rosacea management. J Dtsch Dermatol Ges. 2016;14:29–37. https://doi.org/10.1111/ddg.13141.
Gendron R, Grenier D, Sorsa T, Mayrand D. Inhibition of the activities of matrix metalloproteinases 2, 8, and 9 by chlorhexidine. Clin Diagn Lab Immunol. 1999;6:437–9 ((PMID: 10225852)).
Montagner AF, Sarkis-Onofre R, Pereira-Cenci T, Cenci MS. MMP inhibitors on dentin stability: a systematic review and meta-analysis. J Dent Res. 2014;93:733–43. https://doi.org/10.1177/0022034514538046.
Hamdan-Nassar T, Bellot-Arcís C, Paredes-Gallardo V, García-Sanz V, Pascual-Moscardó A, Almerich-Silla JM. Effect of 2% chlorhexidine following acid etching on microtensile bond strength of resin restorations: a meta-analysis. Medicina. 2019;55:769. https://doi.org/10.3390/medicina55120769.
Sabatini C, Pashley DH. Mechanisms regulating the degradation of dentin matrices by endogenous dentin proteases and their role in dental adhesion. A review. Am J Dent. 2014;27:203–14 ((PMID: 25831604)).
Sabatini M, Lesur C, Thomas M, Chomel A, Anract P, de Nanteuil G, Pastoureau P. Effect of inhibition of matrix metalloproteinases on cartilage loss in vitro and in a guinea pig model of osteoarthritis. Arthritis Rheumat. 2005;52:171–80. https://doi.org/10.1002/art.20900.
Moon PC, Weaver J, Brooks CN. Review of matrix metalloproteinases’ effect on the hybrid dentin bond layer stability and chlorhexidine clinical use to prevent bond failure. Open Dent J. 2010;4:147–52. https://doi.org/10.2174/1874210601004010147.
Toledano M, Yamauti M, Osorio E, Osorio R. Zinc-inhibited MMP-mediated collagen degradation after different dentine demineralization procedures. Caries Res. 2012;46:201–7. https://doi.org/10.1159/000337315.
Osorio R, Yamauti M, Osorio E, Ruiz-Requena ME, Pashley D, Tay F, Toledano M. Effect of dentin etching and chlorhexidine application on metalloproteinase-mediated collagen degradation. Eur J Oral Sci. 2011;119:79–85. https://doi.org/10.1111/j.1600-0722.2010.00789.x.
Pavlík V, Sojka M, Mazúrová M, Velebný V. Dual role of iodine, silver, chlorhexidine and octenidine as antimicrobial and antiprotease agents. PLoS One . 2019;14:e0211055. https://doi.org/10.1371/journal.pone.0211055.
Tezvergil-Mutluay A, Agee KA, Uchiyama T, Imazato S, Mutluay MM, Cadenaro M, Breshi Y, Nishitani FR, Tay DH, Pashley DH. The inhibitory effects of quaternary ammonium methacrylates on soluble and matrix-bound MMPs. J Dent Res. 2011;90:535–40. https://doi.org/10.1177/0022034510389472.
Supuran CT. Sulfonamides. Molecules. 2017;22:1642. https://doi.org/10.3390/molecules22101642.
Yousef F, Mansour O, Herbali J (2018) Sulfonamides: Historical discovery development (structure-activity relationship notes). In-vitro In-vivo In-silico Journal 1: 1. https://openaccesspub.org/article/749/iiij-18-2040.pdf
Dax SL (1997) Antibacterial chemotherapeutic agents. Chapter 2: Sulfa antibacterials and arylpyrimidine antifolates. Chapter 2. Blackie Academic and Professional. An Imprint of Chapman and Hall. London, Weinheim, New York, Tokyo, Melbourne, Madras, pp 38–73
Goffin C, Ghuysen JM. Multimodular penicillin-binding proteins: an enigmatic family of orthologs and paralogs. Microbiol Mol Biol Rev. 1998;62:1079–93 ((PMID: 9841666)).
Tripathi LP, Sowdhamini R. Genome-wide survey of prokaryotic serine proteases: analysis of distribution and domain architectures of five serine protease families in prokaryotes. BMC Genomics. 2008;9:549. https://doi.org/10.1186/1471-2164-9-549.
Pallen MJ, Wren BW. The HtrA family of serine proteases. Mol Microbiol. 1997;26:209–21. https://doi.org/10.1046/j.1365-2958.1997.5601928.x.
Ponting CP. Evidence for PDZ domains in bacteria, yeast, and plants. Protein Sci. 1997;6:464–8. https://doi.org/10.1002/pro.5560060225.
Dallegri F, Dapino P, Arduino N, Bertolotto M, Ottllo L. Cefoperaz prevents the inactivation of α1-antitrypsin by activated neutrophils. Antimicrob Agents Chemother. 1999;43:2307–10. https://doi.org/10.1128/AAC.43.9.2307.
Js A, Elphick H, Pettitt E, Everard ML, Evans GS. Colistin stimulates the activity of neutrophil elastase and Pseudomonas aeruginosa elastase. Eur Respir J. 2002;19:1136–41. https://doi.org/10.1183/09031936.02.00230602.
Gupta A, Halve AK. ß-lactams: a mini review of their biological activity. Int J Pharmaceut Sci Res. 2015;6:978–87. https://doi.org/10.13040/IJPSR.0975-8232.6(3).978-87.
Xing B, Rao J, Liu R. Novel beta-lactam antibiotics derivatives: their new applications as gene reporters, antitumor prodrugs and enzyme inhibitors. Mini Rev Med Chem. 2008;8:455–71. https://doi.org/10.2174/1389557084223558.
Smith DM, Kazi A, Smith L, Long TE, Heldreth B, Turos E, Dou QP. A novel β-lactam antibiotic activates tumor cell apoptotic program by inducing DNA damage. Mol Pharmacol. 2002;61:1348–58. https://doi.org/10.1124/mol.61.6.1348.
Konaklieva MI. β-lactams as inhibitors of serine enzymes. Curr Med Chem Anti Infective Agents. 2002;1:215–38. https://doi.org/10.2174/1568012023354910.
Aravind L. An evolutionary classification of the metallo-beta-lactamase fold proteins. Silico Biol. 1999;1:69–91 ((PMID: 11471246)).
Bebr C. Metallo-beta-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem Pharmacol. 2007;74:1686–701. https://doi.org/10.1016/j.bcp.2007.05.021.
Caetano-Anolles G, Caetano-Anolles D. An evolutionarily structured universe of protein architecture. Genome Res. 2003;13:1563–71. https://doi.org/10.1101/gr.1161903.
Pettinati I, Brem J, Lee SY, McHugh PJ, Schofield CJ. The chemical biology of human metallo-β-lactamase fold proteins. Trends Biochem Sci. 2016;41:338–55. https://doi.org/10.1016/j.tibs.2015.12.007.
Lee JH, Takahashi M, Jeon JH, Kang LW, Seki M, Park KS, Kim TY, Karim AM, Lee JH. Dual activity of PNGM-1 pinpoints the evolutionary origin of subclass B3 metallo-β-lactamases: a molecular and evolutionary study. Emerg Microbes Infect. 2019;8:1688–700. https://doi.org/10.1080/22221751.2019.1692638.
Baier F, Tokuriki N. Connectivity between catalytic landscapes of the metallo-β-lactamase superfamily. J Mol Biol. 2014;426:2442–56. https://doi.org/10.1016/j.jmb.2014.04.013.
Redko Y, de la Li Sierra-Gallay I, Condon C. When all’s zed and d: the structure and function of RNase Z in prokaryotes. Nat Rev Microbiol. 2007;5:278–86. https://doi.org/10.1038/nrmicro1622.
Park KS, Kim TY, Kim JH, Lee JH, Jeon JH, Karim AM, Malik SK, Lee SH. PNGM-1, a novel subclass B3 metallo-β-lactamase from a deep-sea sediment metagenome. J Glob Antimicrob Resist. 2018;14:302–5. https://doi.org/10.1016/j.jgar.2018.05.021.
Polianskyte Z, Peitsaro N, Dapkunas A, Liobikas J, Soliymani R, Lalowski M, Speer O, Seitsn J, Butcher S, Cereghetti GM, Linder MD, Merckel M, Thompson J, Eriksson O. LACTB is a filament-forming protein localized in mitochondria. Proc Nat Acad Sci. 2009;106:18960–5. https://doi.org/10.1073/pnas.0906734106.
Peitsaro N, Polianskyte Z, Tuimala J, Pörn-Ares I, Liobikas J, Speer O, Lingholm D, Thompson J, Eriksson O. Evolution of a family of metazoan active-site-serine enzymes from penicillin-binding proteins: a novel facet of the bacterial legacy. BMC Evol Biol. 2008;8:26. https://doi.org/10.1186/1471-2148-8-26.
Keckesova Z, Donaher JL, De Cock J, Freinkman E, Lingrell S, Bachovchin DA, Bierie B, Tischler V, Noske A, Okondo MC, Reinhardt F, Thiru P, Golub TR, Vance JE, Weinberg RA. LACTB is a tumour suppressor that modulates lipid metabolism and cell state. Nature. 2017;543:681–6. https://doi.org/10.1038/nature21408.
Eriksson O, Lalowski M, Lindholm D. Commentary: LACTB is a tumour suppressor that modulates lipid metabolism and cell state. Front Physiol. 2017;8:396. https://doi.org/10.3389/fphys.2017.00396.
