
Abstract
Localized surface plasmons can modify linear optical responses of materials located in their vicinity. In particular, rare earth ions luminescence can be enhanced by gold nanoparticles. Luminescence exaltation is a complex phenomenon that depends on multiple parameters, a critical one being the coupling distance between the emitting species and the plasmonic core. An original multilayer nanostructure designed to precisely control the distance between the gold cores and the luminescent ions, and to study its effect on the luminescent properties is presented here. Homogeneous silica shells with controlled thicknesses adjustable from 2 to 50 nm and rare earth ion doping rates up to 2 × 1020 Eu/cm3 of silica were deposited onto gold nanospheres. These original nanostructures are then incorporated into densified sol–gel silica composites with high-optical quality. Luminescence properties are studied for increasing gold–europium (III) distances. Strong luminescence quenching is evidenced for coupling distances up to 28 nm.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Gold nanoparticles interact with light and exhibit strong and tunable surface plasmon resonance (SPR). These localized surface plasmons originating from gold nanoparticles in various media can enhance linear and non linear optical responses of materials located in their vicinity, mainly due to electromagnetic field enhancement. Recent studies have highlighted the possible coupling between gold nanostructures and quantum dots [1], organic fluorophores [2, 3], silicon carbide [4], or lanthanide ions [5].
In the case of lanthanide ions, the quantum yield, which is the probability for a photon to be emitted after one photon has been absorbed, is close to 1 [6]. However, the excitation process efficiency is limited by their weak absorption cross sections, due to parity-forbidden transitions. This is usually overcome by energy transfer from sensitizer ions [7, 8], ligands [7, 9], or host matrix [10], but absorption exaltation through coupling to plasmonic nanostructures could yield to even better sensibilization [5, 11].
Efficient coupling requires the surface plasmon resonance and the quantum emitter absorption band to be correctly matched in wavelength [12]. It also highly depends on the distance between the emitter and the gold core. For short distances around 1 nm, the luminescence is mainly quenched due to non-radiative decays [13, 14]. However, the radiative decay rate may be amplified by several orders of magnitude [4] for theoretical spacing distances predicted around 10 nm [15]. This optimum coupling distance has to be determined for each sensitizer/emitter couple.
Most of the exaltation studies were conducted on planar configurations [6, 16]. Pillonnet et al. [6] demonstrated limited exaltation factors around three for Eu (III) ions coupled to silver nanoparticles, with an optimum coupling distance around 15 nm. Recently, three-dimensional nanoparticles systems, coupling metallic nanoparticles with either lanthanide complexes [17] or fluorophores [18], revealed strong luminescence quenching for spacing distances up to 40 nm, showing the more complex behavior of non planar nanostructures. The nanoparticle approach, based on versatile synthetics procedures [3, 19, 20], allows for a fine control of each synthetic parameter in a wide range, and opens new applications in the field of optics. More specifically, rare earth ion incorporation into silica matrices could bring new developments for optical amplifiers [21].
In this work, Au@SiO2@SiO2:Eu(III) core-shell nanoparticles, exhibiting adequate absorption and emission spectral overlap, are synthesized via a sol–gel process allowing fine control of coupling distances and doping rates. These multilayer nanostructures are intended to the study of luminescence exaltation via plasmon-induced local field enhancement. In addition, for a better understanding of the plasmon effect, reference nanoparticles are prepared by gold core dissolution on the final multilayer nanostructures. Europium (III) photoluminescence intensities, excitation spectra, and lifetime decay profiles were analyzed to study the plasmon-induced luminescence properties.
Experimental details
Au@SiO2@SiO2:Eu(III) multilayer nanostructures, as illustrated on Fig. 1, are synthesized by the multi-step process described below. They are then incorporated into reference silica matrices for optical measurements.

Multilayer nanostructures; NP metallic nanoparticle
Reagents
All chemicals were purchased from Sigma-Aldrich and used as received; hydrogen tetrachloroaurate (III) trihydrate (99.9 %), 3-mercaptopropyltrimethoxysilane (MPS, 95 %), sodium silicate solution (27 wt.-% SiO2), europium (III) chloride hexahydrate (99.9 %), ammonia (28 wt.-%).
