
Abstract
Background and Objectives
The recombinant human interleukin-1 receptor antagonist (rhIL-1Ra) GR007 is a candidate drug with the potential to prevent the toxicity induced by chemotherapy agents by blocking the IL-1 signaling pathway. The aim of this study was to assess the pharmacokinetics and safety of GR007 in healthy Chinese subjects.
Methods
Thirty subjects received a single intramuscular injection of 30 mg (n = 10), 90 mg (n = 10), or 150 mg (n = 10) GR007. After administration, the pharmacokinetic characteristics and safety were evaluated.
Results
No serious adverse events were reported in this study, and the adverse events reported showed no dose dependency. Pharmacokinetic analysis showed that the median time to maximum concentration (Tmax) of GR007 in the three groups was between 2.75 and 4.00 h and the geometric mean elimination half-life (T1/2) for each group was 2.38, 2.22, and 3.29 h, respectively. The area under the concentration vs time curve (AUC), but not the maximum concentration (Cmax), increased in a dose-proportional manner.
Conclusions
The results showed that a single intramuscular injection of 30–150 mg GR007 had good safety and tolerability in healthy Chinese subjects. The results of the evaluation of the safety and pharmacokinetics of GR007 performed in this study support its use as a repeated daily injection in ongoing clinical trials focusing on patients with cancer.
Similar content being viewed by others

This is the first pharmacokinetic study of the recombinant human interleukin-1 receptor antagonist (rhIL-1Ra) GR007 in Chinese subjects. This study evaluated the pharmacokinetics of single ascending doses of GR007 |
AUC increased in a dose-proportional manner from 30 to 150 mg in healthy Chinese subjects. The changes in Cmax were not dose proportional, but this may be related to the injection volume |
There was no significant difference in the exposure to GR007 for either sex in the 30 mg and 150 mg groups |
1 Introduction
The interleukin-1 receptor antagonist (IL-1Ra) was first discovered in 1984 in the urine and serum of patients with leukemia and in the supernatant of cultured monocytes [1]. IL-1Ra was so named because it competitively binds the IL-1 receptor and antagonizes its function [2]. Human IL-1Ra is composed of 153 amino acid residues with a molecular weight of 17.3 kDa in the absence of glycosylation. IL-1Ra is highly evolutionarily conserved, with 77% homology between the human and mouse IL-1Ra amino acid sequences [3]. As the biological effects of IL-1Ra are not species specific, human IL-1Ra has been tested widely in various experimental animal models to verify its inhibitory effect on IL-1 activity [4,5,6,7]. A high level of IL-1 was detected in the serum and synovial fluid of patients with rheumatoid arthritis (RA) [8]. IL-1 was found to be involved in the pathological process of articular cartilage destruction in RA through multiple pathologies, including inducing the rapid decay of proteoglycans and the degradation of cartilage [1]. IL-1Ra competitively inhibits the binding of IL-1 to the active IL-1 receptor I (IL-1RI), blocking the biological activity of IL-1 in various human tissues and organs [4, 9]. However, the natural endogenous IL-1Ra in the synovium and synovial fluid of patients with RA is not sufficient to antagonize the receptor in the presence of increased IL-1 in the local tissue [8, 10, 11]. Therefore, the exogenous administration of IL-1Ra could effectively relieve the signs and symptoms of RA in patients. The safety and efficacy of recombinant IL-1Ra were examined in a series of clinical trials of inflammatory-related diseases, such as RA [12,13,14] and neonatal multisystem inflammatory diseases [15, 16], and the results led to the approval of recombinant IL-1Ra (chemical name: anakinra, commercial name: Kineret) by the Food and Drug Administration (FDA) in December 2001 for the treatment of RA and in December 2012 for the treatment of neonatal multisystemic inflammatory diseases. Anakinra was also approved by the European Medicines Agency (EMA) in March 2002 for the treatment of RA.
The investigational drug GR007 used in this study was produced by the E. coli expression system, and its amino acid sequence was consistent with that of anakinra. Pre-clinical pharmacology studies showed that rhIL-1Ra had the ability to bind the IL-1 receptor, IL-1RI. By blocking the IL-1 signaling pathway, rhIL-1Ra was found to specifically and reversibly increase the expression of the cell cycle inhibitory factor p21 in normal cells, and to inhibit the rapid proliferation of cells such as bone marrow hematopoietic cells and intestinal mucosal crypt cells [17], thus decreasing their sensitivity to chemotherapeutic agents, protecting them from the toxicity of chemotherapeutic agents and reducing the incidence and severity of chemotherapy-induced neutropenia (CIN) and chemotherapy-related diarrhea (CID) [18,19,20]. GR007 therefore has the potential to effectively prevent CIN and CID and to significantly reduce their incidence and severity. The study reported in the present paper was a phase I clinical trial to evaluate the safety and pharmacokinetics of GR007 in healthy Chinese subjects; the results provide a foundation for follow-up clinical trials.
2 Subjects and Methods
2.1 Subjects
The subjects enrolled in this study were healthy Chinese male and female volunteers who met the following requirements: 18–45 years of age; body weight ≥ 50 kg; body mass index (BMI) between 19 and 24 kg/m2; good health, as judged by physical examination, medical history, vital signs, electrocardiograms, and clinical laboratory examinations; no clinical abnormalities or abnormalities in any of the laboratory tests; and no history or evidence of alcohol or drug abuse. The key exclusion criteria included a white blood cell count (WBC) of < 4.0 × 109/L, a neutrophil count (ANC) of < 2.0 × 109/L, blood donation within 3 months of the first dose; serologically positive results for hepatitis B, hepatitis C virus (HCV), syphilis, or human immunodeficiency virus (HIV); use of any long-acting biologics in the 6 months prior to the study; use of any medications known to be strong inducers or inhibitors of cytochrome enzymes within 30 days of testing; use of prescriptions, over-the-counter drugs, or nutraceutical products in the 2 weeks before this trial; participation in another clinical trial in the 3 months prior to enrollment in this study; pregnant women or lactating women; and women or men of childbearing potential who were reluctant to adopt reliable contraceptive measures during the trial and for 6 months after the end of the trial. The study was conducted in accordance with the Declaration of Helsinki and good clinical practice guidelines. Subjects were also required to be fully informed of the study and to freely sign an informed consent form prior to the trial.
2.2 Study Design
This study was an open-label, single-center, single-dose phase I clinical trial conducted at the Clinical Drug Testing Institute of Peking University First Hospital. The protocol and informed consent documentation were reviewed and approved by the ethics committee of Peking University First Hospital (China). The study was registered at the Chinese Clinical Trial Registry Platform (ICTRP): ChiCTR1800015222. The drug (GR007) tested in this study came in the form of a freeze-dried powder injection that was manufactured under GMP conditions by General Regeneratives (Shanghai) Limited, located at No. 6 Lane 898, Halei Road, Shanghai, P.R. China. The subjects enrolled in this study were divided into three dose groups who received a single intramuscular injection of 30, 90, or 150 mg GR007. The starting dose (30 mg GR007) was selected based on the no observed adverse effect level (NOEAL) exposure gauged from rat and cynomolgus monkey studies, according to regulatory guidance. Each group contained ten subjects. During the hospitalization period, unified diets were adopted. The subjects were given breakfast before administration on the first day (day 1) and remained under observation at the clinical trial center for 72 h after the administration of the study drug. They left with the permission of the investigator after all the biological samples had been collected and all study assessments were completed. A clinical follow-up visit was performed on day 7 in the fasting state for the safety assessments, which included vital signs, a 12-lead electrocardiogram (ECG), clinical laboratory evaluations, and a physical examination. Subjects also received a follow-up phone call on approximately day 30, and they were considered to have completed this study if the investigator confirmed that they had no clinically significant abnormalities in the laboratory tests or clinical observations.
2.3 Safety Assessment
During this study, the safety-related outcomes of the subjects—including vital signs, physical examinations, laboratory tests (blood routine, urine routine, blood biochemistry, coagulation function, C-reactive protein, immune function), electrocardiograms, injection site reactions, and adverse events—were collected and evaluated. All adverse events (AEs) experienced by the subjects were graded according to the National Institutes of Health—Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 4.03. The relationship of each AE to the study drug was graded as either positively related, probably related, possibly related, probably not related, or not related.
2.4 Pharmacokinetic Assessment
A series of venous blood samples for the measurement of GR007 serum levels were collected in 3.5-mL blood collection tubes before dosing and at 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 16, and 24 h after GR007 administration to detect the serum concentration of GR007 by electrochemiluminescence immunoassay; from these measurements, the pharmacokinetic parameters of GR007 were estimated.
The characteristic pharmacokinetic parameters included maximum concentration (Cmax), area under the concentration-time curve from time zero to last quantifiable concentration (AUC0–t) and from time zero to infinity (AUC0–∞), time to reach Cmax, eliminatin half-life (T1/2), clearance rate (CL/F), apparent volume of distribution (Vd/F), the terminal rate constant (λz), and the elimination rate constant (Kel).
2.5 Bioanalytical Assay
In this study, human serum samples were analyzed by electrochemiluminescence immunoassay. Briefly, the human IL-1Ra/IL-1F3 capture antibody (R&D) was first coated onto the microtiter plate, and the tested serum sample was added and diluted fourfold with 10% human blank serum/1% BSA mix solution to bind the rhIL-1Ra. Subsequently, the human IL-1ra/IL-1F3 detection antibody (R&D) solution, SULFO-TAG™ streptavidin solution (Meso Scale Diagnostics), and finally the MSD® read buffer (Meso Scale Diagnostics) were added. The signal value was read using a MESO® SECTOR S600 electrochemical meter. The standard curve was fitted using a four-parameter regression model, and the sample concentration was computed using Discovery Workbench® software 4.0. The measurement range was 10–640 ng/mL.
2.6 Statistical Analysis
The safety analysis was performed on the safety data set. AEs in different groups were coded and counted in accordance with the system organ classification (SOC) and preferred terminology (PT) from MedDRA (version 20.0). The AEs and drug-related AEs were also classified and summarized based on the SOC and PT. The number and incidence of AEs were recorded, as were the subjects who experienced the AEs. The safety evaluation was statistically analyzed using SAS software (version 9.4).
Pharmacokinetic parameters were calculated using the noncompartmental model (NCA) and the linear-log trapezoidal method based on the log blood concentration data from each subject. The calculations were performed and a descriptive statistical summary of the PK parameters was obtained using Phoenix WinNonlin v.6.4. Statistical comparisons of the groups conducted by using IBM SPSS Statistics v.21. Differences in the exposure-related pharmacokinetic parameters of GR007 (such as Cmax, AUC0–t, and AUC0–∞) between the male and female subjects were checked for significance using the t test unless the number of male and female subjects was less than three in any dose group. Data from multiple groups were compared using one-way analysis of variance (ANOVA), and a pairwise comparison was performed after examining the homogeneity of variance. The dose proportionality of exposure was analyzed using the power model Y = α × Dβ, where Y represents the pharmacokinetic parameter (AUC or Cmax), D represents the dose, α represents the intercept, and β represents the slope. When the 90% confidence interval of β was within 80–125%, the exposure was judged to be proportional to the dose.
3 Results
3.1 Demographic and Baseline Characteristics
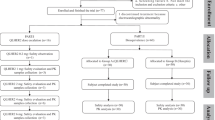
In total, 135 volunteers were screened in the study; 38 were eligible volunteers and 30 were enrolled in the study, and equal numbers were assigned to the three different dose groups. One subject dropped out owing to noncompletion of the telephone follow-up at the end of day 30, but this did not influence the PK analysis and safety evaluation. Data collected from all 30 subjects were included in the PK analysis set and safety analysis set (Fig. 1).

Subject screening and distribution flow chart. PK pharmacokinetic
The demographic and baseline characteristics were comparable across all dose groups. The detailed demographic characteristics of the enrolled subjects are summarized in Table 1. Eight female subjects were enrolled in this study, but only one female subject was allocated to the 90 mg group.
3.2 Pharmacokinetic Properties
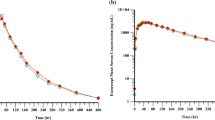
In total, 30 volunteers were included in the pharmacokinetic analysis. After detecting and analyzing the serum concentration of GR007 in each blood sample, the mean serum concentration vs time curve of each group was constructed (Fig. 2).

Mean (SD) serum concentration vs time curve of GR007 for each group
Pharmacokinetic analysis showed that GR007 was absorbed slowly, with peak exposure occurring between 2.75 and 4.00 h. The elimination of GR007 was relatively rapid, with a geometric mean terminal T1/2 of 2.38, 2.22, and 3.29 h in the 30, 90, and 150 mg dose groups, respectively. The geometric mean CL/F at the three dose levels averaged between 9.21 and 10.7 L/h, the Vz/F averaged between 29.5 and 51.0 L, and the Kel averaged between 0.211 and 0.312 1/h. The key pharmacokinetic parameters are shown in Table 2.
The results showed that there was no significant difference in GR007 exposure between the sexes in the 30 mg and 150 mg groups (p > 0.05). No comparison was made between the sexes in the 90 mg group because there was only one female subject in this group. Pairwise comparisons of the major pharmacokinetic parameters between the different groups were performed by ANOVA analysis. The results suggested that the exposure-related parameters such as Cmax, AUC0–t, and AUC0–∞ differed significantly between the groups over the dose range of 30–150 mg (p < 0.01). For the parameters related to distribution and elimination, statistical analysis showed that Kel, T1/2, and Vz/F differed significantly between the three dose groups (p < 0.01).
Based on the power model assessment, for the three doses, the 90% confidence interval for the value of the fitting parameter β was 0.877–1.047 for AUC0–t and 0.626–0.982 for Cmax. Assuming that the exposure is dose proportional when the 90% confidence interval of β lies within 80–125%, it was judged that AUC0–t (but not Cmax) was dose proportional across all of the studied doses. The increase in Cmax was less than dose proportional over the dose range of 90–150 mg. For the low and medium doses, the 90% confidence interval of β was 0.933–1.192 for AUC0–t and 0.945–1.350 for Cmax; thus, again, AUC0–t but not Cmax was dose proportional for the low and medium doses.
3.3 Safety Outcomes
Thirty eligible subjects were enrolled in the study and were included in the safety analysis set; however, one subject in the 150 mg dose group did not complete the day 30 telephone follow-up. There were no clinically significant changes in vital signs, physical examinations, electrocardiograms, or injection-site reactions throughout the trial. Eight mild adverse events were reported by seven (23.3%) subjects across the whole trial. Of these AEs, two were considered to be possibly related to the study drug: a decrease in the CD4/CD8 ratio (3.33%) in the 150 mg group and a decrease in the T-lymphocyte count (3.33%), both reported by the same subject. In the 150 mg GR007 group, the levels of WBC and ANC in healthy subjects were slightly reduced (ANC, 3.50 ± 0.76 × 109/L at day 7 after GR007 administration vs. 4.02 ± 1.37 × 109/L at baseline, p > 0.05, Fig. 3; WBC, 5.37 ± 1.00 × 109/L on day 7 after GR007 administration vs. 6.23 ± 1.55 × 109/L at baseline, p > 0.05, Fig. 4). However, there was no significant difference between the three dose groups. Four AEs were judged to be probably unrelated to the study drug: one case of increased uric acid in the 30 mg group and three cases of triglyceride elevation, one in each dose group. No subject was discontinued as a result of an AE, and no dose-related trends were observed with regard to AE incidence or severity.

Absolute neutrophil count (ANC) after the administration of 150 mg GR007 to healthy subjects

White blood cell count (WBC) after the administration of 150 mg GR007 to healthy subjects
4 Discussion
Human IL-1Ra is a natural endogenous glycoprotein that competes with IL-1α or IL-1β to bind the receptor IL-1RI, blocking its downstream signaling pathways and specifically antagonizing its biological function. The binding of IL-1Ra to IL-1RI did not activate the receptor or the downstream signaling pathways [21]; thus, theoretically, IL-1Ra is very safe. The safety of rhIL-1Ra (anakinra) in clinical applications has been confirmed by a long-term safety evaluation [16]. In this study, we evaluated the safety of GR007 in healthy Chinese subjects and confirmed that GR007 had good safety and tolerability after the injection of a single dose of between 30 and 150 mg to the gluteus maximus.
The most common adverse reaction to the long-term use of subcutaneously injected anakinra as a rhIL-1Ra is an injection site reaction [22]. However, no injection site response was observed in this study, which may be because only a single injection of GR007 was administered intramuscularly. In the study, the AEs associated with GR007, as judged by the investigators, were one case of a mild decrease in the CD4/CD8 ratio and one case of a slight decrease in T-lymphocyte count in the 150 mg group. It was reported that nebulous neutropenia occurred in 2.1% of RA patients who used anakinra over a long period [14]. In a phase 2 clinical trial focusing on patients with acute stroke [23], 72 h of continuous intravenous infusion of anakinra at a dose of 2 mg/kg/h led to a 45% decrease in neutrophils and a reversible and transient 27% decrease in white blood cells. Interestingly, the adverse events associated with anakinra use—such as neutropenia—reflected the potential action of rhIL-1Ra to prevent the toxic and side effects of chemotherapy, which are mediated by blocking the cell cycle of rapidly proliferating cells such as bone marrow hematopoietic cells and intestinal crypt cells, thus arresting the cycle in the G0/G1 phase before chemotherapy [19]. This means that rhIL-1Ra can reduce the levels of leukocytes and neutrophils, and indicates that WBC and ANC are pharmacodynamic indicators. There was no significant difference in pharmacodynamic indicators between the three dose groups, which may be related to the fact that only a single injection of GR007 was carried out. According to our previous laboratory study, GR007 is most effectively administered for the treatment of toxic side effects of chemotherapy through continuous daily injection for 3–5 days before the chemotherapy [19, 20]. Further trials should monitor the pharmacodynamic indicators WBC and ANC after continuous multiple administration of GR007.
Pharmacokinetic data for anakinra (Kineret) in Western populations are already available, as it has been marketed overseas for many years. However, corresponding pharmacokinetic data for Chinese populations are yet to be reported. One of the main purposes of this study was to evaluate the pharmacokinetic parameters of GR007 following a single injection in Chinese healthy subjects. In the study, the median peak exposure times of rhIL-1Ra in the 30, 90, and 150 mg dose groups were 3.50, 2.75, and 4.00 h, respectively, and the geometric mean elimination half-lives were 2.38, 2.22, and 3.29 h, respectively. Based on the manufacturer’s description of anakinra [24], after a single subcutaneous injection of 1–2 mg/kg of anakinra into patients with RA, the plasma concentration of anakinra peaked at 3–7 h and the elimination half-life was 4–6 h. In this study, the peak exposure time and half-life of GR007 in healthy Chinese subjects were less than those in Western subjects. A possible explanation for this may be differences between the dosage forms and the modes of administration of GR007 and anakinra, as the former was administered via intramuscular injection of freeze-dried powder, whereas anakinra was supplied in prefilled glass syringes and administered as a subcutaneous injection. It is not yet clear if the ethnic differences between Chinese and Western populations play a role in the differences between GR007 and anakinra in peak time and elimination half-life. According to Yang [25], the elimination half-life of anakinra after single intravenous administration of 1 mg/kg was 2.64 h in healthy subjects, which is not much different to that of GR007. The impact of ethnic differences on the pharmacokinetics of GR007 requires further evaluation in a global clinical trial.
In the exposure analysis based on the power model assessment, AUC but not Cmax was found to be dose proportional at all of the studied doses. AUC0–t was dose proportional in human subjects, which was consistent with the pharmacokinetics of this drug in an animal model. Pre-clinical pharmacokinetic studies of GR007 showed that after a single intramuscular injection of GR007 in rats (1–16 mg/kg) and cynomolgus monkeys (0.5–4.5 mg/kg), AUC and Cmax were increased in proportion to the dose, suggesting that the single intramuscular injection of GR007 in rats and cynomolgus monkeys presented linear elimination kinetics (Zhang Y, unpublished data). In this study, Cmax was not dose proportional across all of the studied doses, as the geometric mean Cmax of GR007 in the 150 mg dose group (1530 ng/mL) was slightly lower than that in the 90 mg dose group (1640 ng/mL). A possible explanation for this behavior is that the injection volume of this group was 5 mL, which was much more than the injection volume of the 90 mg dose group (3 mL). The extra injection volume resulted in a temporary pool of GR007 in the muscle and the slow elimination of GR007. This explanation is consistent with the higher Tmax and half-life values of the GR007 in the 150 mg dose group than in the 90 mg dose group (4.00 h vs. 2.75 and 3.29 h vs. 2.22 h).
Overall, single intramuscular injection of 30, 90, or 150 mg GR007 in healthy Chinese subjects showed good safety and tolerability, and the pharmacokinetic results indicate that multiple daily injection of GR007 should be examined in subsequent clinical trials.
5 Conclusion
Single intramuscular injection of 30, 90, or 150 mg GR007 showed good safety and tolerability in healthy Chinese subjects, and the AUC was observed to be proportional to the dosage. There was no significant difference in exposure to GR007 between the male and female subjects in the 30 mg and 150 mg GR007 groups.
References
Arend WP. The balance between IL-1 and IL-1Ra in disease. Cytokine Growth Factor Rev. 2002;13(4–5):323–40.
Arend WP, Joslin FG, Massoni RJ. Effects of immune complexes on production by human monocytes of interleukin 1 or an interleukin 1 inhibitor. J Immunol. 1985;134(6):3868–75.
Kirisawa R, Fukuda T, Yamanaka H. Enzymatic amplification and expression of bovine interleukin-1 receptor antagonist cDNA. Vet Immunol Immunopathol. 1998;62(3):197–208.
Hannum CH, Wilcox CJ, Arend WP. Interleukin-1 receptor antagonist activity of a human interleukin-1 inhibitor. Nature. 1990;343(6256):336–40.
Eisenberg SP, Evans RJ, Arend WP. Primary structure and functional expression from complementary DNA of a human interleukin-1 receptor antagonist. Nature. 1990;343(6256):341–6.
Gabay C, Porter B, Fantuzzi G. Mouse IL-1 receptor antagonist isoforms: complementary DNA cloning and protein expression of intracellular isoform and tissue distribution of secreted and intracellular IL-1 receptor antagonist in vivo. J Immunol. 1997;159(12):5905–13.
Dinarello CA. Therapeutic strategies to reduce IL-1 activity in treating local and systemic inflammation. Curr Opin Pharmacol. 2004;4(4):378–85.
Deleuran BW, Chu CQ, Field M. Localization of interleukin-1 alpha, type 1 interleukin-1 receptor and interleukin-1 receptor antagonist in the synovial membrane and cartilage/pannus junction in rheumatoid arthritis. Br J Rheumatol. 1992;31(12):801–9.
Dinarello CA. Biologic basis for interleukin-1 in disease. Blood. 1996;87(6):2095–147.
Firestein GS, Boyle DL, Yu C. Synovial interleukin-1 receptor antagonist and interleukin-1 balance in rheumatoid arthritis. Arthritis Rheum. 1994;37(5):644–52.
Chomarat P, Vannier E, Dechanet J. Balance of IL-1 receptor antagonist/IL-1 beta in rheumatoid synovium and its regulation by IL-4 and IL-10. J Immunol. 1995;154(3):1432–9.
Bresnihan B, Alvaro GJM, Cobby M. Treatment of rheumatoid arthritis with recombinant human interleukin-1 receptor antagonist. Arthritis Rheum. 1998;41:2196–204.