Diene SM, Pinault L, Keshri V, Armstrong N, Khelaifia S, Chabrière E, Caetano-Anolles G, Colson P, LaScola B, Rolain JM, Pontarotti P. Human metallo-β-lactamase enzymes degrade penicillin. Sci Rep. 2019;9:1–7. https://doi.org/10.1038/s41598-019-48723-y.
Lee SY, Brem J, Pettinati I, Claridge TD, Gileadi O, Schofield CJ, McHugh PJ. Cephalosporins inhibit human metallo β-lactamase fold DNA repair nucleases SNM1A and SNM1B/apollo. Chem Commun. 2016;52:6727–30. https://doi.org/10.1039/C6CC00529B.
Hafkemeyer P, Neftel KA, Hübscher U. HIV-reverse transcriptase and human DNA polymerase alpha share amino acid sequence homologies to bacterial penicillin-binding proteins. Methods Find Exp Clin Pharmacol. 1990;12:43–6 ((PMID: 1690323)).
Neftel KA, Hübscher U. Effects of beta-lactam antibiotics on proliferating eucaryotic cells. Antimicrob Agents Chemother. 1987;31:1657–61. https://doi.org/10.1128/AAC.31.11.1657.
Do UH, Neftel KA, Spadari S, Hübscher U. Betalactam antibiotics interfere with eukaryotic DNA-replication by inhibiting DNA polymerase α. Nucleic Acids Res. 1987;15:10495–506. https://doi.org/10.1093/nar/15.24.10495.
Huegin AW, Cerny A, Zinkernagel RM, Neftel KA. Suppressive effects of ß-lactam-antibiotics on in vitro generation of cytotoxic T-cells. Int J Immunopharmacol. 1986;8:723–9. https://doi.org/10.1016/0192-0561(86)90008-1.
Cottagnoud P, Neftel KA. Beta-lactams act on DNA synthesis in K-562 cells. Cell BiolToxicol. 1986;2:523–9. https://doi.org/10.1007/bf00117854.
Weston BJ, Spackman VM, Dewdney JM. Effect of beta-lactam antibiotics on a human myeloid cell line: investigation of potential in vivo correlates in the mouse. Cell Biol Toxicol. 1986;2:549–57. https://doi.org/10.1007/bf00117857.
Rasmussen BA, Baron DA, Kim JK, Unterwald EM, Rawls SM. β-lactam antibiotic produces a sustained reduction in extracellular glutamate in the nucleus accumbens of rats. Amino Acids. 2011;40:761–4. https://doi.org/10.1007/s00726-010-0589-0.
Rawls SM, Tallarida R, Robinson W, Amin M. The beta-lactam antibiotic, ceftriax, attenuates morphine-evoked hyperthermia in rats. Brit J Pharmacol. 2007;151:1095–102. https://doi.org/10.1038/sj.bjp.0707309.
Rawls SM, Cavallo F, Capasso A, Ding Z, Raffa RB. The β-lactam antibiotic ceftriax inhibits physical dependence and abstinence-induced withdrawal from cocaine, amphetamine, methamphetamine, and clorazepate in planarians. Eur J Pharmacol. 2008;584:278–84. https://doi.org/10.1016/j.ejphar.2008.02.018.
Rawls SM, Zielinski M, Patel H, Sacavage S, Baron DA, Patel D. Beta-lactam antibiotic reduces morphine analgesic tolerance in rats through GLT-1 transporter activation. Drug Alcohol Depend. 2010;107:261–3. https://doi.org/10.1016/j.drugalcdep.2009.10.010.
Rawls SM, Karaca F, Madhani I, Bhojani V, Martinez RL, Abou-Gharbia RRB. β-lactamase inhibitors display anti-seizure properties in an invertebrate assay. Neuroscience. 2010;169:1800–4. https://doi.org/10.1016/j.neuroscience.2010.06.041.
Lipski J, Wan CK, Bai JZ, Pi R, Li D, Donnelly D. Neuroprotective potential of ceftriax in in vitro models of stroke. Neuroscience. 2007;146:617–29. https://doi.org/10.1016/j.neuroscience.2007.02.003.
Nizzardo M, Nardini M, Ronchi D, Salani S, Donadoni C, Fortunato F, Colgiago GP, Falc M, Sim C, Riboldi G, Govoni A, Bresolin N, Comi GP, Corti S. Beta-lactam antibiotic offers neuroprotection in a spinal muscular atrophy model by multiple mechanisms. Exp Neurol. 2011;229:214–25. https://doi.org/10.1016/j.expneurol.2011.01.017.
Tai CH, Bellesi M, Chen AC, Lin CL, Li HH, Liao WC, Hung CS, Schwarting RK, Yl Ho. A new avenue for treating neuronal diseases: Ceftriax, an old antibiotic demonstrating behavioral neuronal effects. Behav Brain Res. 2019;364:149–56. https://doi.org/10.1016/j.bbr.2019.02.020.
Fontana AC. Current approaches to enhance glutamate transporter function and expression. J Neurochem. 2015;134:982–1007. https://doi.org/10.1111/jnc.13200.
Yimer EM, Hishe HZ, Tuem KB. Repurposing of the β-lactam antibiotic, Ceftriax for neurological disorders: a review. Front Neurosci. 2019;13:236. https://doi.org/10.3389/fnins.2019.00236.
Amiri M, Taherian R, Nazari H, Taherian M. Ceftriax decreases MPTP-induced behavioral disturbances in animal model of Parkinson’s disease. Int Clin Neuroscience J. 2016;3:206–13. https://doi.org/10.22037/icnj.v3i4.14365.
Amin B, Hajhashemi V, Abnous K, Hooseinzadeh H. Ceftriax, a beta-lactam antibiotic, modulates apoptosis pathways and oxidative stress in a rat model of neuropathic pain. Biomed Res Int. 2014. https://doi.org/10.1155/2014/937568 ((Article ID 937568)).
Wei J, Pan X, Pei Z, Wang W, Qiu W, Shi Z, Xiao G. The beta-lactam antibiotic, ceftriax, provides neuroprotective potential via anti-excitotoxicity and anti-inflammation response in a rat model of traumatic brain injury. J Trauma Acute Care Surg. 2012;73:654–60. https://doi.org/10.1097/TA.0b013e31825133c0.
Tikhonova MA, Ho SC, Akopyan AA, Kolosova NG, Weng JC, Meng WY, Lin CL, Amstislavskaya TG, Ho YJ. Neuroprotective effects of ceftriax treatment on cognitive and neuronal deficits in a rat model of accelerated senescence. Behav Brain Res. 2017;330:8–16. https://doi.org/10.1016/j.bbr.2017.05.002.
Tikhonova MA, Amstislavskaya TG, Belichenko VM, Fedoseeva LA, Kovalenko SP, Pisareva EE, Avdeeva AS, Kolosova NG, Belyaev ND, Aftanas LI. Modulation of the expression of genes related to the system of amyloid-beta metabolism in the brain as a novel mechanism of ceftriax neuroprotective properties. BMC Neurosci. 2018;19:13. https://doi.org/10.1186/s12868-018-0412-5.
McKhann GM. Beta-lactam antibiotics offer neuroprotection. Neurosurgery. 2005;56:N9. https://doi.org/10.1227/01.NEU.0000308742.63129.E3.
Rothstein JD, Patel S, Regan MR, Haenggeli C, Huang YH, Bergles DE, Jin L, Hoberg MD, Vidensky S, Chung DS, Toan SV, Bruijn LJ, Su ZS, Gupta P, Fisher PB. β-Lactam antibiotics offer neuroprotection by increasing glutamate transporter expression. Nature. 2005;433:73–7. https://doi.org/10.1038/nature03180.
Mao J. Glutamate transporter: an unexpected target for some antibiotics. Mol Pain. 2005;1:1744–8069. https://doi.org/10.1186/1744-8069-1-5.
Retzlaff CL, Kussrow A, Schorkopf T, Saetear P, Bornhop DJ, Hardaway JA, Sturgeon SM, Wright J, Blakely RD. Metallo-β-lactamase domain-containing protein 1 (MBLAC1) is a specific, high-affinity target for the glutamate transporter Inducer ceftriax. ACS Chem Neurosci. 2017;8:2132–8. https://doi.org/10.1021/acschemneuro.7b00232.
Mormile R, De Michele M, Squarcia U, Vittori G. Ceftriax-induced neuroprotection in glutamate excitotoxicity: more reason to treat bacterial meningitis with it ? Pediat Infect Dis J. 2012;31:1212–3. https://doi.org/10.1097/INF.0b013e3182635e0c.
Zhang Y, Zhang X, Qu S. Ceftriax protects astrocytes from MPP+ via suppression of NF-κB/JNK/c-Jun signaling. Mol Neurobiol. 2015;52:78–92. https://doi.org/10.1007/s12035-014-8845-z.
Lee SG, Su ZZ, Emdad L, Gupta P, Sarkar D, Borjabad A, Volsky DJ, Fisher PB. Mechanism of ceftriax induction of excitatory amino acid transporter-2 expression and glutamate uptake in primary human astrocytes. J Biol Chem. 2008;283:13116–23. https://doi.org/10.1074/jbc.M707697200.
Feng D, Wang W, Dong Y, Wu L, Huang J, Ma Y, Zhang Z, Wu S, Gao G, Qin H. Ceftriax alleviates early brain injury after subarachnoid hemorrhage by increasing excitatory amino acid transporter 2 expression via the PI3K/Akt/NF-κB signaling pathway. Neuroscience. 2014;268:21–32. https://doi.org/10.1016/j.neuroscience.2014.02.053.
Brooks BM, Flanagan BF, Thomas AL, Coleman JW. Penicillin conjugates to interferon-γ and reduces its activity: a novel drug–cytokine interaction. Biochem Biophys Res Commun. 2001;288:1175–81. https://doi.org/10.1006/bbrc.2001.5896.
Brooks BM, Thomas AL, Coleman JW. Benzylpenicillin differentially conjugates to IFN-γ, TNF-α, IL-1β, IL-4 and IL-13 but selectively reduces IFN-γ activity. Clin Exp Immunol. 2003;131:268–74. https://doi.org/10.1046/j.1365-2249.2003.02069.x.
Brooks BM, Hart CA, Coleman JW. Differential effects of β-lactams on human IFN-γ activity. J Antimicrob Chemother. 2005;56:1122–5. https://doi.org/10.1093/jac/dki373.
Meng X, Al-Attar Z, Yaseen FS, Jenkins R, Earnshaw C, Whitaker P, Peckham D, French NS, Naisbitt DJ, Park BK. Definition of the nature and hapten threshold of the β-lactam antigen required for T cell activation in vitro and in patients. J Immunol. 2017;198:4217–27. https://doi.org/10.4049/jimmunol.1700209.
Mor F, Cohen IR. Beta-lactam antibiotics modulate T-cell functions and gene expression via covalent binding to cellular albumin. Proc Nat Acad Sci. 2013;110:2981–6. https://doi.org/10.1073/pnas.1215722110.
Li X, Li H, Li S, Zhu F, Kim DJ, Xie H, Li Y, Nadas J, Oi N, Zykova TA, Yu DH, Lee MH, Kim MO, Wang L, Ma W, Lubet RA, Bode AM, Dong Z, Dong Z. Ceftriax, an FDA-approved cephalosporin antibiotic, suppresses lung cancer growth by targeting Aurora B. Carcinogenesis. 2012;33:2548–57. https://doi.org/10.1093/carcin/bgs283.
Stock ML, Fiedler KJ, Acharya S, Lange JK, Mlynarczyk GS, Anderson SJ, McCormack GR, Kanuri SH, Kondru NC, Brewer MT, Carlson SA. Antibiotics acting as neuroprotectants via mechanisms independent of their anti-infective activities. Neuropharmacology. 2013;73:174–82. https://doi.org/10.1016/j.neuropharm.2013.04.059.
Yulug B, Hanoglu L, Kilic E, Schabitz WR. Rifampicin: an antibiotic with brain protective function. Brain Res Bull. 2014;107:37–42. https://doi.org/10.1016/j.brainresbull.2014.05.007.
Fan-Minogue H, Bedwell DM. Eukaryotic ribosomal RNA determinants of aminoglycoside resistance and their role in translational fidelity. RNA. 2008;14:148–57. https://doi.org/10.1261/rna.805208.
Fong DH, Xiong B, Hwang J, Berghuis AM. Crystal structures of two aminoglycoside kinases bound with a eukaryotic protein kinase inhibitor. PLoS One . 2011;6:e19589. https://doi.org/10.1371/journal.pone.0019589.
Prokhorova I, Altman RB, Djumagulov M, Shrestha JP, Urzhumtsev A, Ferguson A, Chang CWT, Yusupov M, Blanchard SC, Yusupova G. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc Nat Acad Sci. 2017;114:E10899–908. https://doi.org/10.1073/pnas.1715501114.
Palmer E, Wilhelm JM, Sherman F. Phenotypic suppression of nonsense mutants in yeast by aminoglycoside antibiotics. Nature. 1979;277:148–50. https://doi.org/10.1038/277148a0.
Lee HL, Dougherty JP. Pharmaceutical therapies to recode nonsense mutations in inherited diseases. Pharmacol Ther. 2012;136:227–66. https://doi.org/10.1016/j.pharmthera.2012.07.007.
Manuvakhova M, Keeling K, Bedwell DM. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translationsystem. RNA. 2000;6:1044–55. https://doi.org/10.1017/S1355838200000716.
Keeling KM, Xue X, Gunn G, Bedwell DM. Therapeutics based on stop codon readthrough. Annu Rev Genomics Hum Genet. 2014;15:371–94. https://doi.org/10.1146/annurev-genom-091212-153527.
Malik V, Rodino-Klapac LR, Violett L, Mendell JR. Aminoglycoside-induced mutation suppression (stop codon readthrough) as a therapeutic strategy for Duchenne muscular dystrophy. Ther Adv Neurol Disorders. 2010;3:379–89. https://doi.org/10.1177/17562856120388693.
Nagel-Wolfrum K, Möller F, Penner I, Baasov T, Wolfrum U. Targeting nonsense mutations in diseases with translational read-through-inducing drugs (TRIDs). BioDrugs. 2016;30:49–74. https://doi.org/10.1007/s40259-016-0157-6.
Dabrowski M, Bukowy-Bieryllo Z, Zietkiewicz E. Advances in therapeutic use of a drug-stimulated translational readthrough of premature termination codons. Mol Med. 2018;24:25. https://doi.org/10.1186/s10020-018-0024-7.
Baradaran-Heravi A, Niesser J, Balgi AD, Choi K, Zimmerman C, South AP, Anderson HJ, Strynadka NC, Bally MB, Roberge M. Gentamicin B1 is a minor gentamicin compnt with major nonsense mutation suppression activity. Proc Nat Acad Sci. 2017;114:3479–84. https://doi.org/10.1073/pnas.1620982114.
Friesen WJ, Johnson B, Sierra J, Zhuo J, Vazirani P, Xue X, Tomizawa Y, Baiazitov R, Morrill C, Ren H, Babu S, Moon YC, Branstrom A, Mollin A, Hedrick J, Sheedy J, Elfring G, Weetall M, Colacino JM, Welch EM, Peltz SW. The minor gentamicin complex compnt, X2, is a potent premature stop codon readthrough molecule with therapeutic potential. PLoS One . 2018;13:e0206158. https://doi.org/10.1371/journal.pone.0206158.
Howard MT, Shirts BH, Petros LM, Flanigan KM, Gesteland RF, Atkins JF. Sequence specificity of aminoglycoside-induced stop codon readthrough: Potential implications for treatment of Duchenne muscular dystrophy. Ann Neurol. 2000;48:164–9. https://doi.org/10.1002/1531-8249(200008)48:2%3c164::AID-ANA5%3e3.0.CO;2-B.
Bidou L, Hatin I, Perez N, Allamand V, Panthier JJ, Rousset JP. Premature stop codons involved in muscular dystrophies show a broad spectrum of readthrough efficiencies in response to gentamicin treatment. Gene Ther. 2004;11:619–27. https://doi.org/10.1038/sj.gt.3302211.
Floquet C, Hatin I, Rousset JP, Bidou L. Statistical analysis of readthrough levels for nonsense mutations in mammalian cells reveals a major determinant of response to gentamicin. PLoS One Genet. 2012;8:e1002608. https://doi.org/10.1371/journal.pgen.1002608.
Dabrowski M, Bukowy-Bieryllo Z, Zietkiewicz E. Translational readthrough potential of natural termination codons in eucaryotes—the impact of RNA sequence. RNA Biol. 2015;12:950–8. https://doi.org/10.1080/15476286.2015.1068497.
Goodenough E, Robinson TM, Zook MB, Flanigan KM, Atkins JF, Howard MT, Eisenlohr LC. Cryptic MHC class I-binding peptides are revealed by aminoglycoside-induced stop codon read-through into the 3′ UTR. Proc Nat Acad Sci. 2014;111:5670–5. https://doi.org/10.1073/pnas.1402670111.
Hobbie SN, Akshay S, Kalapala SK, Bruell CM, Shcherbakov D, Böttger EC. Genetic analysis of interactions with eukaryotic rRNA identify the mitoribosome as target in aminoglycoside ototoxicity. Proc Nat Acad Sci. 2008;105:20888–93. https://doi.org/10.1073/pnas.0811258106.
Shulman E, Belakhov V, Wei G, Kendall A, Meyron-Holtz EG, Ben-Shachar D, Schacht J, Baasov T. Designer aminoglycosides that selectively inhibit cytoplasmic rather than mitochondrial ribosomes show decreased ototoxicity a strategy for the treatment of genetic diseases. J Biol Chem. 2014;289:2318–30. https://doi.org/10.1074/jbc.M113.533588.
Nguyen HL, Pham DL, O’Brien EP, Li MS. Erythromycin leads to differential protein expression through differences in electrostatic and dispersion interactions with nascent proteins. Sci Rep. 2018;8:6460. https://doi.org/10.1038/s41598-018-24344-9.
Kannan K, Kanabar P, Schryer D, Florin T, Oh E, Bahroos N, Tenson T, Weissman JS, Mankin AS. The general mode of translation inhibition by macrolide antibiotics. Proc Nat Acad Sci. 2014;111:15958–63. https://doi.org/10.1073/pnas.1417334111.
Vázquez-Laslop N, Mankin AS. How macrolide antibiotics work. Trends Biochem Sci. 2018;43:668–84. https://doi.org/10.1016/j.tibs.2018.06.011.
Vázquez-Laslop N, Ramu H, Klepacki D, Kannan K, Mankin AS. The key function of a conserved and modified rRNA residue in the ribosomal response to the nascent peptide. EMBO J. 2010;29:3108–17. https://doi.org/10.1038/emboj.2010.180.
Kannan K, Vázquez-Laslop N, Mankin AS. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell. 2012;151:508–20. https://doi.org/10.1016/j.cell.2012.09.018.
Davis AR, Gohara DW, Yap MNF. Sequence selectivity of macrolide-induced translational attenuation. Proc Nat Acad Sci. 2014;111:15379–84. https://doi.org/10.1073/pnas.1410356111.
Thompson J, Pratt CA, Dahlberg AE. Effects of a number of classes of 50S inhibitors on stop codon readthrough during protein synthesis. Antimicrob Agents Chemother. 2004;48:4889–91. https://doi.org/10.1128/AAC.48.12.4889-4891.2004.
Douthwaite S, Hansen LH, Mauvais P. Macrolide-ketolide inhibition of MLS-resistant ribosomesis improved by alternative drug interaction with domain II of 23S rRNA. Mol Microbiol. 2000;36:183–93. https://doi.org/10.1046/j.1365-2958.2000.01841.x.
Xiong L, Korkhin Y, Mankin AS. Binding site of the bridged macrolides in the Escherichia coliribosome. Antimicrob Agents Chemother. 2005;49:281–8. https://doi.org/10.1128/AAC.49.1.281-288.2005.
Bommakanti AS, Lindahl L, Zengel JM. Mutation from guanine to adenine in 25S rRNA at the position equivalent to E. coli A2058 does not confer erythromycin sensitivity in Sacchromyces cerevisae. RNA. 2008;14:460–4. https://doi.org/10.1261/rna.786408.
Mathis A, Wild P, Boettger EC, Kapel CM, Deplazes P. Mitochondrial ribosome as the target for the macrolide antibiotic clarithromycin in the helminth Echinococcus multilocularis. Antimicrob Agents Chemother. 2005;49:3251–5. https://doi.org/10.1128/AAC.49.8.3251-3255.2005.
Roll M (2014) Erythromycin treatment for readthrough of APC genes top codon mutations in familial adenomatous polyposis. ClinicalTrials.gov Identifier: NCT02175914
Ng MY, Zhang H, Weil A, Singh V, Jamiolkowski R, Baradaran-Heravi A, Roberge M, Jycobson A, Friesen W, Welch E, Goldman YF, Cooperman BS. New in vitro assay measuring direct interaction of nonsense suppressors with the eukaryotic protein synthesis machinery. ACS Med Chem Lett. 2018;9:1285–91. https://doi.org/10.1021/acsmedchemlett.8b00472.
Caspi M, Firsow A, Rajkumar R, Skalka N, Moshkovitz I, Munitz A, Pasmanik-Chor M, Greif H, Megido D, Rosenberg DW, Rosin-Arbesfeld R. A flow cytometry-based reporter assay identifies macrolide antibiotics as nonsense mutation read-through agents. J Mol Med. 2016;94:469–82. https://doi.org/10.1007/s00109-015-1364-1.
Osman EY, Washington CW III, Simon ME, Megiddo D, Greif H, Lorson CL. Analysis of azithromycin monohydrate as a single or a combinatorial therapy in a mouse model of severe spinal muscular atrophy. J Neuromuscul Dis. 2017;4:237–49. https://doi.org/10.3233/JND-170230.
Zilberberg A, Lahav L, Rosin-Arbesfeld R. Restoration of APC gene function in colorectal cancer cells by aminoglycoside-and macrolide-induced read-through of premature termination codons. Gut. 2010;59:496–507. https://doi.org/10.1136/gut.2008.169805.
Kariv R, Caspi M, Fliss-Isakov N, Shroer Y, Shor Y, Rosner G, Brazowski E, Beer G, Cohen S, Rosin-Arbesfeld R. Resorting the function of the colorectal cancer gatekeeper adenomatous polyopsis coli. Int J Cancer. 2020;146:1064–74. https://doi.org/10.1002/ijc.32557.
Kariv R, Fliss-Isacov N, Caspi M, Arbesfeld R. Erythromycin readthrough of APC nonsense stop codon mutation in familial adenomatous polyposis. Ann Oncol. 2018;29:mdy047.075. https://doi.org/10.1093/annonc/mdy047.075.
Catnach SM, Fairclough PD. Erythromycin and the gut. Gut. 1992;33:397–401. https://doi.org/10.1136/gut.33.3.397.
Coulie B, Tack J, Peeters T, Janssens J. Involvement of two different pathways in the motor effects of erythromycin on the gastric antrum in humans. Gut. 1998;43:395–400. https://doi.org/10.1136/gut.43.3.395.
Deloose E, Vos R, Janssen P, Van den Bergh O, Van Oudenhove L, Depoortere I, Tack J. The motilin receptor agonist erythromycin stimulates hunger and food intake through a cholinergic pathway. Am J Clin Nutr. 2016;103:730–7. https://doi.org/10.3945/ajcn.115.113456.
Doherty WL, Winter B. Prokinetic agents in critical care. Crit Care. 2003;7:206–8. https://doi.org/10.1186/cc1849.
Chini P, Toskes PP, Waseem S, Hou W, McDonald R, Moshiree B. Effect of azithromycin on small bowel motility in patients with gastrointestinal dysmotility. Scand J Gastroenterol. 2012;47:422–7. https://doi.org/10.3109/00365521.2012.654402.
Galligan JJ, Vanner S. Basic and clinical pharmacology of new motility promoting agents. Neurogastroenterol Motil. 2005;17:643–53. https://doi.org/10.1111/j.1365-2982.2005.00675.x.
Ishii A, Kumasaka M, Nagashima Y, Nakajima Y, Kuramochi K, Sugawara F, Narukawa M, Kamakura T. A eukaryotic molecular target candidate of roxithromycin: fungal differentiation as a sensitive drug target analysis system. Biosci Biotechnol Biochem. 2013;77:1539–47. https://doi.org/10.1271/bbb.13021.
Qiu L, Wu J, Pan C, Tan X, Lin J, Liu R, Chen S, Geng R, Huang W. Downregulation of CDC27 inhibits the proliferation of colorectal cancer cells via the accumulation of p21Cip1/Waf1. Cell Death Dis. 2016;7:e2074. https://doi.org/10.1038/cddis.2015.402.
Chloramphenicol PS. In: Corcoran JW, Hahn FE, Snell JF, Arora KL, editors. Antibitoics: mechanism of action of antimicrobial and antitumor agents. Berlin, Heidelberg, New York: Springer-Verlag; 1975. p. 370–95.
Dunkle JA, Xiong L, Mankin AS, Cate JH. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc Nat Acad Sci. 2010;107:17152–7. https://doi.org/10.1073/pnas.1007988107.
Bulkley D, Innis CA, Blaha G, Steitz TA. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc Nat Acad Sci. 2010;107:17158–63. https://doi.org/10.1073/pnas.1008685107.
Tereshchenkov AG, Dobosz-Bartoszek M, Osterman IA, Marks J, Sergeeva VA, Kasatsky P, Komarova ES, Stavrianidi AN, Rodin IA, Kvega AL, Sergiev PV, Sumbatyan NV, Mankin AS, Bogdanov AA, Polikanov YS. Binding and action of amino acid analogs of chloramphenicol upon the bacterial ribosome. J Mol Biol. 2018;430:842–52. https://doi.org/10.1016/j.jmb.2018.01.016.
Barnhill AE, Brewer MT, Carlson SA. Adverse effects of antimicrobials via predictable or idiosyncratic inhibition of host mitochondrial compnts. Antimicrob Agents Chemother. 2012;56:4046–51. https://doi.org/10.1128/AAC.00678-12.
Singh R, Sripada L, Singh R. Side effects of antibiotics during bacterial infection: mitochondria, the main target in host cell. Mitochondrion. 2014;16:50–4. https://doi.org/10.1016/j.mito.2013.10.005.
Js CN, Miller C, Tenenbaum A, Spremulli LL, Saada A. Antibiotic effects on mitochondrial translation and inpatients with mitochondrial translational defects. Mitochondrion. 2009;9:429–37. https://doi.org/10.1016/j.mito.2009.08.001.
Ibrahim NG, Burke JP, Beattie DS. The sensitivity of rat liver and yeast mitochondrial ribosomes to inhibitors of protein synthesis. J Biol Chem. 1974;249:6806–11 ((PMID: 4609092)).
Lamb AJ, Clark-Walker GD, Linnane AW. The biogenesis of mitochondria 4 The differentiation of mitochondrial and cytoplasmic protein synthesizing systems in vitro by antibiotics. Biochim Biophys Acta. 1968;161:415–27. https://doi.org/10.1016/0005-2787(68)90119-6.
Thompson J, O’Connor M, Mills JA, Dahlberg AE. The protein synthesis inhibitors, oxazolidins and chloramphenicol, cause extensive translational inaccuracy in vivo. J Mol Biol. 2002;322:273–9. https://doi.org/10.1016/S0022-2836(02)00784-2.
Tobe R, Naranjo-Suarez S, Everley RA. High error rates in selenocysteine insertion in mammalian cells treated with the antibiotic doxycycline, chloramphenicol, or geneticin. J Biol Chem. 2013;288:14700–15. https://doi.org/10.1074/jbc.M112.446666.
Mayer FQ, Artigalas OA, Lagranha VL, Baldo G, Schwartz IV, Matte U, Giugliani R. Chloramphenicol enhances IDUA activity on fibroblasts from mucopolysaccharidosis I patients. Curr Pharm Biotechnol. 2013;14:194–8. https://doi.org/10.2174/138920113805219467.
Fan Y, Evans CR, Barber KW, Banerjee K, Weiss KJ, Margolin W, Igoshin OA, Rinehart J, Ling J. Heterogeneity of stop codon readthrough in single bacterial cells and implications for population fitness. Mol Cell. 2017;67:826–36. https://doi.org/10.1016/j.molcel.2017.07.010.