Au@SiO2 synthesis
Spherical gold nanoparticles (NP) 50 nm in diameter were obtained by the well-known Türkevich method [22–24]. The citrate/gold molar ratio, that controls the nanospheres diameter, was set to 1. 50 mL of a 2.5 mmol L−1 HAuCl4 aqueous solution were added to 400 mL of boiling Milli-Q water. Once the boiling started again, 50 mL of a 2.5 mmol L−1 sodium citrate aqueous solution were quickly added under vigorous magnetic stirring. The heating and stirring were extended for 5 more minutes after the suspension turned violet-red.
Multiple silica shells were then successively deposited on the gold NP through optimization of the Liz-Marzan process [19, 25]. Two hundred microliters of 1 mol L−1 MPS solution was first added to 250 mL of the gold NP solution, corresponding to 60 % of total surface coverage based on 40-Å2 coverage per MPS molecule. The suspension was magnetically stirred for 15 min to allow for surface functionalization. Then, 10 mL of 0.34 wt.-% (77.3 mmol L−1) sodium silicate solution, previously activated at pH = 10.4 with Amberlite IRN77 cation exchange resin, were added to the mixture. The suspension was maintained 5 days at 40 °C in a stove.
At this stage, the silica-coated nanoparticles are stable into water/ethanol mixtures. Homogeneous 2-nm thick silica shells are obtained, suggesting an uncompleted silica precursor heterocondensation. Indeed, upon ammonia addition in the suspension, a 25-nm homogeneous silica shell is obtained by condensation of excess unreacted silicates.
To grow silica shells with tailored intermediate thicknesses, the 2-nm silica-coated particles are first washed with same pH water (pH ~9) to remove unreacted silicates and concentrated up to 1.8 × 1014 NP/L using an ultrafiltration 30-kDa membrane. The suspension is then diluted in four volumes of ethanol. Controlled amounts of 0.02 mol L−1 tetraethyl orthosilicate (TEOS) and 40 μL of ammonia per milliliter of suspension are successively added under vigorous magnetic stirring. The required TEOS amount can be predicted according to the targeted shell thickness.
Au@SiO2@SiO2:Eu(III) synthesis
Lanthanide (Ln (III)) doping of the shells is obtained following the same procedure as for pure silica shells, except for the addition of a defined volume of 1 mmol L−1 lanthanide trichloride solution into the mixture just before ammonia. The suspension is magnetically stirred during 24 h before the next doped silica layer coating step. Typically, the addition of 420 μL of TEOS solution, 42 μL of Eu (III) solution and 800 μL of 28 wt.-% ammonia into 20 mL of suspension gives 12-nm silica shells doped with 1020 Eu/cm3 of silica.
When the desired multilayer nanostructure is reached, the suspension is washed with water/ethanol mixture (1:4 v/v) by 3 cycles of 30-min centrifugation at 29,220g.
Gold core dissolution for reference nanoparticles synthesis
The gold cores were dissolved via a cyanide-etching process adapted from [26]. Typically, 1 mL of core-shell suspension was diluted into 29 mL of water/ethanol mixture and 755 μL of 0.1 mol L−1 KCN were added (CN/Au = 50). The dissolution was carried on for 5 days under magnetic stirring. The suspension was then washed with water/ethanol mixture by 3 cycles of 30-min centrifugation at 29,220g.
Incorporation into silica matrices
The nanostructures were incorporated into sol–gel silica monoliths to study the luminescence properties of the samples. The condensation process was optimized to minimize organic and silanols residuals into the final densified silica.
Equimolar TEOS to triethoxysilane (TREOS) mixtures [27] were diluted into ethanol (TEOS/TREOS/EtOH molar ratios are 1:1:8.6) and prehydrolyzed with HCl at pH = 1 (25 μL for 1.82 mmol of TEOS) during 2 h under vigorous magnetic stirring. Core-shell suspension and water were then successively added and the mixture was kept stirring for 1 h to ensure sol homogeneity. Then, 0.75 mL of the sol was cast onto a 5-mL polypropylene vessel with pierced lid. After 5 days of condensation at room temperature, the gel is dried for 5 more days in a 40 °C stove. Thermal densification is then performed in a furnace up to 600 °C for 12 h.
Densified silica monoliths (1 cm2 × 1 mm) with good optical quality are thus obtained. For the sake of reproducibility in luminescence emission measurements, the monoliths are then grinded into fine powder.
Characterizations
UV-visible absorption spectra were recorded from 400 to 800 nm with 1-nm steps on a SECOMAM UVIKON XL spectrophotometer.
