
Abstract
This study is unique as it compares traditional and high-resolution culture-independent approaches using the same set of samples to study the saprotrophic fungi on Vitis vinifera. We identified the saprotrophic communities of table grape (Red Globe) and wine grape (Carbanate Gernischet) in China using both traditional and culture-independent techniques. The traditional approach used direct observations based on morphology, single spore isolation and phylogenetic analysis yielding 45 taxa which 19 were commonly detected in both cultivars. The same set of samples were then used for Illumina sequencing which analyzed ITS1 sequence data and detected 226 fungal OTUs, of which 176 and 189 belong to the cultivars Carbanate Gernischet and Red Globe, respectively. There were 139 OTUs shared between the two V. vinifera cultivars and 37 and 50 OTUs were specific to Carbanate Gernischet and Red Globe cultivars respectively. In the Carbanate Gernischet cultivar, Ascomycota accounted for 77% of the OTUs and in Red Globe, almost all sequenced were Ascomycota. The fungal taxa overlap at the genus and species level between the traditional and culture-independent approach was relatively low. In the traditional approach we were able to identify the taxa to species level, while in the culture-independent method we were frequently able to identify the taxa to family or genus level. This is remarkable as we used the same set of samples collected in China for both approaches. We recommend the use of traditional techniques to accurately identify taxa. Culture-independent method can be used to get a better understanding about the organisms that are present in a host in its natural environment. We identified primary and secondary plant pathogens and endophytes in the saprotrophic fungal communities, which support previous observations, that dead plant material in grape vineyards can be the primary sources of disease. Finally, based on present and previous findings, we provide a worldwide checklist of 905 fungal taxa on Vitis species, which includes their mode of life and distribution.
Similar content being viewed by others

Introduction
Vitis (family Vitaceae) is a plant genus that includes the economically important grapes, and thus because of its importance, its pathogens have received a considerable amount of attention during the past decade (Yan et al. 2015; Chethana et al. 2017). The importance of this fruit is associated with its multiple uses; as a source of nutrition, health and medicinal value, as well as its high economical significance (Dohadwala and Vita 2009; Bokulich et al. 2014). About 90% of cultivated grapes in the world are V. vinifera, which comprises wine, as well as table grapes. This genus comprises 79 accepted species of perennial woody and herbaceous vines. There are thousands of cultivars of V. vinifera that has been grown successfully around the globe (Terral et al. 2009). Species of Vitis are valued for their decorative foliage providing ornamental value for the genus. Their ability to cover walls and arches, as well as providing shade has made them important in domestic cultivation.
Numerous diseases of grapes have been identified which reduce the yield and quality of this fruit crop (Úrbez-Torres et al. 2009). Among various pathogens known on grapevine, the damage caused by fungi is significant (Úrbez-Torres 2011). Most studies on fungal pathogens in grape have focused on their pathogenic phase, which relies on direct observation and isolations of fungal pathogens from infected grape material. Fungi that may live within the host tissue are known as endophytes and are considered to cause symptomless infections (Lane and Kirk 2012). Plant pathogenic fungi can survive by changing their biotrophic mode from pathogenic to saprotrophic or at least can remain dormant on the decaying plant materials and become active when suitable conditions for infection exist (Hoppe et al. 2016; Purahong et al. 2018). For example, Botrytis cinerea causing gray mold disease in grape is able to live as a parasite in green tissues and as a saprotroph in dead plant material (Armijo et al. 2016). Unfortunately, our knowledge on saprotrophic fungal community associated with V. vinifera is limited, especially those obtained by high-resolution culture-independent techniques. The percentage of potential fungal pathogens hidden in saprotrophic community is still unclear.
Saprotrophs are organisms that derive nourishment from dead or decaying organic matter (Hyde et al. 2007). Saprotrophs are heterotrophic organisms that break down the complex compounds of dead organisms (Deighton 2016). They play an important role as decomposers of dead organic matter in natural ecosystems by releasing enzymes from hyphal tips (Duarte et al. 2006; Bucher et al. 2004). Saprotrophic fungi can be either macrofungi (Agaricus sp., Phallus sp.) (García et al. 1998) or microfungi (Aspergillus sp., Dothiorella sp., Mucor sp., Neomassaria sp., Rhizopus sp.) (Vohník et al. 2011; Hyde et al. 2016). Hyde et al. (2007) and Purahong et al. (2018), provided evidence that some plants accommodate large numbers of saprotrophic taxa and that some might be host- or organ-specific. Other than being decomposers, saprotrophs can also provide other eco-system services, such as soil formation, defence against pathogens, as a food source and modification of pollutants (Deighton 2016). Promputtha et al. (2007, 2010) provided evidence that not only fungal pathogens, but also fungal endophytes, can switch lifestyles to saprotrophs. Thus, the study of saprotrophic fungal communities associated with V. vinifera can provide information on strict saprotrophism as well as potential endophytes and pathogens with saprotrophic ability.
Fungal species identification has traditionally been based on direct observation, microscope examination, culture dependent isolation and phylogenetic analysis (Cai et al. 2011; Hyde et al. 2010, 2017; Rastogi and Sani 2011; Fadrosh et al. 2014, Tibpromma et al. 2017). Such studies have investigated the microbial ecology of various environments (Rastogi and Sani 2011). However, it has been recognized that the actual number of microbes in nature, often exceed the number of microbes identified by traditional methods (Fadrosh et al. 2014). Traditional approaches rely on growing the organisms in media, but many of the microbes in the environment may not be cultivatable (Stewart 2012). Artificial medium typically allows growth of only a small fraction of, often fast growing organisms. Therefore, traditional techniques do not provide a total community resolution (Hoppe et al. 2016).
During the past decade, microbial research made a shift from phylogenetic analyses to experimental characterization of communities through the use of complex experimental designs (Kozich et al. 2013). This shift focused on relatively inexpensive next-generation sequencing approaches (NGS) and powerful bio-informatics tools to analyse the microbial ecology (Carraro et al. 2011; Rastogi and Sani 2011). High-throughput DNA sequencing allows us to understand the presence of microbes and how their communities are structured in complex ecosystems. Microbiome analysis is a culture-independent technique which requires a low quantity of sample, but with a high sequencing depth. The term microbiome refers to the entire habitat including the microorganisms, their genomes and the surrounding environment. It is characterized by the application of one or combinations of metagenomics, metabonomics, metatranscriptomics and metaproteomics (Marchesi and Ravel 2015). Analysis of the plant microbiome involves linking microbial ecology and the plants biology and functions, and at the same time viewing microorganisms as a reservoir of additional genes and functions for their host (Vandenkoornhuyse et al. 2015). Mycobiome refers to the fungal component in a habitat (Underhill and Iliev 2014). However, these techniques may also have disadvantages. For example, the conditions that we select to do the PCR can give us biased results. Sometimes it is difficult to understand whether the fungi identified by this technique actually exist in the natural system (Mitchell and Zuccaro 2006). Therefore, in order to obtain a better resolution in species identification, richness and distribution patterns of microbes, a combination of both approaches (i.e. traditional and culture-independent) are needed. However, we are aware of no studies that have been conducted using both these approaches.
There have been many studies of major pathogens from Vitis using both morphology and phylogeny (Úrbez-Torres et al. 2012, 2013a, b; Dissanayake et al. 2015; Jayawardena et al. 2015, 2016a; Chethana et al. 2017). Even though a number of sexual and asexual fungi have been reported on Vitis species, updated information of the taxa present in this genus is lacking. Only some have good illustrations and gene sequence data. Our knowledge on saprotrophic fungal communities associated with V. vinifera is limited and data based on high-resolution culture-independent technique is lacking. Besides the percentage of potential fungal pathogens hidden in saprotrophic community is still unknown.
In this study, we aim to (i) provide taxonomic information on the saprotrophic microfungi collected from China, Italy, Thailand and Russia, (ii) compare traditional and culture-independent approaches for characterizing the saprotrophic fungal communities associated with two cultivars of V. vinifera in China, (iii) quantify plant pathogens and endophytes hidden in the saprotrophic fungal community and (iv) provide a worldwide checklist for the fungi on Vitis species based on previous and current research.
Materials and methods
Study site, sampling and isolation of fungi
Fungal species associated with Vitis sp. were collected from China (Beijing, Sichuan and Yunnan Province), Italy (Province of Forlì-Cesena), Russia (Rostov Region) and Thailand (Chiang Sean) (Tables 1, 2). Shoots, leaves, inflorescences, bark and root samples of Vitis vinifera were used for isolation. The same set of samples from Beijing (Red Globe cultivar) and Yunnan Province (Carbanate Gernischet cultivar) were used for the mycobiome analysis to establish the fungal communities (Fig. 1). The sample sets were randomly split into two subsamples to employ the two approaches at the same time. Specimens were incubated in a moist chamber for 3–7 days at 25 °C, if they did not sporulate. Fungi were isolated by a modified single spore/conidial isolation method (Chomnunti et al. 2014) from the samples. Growth rate, colony characteristics and sexual/asexual morph morphology were determined from cultures grown on potato-dextrose agar (PDA) at 25 °C, under 12 h light/12 h dark. Fungal mycelia and spores were examined by differential interference contrast (DIC) and photographed with an axio Imager Z2 photographic Microscope (Carl Zeiss Microscopy, Germany) (Supplementary Figs. S1a–S1d, S2). Forty conidial measurements were taken for each isolate. All microscopic measurements were recorded with ZeM PRo 2012 software. Representative herbariums are deposited in the herbarium of Mae Fah Luang University, Chiang Rai, Thailand (MFU) and in Kunming, China (KIB). Representative cultures were deposited at Mae Fah Luang Culture Collection (MFLUCC), Beijing Academy of Agriculture and Forestry Sciences, China (JZB) and Kunming Culture Collection (KUMCC).

Dead V. vinifera samples at collection sites
DNA extraction, PCR amplification, sequencing and phylogenetic analysis
The methods used are presented in detail in Jayawardena et al. (2018).
Culture-independent approach: mycobiome analysis
Two cultivars were selected for this analysis; Red Globe being the table grape cultivar and Carbanate Gernischet being the Wine grape cultivar. Samples of Red Globe (RG) were collected from Yanqin District of Beijing while samples of Carbanate Gernischet (CG) were collected from Yunnan Province were used for this analysis. For each cultivar, three representative grapevine plants were sampled. The root, bark, shoot, inflorescence and leaves were homogenized and sub-sampled. For culture-independent technique of fungal communities, total DNA extraction was performed using 1 g of ground specimens using 2× CTAB method. All the DNA samples were quantitated and quality checked with the NanoDrop ND-2000C spectrophotometer (ThermoFisher Scientific, Dreieich, Germany). DNA extracts were then stored at − 20 °C for further analysis. For fungal Illumina sequencing, we targeted the Internal Transcribed Spacer 1 (ITS1) region of ribosomal RNA gene cluster. ITS1 was amplified with the forward primer ITS5-1737 (GGAAGTAAAAGTCGTAACAAGG) and reverse primer ITS2-2043R (GCTCGCTTCTTCATCGATGC) (White et al. 1990). The PCR reaction was performed in a 50 ml volume that contained approximately 10 ng of DNA, ExTaq buffer, 0.2 mM of dNTPs, 0.2 mM of each primer, and 2 units of ExTaq DNA polymerase. The cycling consisted of an initial denaturing at 94 °C for 30 s, followed by 25 cycles of denaturing at 94 °C for 30 s, annealing at 54 °C for 1 min and extension at 72 °C for 2 min, and a final extension at 72 °C for 8 min. All PCR reactions were carried out with Phusion® High-Fidelity PCR Master Mix (New England Biolabs Inc. Ipswich, MA, USA). The PCR products were mixed with same volume of 1× loading buffer (contained SYB green) and then operated electrophoresis on 2% agarose gel for quality detection. Only samples with bright main strip between 400–450 bp were chosen for further experiments. The qualified PCR products were mixed in equidensity ratios. Then, mixture PCR products were purified with Qiagen Gel Extraction Kit (Qiagen, Germany) following the manufacture’s protocol.
Sequencing libraries were generated using TruSeq® DNA PCR-Free Sample Preparation Kit (Illumina, San Diego, CA, USA) following manufacturer’s recommendations and index codes were added. The library quality was assessed on the Qubit® 2.0 Fluorometer (Thermo Scientific) and Agilent Bioanalyzer 2100 system. At last, the library was sequenced on an IlluminaHiSeq2500 platform and 250 bp paired-end reads were generated.
Paired-end reads were assigned to samples based on their unique barcode and truncated by cutting off the barcode and primer sequence. Paired-end reads were merged using FLASH (V1.2.7, http://ccb.jhu.edu/software/FLASH/) (Magoč and Salzberg 2011). Quality filtering on the raw tags was performed under specific filtering conditions to obtain the high-quality clean tags (Bokulich et al. 2013) according to the QIIME (V1.7.0, http://qiime.org/index.html) quality controlled process (Caporaso et al. 2010). The tags were compared with the reference database (Unite Database, https://unite.ut.ee/) using UCHIME algorithm (UCHIME Algorithm, http://www.drive5.com/usearch/manual/uchime_algo.html) to detect chimera sequences (Edgar et al. 2011), and then the chimera sequences were removed (Haas et al. 2011). Then the Effective Tags were finally obtained. Sequences analysis was performed by Uparse software (Uparse v7.0.1001, http://drive5.com/uparse/) (Edgar 2013). Sequences with ≥ 97% similarity were assigned to the same OTUs. Representative sequence for each OTU was screened for further annotation. For each representative sequence, the Unite Database (https://unite.ut.ee/) (Kõljalg et al. 2013) was used to annotate taxonomic information based on Blast algorithm, which was calculated by QIIME software (Version 1.7.0) (http://qiime.org/scripts/assign_taxonomy.html). In order to study phylogenetic relationship of different OTUs, and the difference of the dominant species in different samples (groups), multiple sequence alignment were conducted using the MUSCLE software (Version 3.8.31, http://www.drive5.com/muscle/) (Edgar 2004).
All OTU abundance information was normalized using a standard of sequence number corresponding to the sample with the least sequences (45, 246 sequences). From the data set, rare OTUs (singletons), which could have potentially originated from sequencing errors (Kunin et al. 2010), were removed. We used a Mantel test based on Bray–Curtis distance measure with 999 permutations to assess the correlation between the whole matrix and a matrix excluding the rare OTUs (Hammer et al. 2001; Hoppe et al. 2016). The results indicated that the removal of rare OTUs from the fungal communities had no effect (RMantel = 1.000, P = 0.002). Subsequent analysis of alpha diversity and community composition were all performed basing on these normalized rare OTUs removal data. The fungal ITS rDNA genes Illumina sequencing data are deposited in the NCBI under the BioProject No PRJNA437133.
Statistical analysis
Mycobiome analysis
All datasets related to fungal OTU richness were tested for normality and equality of variances using the Jarque–Bera test. To assess the coverage of the sequencing depth, individual rarefaction analysis was performed for each sample using the “diversity” function in PAST (Hammer et al. 2001). In this work we used observed fungal OTU richness and Shannon diversity index as the measures for fungal diversity. The difference in fungal OTU richness between the two deadwood species was compared using a two-sample t test in PAST. To visualize the fungal community compositions, we used non-metric multidimensional scaling (NMDS) analysis based on the Bray–Curtis dissimilarity index calculated PAST. Similarity Percentages (SIMPER) analysis using PAST was used to obtain the identity and relative abundances of the fungal taxa that contributed to 92.92% of the observed pair-wise variation in the fungal community composition due to different V. vinifera cultivars. To accounting for the effect of locations when compared the fungal community compositions of the two grape cultivars which were collected from different locations, we eliminated all location specific fungal OTUs (87 OTUs). We finally retained 139 OTUs for the community composition analysis using NMDS based on the Bray–Curtis dissimilarity. The results from these reduced datasets were highly consistent compared with the total datasets (Supplementary Fig. S3a, b). Potential fungal functional groups were identified using the online Guilds application tool: FUNGuildb (Nguyen et al. 2015). The ITS1 fragments were extracted from both the Sanger sequencing (traditional) and Illumina sequencing datasets using ITS1. The output showed that both datasets have the ITS1 region except culture sequences of the genus Neopestalotiopsis (9 sequences). The sequence similarity based comparison was performed using the cd-hit-est-2d algorithm at 90% similarity level for a genus level comparison.
Diversity analysis
Taxa were recorded as either present or absent from each sample. Occurrence of a fungus was designated based on the presence of a particular fungus on the host samples. Percentage occurrence of a taxon on one sample was calculated using the following formula (Tsui et al. 2001; Yanna and Hyde 2002; Wang et al. 2008):
Following diversity indices were calculated using the R software for the two cultivars and the habits.
-
(i)
$$ {\text{Shannon}}{-}{\text{Wiener's Index (H)}} = \sum p_{i} \ln p_{i},$$
where p i is the frequency of fungal species I occurring on a specific sample (Begon et al. 1993; Wong and Hyde 2001; Wang et al. 2008).
-
(ii)
$$ {\text{S}}{\o}{\text{rensen's index of similarity (S)}} = 2c/(a + b), $$
where a is the total number of species on host A, b is the total number of species on host B and c is the number of species on both host. Similarity is expressed with values between 0 (no similarity) and 1 (absolute similarity) (Wang et al. 2008).
-
(ii)
Compiling the checklist
The checklist is based on, articles in referred journals, Index to Saccardo’s Sylloge fungorum, Petrak’s Lists, Index of Fungi, graduate student theses, books, and web-based resources such as annual reports on this host and the SMML database (https://nt.ars-grin.gov/fungaldatabases/) (latest accessed 14-9-2017). The mode of life, such as pathogen, endophyte or saprotroph is listed. The checklist includes species names, family, life modes, disease name if any and locality. The current name is used according to Index Fungorum (2018) and Wijayawardene et al. (2017) and the classification follows Wijayawardene et al. (2018). Genera and species are listed in alphabetical order. Identification confirmed by molecular data is marked with an asterisk (*). In some cases, the host name given in the original citation was changed to be consistent with current taxonomy. In a few cases, neither the species cited nor a proper synonym was identified and the species name was used as originally cited.
Results
Species identified from fresh collections based on morphology and phylogeny (traditional method)
Fungal saprophytic diversity and community composition of the two grape cultivars: traditional method
Examination of decaying leaves, shoots, inflorescence, berries, root and bark of two cultivars of V. vinifera from China yielded 461 collections for the Red Globe variety and 180 collections for Carbanate Gernischet. The Red Globe variety had higher species richness (41) than the Carbanate Gernischet variety (23), however the Shannon diversity was not significantly different (Table 3). The majority of the culturable saprotrophic fungi were ascomycetes. However, there were two species belonging to Agaricomycetes and one species belonging to Oomycota incertae sedis. Thirty genera and 45 taxa were identified based on morphology and phylogenetic sequence data. From the identified isolates, 32.6% were Sordariomycetes, 26.1% Dothideomycetes 19.7% Eurotiomycetes, 6.5% Mucoromycetes, 4.4% Agaricomycetes, 2.2% Leotiomycetes and 2.2% of Oomycota incertae sedis. There were four taxa belonging to Zygomycota incertae sedis, which we were unable to identify. The identified Sordariomycetes belonged to Bionectriaceae (6.7%), Diaporthaceae (6.7%), Glomerellaceae (20%), Hypocreaceae (20%), Nectriaceae (13.3%), Schizoparmaceae (6.7%), Stachybotryaceae (13.3%) and Sporocadaceae (13.3%). Dothideomycete isolates belonged to Botryosphaeriaceae (16.7%), Cladosporiaceae (16.7%), Didymellaceae (33.3%) and Pleosporaceae (33.3%). The rest of the isolates belong to Mucoraceae (6.5%), Peniophoraceae (2.2%), Pythiaceae (2.2%), Rhizopodaceae (2.2%), Sclerotiniaceae (2.2%) and Trichocomaceae (17.4%). Among those 45 taxa, we found 19 species that were common on both cultivars: Actinomucor elegans, Alternaria alternata, Aspergillus niger, A. aculeatus, Cladosporium cladosporioides, Cladosporium sp., Clonostachys rosea, Coniella vitis, Diaporthe eres, Fusarium oxysporum, Mucor racemosus, Penicillium terrigenum, Phoma medicaginis, Rhizopus oryzae, