
Abstract
In vivo and ex vivo models of reoviral encephalitis were utilized to delineate the contribution of type I interferon (IFN) to the host’s defense against local central nervous system (CNS) viral infection and systemic viral spread. Following intracranial (i.c.) inoculation with either serotype 3 (T3) or serotype 1 (T1) reovirus, increased expression of IFN-α, IFN-β, and myxovirus-resistance protein (Mx1; a prototypical IFN stimulated gene) was observed in mouse brain tissue. Type I IFN receptor deficient mice (IFNAR−/−) had accelerated lethality, compared to wildtype (B6wt) controls, following i.c. T1 or T3 challenge. Although viral titers in the brain and eyes of reovirus infected IFNAR−/− mice were significantly increased, these mice did not develop neurologic signs or brain injury. In contrast, increased reovirus titers in peripheral tissues (liver, spleen, kidney, heart, and blood) of IFNAR−/− mice were associated with severe intestinal and liver injury. These results suggest that reovirus-infected IFNAR−/− mice succumb to peripheral disease rather than encephalitis per se. To investigate the potential role of type I IFN in brain tissue, brain slice cultures (BSCs) were prepared from IFNAR−/− mice and B6wt controls for ex vivo T3 reovirus infection. Compared to B6wt controls, reoviral replication and virus-induced apoptosis were enhanced in IFNAR−/− BSCs indicating that a type I IFN response, initiated by resident CNS cells, mediates innate viral immunity within the brain. T3 reovirus tropism was extended in IFNAR−/− brains to include dentate neurons, ependymal cells, and meningeal cells indicating that reovirus tropism within the CNS is dependent upon type I interferon signaling.
Similar content being viewed by others

Introduction
Innate immune responses, including inflammatory cytokine and chemokine production, mark the earliest phase of the host response to an invading organism. The type I interferon (IFN) genes, IFN-α and IFN-β, are induced in response to viral infection via activation of IFN regulatory factor 3 (IRF3) and nuclear factor-kappa B (NF-κB) transcription factors (Randall and Goodbourn 2008). These cytokines are then secreted from cells to act in an autocrine and paracrine manner through a common heterodimeric type I interferon receptor (IFNAR). Agonist–receptor binding results in activation of transcription factors STAT1 and STAT2 (signal transducer and activator of transcription) which, along with IRF9, form a heterotrimeric transcription factor complex known as interferon-stimulated gene factor 3 (ISGF3). ISGF3 translocates to the nucleus where it binds to IFN-stimulated response elements to induce transcription of hundreds of IFN-stimulated genes (ISGs). The cellular “antiviral state” is established through the combined functionality of ISG gene products. Several ISGs have direct antiviral functions while others modulate the immune system by activating effector cells and promoting the development of an acquired immune response.
Mammalian orthoreoviruses (reoviruses) are non-enveloped, icosahedral viruses with a genome consisting of ten double-stranded RNA segments (Tyler et al. 2001). Reoviruses have the capacity to induce apoptosis in both cultured cells in vitro (Clarke and Tyler 2003) and target organs in vivo (Clarke and Tyler 2009). Reoviral infection has served as a versatile model system for the study of viral pathogenesis and antiviral immunity in the brain (encephalitis; Clarke et al. 2005), spinal cord (myelitis; Goody et al. 2008), and heart (myocarditis; Sherry 2002). The potential with which a given reovirus strain induces myocarditis is negatively correlated to its induction of, and sensitivity to, IFN-α/β (Sherry et al. 1998). Furthermore, the IFN-α/β response is critical for myocardiocyte protection against reovirus replication both in vitro and in vivo (Sherry 2009). The importance of the IFN response in limiting reovirus replication and pathogenesis is also evidenced by in vivo models of peroral inoculation with reovirus serotype 1 strain Lang (T1L). For instance, systemic T1L infection and animal lethality is inhibited via IFN-α/β release from intestinal dendritic cells residing in Peyer’s patches (Johansson et al. 2007). Similarly, IRF3 plays a role in restricting systemic T1L replication, limiting pathological changes in the heart, and preventing associated mortality (Holm et al. 2010).
In contrast to the heart, the role of IFN in reovirus infections of the central nervous system (CNS) is poorly understood. Reovirus infection of the brain (Tyler et al. 2010) and spinal cord (S.A.S., personal communication) is associated with increased expression of ISGs. The profile of ISGs induced in the brain during reovirus encephalitis is remarkably similar to that seen following infection of the brain with other neurotropic viruses, including Venezuelan equine encephalitis virus (VEEV) (Sharma et al. 2008) and dengue virus type 1 (Bordignon et al. 2008), suggesting a consistent host response to viral infections of the brain. We have previously demonstrated that, in brains infected with reovirus serotype 3 strain Abney (T3A), (STAT1) is phosphorylated at sites critical for its transcriptional activity (Goody et al. 2007). Mice lacking STAT1 demonstrate increased susceptibility to reovirus infection, increased mortality, and higher viral titers in the brain compared to wildtype controls. However, the role of IFN in virus-induced brain injury and disease was not described in these studies and the cause of death was not established. In the present study, we sought to directly assess the role of IFN in the virally infected host in terms of CNS-specific and -systemic viral replication/pathogenesis by utilizing ex vivo and in vivo models of reovirus infection.
Herein, we demonstrate that reovirus infection induces CNS expression of type I interferon genes (i.e., IFN-α/β) and the interferon-stimulated gene, myxovirus-resistance protein (Mx1). Mice lacking functional IFN-α/β receptor (IFNAR−/−) have accelerated mortality and markedly increased viral titers in both brain and peripheral organs following reovirus infection. IFNAR−/− mice infected with T3 reovirus show an extended pattern of viral tropism in the CNS compared to congenic wildtype (B6wt) mice, as evidenced by the previously unreported capacity of the virus to infect neurons of the dentate gyrus and both ependymal and meningeal cells. We utilized a novel ex vivo brain slice culture system to reveal that IFN is produced locally in the CNS and does not depend upon extraneuronal aspects of viral infection (e.g., infiltrating inflammatory cells). This brain-derived IFN limits viral replication and reduces viral-induced apoptotic injury within the CNS. Overall, these data indicate that reovirus infection (both in vivo and ex vivo) induces a robust IFN response, which serves to restrict viral tropism, limit viral growth, and reduce consequent apoptosis within the CNS.