Transmission electron microscopy (TEM) grids were prepared by evaporating one drop of core-shell suspension onto a carbon-coated copper TEM grid. The samples were then characterized with a Tecnai G2 Spirit apparatus operating at 120 kV. Particle size distributions were evaluated from 200 nanoparticles.
Lifetime measurements were conducted directly on the monoliths. Photoluminescence (PL) emission spectra were measured on grinded monoliths. A tunable optical parametric oscillator pumped with the third harmonic of a Nd:YAG Q-switched laser (Ekspla NT-342B-SH, 10-Hz repetition frequency, 6-ns pulse width) was used as excitation source. The luminescence was analyzed with a Jobin Yvon HR250 monochromator coupled to a Roper ICCD camera. With the ICCD camera, integrated and delay times can be adjusted to the emission ion. For europium (III), a typical set-up is 5-ms integrated time and 5-μs delay time. Lifetime measurements were performed during a total time of 7 ms with 1-μs increment in the delay time.
Materials description
Metallic core
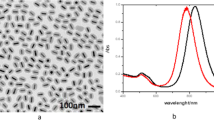
Gold salt reduction with citrate ions in water yields a stable suspension of roughly spherical gold nanospheres with a diameter of 49.3 ± 5.2 nm (see Fig. 2 and size distribution in Online Resource 1). The monodispersity level reached is not perfect but good enough for the targeted application. A diameter of 50 nm is estimated to be a good compromise between the enhancement of local electromagnetic field by larger nanoparticles and the problem of light diffusion when particles size is too large [15].

Transmission electron micrograph of the 50-nm roughly spherical gold nanoparticles
The gold nanosphere suspension exhibits a surface plasmon resonance extinction spectrum (Fig. 3b), characterized by a strong absorption band in the 450–600-nm spectral range and a maximum of 535 nm, as evidenced by the red-wine color of the suspension. The europium (III) excitation and emission spectra are reported in Fig. 3. The 7F0 → 5D1 absorption band is perfectly matched with the SPR maximum, thus allowing coupling at absorption. The 7F0 → 5D2 absorption band is shifted towards shorter wavelengths compared to the gold surface plasmon resonance, enabling non resonant excitation at 465 nm.

Intensities of a) europium (III) excitation spectra recorded at 615 nm; b) gold nanospheres suspension extinction spectra; and c) europium (III) emission spectra for excitation at 532 nm. For comparison sake, intensities are normalized to 1
As for the 5D0 → 7F2 europium (III) emission band, the 80-nm red-shift compared to the plasmon maximum is larger than the usual exaltation studies conducted with organic dyes [2].
Pure silica buffer layers with controlled thicknesses
The vitreophobic surface of gold nanoparticles is first functionalized with MPS to promote silica heterogeneous condensation [19]. A 60 % theoretical coverage of the NP surface is required to allow further coating, while excess MPS molecules would induce gold nanospheres aggregation, eventually leading to precipitation.
By condensation of sodium silicates, silica shells as thin as 2 nm (see Fig. 4a1, a2) are obtained in water. All the gold cores are totally coated. This coating step does not impact the UV-visible absorption spectra (not shown), indicating the formation of a very thin silica layer and a good nanoparticle dispersion. This will be further confirmed by the next coating steps, leading to only a very few multi-core nanostructures.

Transmission electron micrographs of Au@SiO2 nanostructures coated with a 2 nm, b 10 nm, and c 35-nm pure silica shells. d Nanostructures with 25-nm pure silica shells after etching with cyanide ions for 5 days
The 2-nm silica-coated nanoparticles are stable into water/ethanol 1:4 v/v mixtures. To grow silica shells with tailored intermediate thicknesses, removal of excess unreacted silicates is needed; otherwise, silica condensation preferently occurs on small gold-free pure silica nuclei. Washing was performed by ultrafiltration to avoid any aggregation that may occur during centrifugation or dialysis [19].
Pure silica shell growth is performed through ammonia-catalyzed sol–gel condensation [28]. Ammonia concentration has to be controlled to develop a coating process compatible with potential further doping with lanthanide ions. In consequence, concentrated gold suspensions were used. Ultrafiltration allows concentration of the sols up to 6 × 1014 NP/L, but the suspension stability towards aggregation is then strongly affected. The optimum concentration was determined to be 1.8 × 1014 NP/L. In these conditions, the optimum ammonia concentration was 0.569 mol/L.