Nozaka A, Nishiwaki A, Nagashima Y, Endo S, Kuroki M, Nakajima M, Narukawa M, Kamisuki S, Arazoe T, Taguchi H, Sugawara F, Kamakura T. Chloramphenicol inhibits eukaryotic Ser/Thr phosphatase and infection-specific cell differentiation in the rice blast fungus. Sci Rep. 2019;9:9283. https://doi.org/10.1038/s41598-019-41039-x.
Slee AM, Wuonola MA, McRipley RJ, Zajac I, Zawada MJ, Bartholomew PT, Gregory WA, Forbes M. Oxazolidins, a new class of synthetic antibacterial agents: in vitro and in vivo activities of DuP 105 and DuP 721. Antimicrob Agents Chemother. 1987;31:1791–17970. https://doi.org/10.1128/AAC.31.11.1791.
Daly JS, Eliopoulos GM, Reiszner E, Moellering RC Jr. Activity and mechanism of action of DuP 105 and DuP 721, new oxazolidin compounds. J Antimicrob Chemother. 1988;21:721–30. https://doi.org/10.1093/jac/21.6.721.
Eustice DC, Feldman PA, Zajac I, Slee AM. Mechanism of action of DuP 721: inhibition of an early event during initiation of protein synthesis. Antimicrob Agents Chemother. 1988;32:1218–22. https://doi.org/10.1128/aac.32.8.1218.
Matassova NB, Rodnina MV, Endermann R, Kroll HP, Pleiss U, Wild H, Wintermeyer W. Ribosomal RNA is the target for oxazolidins, a novel class of translational inhibitors. RNA. 1999;5:939–46. https://doi.org/10.1017/S1355838299990210.
Wilson DN, Schluenzen F, Harms JM, Starosta AL, Connell SR, Fucini P. The oxazolidin antibiotics perturb the ribosomal peptidyl-transferase center and effect tRNA positioning. Proc Nat Acad Sci. 2008;105:13339–44. https://doi.org/10.1073/pnas.0804276105.
Ippolito JA, Kanyo F, Wang D, Franceschi FJ, Moore PB, Steitz TA, Duffy EM. Crystal structure of the oxazolidin antibiotic linezolid bound to the 50S ribosomal subunit. J Med Chem. 2008;51:3353–6. https://doi.org/10.1021/jm800379d.
Lin AH, Murray RW, Vidmar TJ, Marotti KR. The oxazolidin eperezolid binds to the 50S ribosomal subunit and competes with binding of chloramphenicol and lincomycin. Antimicrob Agents Chemother. 1997;41:2127–31. https://doi.org/10.1128/aac.41.10.2127.
Marks J, Kannan K, Roncase EJ, Klepacki D, Kefi A, Orelle C, Vázquez-Laslop N, Mankin AS. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc Nat Acad Sci. 2016;113:12150–5. https://doi.org/10.1073/pnas.1613055113.
Saini JS, Homeyer N, Fulle S, Gohlke H. Determinants of the species selectivity of oxazolidin antibiotics targeting the large ribosomal subunit. Biol Chem. 2013;394:1529–41. https://doi.org/10.1515/hsz-2013-0188.
Skripkin E, McConnell TS, DeVito J, Lawrence L, Ippolito JA, Duffy EM, Sutcliffe J, Franceschi F. R chi-01, a new family of oxazolidins that overcome ribosome-based linezolid resistance. Antimicrob Agents Chemother. 2008;52:3550–7. https://doi.org/10.1128/AAC.01193-07.
Zhu Y, Weldon JE. Evaluating the influence of common antibiotics on the efficacy of a recombinant immunotoxin in tissue culture. BMC Res Notes. 2019;12:293. https://doi.org/10.1186/s13104-019-4337-6.
Esner M, Graifer D, Lleonart ME, Lyakhovich A. Targeting cancer cells through antibiotics-induced mitochondrial dysfunction requires autophagy inhibition. Cancer Lett. 2017;384:60–9. https://doi.org/10.1016/j.canlet.2016.09.023.
Abad E, García-Mayea Y, Mir C, Sebastian D, Zorzano A, Potesil D, Zdrahal Z, Lyakhovich A, Lleonart ME. Common metabolic pathways implicated in resistance to chemotherapy point to a key mitochondrial role in breast cancer. Mol Cell Proteom. 2019;18:231–44. https://doi.org/10.1074/mcp.RA118.001102.
Sharon D, Cathelin S, Mirali S, Di Trani JM, Yanofsky DJ, Keon KA, Rubinstein JL, Schimmer AD, Ketela T, Chan SM. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci Transl Med 2019; 11(516). https://doi.org/10.1126/scitranslmed.aax2863
Kaku N, Yanagihara K, Morinaga Y, Yamada K, Harada Y, Migiyama Y, Nagaoka K, Nakamura S, Izumikawa K, Kohno S. Immunomodulatory effect of linezolid on methicillin-resistant Staphylococcus aureus supernatant-induced MUC5AC overexpression in human airway epithelial cells. Antimicrob Agents Chemother. 2014;58:4131–7. https://doi.org/10.1128/AAC.02811-13.
Wang J, Xia L, Wang R, Cai Y. Linezolid and its immunomodulatory effect: in vitro and in vivo evidence. Front Pharmacol. 2019;10:1389. https://doi.org/10.3389/fphar.2019.01389.
Yamada K, Morinaga Y, Yanagihara K, Kaku N, Harada Y, Uno N, Nakamura S, Imamura Y, Hasegawa H, Miyazaki T, Izumikawa K, Kakeya H, Mikamo H, Kohno S. Azithromycin inhibits MUC5AC induction via multidrug-resistant Acinetobacter baumannii in human airway epithelial cells. Pulm Pharmacol Ther. 2014;28:165–70. https://doi.org/10.1016/j.pupt.2014.05.006.
Chopra I, Roberts M. Tetracycline antibiotics: mode of action, applications, molecular biology, and epidemiology of bacterial resistance. Microbiol Mol Biol Rev. 2001;65:232–60. https://doi.org/10.1128/MMBR.65.2.232-260.2001.
Grossman TH. Tetracycline antibiotics and resistance. CSH Perspect Med. 2016;6:a025387. https://doi.org/10.1101/cshperspect.a025387.
Mortison JD, Schen M, Myers JA, Zhang Z, Chen L, Ciarlo C, Comer E, Natchiar SK, Carr SA, Klaholz BP, Myers AG. Tetracyclines modify translation by targeting key human rRNA substructures. Cell Chem Biol. 2018;25:1506–18. https://doi.org/10.1016/j.chembiol.2018.09.010.
Boer RE, Schneekloth JS Jr. Targeting mammalian translational inhibition with tetracyclines. Cell Chem Biol. 2018;25:1437–8. https://doi.org/10.1016/j.chembiol.2018.12.006.
O’Dell JR, Elliott JR, Mallek JA, Mikuls TR, Weaver CA, Glickstein S, Blakely KM, Hausch R, Leff RD. Treatment of early seropositive rheumatoid arthritis: doxycycline plus methotrexate versus methotrexate al. Arthritis Rheum. 2006;54:621–7. https://doi.org/10.1002/art.21620.
Brandt KD, Mazzuca SA, Katz BP, Lane KA, Buckwalter KA, Yocum DE, Wolfe F, Schnitzer TJ, Moreland LW, Manzi S, Sharma L, Oddis CV, Hugenberg ST, Heck LW. Effects of doxycycline on progression of osteoarthritis: results of a randomized, placebo-controlled, double-blind trial. Arthritis Rheum. 2005;52:2015–25. https://doi.org/10.1002/art.21122.
Walker C, Puumala S, Golub LM, Str JA, Reinhardt RA, Lee HM, Payne JB. Subantimicrobial dose doxycycline effects on osteopenic b loss: microbiologic results. J Periodontol. 2007;78:1590–601. https://doi.org/10.1902/jop.2007.070015.
Leigh MJ, Nguyen DV, Mu Y, Winarni TI, Schneider A, Chechi T, Polussa J, Doucet P, Tass F, Rivera SM, Hesse D, Hagerman RL. A randomized double-blind, placebo-controlled trial of minocycline in children and adolescents with fragile x syndrome. J Dev Behav Pediatr. 2013;34:147–55. https://doi.org/10.1097/DBP.0b013e318287cd17.
Dodd BR, Spence RA. Doxycycline inhibition of abdominal aortic aneurysm growth—a systematic review of the literature. Curr Vasc Pharmacol. 2011;9:471–8. https://doi.org/10.2174/157016111796197288.
Abdul-Hussien H, Hanemaaijer R, Verheijen JH, van Bockel JH, Geelkerken RH, Lindeman JH. Doxycycline therapy for abdominal aneurysm: Improved proteolytic balance through reduced neutrophil content. J Vasc Surg. 2009;49:741–9. https://doi.org/10.1016/j.jvs.2008.09.055.
Baxter BT, Matsumura J, Curci JA, McBride R, Larson LA, Blackwelder W, Lam D, Wijesinha M, Terrin M. Effect of doxycycline on aneurysm growth among patients with small infrarenal abdominal aortic aneurysms: a randomized clinical trial. JAMA. 2020;323:2019–38. https://doi.org/10.1001/jama.2020.5230.
Meijer CA, Stijnen T, Wasser MN, Hamming JF, van Bockel JH, Lindeman JH. Doxycycline for stabilization of abdominal aortic aneurysms: a randomized trial. Ann Intern Med. 2013;159:815–23. https://doi.org/10.7326/0003-4819-159-12-201312170-00007.