Results
IFN signaling occurs in reovirus-infected brain tissue
To determine whether reovirus induces a type I IFN response in the brain, outbred 2-day-old Swiss Webster mice were intracranially (i.c.) injected with T3A (1,000 pfu), T1L (1,000 pfu), or phosphate buffered saline (PBS) only (mock) and total brain RNA was isolated for reverse transcription polymerase chain reaction (RT-PCR) amplification of IFN-α/β transcripts. Infection with either reovirus serotype induced significant upregulation of IFN-α (Fig. 1a) and IFN-β (Fig. 1b) transcripts in comparison to mock animals at 8 days post-infection (dpi). Compared to T1L-infected brains at 8 dpi, IFN-α/β induction was significantly greater (p ≤ 0.027) in T3A-infected brains. Mx1 is a prototypical ISG, thus its expression serves as an indicator of functional IFN signaling. Mx1 mRNA was significantly elevated (p ≤ 0.044) in T3A-infected brains, in comparison to either T1L or mock samples at multiple time points (Fig. 1c). Compared to mock-infected brains, T1L-infected brains had significantly greater (p ≤ 0.036) expression of Mx1 only at 8 dpi. We previously demonstrated IFN and ISG induction by oligonucleotide arrays performed on RNA extracted from the brains of mice infected i.c. with reovirus serotype 3 strain Dearing (T3D; Tyler et al. 2010). Specifically, T3D infection induced expression of 8.7-fold greater IFN-β, 9.9-fold greater IFN-α, and 84.2-fold greater Mx1 in comparison to mock controls at 8 dpi. The magnitude of these effects is comparable to those seen following infection with T3A (Fig. 1a–c). Taken together, these findings indicate that both T3 and T1 reoviruses initiate a functional IFN response in the host brain.

IFN signaling occurs in reovirus-infected brain tissue. Total RNA was purified from the brains of mock-, T1L-, or T3A-injected Swiss Webster mice at indicated time points (N = 3–4) and reverse transcribed to cDNA. qPCR revealed a significant upregulation (p ≤ 0.028) of IFN-α (a) and IFN-β (b) mRNA induced by both T3A and T1L, in comparison to mock infection at 8 dpi. qPCR also revealed a significant upregulation of Mx1 mRNA in T3A (p ≤ 0.044)- and T1L-infected brains (p = 0.036) (c)
IFNAR−/− mice rapidly succumb to intracranial reovirus infection
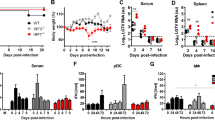
Having shown that T3 and T1 strains of reovirus induce a type I IFN response in the brains of infected mice, we next investigated whether this response influenced virus-induced disease. IFNAR−/− and B6wt mice that had been infected with reovirus were monitored for up to 21 days to assess the importance of IFN signaling in limiting virus lethality. IFNAR−/− mice had enhanced mortality following infection with T1L (Fig. 2a), T3A (Fig. 2b), and T3D (Fig. 2c). Following T1L injection, all IFNAR−/− mice succumbed to viral disease (\( {\text{MDD}} = {4}.{3}\pm 0.5 \); N = 8), whereas only two of 14 B6wt mice died (\( {\text{MDD}} = 13\pm {2}.{8} \); N = 8). Following T3A injection, IFNAR−/− mice (\( {\text{MDD}} = 4.8\pm 0.{9} \); N = 8) lived fewer days than B6wt mice (\( {\text{MDD}} = 11.1\pm {2}.{1} \); N = 9). Similarly, IFNAR−/− mice (\( {\text{MDD}} = 4.6\pm 0.{7} \); N = 16) lived fewer days than B6wt mice (\( {\text{MDD}} = {1}0.5\pm 0.9 \); N = 20) following T3D injection. Survival distributions were compared by log-rank (Mantel–Cox) test. Highly significant (p < 0.0001) differences existed between IFNAR−/− and B6wt survival distributions, regardless of which virus strain was injected. These results unequivocally demonstrate that the type I IFN response is protective following reovirus infection.

IFNAR−/− mice rapidly succumb to intracranial reovirus infection. B6wt and IFNAR−/− were inoculated i.c. with either T1L (a), T3A (b), or T3D (c) reovirus and survival was monitored until death or for 21 days (N ≥ 8). IFNAR−/− and B6wt survival distributions were significantly different (p < 0.0001), regardless of the reovirus serotype injected. All virus serotypes were 100% lethal in IFNAR−/− animals by day 6. In contrast, 87% of T1L-infected B6wt animals survived to the study endpoint and T3-infected B6wt animals lived until ∼11 dpi
Reovirus titers are increased in the CNS and peripheral organs of IFNAR−/− mice
To assess relative viral replication and spread after i.c. inoculation, CNS and peripheral organs were harvested immediately prior to IFNAR−/− animal death (i.e., 4 dpi) and viral titer was determined by plaque assay. All CNS (brains and eyes) and peripheral (liver, spleen, kidneys, heart, and blood) tissues contained significantly higher (p ≤ 0.047) viral titers in IFNAR−/− mice, as compared to B6wt mice, following infection with T1L (Fig. 3a), T3A (Fig. 3b), and T3D (Fig. 3c). T3D is a neurotropic reovirus strain that is not extensively spread through the hematogenous route. However, in the absence of IFNAR, T3D enters the blood and replicates to high titers in several organs. Overall, this data indicates that viral replication and spread is controlled by the host IFN response.

Reovirus titers are increased in the CNS and peripheral organs of IFNAR−/− mice. B6wt and IFNAR−/− were inoculated i.c. with either T1L (a), T3A (b), or T3D (c) reovirus. Tissues were harvested at 4 dpi and processed for viral titer determination by plaque assay on L929 mouse fibroblasts. Both CNS and peripheral organ titers were significantly greater in IFNAR−/− animals. Each dot represents an organ taken from an individual animal. Horizontal lines represent group means
Reovirus induces acute liver and intestinal injury but does not accelerate neuronal injury in IFNAR−/− mice
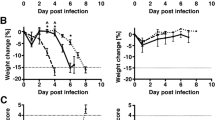
At 4 dpi, grossly abnormal liver (Fig. 4a, top panel) and intestinal (Fig. 4b, bottom panel) tissue was harvested from a subpopulation of T3-infected IFNAR−/− mice. Such injury was not seen in B6wt animals at any time post-infection (data not shown). Hematoxylin and eosin (H&E) staining confirmed that virus-infected IFNAR−/− mice, but not B6wt mice, develop virus-induced hepatic and intestinal injury. At its most severe, this liver injury is characterized by hyperemia and hepatocyte enlargement (Fig. 4a, bottom panel). Intestinal injury in IFNAR−/− mice is characterized by mucosal perforation accompanied by hemorrhage into the intestinal lumen (Fig. 4b, bottom panel). Not surprisingly in the setting of intestinal injury, bacterial (i.e., Enterococcus and Staphylococcus) species were cultured from the spleens of several IFNAR−/− mice (data not shown). In contrast to findings in liver and intestine, histological analysis revealed that the brains of T3-infected IFNAR−/− mice appeared normal immediately prior to death (Fig. 4c) and had no evidence of apoptosis activation (data not shown). Infected IFNAR−/− mice did not develop neurologic symptoms prior to death despite the fact that these experiments entailed direct brain inoculation of T3 reovirus, a highly neuropathogenic virus. B6wt mice show significant injury at late times post-infection, but only minimal pathology at 4dpi. In aggregate, these data suggest that IFNAR−/− mice do not succumb to encephalitis, but instead develop terminal liver and intestinal injury, accompanied by secondary bacterial infection and sepsis.

Reovirus induces acute liver and intestinal injury, but does not accelerate neuronal injury in IFNAR−/− mice. Livers and intestines harvested from B6wt and IFNAR−/− animals at 4 dpi (N ≥ 4) were fixed in 10% formalin overnight and photographed prior to being paraffin embedded, sectioned at 6 μm, and H&E stained. Photographs of IFNAR−/− livers (a; top panels) and intestines (b; top panels) demonstrate gross pathology induced by reoviral infection in the absence of the type I IFN response. Severe virus-induced hepatic injury in IFNAR−/− mice is characterized by hepatocyte enlargement and hyperemia (a; bottom panels). Severe virus-induced intestinal injury in IFNAR−/− animals is characterized by perforation of mucosa and hemorrhage into the intestinal lumen (b; bottom panels). At indicated time points, IFNAR−/− and B6wt brains were harvested, fixed, sectioned, and H&E stained to access for injury in the CA3 region of the hippocampus. IFNAR−/− mice did not suffer accelerated tissue injury in the brain (c)
In brain slices prepared from IFNAR−/− mice, T3 reovirus replication and viral-induced apoptosis are enhanced
Early lethality (associated with extra-CNS disease) of reovirus-infected IFNAR−/− mice precluded our investigation of the specific roles IFN might have in reovirus infected brain tissue. A brain slice culture (BSC) model of viral encephalitis was recently characterized for the study of brain tissue-specific immune responses in isolation from the whole animal (Dionne et al. 2011). Utilization of this system allowed us to study the potential role of IFN in the brain’s innate immunity against reovirus, independent from events occurring outside of the CNS. To confirm that T3A-induced IFN-β expression occurred ex vivo, BSCs were prepared from Swiss Webster mice and immediately infected with T3 reovirus. Media was changed ∼12 h later and every 2 days thereafter. Media, from experimentally similar wells, was pooled at 5 or 9 dpi for three independent experiments and serially diluted for enzyme-linked immunosorbent assay (ELISA)-based quantification of IFN-β release by mock- or T3A-infected BSCs. In T3A-infected samples, IFN-β protein was detected in 7–9 dpi media (19.98 ± 20.34 pg/mL), but not 3–5 dpi media. IFN-β was not detected in the media from mock samples suggesting that BSCs release little to no IFN-β at baseline. This study confirmed that cells, resident to the CNS, produce IFN-β at the protein level in response to T3A reovirus.
We next sought to investigate, across an extended time frame, whether viral replication and apoptosis would be more severe in reovirus-infected slices prepared from IFNAR−/− mice as compared to B6wt controls. Quantification of the reovirus L1 gene segment by RT-PCR revealed significantly elevated viral load in T3A- and T3D-infected IFNAR−/− slices, compared to B6wt slices by 7 dpi (Fig. 5a). T3A-infected IFNAR−/− BSCs had 4.3-, 4.5 (p = 0.009)-, and 7 (p = 0.024)-fold greater expression of the viral L1 gene segment at 5, 7, and 9 dpi respectively, compared to T3A-infected B6wt BSCs. In T3D experiments, virus-infected IFNAR−/− BSCs had 5.0-, 6.5 (p = 0.015)-, and 17 (p = 0.036)-fold greater viral gene expression at the same time points. Overall, these data indicate that innate IFN signaling plays a key role in restricting viral replication in brain tissue.

In brain slices prepared from IFNAR−/− mice, T3 reovirus replication and viral-induced apoptosis are enhanced. Brain slice cultures (BSCs) were prepared from 2- to 3-day-old B6wt and IFNAR−/− mice. Cultures were infected with mock or T3 reovirus immediately upon plating. At indicated times post-infection, total RNA was purified from BSCs (N = 3–4) and the reovirus L1 gene was quantified by RT-PCR. At all time points, IFNAR−/− slices showed increased viral burden when compared to B6wt brain slices (a). Increased viral replication in IFNAR−/− ultimately resulted in increased apoptosis at 11 dpi, as assessed by fluorogenic Caspase 3 activity assay (b)
Virus-induced tissue apoptosis can be quantitatively measured in BSC slices via fluorogenic Caspase 3 activity assay of BSC lysates (Dionne et al. 2011). Utilizing this methodology, virus-induced apoptosis was found to be more pronounced in IFNAR−/− BSCs at 7, 9, and 11 dpi, with a significant difference (p = 0.01) occurring between mock and IFNAR−/− samples at 11 dpi (Fig. 5b). These data reveal that local IFN signaling within brain tissue effectively limits reovirus-induced apoptosis in the CNS.