With this procedure, pure silica shells with controlled thicknesses ranging from 10 to 35 nm (see Fig. 4b1–2, c1–2) are obtained in one single TEOS addition step. To grow even thicker shells, multiple TEOS addition steps can be performed. As illustrated in Fig. 4, the silica shells are very homogeneous and only a few (<1 %) multi-core nanostructures are obtained, confirming the high stability of this colloidal suspension at each step.
Despite the washing step, some core-free pure silica nanoparticles are systematically formed. This leads to a bias of about 5 nm between the predicted and observed silica shell thicknesses. These core-free particles will also contribute to the luminescence baseline, not affected by the plasmon, when doped with europium ions.
As the silica shell thickened, the SPR is slightly red-shifted (see Online Resource 2). This is correlated to an increase in the local refractive index around the gold core [19]. The maximum shift observed in our samples for 35-nm silica shells was 8 nm. This still allows good coupling in absorption with the 7F0 → 5D1 europium (III) absorption band presented in Fig. 3.
Silica shells doped with lanthanide ions
Once the gold nanoparticles have been coated with a first 2-nm pure silica shell, any of the subsequent shells can be doped with lanthanide (III) ions, even prior to washing of excess unreacted silicates.
Ammonia-catalyzed condensation occurring in basic conditions, electrostatic interactions can favor lanthanide (III) incorporation into the condensing negatively charged silica network. For europium (III) ions, the maximum accessible doping rate was determined to be 2 × 1020 Eu/cm3 of silica. For higher doping rates, the silica network condensation is highly disturbed (see Online Resource 3). Bright-contrasted nuclei, attributed to europium hydroxides [29], appear and promote the formation of a high number of core-free pure silica nanoparticles, leading to silica shell thicknesses being significantly lower than the predicted ones. For rare earth concentration in the shell larger than 1 × 1017 Eu/cm3 of silica, clustering of the Eu(III) ions may occur but its consequences on the luminescence properties is compensated by the use of the appropriate reference samples, as described below.
Due to the low-doping rates in the final nanostructures and silica matrices, no quantitative measurement by chemical analysis could confirm the luminescent ion incorporation efficiency. The doping is, however, confirmed by the luminescence studies presented below.
Gold core dissolution for reference nanoparticles
In-depth study of the possibility to enhance lanthanide ions emission through plasmonic exaltation, requires reference samples to study the gold core effect on optical properties. Two main strategies emerge from the literature: the use of very thick spacer layers to get rid of any coupling [13, 17], or the removal of the plasmonic part [3, 6]. Due to the potential long-range coupling with gold [17, 18], the latter was favored in this study and achieved by dissolution of the plasmonic core.
Gold core dissolution can be achieved by etching with either cyanide ions [26, 30] or aqua regia [31]. Aqua regia, being a highly acidic medium, might promote lanthanide (III) ions release from the silica network. Dissolution through cyanide ions complexation was therefore applied. It can be summarized by the following equation:
Complete gold core dissolution proceeds by inward diffusion of cyanide ions and outward diffusion of gold-cyanide complexes through the all silica shells thickness [26]. This process is diffusion-limited and requires a few reaction days. Potassium counter-ions are known as silica network modifiers. They can lead to partial silica shell dissolution. Moreover, during the dissolution reaction, released HO− anions induce a small increase in pH that may also promote silica dissolution. An optimum between the dissolution rate and the silica integrity was experimentally found for a cyanide/gold molar ratio of 50. After 5 days of etching, more than 90 % of the gold cores are dissolved, as proved by the decrease in SPR intensity (see Online Resource 4).
Typical multilayer nanostructures after etching with cyanide ions are presented in Fig. 4d1–2. Silica shells' thicknesses and homogeneity are preserved. In some cases, the external part of the shells is partially dissolved during etching [30], leading to a shrinking of the shell and a partial release of lanthanide ions. To get perfect reference nanoparticles, the cyanide to gold molar ratio should be optimized for each shell thickness.
Composites silica matrices
After incorporation of the nanostructures into optical quality-densified silica monoliths involving a heating treatment at 600 °C under air, the SPR extinction spectrum exhibits the same shape as the gold suspensions, indicating that the nanostructures are well dispersed inside the matrix.
Luminescence properties
Luminescence excitation measurements conducted for gold-loaded and dissolved nanostructures with only 2-nm pure silica buffer layer and 25 nm of Eu (III)-doped silica are presented in Fig. 5. The excitation spectra is not modified by the presence of the gold core compared to europium (III) ions dispersed in a pure silica matrix. A strong decrease in the emission intensity, by about 50 %, is observed for the gold-loaded sample. Surprisingly, the europium (III) emission is quenched for excitation wavelengths in transitions both resonant (532 nm) and non resonant (465 nm) with the gold SPR.

a Excitation spectra recorded at 615 nm and b–c decay curves for excitation at b 465 nm and c 532 nm for either gold-loaded samples (gray closed squares) or gold-dissolved references (black closed triangles) with 2-nm pure silica buffer layer and 25 nm of Eu (III)-doped silica
This gold-induced luminescence quenching is further confirmed by lifetime measurements. Luminescence decays were found to be exponential in both cases. Dissolved nanostructures systematically exhibit a longer lifetime than gold-loaded ones, arising from the disappearance of the plasmon-induced non-radiative decay channel. Lifetime values are reduced by ~10 % in these strong quenching conditions. Such weak lifetime variations have already been reported [3, 17].
This strong luminescence quenching for very short coupling distances is in good agreement with theoretical predictions [14]. Quenching may come from luminescence re-absorption by the gold nanoparticles [13, 32], especially for non resonant excitation. Figure 3 shows that the SPR is still quite intense at the europium (III) emission wavelength, and this is amplified by the difference in oscillator strengths [33, 34].
Figure 6 shows photoluminescence intensities recorded under excitation at 532 nm for samples with increasing buffer layer thicknesses. For this study, after pure silica layers condensation, the same amounts of TEOS and europium (III) salt were added to all the samples. The shells are doped with 1020 Eu/cm3 of silica and have different thicknesses ranging from 20 nm for the sample with 2-nm buffer layer, to 9 nm for the sample with 28-nm buffer layer.

Integrated PL intensities (excitation at 532 nm) for either gold-loaded samples (gray closed squares) or gold-dissolved references (black closed triangles) with increasing pure silica buffer thicknesses and identical Eu (III)-doping. The best reference, defined in the text, is identified as a dotted line. Error bars are estimated from three measurements
Reference samples gave unexpectedly different signal intensities depending on the buffer layer thickness. This may be explained by a partial dissolution of the external silica shell, releasing some luminescent ions; for similar etching conditions, as the final silica thickness increases, one can predict that a higher percentage of europium (III) ions are released, leading to photoluminescence decrease. The best reference sample would thus be the one with the thinner buffer layer and the higher luminescence intensity.
Compared to this best reference, identified as a dotted line on Fig. 6, luminescence quenching is observed for all studied buffer thicknesses. The quenching intensity diminishes as the buffer thickness increases: 70 % at 2 nm and 46 % at 28 nm. Same behavior is observed for excitation at 465 nm. Quenching at such long distances is not predicted theoretically [14] but has been sometimes observed experimentally [17, 18]. It is usually attributed to non-radiative energy transfer towards gold nanoparticles, but a possible modification of the efficiency to populate the Eu(III) 5D0 emitting level has also been reported [17]. No clear lifetime difference was evidenced among the samples, but because of the low-doping rates, the luminescence decays (not shown) signals are very weak.
Once again, we believe that this unexpected quenching at long distance may come from luminescence re-absorption by the gold cores. Compared to oriented planar configurations, systems reported here favor interactions between gold nanoparticles and rare earth ions. Indeed, in bulk samples, europium (III) emission has to come out through the whole sample thickness. Interactions with multiple gold nanoparticles with large absorption cross sections may occur, especially as emission bands partially overlap with gold SPR.
Conclusion
Multilayer nanostructures made of a spherical 50-nm gold core coated with silica shells, either pure or europium (III)-doped, were successfully prepared. Optical reference nanoparticles were obtained by gentle gold core dissolution with cyanide ions. A systematic study of the effect of the coupling distance between the gold core and the luminescent ions was conducted. Strong quenching is evidenced for coupling distances up to 28 nm, the quenching intensity increasing as the pure silica buffer layer diminishes.
This quenching is attributed to a re-absorption of the emitted luminescence by the gold cores. Emitting species exhibiting larger red-shifts compared to the SPR wavelength could probably help overcome this problem [35]. In this particular study, no efficient coupling is achieved between the metallic core and the luminescent ions. This may arise from the thickness of the doped shells; only a very small portion of the luminescent ions is actually in an optimal coupling configuration. Anisotropic nanoparticles, such as gold nanorods [36–38], could also yield more efficient coupling [35].