Cohen S, Hurd E, Cush J. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46(3):614–24.
Fleischmanna RM, Schechtman JB, Bennett R. Anakinra, a recombinant human interleukin-1 receptor antagonist (r-metHuIL-1ra), in patients with rheumatoid arthritis: a large, international, multicenter, placebo-controlled trial. Arthritis Rheum. 2003;48(4):927–34.
Mansky MD, Natalie J, Dailey MD. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med. 2006;355(6):581–92.
Neven B, Marvillet I, Terrada C. Long-term efficacy of the interleukin-1 receptor antagonist anakinra in ten patients with neonatal-onset multisystem inflammatory disease/chronic infantile neurologic, cutaneous, articular syndrome. Arthritis Rheum. 2010;62(1):258–67.
Wang X, Zhu S, Qian L. IL-1Ra selectively protects intestinal crypt epithelial cells, but not tumor cells, from chemotoxicity via p53-mediated upregulation of p21(WAF1) and p27(KIP1). Pharmacol Res. 2014;82:21–33.
Xiang D, Guo Y, Zhang J. Interleukin-1 receptor antagonist attenuates cyclophosphamide-induced mucositis in a murine model. Cancer Chemother Pharmacol. 2011;67(6):1445–53.
Qian L, Xiang D, Zhang J. Recombinant human interleukin-1 receptor antagonist reduces acute lethal toxicity and protects hematopoiesis from chemotoxicity in vivo. Biomed Pharmacother. 2013;67(2):108–15.
Wang X, Gao J, Qian L. Exogenous IL-1Ra attenuates intestinal mucositis induced by oxaliplatin and 5-fluorouracil through suppression of p53-dependent apoptosis. Anticancer Drugs. 2015;26(1):35–45.
Arend WP, Gabay C. Physiologic role of interleukin-1 receptor antagonist. Arthritis Res. 2000;2(4):245–8.
Fleischmann RM, Tesser J, Schiff MH. Safety of extended treatment with anakinra in patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65(8):1006–12.
Emsley HC, Smith CJ, Georgiou RF. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76(10):1366–72.
Amgen Inc. Description of Kineret. Thousand Oaks, CA: Amgen Inc.; 2001..
Yang BB, Baughman S, Sullivan JT. Pharmacokinetics of anakinra in subjects with different levels of renal function. Clin Pharmacol Ther. 2003;74(1):85–94.
Acknowledgements
The authors thank all of the subjects enrolled in this study. We are grateful to the staff of the clinical ward in Peking University First Hospital. We also thank Dr. Xi Zhu and Pengju Wang from Shanghai Innostar Biotech Co. for the serum drug concentration measurements and PK analysis.
Author information
Authors and Affiliations
Contributions
YC, XZ, YZ, WH, and YY contributed to the design of the study. YC, XZ, XW, RX, NZ, and SZ performed the research and interpreted the data. Initial manuscript preparation was carried out by YZ and RX. The manuscript was revised and approved by RX, YZ, NZ, SZ, XW, WH, YY, XZ, and YC. YZ and WH are part-time employees of General Regeneratives (Shanghai) Limited.
Corresponding authors
Ethics declarations
Funding
This work was supported by the Shanghai Association for Science and Technology (award number: 17431901100).
Conflict of Interest
Yang Zhang and Wei Han are part-time employees of General Regeneratives (Shanghai) Limited, which sponsored the clinical trial. The other authors have no relevant conflict of interest.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Declaration of Helsinki and good clinical practice guidelines. The study was approved by the ethics committee of Peking University First Hospital (China).
Informed Consent
All subjects provided written informed consent prior to participation.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Xie, R., Zhang, Y., Zhao, N. et al. Pharmacokinetics and Safety of Recombinant Human Interleukin-1 Receptor Antagonist GR007 in Healthy Chinese Subjects. Eur J Drug Metab Pharmacokinet 44, 353–360 (2019). https://doi.org/10.1007/s13318-018-0523-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13318-018-0523-5