Sironi M, Bandi C, Sacchi L, Di Sacco B, Damiani G, Genchi C. Molecular evidence for a close relative of the arthropod endosymbiont Wolbachia in a filarial worm. Mol Biochem Parasitol. 1995;74:223–7. https://doi.org/10.1016/0166-6851(95)02494-8.
Rao RU, Huang Y, Abubucker S, Heinz M, Crosby SD, Mitreva M, Weil GJ. Effects of Doxycycline on gene expression in Wolbachia and Brugia malayi adult female worms in vivo. J Biomed Sci. 2012;19:21. https://doi.org/10.1186/1423-0127-19-21.
Dolezal P, Smíd O, Rada P, Zubácová Z, Bursać D, Suták R, Nebesárová J, Lithgow T, Tachezy J. Giardia mitosomes and trichomonad hydrogenosomes share a common mode of protein targeting. Proc Nat Acad Sci. 2005;102:10924–9. https://doi.org/10.1073/pnas.0500349102.
Huang KY, Ku FM, Cheng WH, Lee CC, Huang PJ, Chu LJ, Fang YK, Wu HH, Tang P. Novel insights into the molecular events linking to cell death induced by tetracycline in the amitochondriate protozoan Trichomonas vaginalis. Antimicrob Agents Chemother. 2015;59:6891–903. https://doi.org/10.1128/AAC.01779-15.
Lin Q, Katakura K, Suzuki M. Inhibition of mitochondrial and plastid activity of Plasmodium falciparum by minocycline. FEBS Lett. 2002;515:71–4. https://doi.org/10.1016/S0014-5793(02)02437-7.
Chukwudi CU, Good L. Interaction of the tetracyclines with double-stranded RNAs of random base sequence: new perspectives on the target and mechanism of action. J Antibiot. 2016;69:622–30. https://doi.org/10.1038/ja.2015.145.
Lambs L, Venturini M, Decock-Le Reverend B, Kozlowski H, Berthon G. Metal ion-tetracycline interactions in biological fluids. Part 8. Potentiometric and spectroscopic studies on the formation of Ca(II) and Mg(II) complexes with 4-dedimethylamino-tetracycline and 6-desoxy-6-demethyl-tetracycline. J Inorg Biochem. 1988;33:193–210. https://doi.org/10.1016/0162-0134(88)80049-7.
Grenier D, Huot MP, Mayrand D. Iron-chelating activity of tetracyclines and its impact on the susceptibility of Actinobacillus actinomycetemcomitans to these antibiotics. Antimicrob Agents Chemother. 2000;44:763–6. https://doi.org/10.1128/AAC.44.3.763-766.2000.
Caswell AH, Hutchison JD. Selectivity of cation chelation to tetracyclines: evidence for special conformation of calcium chelate. Biochem Biophysical Res Commun. 1971;43:625–30. https://doi.org/10.1016/0006-291X(71)90660-7.
Brion M, Lambs L, Berthon G. Metal ion-tetracycline interactions in biological fluids. Part 5. Formation of zinc complexes with tetracycline and some of its derivatives and assessment of their biological significance. Agents Actions. 1985;17:229–42. https://doi.org/10.1007/bf01966597.
Fuoco D. Classification framework and chemical biology of tetracycline-structure-based drugs. Antibiotics. 2012;1:1. https://doi.org/10.3390/antibiotics1010001.
Griffin MO, Fricovsky E, Ceballos G, Villarreal F. Tetracyclines: a pleitropic family of compounds with promising therapeutic properties. Review of the literature. Am J Physiol Cell Physiol. 2010b;299:C539–48. https://doi.org/10.1152/ajpcell.00047.2010.
Soory M. A role for non-antimicrobial actions of tetracyclines in combating oxidative stress in periodontal and metabolic diseases: a literature review. Open Dent J. 2008;2:5–12. https://doi.org/10.2174/1874210600802010005.
Joseph BB. Subantimicrobial dose doxycycline for acne and rosacea. SKINmed. 2003;2:234–46. https://doi.org/10.1111/j.1540-9740.2003.03014.x.
Sapadin AN, Fleischmajer R. Tetracyclines: nonantibiotic properties and their clinical implications. J Am Acad Dermatol. 2006;54:258–65. https://doi.org/10.1016/j.jaad.2005.10.004.
Payne JB, Golub LM. Using tetracyclines to treat osteoporotic/osteopenic b loss: from the basic science laboratory to the clinic. Pharmacol Res. 2011;63:121–9. https://doi.org/10.1016/j.phrs.2010.10.006.
Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Nat Acad Sci. 1992;89:5547–51. https://doi.org/10.1073/pnas.89.12.5547.
Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–9. https://doi.org/10.1126/science.7792603.
Gossen M, Bujard H. Tetracyclines in the control of gene expression in eukaryotes. In: Nelson M, Hillen W, Greenwald RA, editors. Tetracyclines in Biology, Chemistry and Medicine. Basel: Birkhäuser; 2001. https://doi.org/10.1007/978-3-0348-8306-1_5.
Deuschle U, Meyer WK, Thiesen HJ. Tetracycline-reversible silencing of eukaryotic promoters. Mol Cell Biol. 1995;15:1907–14. https://doi.org/10.1128/MCB.15.4.1907.
Ahler E, Sullivan WJ, Cass A, Braas D, York AG, Bensinger J, Graeber TG, Christofk HR. Doxycycline alters metabolism and proliferation of human cell lines. PLoS One . 2013;8:e64561. https://doi.org/10.1371/journal.pone.0064561.
Das T, Tenenbaum L, Berkhout B. Tet-On systems for doxycycline-inducible gene expression. Curr Gene Ther. 2016;16:156–67. https://doi.org/10.2174/1566523216666160524144041.
Hooper DC, Jacoby GA. Topoisomerase inhibitors: fluoroquinol mechanisms of action and resistance. CSH Perspectives Med. 2016;6:a025320. https://doi.org/10.1101/cshperspect.a025320.
Thu DM, Ziora ZM, Blaskovich MAT. Quinol antibacterials. Med. Chem Commun. 2019;10:1719–39. https://doi.org/10.1039/c9md00120d.
Aldred KJ, Kerns RJ, Osheroff N. Mechanism of quinol action and resistance. Biochemistry. 2014;53:1565–74. https://doi.org/10.1021/bi5000564.
Fief CA, Hoang KG, Phipps SD, Wallace JL, Deweese JE. Examining the impact of antimicrobial fluoroquinols on human DNA topoisomerase IIα and IIβ. ACS Omega. 2019;4:4049–55. https://doi.org/10.1021/acsomega.8b03428.
Aldred KJ, McPherson SA, Turnbough CL Jr, Kerns RJ, Osheroff N. Topoisomerase IV-quinol interactions are mediated through a water-metal ion bridge: mechanistic basis of quinol resistance. Nucleic Acids Res. 2013;41:4628–39. https://doi.org/10.1093/nar/gkt124.
Aldred KJ, Breland EJ, Vlčková V, Strub MP, Neuman KC, Kerns RJ, Osheroff N. Role of the water–metal ion bridge in mediating interactions between quinols and Escherichia coli topoisomerase IV. Biochemistry. 2014;53:5558–67. https://doi.org/10.1021/bi500682e.
Williams GM, Brunnemann KD, Smart DJ, Molina D, Jeffrey AM, Duan JD, Krebsfaenger N, Kampkoetter A, Schmuck G. Relationship of cellular topoisomerase IIα inhibition to cytotoxicity and published genotoxicity of fluoroquinol antibiotics in V79 cells. Chem Biol Interact. 2013;203:386–90. https://doi.org/10.1016/j.cbi.2013.01.003.
Kloskowski T, Gurtowska N, Olkowska J, Nowak JM, Adamowicz J, Tworkiewicz JD, Ebski R, Grzanka A, Drewa T. Ciprofloxacin is a potential topoisomerase II inhibitor for the treatment of NSCLC. Int J Oncol. 2012;41:1943–9. https://doi.org/10.3892/ijo.2012.1653.
Smart DJ, Lynch AM. Evaluating the genotoxicity of topoisomerase-targeted antibiotics. Mutagenesis. 2012;27:359–65. https://doi.org/10.1093/mutage/ger089.
Hangas A, Aasumets K, Kekäläinen NJ, Paloheinä M, Pohjoismäki JL, Gerhold JM, Goffart S. Ciprofloxacin impairs mitochondrial DNA replication initiation through inhibition of Topoisomerase 2. Nucleic Acids Res. 2018;46:9625–36. https://doi.org/10.1093/nar/gky793.
Lawrence JW, Darkin-Rattray S, Xie F, Neims AH, Rowe TC. 4-Quinols cause a selective loss of mitochondrial DNA from mouse L1210 leukemia cells. J Cell Biochem. 1993;51:165–74. https://doi.org/10.1002/jcb.240510208.
Lawrence JW, Claire DC, Weissig V, Rowe TC. Delayed cytotoxicity and cleavage of mitochondrial DNA in ciprofloxacin-treated mammalian cells. Mol Pharmacol. 1996;50:1178–88 ((PMID: 8913349)).
Ghaly H, Jörns A, Rustenbeck I. Effect of fluoroquinols on mitochondrial function in pancreatic beta-cells. Eur J Pharm Sci. 2014;52:206–14. https://doi.org/10.1016/j.ejps.2013.11.011.
Hsiao CJJ, Younis H, Boelsterli UA. Trovafloxacin, a fluoroquinol antibiotic with hepatotoxic potential, causes mitochondrial peroxynitrite stress in a mouse model of underlying mitochondrial dysfunction. Chem Biol Interact. 2010;188:204–13. https://doi.org/10.1016/j.cbi.2010.07.017.