In T3-infected IFNAR−/− brains, viral tropism is extended to new neuronal populations, meningeal cells, and ependymal cells
Having shown that interference of IFN signaling in brain tissue results in enhanced reoviral growth yet does not correlate to increased tissue injury in vivo, we hypothesized that reovirus tropism was expanded to new cell populations in the brains of IFNAR−/− mice. IFNAR−/− and B6wt brains were harvested 4 days following infection with T3D (1,000 pfu) and the pattern of viral infection was examined by immunofluorescent staining of σ3 viral antigen. We have previously shown that, in wildtype mice, T3 reoviruses specifically infect neurons of the lateral thalamus, cingulate cortex, and CA3 region of the hippocampus (Oberhaus et al. 1997; Richardson-Burns et al. 2002). Consistent with these results, effective T3D infection of the CA3 is seen in B6wt animals by 11 dpi, but not as early as 4 dpi (Fig. 6a). In contrast, viral antigen is found in the CA3 of IFNAR−/− mice by 4 dpi. At this time point, there is also evidence of reovirus antigen throughout the IFNAR−/− cortex, thalamus, and hippocampus. Dentate infection does not occur in T3D-infected B6wt mice even at late times post-infection when viral titer approaches 109 pfu/mL; thus, the presence of reovirus antigen in the dentate gyrus in IFNAR−/− mice is indicative of extended T3D tropism. Dual fluorescence imaging of reovirus antigen and NeuN neuronal marker confirmed that enhancement of viral replication in IFNAR−/− mice occurred specifically in neurons of the CA3 and dentate brain regions (Fig. 6b). These data indicate that IFN serves to limit T3 reovirus neurotropism in the brain. T1L does not normally infect neurons in wildtype mouse brains and neuronal tropism was not observed in IFNAR−/− mice following inoculation of T1L (data not shown).

In reovirus-infected IFNAR−/− brains, viral tropism is extended to new neuronal populations, meningeal cells, and ependymal cells. T3D infected brains were harvested from B6wt and IFNAR−/− animals and fixed in 10% formalin overnight, paraffin embedded, and sectioned at 6 μm prior to immunofluorescence labeling. Reovirus σ3 antigen (monoclonal; red) was detected in CA3 and dentate of IFNAR−/− mice at 4 dpi. In comparison, B6wt mice have reovirus antigen in the CA3 only at late times post-infection (i.e., 11 dpi) and do not develop detectable levels of reovirus antigen in the dentate; ×100 magnification. Insets depict reovirus-positive cells at ×2 original magnification (a). To confirm that the infected IFNAR−/− cells are neurons, histological sections were co-labeled with reovirus (polyclonal; red) and NeuN neuronal marker (monoclonal; green). ×630 magnification (b). Reovirus σ3 antigen (monoclonal; red) was found at high levels in the meninges and ependyma of IFNAR−/− animals. These cell types were not infected at any timepoint in B6wt animals; ×100 magnification (c). GFAP-positive (monoclonal; green) astrocytes (located within the parenchyma deep to meningeal and ependymal cell layers), were not reovirus antigen-positive (polyclonal; red) regardless of mouse genetic background; ×400 magnification (d)
We next hypothesized that the absence of IFN would also render other CNS cell populations in the brain susceptible to infection. We found reovirus antigen in both meningeal and ependymal cells in T3D-infected IFNAR−/− mice, but not B6wt mice (Fig. 6c). Pronounced infection of these cell types was also seen in IFNAR−/−, but not B6wt, mice injected with 100 pfu T3A or 1000 pfu T1L at 4 dpi (data not shown). To investigate whether tropism was also extended to glia, astrocytes were labeled with monoclonal glial fibrillary acidic protein (GFAP) antibody (Fig. 6d). T3D viral antigen was not evident in IFNAR−/− or B6wt astrocytes, suggesting that reovirus tropism is limited in this cell type by a mechanism unrelated to IFN signaling. Overall, these data indicate that T3 reoviruses are genetically capable of infecting ependymal and meningeal cell types in addition to neurons in multiple brain regions and that it is the host IFN response that restricts such tropism.
Discussion
The role of IFN during viral infection of the brain remains incompletely understood. In this report, we demonstrate for the first time that reovirus T1 and T3 strains induce the expression of the type I IFN genes, IFN-α/β, and the ISG Mx1 in the brain following infection (Fig. 1). We also show that i.c. inoculation of IFNAR−/− mice with reovirus results in accelerated lethality (Fig. 2) and is associated with higher viral titers in the CNS and peripheral organs (Fig. 3), indicating that IFN signaling plays a key role in reovirus pathogenesis. Death of infected IFNAR−/− mice likely resulted from hepatic and/or intestinal pathology and subsequent bacterial sepsis (Fig. 4). However, we utilized an ex vivo brain slice model of reovirus infection to demonstrate that IFN is produced locally within the CNS to help control CNS viral replication and tissue injury (Fig. 5). Finally, we show that increased viral titer in the IFNAR−/− brain is associated with extended cellular tropism by demonstrating the presence of T3D within neuronal populations and non-neuronal populations (i.e., ependymal and meninges cells) not typically infected by T3 reoviruses (Fig. 6).
Local production of type I interferon within the CNS has been reported following infection with herpes simplex virus (Menachery et al. 2010), Theiler’s murine encephalitis virus (TMEV; Rubio et al. 2010), La Crosse virus (Delhaye et al. 2006), vesicular stomatitis virus (Detje et al. 2009), Semiliki Forest virus (SFV; Fragkoudis et al. 2007), and the neurotropic coronavirus mouse hepatitis virus (MHV; Roth-Cross et al. 2008). In the present study, we demonstrate that both T3 and T1 reovirus strains induce IFN expression in the brain. The T3 response was more pronounced than the T1 response possibly because the T3 virus replicates to higher titers within the brain. Alternatively, the T1L reovirus strain could have the capacity to subvert the IFN response in the brain by modulating the subcellular localization of IRF9, as was recently described in vitro (Zurney et al. 2009).
Expression of ISGs has also been reported in the brain after infection with several viruses including lymphocytic choriomeningitis virus (Wacher et al. 2007), West Nile virus (WNV; Wacher et al. 2007), dengue virus type-1 (Bordignon et al. 2008), and VEEV (Sharma et al. 2008). These results demonstrate that IFN signaling is activated in the brain following infection with a wide variety of viruses. Our results are also consistent with other reports highlighting the importance of IFN-α/β signaling in the control of viral replication in the CNS (Randall and Goodbourn 2008). For example, increases in brain viral load have been reported following infection of IFNAR-deficient mice with Sindbis virus (Ryman et al. 2000), SFV (Fragkoudis et al. 2007), VEEV (Grieder and Vogel 1999), MHV (Ireland et al. 2008), and WNV (Samuel and Diamond 2005). Deficiencies in the expression of other proteins involved in IFN signaling including STAT1, IRF3, and IRF7 have similar effects on viral replication in the brain (Daffis et al. 2008; Goody et al. 2007; Holm et al. 2010; Menachery et al. 2010).
To investigate the role of IFN in virus-induced disease within the CNS, we utilized an ex vivo model of viral encephalitis. We showed that reovirus growth was significantly increased in slices derived from IFNAR−/− mice compared to slices derived from B6wt controls although the magnitude of this increase was not as dramatic as that seen in vivo. This discrepancy is likely due to viral reseeding that occurs in the intact animal, but not the ex vivo culture system. In addition, we saw an increase in Caspase 3 activity in reovirus-infected IFNAR−/− brain slices compared to infected B6wt controls. We have previously demonstrated that apoptosis is a primary mechanism of neuronal cell death and tissue injury during reovirus encephalitis (Oberhaus et al. 1997; Richardson-Burns et al. 2002). Our results thus suggest that local IFN response within the brain is protective and serves to limit both viral growth and virus-induced tissue injury.
In this report, we demonstrate extended reoviral tropism to peripheral organs of IFNAR−/− mice. Increased viral titers, associated with extended tropism, have also been noted following WNV infection of IFNAR-deficient mice, with increased infection of splenic macrophages, B cells, and T cells (Samuel and Diamond 2005). Extended tissue tropism to the heart, intestine, stomach, lung, and kidney is seen following infection of IFNAR-deficient mice with MHV (Roth-Cross et al. 2008). Similarly, SFV gains tropism to the heart, pancreas, adipose tissue, and muscle of IFNAR-deficient mice (Fragkoudis et al. 2007). In aggregate, these results indicate that the type I IFN response plays an important role in determining tissue tropism.
In the brain, we demonstrate, in the absence of IFNAR, expanded tropism of reovirus to a greater neuronal population, and to cells of the meninges and ependyma. Particularly striking is the extended tropism of T3 reovirus strains to ependymal cells (Fig. 6). Early data suggested that T1L, but not T3 reovirus strains, induce hydrocephalus because T1 strains encode a unique viral hemagglutinin, which mediates high-affinity ependymal cell binding (Tardieu and Weiner 1982; Weiner et al. 1977). Our results demonstrate that in the absence of IFN, T3 reovirus strains can also infect and efficiently replicate within ependymal cells. In contrast, wildtype mice do not support reovirus infection within these cell types even at late time points post-infection when whole brain viral titers are in excess of 108 pfu/mL. Given these results, we conclude that T3 reovirus strains have the capacity to infect ependymal and meningeal cells; however, in the presence of the host IFN response viral replication is effectively inhibited in these cell types. Increased tropism to ependymal cells, in the absence of IFN signaling, has also been seen following infection of the brain with WNV (Samuel and Diamond 2005), Sindbis virus (Ryman et al. 2000), measles virus (Mrkic et al. 1998), TMEV (Fiette et al. 1995), and Semiliki forest virus (Fragkoudis et al. 2007). Taken together, our results and prior studies suggest that type I IFN plays a key role in protecting ependymal cells from infection by neurotropic viruses.
Our studies demonstrate the importance of IFN signaling in the virus-infected organism in general and in the brain in particular. An expanding body of literature now implicates a protective role for IFN during virus-induced CNS disease. IFN is already prescribed for the treatment of peripheral viral infections (i.e., human infection with hepatitis C). The demonstrated importance of IFN-α/β in controlling virus replication and virus-induced injury within the brain suggests that therapeutic IFN could benefit patients suffering from viral infections of the CNS.
Materials and methods
Viral stocks
T1L, T3A, and T3D are laboratory stocks derived via plaque purification and double passage in L929 cells. T3 strains were further purified via cesium chloride density gradient centrifugation.
In vivo studies
Swiss Webster outbred mice were obtained from Harlan Laboratories (Indianapolis, IN, USA). Breeder pairs of type I IFNAR−/− were kindly provided by Ross Kedl (National Jewish Health; Denver, CO, USA). Congenic C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All experiments were approved by the Institutional Animal Care and Use Committee and performed in an Association for Assessment and Accreditation of Laboratory Animal Care International-accredited animal facility.
Two-day-old mice were i.c. inoculated with T1L (1,000 pfu), T3A (1,000 pfu into Swiss Webster mice; 100 pfu into IFNAR−/− and B6wt mice), or T3D (1,000 pfu) diluted in a 10-μl volume of PBS. Mock-infected mice received i.c. vehicle PBS at equal volume.
Preparation and infection of organotypic BSCs
Brain slice cultures were prepared from 2 to 3-day-old B6wt or IFNAR−/− mice in compliance with institutional procedural guidelines. Mice were euthanized by decapitation and brains rapidly removed into ice-cold slicing media (MEM, 10 mM Tris, 28 mM D-glucose, pH 7.2, equilibrated with 95% O2/5% CO2). Under semisterile conditions, the frontal cortex and cerebellum were removed and 400-μm coronal sections cut through cerebral regions containing hippocampus and thalamus using a vibrating-blade microtome (Leica VT1000S; Bannockburn, IL, USA). Four slices from a single animal were carefully positioned onto a single semiporous membrane insert (Millipore #PICMORG50; Billerica, MA, USA) according to the method of Stoppini et al. (1991). Membranes were placed in 35 mm tissue culture dishes containing 1.2 mL serum containing media (Neurobasal supplemented with 10 mM HEPES, 1× B-27, 10% fetal bovine serum (FBS), 400 μM l-glutamine, 600 μM Glutamax, 60 U/mL penicillin, 60 μg/mL streptomycin, and 6 U/mL nystatin), such that slices were positioned at the media–air interface. Immediately after plating, slices were infected with 106 pfu T3A or T3D per slice. Virus was diluted into 20 μl PBS and applied dropwise to the top (air interface) of each slice. Mock infections were performed in a similar manner with PBS alone. Media was refreshed with 5% serum-containing media (5% FBS) approximately 12 h after slicing. All subsequent media changes were made with serum-free media every 2 days (i.e., 3, 5, 7, and 9 dpi). Cultures were maintained in a humidified incubator at 5% CO2 and 36.5°C.
RNA purification from whole brains and BSCs
Mock- and T3A-infected brains were harvested from Swiss Webster mice at appropriate time points and immediately placed into RNAlater (Ambion; Foster City, CA, USA). Total RNA was purified by Dounce homogenization of the whole brain into 5 mL of Qiazol. This emulsion was transferred into a clean Falcon tube and set aside for at least 5 min before the addition of 1 mL chloroform. The mixture was shaken vigorously for 15 s and set aside for 5 min before centrifugation at 5,000×g for 15 min at 4°C. The upper 2 mL aqueous phase was then carefully transferred into a new tube containing 2 mL of 70% ethanol (prepared with diethyl pyrocarbonate-treated water). The solution was mixed and transferred onto an RNeasy Midi spin column (Qiagen; Germantown, MD, USA) and RNA was purified according to the manufacturer's specifications. To prevent degradation, RNase inhibitor was added and the sample was stored at −80°C.