References
Naiki H, Masuhara A, Masuo S et al (2013) Highly controlled plasmonic emission enhancement from metal–semiconductor quantum dot complex nanostructures. J Phys Chem C 117:2455. doi:10.1021/jp305408p
Chen H, Ming T, Zhao L et al (2010) Plasmon–molecule interactions. Nano Today 5:494. doi:10.1016/j.nantod.2010.08.009
Schneider G, Decher G, Nerambourg N et al (2006) Distance-dependent fluorescence quenching on gold nanoparticles ensheathed with layer-by-layer assembled polyelectrolytes. Nano Lett 6:530. doi:10.1021/nl052441s
Sui N, Monnier V, Zakharko Y et al (2012) Plasmon-controlled narrower and blue-shifted fluorescence emission in (Au@SiO2) SiC nanohybrids. J Nanopart Res 14:1004. doi:10.1007/s11051-012-1004-4
Fukushima M, Managaki N, Fujii M et al (2005) Enhancement of 1.54-μm emission from Er-doped sol–gel SiO2 films by Au nanoparticles doping. J Appl Phys 98:024316
Pillonnet A, Berthelot A, Pereira A et al (2012) Coupling distance between Eu3+ emitters and Ag nanoparticles. Appl Phys Lett 100:153115. doi:10.1063/1.3703120
Binnemans K (2009) Lanthanide-based luminescent hybrid materials. Chem Rev 109:4283. doi:10.1021/cr8003983
Eliseeva SV, Bünzli J-CG (2011) Rare earths: jewels for functional materials of the future. New J Chem 35:1165. doi:10.1039/c0nj00969e
Dossing A (2005) Luminescence from lanthanide(3+) ions in solution. Eur J Inorg Chem 8:1425. doi:10.1002/ejic.200401043
Huignard A, Gacoin T, Boilot J-P (2000) Synthesis and luminescence properties of colloidal YVO4:Eu phosphors. Chem Mater 12:1090. doi:10.1021/cm990722t
Chiasera A, Ferrari M, Mattarelli M et al (2005) Assessment of spectroscopic properties of erbium ions in a soda-lime silicate glass after silver–sodium exchange. Opt Mater 27:1743. doi:10.1016/j.optmat.2004.11.044
Thomas M, Greffet J-J, Carminati R, Arias-Gonzalez JR (2004) Single-molecule spontaneous emission close to absorbing nanostructures. Appl Phys Lett 85:3863. doi:10.1063/1.1812592
Dulkeith E, Morteani A, Niedereichholz T et al (2002) Fluorescence quenching of dye molecules near gold nanoparticles: radiative and nonradiative effects. Phys Rev Lett 89:12. doi:10.1103/PhysRevLett.89.203002
Anger P, Bharadwaj P, Novotny L (2006) Enhancement and quenching of single-molecule fluorescence. Phys Rev Lett 96:3. doi:10.1103/PhysRevLett.96.113002
Colas des Francs G, Bouhelier A, Finot E et al (2008) Fluorescence relaxation in the near-field of a mesoscopic metallic particle: distance dependence and role of plasmon modes. Opt Express 16:17654
Kalkman J, Kuipers L, Polman A, Gersen H (2005) Coupling of Er ions to surface plasmons on Ag. Appl Phys Lett 86:041113. doi:10.1063/1.1856133
Lin C, Berry MT, Stanley May P (2010) Influence of colloidal-gold films on the luminescence of Eu(TTFA)3 in PMMA. J Lumin 130:1907. doi:10.1016/j.jlumin.2010.05.005
Huang Y-F, Ma K-H, Kang K-B et al (2013) Core–shell plasmonic nanostructures to fine-tune long “Au nanoparticle-fluorophore” distance and radiative dynamics. Colloids Surf, A 421:101. doi:10.1016/j.colsurfa.2012.12.050
Liz-Marzán LM, Giersig M, Mulvaney P (1996) Synthesis of nanosized gold-silica core-shell particles. Langmuir 12:4329. doi:10.1021/la9601871
Bahadur NM, Furusawa T, Sato M et al (2011) Fast and facile synthesis of silica coated silver nanoparticles by microwave irradiation. J Colloid Interface Sci 355:312. doi:10.1016/j.jcis.2010.12.016
Pastouret A, Gonnet C, Collet C, et al. (2009) Nanoparticle doping process for improved fibre amplifiers and lasers. Proc of Spie. doi: 10.1117/12.808125
Frens G (1973) Controlled nucleation for the regulation of the particle size in monodisperse gold suspensions. Nature Phys Sci 241:20
Turkevich J, Stevenson C, Hillier J (1953) The formation of colloidal gold. J Phys Chem 57:670
Ji X, Song X, Li J et al (2007) Size control of gold nanocrystals in citrate reduction: the third role of citrate. J Am Chem Soc 129:13939. doi:10.1021/ja074447k
Obare SO, Jana NR, Murphy CJ (2001) Preparation of polystyrene- and silica-coated gold nanorods and their use as templates for the synthesis of hollow nanotubes. Nano Lett 1:601. doi:10.1021/nl0156134
Ung T, Liz-Marzán LM, Mulvaney P (1998) Controlled method for silica coating of silver colloids. Influence of coating on the rate of chemical reactions. Langmuir 14:3740
Soraru GD, D’Andrea G, Campostrini R, Babonneau F (1995) Characterization of methyl-substituted silica gels with Si-H functionalities. J Mater Chem 5:1363. doi:10.1039/JM9950501363
Stöber W, Fink A, Bohn E (1968) Controlled growth of monodisperse silica spheres in the micron size range. J Colloid Interface Sci 26:62. doi:10.1016/0021-9797(68)90272-5
Zhao D, Qin W, Zhang J et al (2005) Modified spontaneous emission of europium complex nanoclusters embedded in colloidal silica spheres. Chem Phys Lett 403:129. doi:10.1016/j.cplett.2004.12.113
Giersig M, Ung T, Liz-marzan LM, Mulvaney P (1997) Direct observation of chemical reactions in silica-coated gold and silver nanoparticles. Adv Mater 9:570. doi:10.1002/adma.19970090712
Allabashi R, Stach W, Escosura-Muñiz A et al (2008) ICP-MS: a powerful technique for quantitative determination of gold nanoparticles without previous dissolving. J Nanopart Res 11:2003. doi:10.1007/s11051-008-9561-2
Jian Z (2005) SPR induced quenching of the 5D1 → 7F1 emission of Eu3+ doped gold colloids. Phys Lett A 341:212. doi:10.1016/j.physleta.2005.03.075
Mahato KK, Rai SB, Rai A (2004) Optical studies of Eu3+ doped oxyfluoroborate glass. Spectrochim Acta, Part A 60:979. doi:10.1016/S1386-1425(03)00328-7
Haiss W, Thanh NTK, Aveyard J, Fernig DG (2007) Determination of size and concentration of gold nanoparticles from UV–vis spectra. Anal Chem 79:4215. doi:10.1021/ac0702084
Liaw J-W, Tsai H-Y (2012) Theoretical investigation of plasmonic enhancement of silica-coated gold nanorod on molecular fluorescence. J Quant Spectrosc Radiat Transf 113:470. doi:10.1016/j.jqsrt.2011.12.015
Tréguer-Delapierre M, Majimel J, Mornet S et al (2008) Synthesis of non-spherical gold nanoparticles. Gold Bull 41:195
Sau TK, Murphy CJ (2004) Seeded high yield synthesis of short Au nanorods in aqueous solution. Langmuir 20:6414. doi:10.1021/la049463z
Nikoobakht B, El-Sayed MA (2003) Preparation and growth mechanism of gold nanorods (NRs) using seed-mediated growth method. Chem Mater 15:1957. doi:10.1021/cm020732l
Acknowledgments
This work is supported by the French Agence Nationale de la Recherche (ANR) under the Fenoptic project (ANR-09-NANO-23), part of the Nanosciences, Nanotechnologies and Nanosystems (P3N2009) program. Authors thank Draka – Prysmian Group, the Laboratoire de Physico-Chimie des Matériaux Luminescents and the Institut Carnot de Bourgogne for their collaboration.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(PDF 338 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Bertry, L., Durupthy, O., Aschehoug, P. et al. Experimental evidence of luminescence quenching at long coupling distances in europium (III) doped core-shell gold silica nanoparticles. Gold Bull 46, 349–355 (2013). https://doi.org/10.1007/s13404-013-0112-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13404-013-0112-y