Aranha O, Zhu L, Alhasan S, Wood DP, Kuo TH, Sarkar FH. Role of mitochondria in ciprofloxacin induced apoptosis in bladder cancer cells. J Urol. 2002;167:1288–94. https://doi.org/10.1016/S0022-5347(05)65283-4.
Yu M, Li R, Zhang J. Repositioning of antibiotic levofloxacin as a mitochondrial biogenesis inhibitor to target breast cancer. Biochem Biophys Res Commun. 2016;471:639–45. https://doi.org/10.1016/j.bbrc.2016.02.072.
Heisig P. Type II topoisomerases—inhibitors, repair mechanisms and mutations. Mutagenesis. 2009;24:465–9. https://doi.org/10.1093/mutage/gep035.
Maslowska KH, Makiela-Dzbenska K, Fijalkowska IJ. The SOS system: a complex and tightly regulated response to DNA damage. Environ Mol Mutagen. 2019;60:368–84. https://doi.org/10.1002/em.22267.
Baumann V, Winkler J. miRNA-based therapies: strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med Chem. 2014;6:1967–84. https://doi.org/10.4155/fmc.14.116.
Hawley BR, Lu WT, Wilczynska A, Bushell M. The emerging role of RNAs in DNA damage repair. Cell Death Diff. 2017;24:580–7. https://doi.org/10.1038/cdd.2017.16.
Kian R, Moradi S, Ghorbian S. Role of compnts of microRNA machinery in carcinogenesis. Exp Oncol. 2018;40:2–9 ((PMID: 29600985)).
Shah MY, Ferrajoli A, Sood AK, Lopez-Berestein G, Calin GA. microRNA therapeutics in cancer—an emerging concept. EBioMedicine. 2016;12:34–42. https://doi.org/10.1016/j.ebiom.2016.09.017.
Biswas S. MicroRNAs as therapeutic agents: the future of the battle against cancer. Curr Topics Med Chem. 2018;18:2544–54. https://doi.org/10.2174/1568026619666181120121830.
Zhang Q, Zhang C, Xi Z. Enhancement of RNAi by a small molecule antibiotic enoxacin. Cell Res. 2008;18:1077–9. https://doi.org/10.1038/cr.2008.287.
Li Y, Ji P, Jin P. Probing the microRNA pathway with small molecules. Bioorgan Med Chem. 2013;21:6119–23. https://doi.org/10.1016/j.bmc.2013.05.030.
Shan G, Li Y, Zhang J, Li W, Szulwach KE, Duan R, Faghihi MA, Khalil AM, Lu L, Paroo Z, Chan AWS, Shi Z, Liu Q, Wahlestedt C, He C, Jin P. A small molecule enhances RNA interference and promotes microRNA processing. Nat Biotechnol. 2008;26:933–40. https://doi.org/10.1038/nbt.1481.
Toro EJ, Zuo J, Ostrov DA, Catalfamo D, Bradaschia-Correa V, Arana-Chavez V, Caridad AR, Neubert JK, Wronski TJ, Wallert SM, Holliday LS. Enoxacin directly inhibits osteoclastogenesis without inducing apoptosis. J Biological Chem. 2012;287:17894–904. https://doi.org/10.1074/jbc.M111.280511.
Toro EJ, Zuo J, Guiterrez A, La Rosa RL, Gawron AJ, Bradaschia-Correa V, Arana-Chavez V, Dolce C, Rivera MF, Kesavalu L, Bhattacharyya I, Neubert JK, Holliday LS. Bis-enoxacin inhibits b resorption and orthodontic tooth movement. J Dent Res. 2013;92:925–31. https://doi.org/10.1177/0022034513501876.
Toro E, Ostrov DA, Wronski TJ, Holliday LS. Rational identification of enoxacin as a novel V-ATPase-directed osteoclast inhibitor. Curr Protein Pept Sci. 2012;13:180–91. https://doi.org/10.2174/138920312800493151.
Ostrov DA, Magis AT, Wronski TJ, Chan EK, Toro EJ, Donatelli R, Sajek K, Haroun IN, Nagib MI, Piedrahita A, Harris A, Holliday LS. Identification of enoxacin as an inhibitor of osteoclast formation and b resorption by structure-based virtual screening. J Med Chem. 2009;52:5144–51. https://doi.org/10.1021/jm900277z.
Gioia U, Francia S, Cabrini M, Brambillasca S, Michelini F, Js-Weinert CW, di Fagagna FDA. Pharmacological boost of DNA damage response and repair by enhanced biogenesis of DNA damage response RNAs. Sci Rep. 2019;9:6460. https://doi.org/10.1038/s41598-019-42892-6.
Melo S, Villanueva A, Moutinho C, Davalos V, Spizzo R, Ivan C, Rossi S, Setien F, Casanovas O, Simo-Riudalbas L, Carmona J, Carrere J, Vidal A, Aytes A, Puertas S, Ropero S, Kalluri R, Croce CM, Calin GA, Esteller M. Small molecule enoxacin is a cancer-specific growth inhibitor that acts by enhancing TAR RNA-binding protein 2-mediated microRNA processing. Proc Nat Acad Sci. 2011;108:4394–9. https://doi.org/10.1073/pnas.1014720108.
Sousa EJ, Graça I, Baptista T, Vieira FQ, Palmeira C, Henrique R, Jerónimo C. Enoxacin inhibits growth of prostate cancer cells and effectively restores microRNA processing. Epigenetics. 2013;8:548–58. https://doi.org/10.3390/molecules24081580.
Valianatos G, Valcikova B, Growkova K, Verlande A, Mlcochova J, Radova L, Stetkova M, Vyhnakova M, Slaby O, Uldrijan S. A small molecule drug promoting miRNA processing induces alternative splicing of MdmX transcript and rescues p53 activity in human cancer cells overexpressing MdmX protein. PLoS One . 2017;12:e0185801. https://doi.org/10.1371/journal.pone.0185801.
Bicker S, Schratt G. MicroRNAs in ALS: small pieces to the puzzle. EMBO J. 2015;34:2601–3. https://doi.org/10.15252/embj.201592805.
Emde A, Eitan C, Liou LL, Libby RT, Rivkin N, Magen I, Reichenstein I, Oppenheim H, Eilam R, Siverstorm A, Alajajian B, Ben-Dov IZ, Aebischer J, Savidor A, Levin Y, Sons R, Hammond SM, Ravits JM, Möller T, Hornstein E. Dysregulated mi RNA biogenesis downstream of cellular stress and ALS-causing mutations: a new mechanism for ALS. EMBO J. 2015;34:2633–51. https://doi.org/10.15252/embj.201490493.
Rizzuti M, Filosa G, Melzi V, Calandriello L, Dioni L, Bollati V, Bresolin N, Comi GP, Barabino S, Nizzardo M, Corti S. MicroRNA expression analysis identifies a subset of downregulated miRNAs in ALS motor neuron progenitors. Sci Rep. 2018;8:1–12. https://doi.org/10.1038/s41598-018-28366-1.
Smalheiser NR, Zhang H, Dwivedi Y. Enoxacin elevates microRNA levels in rat frontal cortex and prevents learned helplessness. Front Psychiatry. 2014;5:6. https://doi.org/10.3389/fpsyt.2014.00006.
Harmon FG, DiGate RJ, Kowalczykowski SC. RecQ helicase and topoisomerase III comprise a novel DNA strand passage function: a conserved mechanism for control of DNA recombination. Mol Cell. 1999;3:611–20. https://doi.org/10.1016/S1097-2765(00)80354-8.
Plank J, Hsieh TS. Helicase-appended topoisomerases: new insight into the mechanism of directional strand transfer. J Biol Chem. 2009;284:30737–41. https://doi.org/10.1074/jbc.R109.051268.
Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nature Rev Mol Cell Biol. 2002;3:430–40. https://doi.org/10.1038/nrm831.
Croteau DL, Popuri V, Opresko PL, Bohr VA. Human RecQ helicases in DNA repair, recombination, and replication. Ann Rev Biochem. 2014;83:519–52. https://doi.org/10.1146/annurev-biochem-060713-035428.
Laursen LV, Bjergbaek L, Murray JM, Andersen AH. RecQ helicases and topoisomerase III in cancer and aging. Biogerontology. 2003;4:275–87. https://doi.org/10.1023/a:1026218513772.
Simon N, Bochman ML, Seguin S, Brodsky JL, Seibel WL, Schwacha A. Ciprofloxacin is an inhibitor of the Mcm2-7 replicative helicase. Biosci Rep. 2013;33:e00072. https://doi.org/10.1042/BSR20130083.
Jankowsky E. RNA helicases at work: binding and rearranging. Trends Biochem Sci. 2011;36:19–29. https://doi.org/10.1016/j.tibs.2010.07.008.
Cao S, Sun R, Wang W, Zhang Y, Zhang N, Yang X. RNA helicase DHX9 may be a therapeutic target in lung cancer and inhibited by enoxacin. Am J Transl Res. 2017;9:674–82 ((PMID: 28337295)).
Badal S, Her YF, Maher LJ. Nonantibiotic effects of fluoroquinols in mammalian cells. J Biol Chem. 2015;290:22287–97. https://doi.org/10.1074/jbc.M115.671222.
Miro-Canturri A, Averbe-Algaba R, Smani Y. Drug repurposing for the treatment of bacterial and fungal infections. Front Mircobiol. 2019;10:41. https://doi.org/10.3389/fmicb.2019.00041.
Fedorowicz J, Sączewski J. Modifications of quinols and fluoroquinols: hybrid compounds and dual-action molecules. Monatsh Chem. 2018;149:1199–245. https://doi.org/10.1007/s00706-018-2215-x.
Azam A, Peerzada MN, Ahmad K. Parasitic diarrheal disease: drug development and targets. Front Microbiol. 2015;6:1183. https://doi.org/10.3389/fmicb.2015.01183.
AbouLaila M, Munkhjargal T, Sivakumar T, Ueno A, Nakano Y, Yokoyama M, Yoshinari T, Nagano D, Katayama K, El-Bahy N, Yokoyama N, Igarashi I. Apicoplast-targeting antibacterials inhibit the growth of Babesia parasites. Antimicrob Agents Chemother. 2012;56:3196–206. https://doi.org/10.1128/AAC.05488-11.
Rizk MA, AbouLaila M, El-Sayed SAES, Guswanto A, Yokoyama N, Igarashi I. Inhibitory effects of fluoroquinol antibiotics on Babesia divergens and Babesia microti, blood parasites of veterinary and zoonotic importance. Infect Drug Res. 2018;11:1605–15. https://doi.org/10.2147/IDR.S159519.
Williamson DH, Preiser PR, Wilson RJM. Organelle DNAs: the bit players in malaria parasite DNA replication. Parasitol Today. 1996;12:357–62. https://doi.org/10.1016/0169-4758(96)10053-3.
Mahmoudi N, Ciceron L, Franetich JF, Farhati K, Silvie O, Eling W, Sauerwein R, Danis M, Mazier D, Derouin F. In vitro activities of 25 quinols and fluoroquinols against liver and blood stage Plasmodium spp. Antimicrob Agents Chemother. 2003;47:2636–9. https://doi.org/10.1128/AAC.47.8.2636-2639.2003.
Garcia-Estrada C, Prada CF, Rojo-vazquez F, Balana-Fouce R. DNA topoisomerases in apicomplexan parasites: promising targets for drug discovery. Proc R Soc B. 2010;277:1777–87. https://doi.org/10.1098/rspb.2009.2176.
Pura RS. Anticoccidial drugs used in the poultry: an overview. Sci Int. 2013;1:261–5. https://doi.org/10.3923/sciintl.2013.261.265.
Zuma AA, Cavalcanti DP, Maia MC, de Souza W, Motta MCM. Effect of topoisomerase inhibitors and DNA-binding drugs on the cell proliferation and ultrastructure of Trypanosoma cruzi. Int J Antimicrob Agents. 2011;37:449–56. https://doi.org/10.1016/j.ijantimicag.2010.11.031.
Romero IC, Saravia NG, Walker J. Selective action of fluoroquinols against intracellular amastigotes of Leishmania (Viannia) panamensis in vitro. J Parasitol. 2005;91:1474–80. https://doi.org/10.1645/ge-3489.1.
Sousa MC, Poiares-da-Silva J. The cytotoxic effects of ciprofloxacin in Giardia lamblia trophozoites. Toxicol in vitro. 2001;15:297–301. https://doi.org/10.1016/S0887-2333(01)00026-1.
Didier ES, Bowers L, Stovall ME, Kuebler D. Antimicrosporidial activity of (fluoro) quinols in vitro and in vivo. Folia Parasit. 2005;52:173–81. https://doi.org/10.14411/fp.2005.022.
McFadden GI. The apicoplast. Protoplasma. 2011;248:641–50. https://doi.org/10.1007/s00709-010-0250-5.
McFadden GI. Apicoplast. Curr Biol. 2014;24:R262–3. https://doi.org/10.1016/j.cub.2014.01.024).
Sato S. The apicomplexan plastid and its evolution. Cell Mol Life Sci. 2011;68:1285–96. https://doi.org/10.1007/s00018-011-0646-1.
Bell BG, Schellevis F, Stobberingh E, Goossens H, Pringle M (2014) A systematic review and meta-analysis of the effects of antibiotic consumption on antibiotic resistance. BMC infectious diseases 14:13 https://www.biomedcentral.com/1471-2334/14/13
Costelloe C, Metcalfe C, Lovering A, Mant D, Hay AD. Effect of antibiotic prescribing in primary care on antimicrobial resistance in individual patients: systematic review and meta-analysis. BMJ. 2010;340:c2096. https://doi.org/10.1136/bmj.c2096.
Zhen X, Stålsby-Lundborg C, Sun X, Hu X, Dong H. The clinical and economic impact of antibiotic resistance in China: a systematic review and meta-analysis. Antibiotics. 2019;8:115. https://doi.org/10.3390/antibiotics8030115.
Goosens H, Ferech M, Stichele RV, Elseviers M. Outpatient use in Europe and association with resistance: a cross-national database study. Lancet. 2005;365:579–87. https://doi.org/10.1016/S0140-6736(05)17907-0.
Goossens H. Antibiotic consumption and link to resistance. Clin Microbiol Infect. 2009;15:12–5. https://doi.org/10.1111/j.1469-0691.2009.02725.x.
Garrett JPD, Margolis DJ. Impact of long-term antibiotic use for acne on bacterial ecology and health outcomes: a review of observational studies. Curr Derm Rep. 2012;1:23–8. https://doi.org/10.1007/s13671-011-0001-7.
Anonymos. 2020. Longitude Prize. Effectiveness of cancer treatments threatened by rising antibiotic resistance. https://longitudeprize.org/sites/longitude/files/content/attachments/2020-02-17/Longitude%20Prize%20Report_EFFECTIVENESS%20OF%20CANCER%20TREATMENTS%20THREATENED%20BY%20RISING%20ANTIBIOTIC%20RESISTANCE_FINAL.pdf. Accessed May 29, 2020.
Pacheu-Grau D, Gómez-Durán A, López-Pérez MJ, Montoya J, Ruiz-Pesini E. Mitochondrial pharmacogenomics: barcode for antibiotic therapy. Drug Discov Today. 2010;15:33–9. https://doi.org/10.1016/j.drudis.2009.10.008.
Zhang J, Haines C, Watson AJ, Hart AR, Platt MJ, Pardoll DM, Cosgrove SE, Gebo KA, Sears CL. Oral antibiotic use and risk of colorectal cancer in the United Kingdom, 1989–2012: a matched case–control study. Gut. 2019;68:1971–8. https://doi.org/10.1136/gutjnl-2019-318593.
Elliott RL, Jiang XP, Baucom CC. Antibiotic overusage causes mitochondrial dysfunction which may promote tumorigenesis. J Cancer Treatm Res. 2017;5:62–5. https://doi.org/10.11648/j.jctr.20170504.11.
Elliott RL, Jiang XP, Baucom C, Lomnicka Z (2018) Antibiotics friend and foe: “From wonder drug to causing mitochondrial dysfunction, disrupting human microbiome and promoting tumorigenesis”. Int J Clin Med 9: 182–186. https://www.scirp.org/journal/ijcm
Andrade MJ, Jayaprakash C, Bhat S, Evangelatos N, Brand A, Satyamoorthy K. Antibiotics-induced obesity: a mitochondrial perspective. Public Health Genomics. 2017;20:257–73. https://doi.org/10.1159/000485095.
Stefano G, Sanuel J, Kream RM. Antibiotics may trigger mitochondrial dysfunction inducing psychiatric disorders. Med Sci Monit. 2017;23:101–6. https://doi.org/10.12659/MSM.899478.
Obregon D, Parker-Athill EC, Tan J, Murphy T. Psychotropic effects of antimicrobials and immune modulation by psychotropics: implications for neuroimmune disorders. Neuropsychiatry. 2012;2:331–43. https://doi.org/10.2217/npy.12.41.
Elliott RL, Jiang X-P. The adverse effect of gentamicin on cell metabolism in three cultured mammary cell lines: “Are cell culture data skewed?” PLoS One . 2019;14:e0214586. https://doi.org/10.1371/journal.pone.0214586.
Chemaly RF, Hanmod SS, Jiang Y, Rathod DB, Mulanovich V, Adachi JA, Rolston KV, Raad II, HAchem RY. Tigecycline use in cancer patients with serious infections: a report on 110 cases from a single institution. Medicine. 2009;88:211–20. https://doi.org/10.1097/MD.0b013e3181af01fc.
Arora R, Jain S, Rahimi H. Evaluating the efficacy of Tigecycline to target multiple cancer-types: a review. STEM Fellowsh J. 2019;4:5–11. https://doi.org/10.17975/sfj-2018-002.
Bucaneve G, Micozzi A, Picardi M, Ballanti S, Cascavilla N, Salutari P, Specchia G, Fanci R, Luppi M, Cudillo L, Cantaffa R, Mil G, Bocchia M, Martinelli G, Offidani M, Chierchini A, Fabbiano F, Quarta G, Priomon V, Manna A, Zuffa E, Ferrari A, Gentile G, Foa R, Del Favero A. Results of a multicenter, controlled, randomized clinical trial evaluating the combination of piperacillin/tazobactam and tigecycline in high-risk hematologic patients with cancer with febrile neutropenia. J Clin Oncol. 2014;32:1463–71. https://doi.org/10.1200/JCO.2013.51.6963.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares that there is no conflict of interest.
Additional information
A companion paper entitled “Are antibacterial effects of non-antibiotic drugs random or purposeful because of a common evolutionary origin of bacterial and mammalian targets?” describes effects of non-antibiotic drugs on bacteria.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dalhoff, A. Selective toxicity of antibacterial agents—still a valid concept or do we miss chances and ignore risks?. Infection 49, 29–56 (2021). https://doi.org/10.1007/s15010-020-01536-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s15010-020-01536-y