For RNA purification from BSCs, four experimentally similar slices were washed three times in PBS and triturated in 600 μL RLT buffer (Qiagen; Germantown, MD, USA) containing 1% β-mercaptoethanol. Samples were stored at −80°C until lysate was processed through a QIAshredder (Qiagen; Germantown, MD, USA) and loaded onto an RNeasy Mini spin column (Qiagen; Germantown, MD, USA) for RNA purification according to the manufacturer’s protocol. RNA samples were stored at −80°C until RT-PCR analysis.
RT-PCR quantification of gene transcripts
RT-PCR was utilized to quantify reovirus transcript in BSC-derived total RNA samples. Two primers designated RV-3 (5′ CAT ATG ACT ACC ACT TTC CCG 3′) and RV-4 (5′ GCT ATG TCA TAT TTC CAT CCG 3′) were synthesized (Invitrogen; San Diego, CA, USA) to amplify a 298-bp segment of the reovirus L1 gene (Tyler et al. 1998). Determination of viral burden, relative to a housekeeping gene, was achieved by concurrent amplification of mouse β-actin (SABiosciences primer #PPM02945A; Frederick, MD, USA). Purified RNA template, primers, RT-PCR master mix, and reverse transcriptase (iScript One-Step RT-PCR Kit with SYBR Green; Bio-Rad; Hercules, CA, USA) were mixed into a total volume of 20 μL. Forty cycles of PCR amplification were performed on a Bio-Rad CFX96 thermocycler (Hercules, CA, USA) as follows: cDNA synthesis at 50°C for 10 min, reverse transcriptase inactivation at 95°C for 5 min, denaturation at 95°C for 10 s, and annealing/extension at 60°C for 30 s. Melt curve analysis confirmed absence of non-specific products and primer-dimers. C(t) values were converted to relative expression values on Bio-Rad CFX Manager analysis software.
IFN-α, IFN-β, and Mx1 transcripts were quantified in whole brain-derived total RNA samples via qPCR. Purified RNA (2 μg) was first reverse transcribed to cDNA using the Applied Biosystems High Capacity cDNA Kit per manufacture directions (Carlsbad, CA, USA). IFN-α (#PPM03549E), IFN-β (#PPM03594B), and Mx1 (#PPM05520A) primers were purchased from SABiosciences (Frederick, MD, USA). Quantification of IFN-α, IFN-β, or Mx1 relative to a housekeeping gene was achieved by concurrent amplification of mouse β-actin (SABiosciences primer #PPM02945A; Frederick, MD, USA). cDNA (diluted 1:10), primers, and 2× Real Time SYBR green mix (SAbiosciences; Frederick, MD, USA) were mixed into a total volume of 25 μL. Forty cycles of PCR amplification were performed on a Bio-Rad CFX96 thermocycler (Hercules, CA, USA) as follows: Taq activation at 95°C for 10 min, denaturation at 95°C for 15 s, and annealing/extension at 60°C for 1 min. Melt curve analysis confirmed absence of non-specific products and primer dimers. C(t) values were converted to relative expression values on Bio-Rad CFX Manager analysis software.
Histological studies
Brain, liver, and intestinal tissue was immersed in 10% buffered formalin for no less than 12 h, embedded in paraffin, and serially sectioned into 6 μm-thick sections, and mounted onto slides. Tissue injury was assessed by H&E staining followed by light microscopy. To visualize infected cells, sections were desiccated at 50°C for 15 min then rehydrated in PBS over 30 min. Tissue was subjected to antigen retrieval (antigen unmasking solution; Vector Laboratories; Burlingame, CA, USA) and permeabilization/blocking (5% fetal bovine serum and 5% normal goat serum in 0.3% Triton/PBS) prior to incubation with primary antibodies: monoclonal reovirus σ3 antibody (4F2; 1:100), polyclonal reovirus antibody (1: 500), mouse anti-neuronal nuclear antibody (NeuN; 1:100; Millipore; Billerica, MA, USA), and/or mouse anti-GFAP (1:300; Sigma-Aldrich; St. Louis, MO, USA). Sections were then incubated with secondary antibodies: Alexa Fluor 488-conjugated goat anti-mouse or anti-rabbit IgG (1:200; Invitrogen; Carlsbad, CA, USA) and/or Cy3-conjugated AffiniPure goat anti-rabbit or anti-mouse IgG (1:300; Jackson ImmunoResearch; West Grove, PA, USA). For consistency, reovirus antigen was always labeled with Cy3. Nuclei were Hoechst stained (10 μg/mL; Immunochemistry Technology; Bloomington, MN, USA) prior to mounting sections with VectorShield Mounting Medium (Vector Laboratories; Burlingame, CA, USA). Slides were imaged on a Marianas fluorescence imaging workstation (Intelligent Imaging Innovation Inc, Denver, CO, USA) based on a Zeiss 300 M inverted microscope and CoolSnap HQ2 CCD camera, all controlled by Slidebook 4.2 software (Intelligent Imaging Innovation). Images are presented as obtained in Slidebook without changing “gamma” settings and without cropping.
IFN-β ELISA
BSC media was screened for IFN-β protein synthesis/release with a Mouse Interferon Beta ELISA Kit (PBL Biomedial Laboratories, Piscataway NJ, USA) according to the manufacturer’s protocol. Briefly, media from experimentally similar wells for individual BSC preps (N ≥ 3) was pooled and pipetted into ELISA plates for a 2-h binding incubation. The plates were washed three times prior to 1-h incubation with detection antibody. Following another series of three washes, bound secondary antibody was detected by streptavidin-horseradish peroxidase and quantified on a microplate reader (Molecular Devices Emax; Sunnyvale, CA, USA). Absorbance was read at 450 nm (with wavelength correction at 560 nm). Quantification of IFN-β concentration was performed by placing absorbance values upon a standard curve (R 2 = 0.996).
Viral titer plaque assays
Viral titers were determined by plaque assays as previously described (Debiasi et al. 1999). Briefly, L929 mouse fibroblasts, used for viral titer assays, were maintained in DMEM medium (supplemented with 10% heat-inactivated FBS and 4 mM l-glutamine). Brain and liver were washed three times with PBS and placed into 1 mL PBS, before being subjected to three freeze–thaw cycles and brief sonication with an ultrasonic processor set at 35% amplitude (Cole-Parmer; Vernon Hills, IL, USA). Eyes, heart, spleen, intestine, kidneys, and blood were placed in 200 μL PBS before being subjected to three freeze–thaw cycles and brief sonication. Serial dilutions of the organ homogenate were prepared in gel saline to a total inoculum volume of 200 μL. Viral adsorption proceeded for 1 h before L929s were overlayed with 1.5% Agar/2×199 media. Cells were overlayed again at 4 dpi. On 7 dpi, both layers of agar/media were gently removed and remaining cells fixed and stained with 0.3% methylene blue in 5% formalin. Plaques were counted and the reported pfu per milliliter value was calculated by correcting for inoculum volume and dilution factors.
Lactate dehydrogenase assay
Lactate dehydrogenase (LDH) efflux is commonly used as a measurement of injury in organotypic slices (Bruce et al. 1995; Noraberg et al. 1999). BSC media was collected at various times post-infection and triplicate 10-μl samples were analyzed with the LDH-Cytotoxicity Assay Kit II (Biovision; Mountain View, CA, USA) in which water was used as a blanking control and purified LDH used as a positive control. LDH activity was quantified on a microplate reader (Molecular Devices Emax; Sunnyvale, CA, USA) as a function of the optical density measured at 450 nm with wavelength correction at 650 nm.
Fluorogenic Caspase 3 activity assay
Caspase 3 activity was determined by utilizing a fluorogenic assay (B&D Biosciences; San Jose, CA, USA). Activated Caspase 3 hydrolyzes the synthetic peptide substrate Ac-DEVD-7-amino-4-trifluoromethyl coumarin (AFC) resulting in the generation of a fluorescent reporter molecule AFC. The reaction product was measured on a Cytofluor Series 4000 spectrofluorometer (Applied Biosystems; Carlsbad, CA, USA) at excitation wavelength of 450 nm and emission wavelength of 530 nm.
Statistical analysis
All bar graphs are presented as mean ± SD. Numbers of independent experiments are indicated by the N value. All statistical analyses were performed using Instat and Prism software (GraphPad Software Inc., San Diego, CA, USA). Statistical comparisons between two groups (i.e., mock vs. infected or B6wt vs. IFNAR−/−) were made using a two-tailed, unpaired t test with Welch correction. For survival studies, the mean day of death ± standard deviation was calculated for each group of mice. Differences in survival were calculated using a logrank (Mantel–Cox) test. Probability values of p < 0.05 were considered significant differences and denoted as such with asterisks.
Abbreviations
- IFN:
-
Interferon
- IFNAR:
-
Type I interferon receptor
- B6wt:
-
C57BL/6 wildtype mouse
- IFNAR−/− :
-
Type I interferon receptor knockout mouse
- ISG:
-
Interferon-stimulated gene
- T1:
-
Serotype 1 reovirus
- T1L:
-
Reovirus serotype 1 strain Lang
- T3:
-
Serotype 3 reovirus
- T3A:
-
Reovirus serotype 3 strain Abney
- T3D:
-
Reovirus serotype 3 strain Dearing
- pfu:
-
Plaque-forming units
- i.c.:
-
Intracranial
- dpi:
-
Days post-infection
- BSC:
-
Brain slice culture
- CNS:
-
Central nervous system
- Mx1:
-
Myxovirus-resistance protein
- MDD:
-
Mean day of death
References
Bordignon J, Probst CM, Mosimann AL, Pavoni DP, Stella V, Buck GA, Satproedprai N, Fawcett P, Zanata SM, de Noronha L, Krieger MA, Duarte Dos Santos CN (2008) Expression profile of interferon stimulated genes in central nervous system of mice infected with dengue virus type-1. Virology 377:319–329
Bruce AJ, Sakhi S, Schreiber SS, Baudry M (1995) Development of kainic acid and N-methyl-d-aspartic acid toxicity in organotypic hippocampal cultures. Exp Neurol 132:209–219
Clarke P, Tyler KL (2003) Reovirus-induced apoptosis: a minireview. Apoptosis 8:141–150
Clarke P, Tyler KL (2009) Apoptosis in animal models of virus-induced disease. Nat Rev Microbiol 7:144–155
Clarke P, DeBiasi RL, Goody R, Hoyt CC, Richardson-Burns S, Tyler KL (2005) Mechanisms of reovirus-induced cell death and tissue injury: role of apoptosis and virus-induced perturbation of host-cell signaling and transcription factor activation. Viral Immunol 18:89–115
Daffis S, Samuel MA, Suthar MS, Keller BC, Gale M Jr, Diamond MS (2008) Interferon regulatory factor IRF-7 induces the antiviral alpha interferon response and protects against lethal West Nile virus infection. J Virol 82:8465–8475
Debiasi RL, Squier MK, Pike B, Wynes M, Dermody TS, Cohen JJ, Tyler KL (1999) Reovirus-induced apoptosis is preceded by increased cellular calpain activity and is blocked by calpain inhibitors. J Virol 73:695–701
Delhaye S, Paul S, Blakqori G, Minet M, Weber F, Staeheli P, Michiels T (2006) Neurons produce type I interferon during viral encephalitis. Proc Natl Acad Sci USA 103:7835–7840
Detje CN, Meyer T, Schmidt H, Kreuz D, Rose JK, Bechmann I, Prinz M, Kalinke U (2009) Local type I IFN receptor signaling protects against virus spread within the central nervous system. J Immunol 182:2297–2304
Dionne KR, Leser JS, Lorenzen KA, Beckham JD, Tyler KL (2011) A brain slice culture model of viral encephalitis reveals an innate CNS cytokine response profile and the therapeutic potential of caspase inhibition. Exp Neurol 228:222–231
Fiette L, Aubert C, Muller U, Huang S, Aguet M, Brahic M, Bureau JF (1995) Theiler's virus infection of 129 Sv mice that lack the interferon alpha/beta or interferon gamma receptors. J Exp Med 181:2069–2076
Fragkoudis R, Breakwell L, McKimmie C, Boyd A, Barry G, Kohl A, Merits A, Fazakerley JK (2007) The type I interferon system protects mice from Semliki Forest virus by preventing widespread virus dissemination in extraneural tissues, but does not mediate the restricted replication of avirulent virus in central nervous system neurons. J Gen Virol 88:3373–3384
Goody RJ, Beckham JD, Rubtsova K, Tyler KL (2007) JAK-STAT signaling pathways are activated in the brain following reovirus infection. J Neurovirol 13:373–383
Goody RJ, Schittone SA, Tyler KL (2008) Experimental reovirus-induced acute flaccid paralysis and spinal motor neuron cell death. J Neuropathol Exp Neurol 67:231–239
Grieder FB, Vogel SN (1999) Role of interferon and interferon regulatory factors in early protection against Venezuelan equine encephalitis virus infection. Virology 257:106–118
Holm GH, Pruijssers AJ, Li L, Danthi P, Sherry B, Dermody TS (2010) Interferon regulatory factor 3 attenuates reovirus myocarditis and contributes to viral clearance. J Virol 84:6900–6908
Ireland DD, Stohlman SA, Hinton DR, Atkinson R, Bergmann CC (2008) Type I interferons are essential in controlling neurotropic coronavirus infection irrespective of functional CD8 T cells. J Virol 82:300–310
Johansson C, Wetzel JD, He J, Mikacenic C, Dermody TS, Kelsall BL (2007) Type I interferons produced by hematopoietic cells protect mice against lethal infection by mammalian reovirus. J Exp Med 204:1349–1358
Menachery VD, Pasieka TJ, Leib DA (2010) Interferon regulatory factor 3-dependent pathways are critical for control of herpes simplex virus type 1 central nervous system infection. J Virol 84:9685–9694
Mrkic B, Pavlovic J, Rulicke T, Volpe P, Buchholz CJ, Hourcade D, Atkinson JP, Aguzzi A, Cattaneo R (1998) Measles virus spread and pathogenesis in genetically modified mice. J Virol 72:7420–7427
Noraberg J, Kristensen BW, Zimmer J (1999) Markers for neuronal degeneration in organotypic slice cultures. Brain Res Brain Res Protoc 3:278–290
Oberhaus SM, Smith RL, Clayton GH, Dermody TS, Tyler KL (1997) Reovirus infection and tissue injury in the mouse central nervous system are associated with apoptosis. J Virol 71:2100–2106
Randall RE, Goodbourn S (2008) Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol 89:1–47
Richardson-Burns SM, Kominsky DJ, Tyler KL (2002) Reovirus-induced neuronal apoptosis is mediated by caspase 3 and is associated with the activation of death receptors. J Neurovirol 8:365–380
Roth-Cross JK, Bender SJ, Weiss SR (2008) Murine coronavirus mouse hepatitis virus is recognized by MDA5 and induces type I interferon in brain macrophages/microglia. J Virol 82:9829–9838
Rubio N, Palomo M, Alcami A (2010) Interferon-alpha/beta genes are up-regulated in murine brain astrocytes after infection with Theiler's murine encephalomyelitis virus. J Interferon Cytokine Res 30:253–262
Ryman KD, Klimstra WB, Nguyen KB, Biron CA, Johnston RE (2000) Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol 74:3366–3378
Samuel MA, Diamond MS (2005) Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol 79:13350–13361
Sharma A, Bhattacharya B, Puri RK, Maheshwari RK (2008) Venezuelan equine encephalitis virus infection causes modulation of inflammatory and immune response genes in mouse brain. BMC Genomics 9:289
Sherry B (2009) Rotavirus and reovirus modulation of the interferon response. J Interferon Cytokine Res 29:559–567
Sherry B (2002) The role of interferon regulatory factors in the cardiac response to viral infection. Viral Immunol 15:17–28
Sherry B, Torres J, Blum MA (1998) Reovirus induction of and sensitivity to beta interferon in cardiac myocyte cultures correlate with induction of myocarditis and are determined by viral core proteins. J Virol 72:1314–1323
Stoppini L, Buchs PA, Muller D (1991) A simple method for organotypic cultures of nervous tissue. J Neurosci Methods 37(2):173–182
Tardieu M, Weiner HL (1982) Viral receptors on isolated murine and human ependymal cells. Science 215:419–421
Tyler KL, Sokol RJ, Oberhaus SM, Le M, Karrer FM, Narkewicz MR, Tyson RW, Murphy JR, Low R, Brown WR (1998) Detection of reovirus RNA in hepatobiliary tissues from patients with extrahepatic biliary atresia and choledochal cysts. Hepatology 27(6):1475–1482
Tyler KL, Clarke P, DeBiasi RL, Kominsky D, Poggioli GJ (2001) Reoviruses and the host cell. Trends Microbiol 9:560–564
Tyler KL, Leser JS, Phang TL, Clarke P (2010) Gene expression in the brain during reovirus encephalitis. J Neurovirol 16:56–71
Wacher C, Muller M, Hofer MJ, Getts DR, Zabaras R, Ousman SS, Terenzi F, Sen GC, King NJ, Campbell IL (2007) Coordinated regulation and widespread cellular expression of interferon-stimulated genes (ISG) ISG-49, ISG-54, and ISG-56 in the central nervous system after infection with distinct viruses. J Virol 81:860–871
Weiner HL, Drayna D, Averill DR Jr, Fields BN (1977) Molecular basis of reovirus virulence: role of the S1 gene. Proc Natl Acad Sci USA 74:5744–5748
Zurney J, Kobayashi T, Holm GH, Dermody TS, Sherry B (2009) Reovirus mu2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J Virol 83:2178–2187
Acknowledgments
This work was supported by RO1NS050138 (KLT), RO1NS051403 (KLT), and a VA Merit Grant (KLT). KRD was supported by an institutional MST Program training grant (T32 GM008497) and a National Research Service Award for Individual Predoctoral MD/PhD Fellows (F30 NS071630). SAS was supported by a National Research Service Award for Individual Predoctoral Fellows (F31 NS06258303). The authors extend their gratitude to J. Smith Leser for technical support. We thank Lai Kuan Goh for her critical reading of the manuscript.
Conflict of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dionne, K.R., Galvin, J.M., Schittone, S.A. et al. Type I interferon signaling limits reoviral tropism within the brain and prevents lethal systemic infection. J. Neurovirol. 17, 314–326 (2011). https://doi.org/10.1007/s13365-011-0038-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13365-011-0038-1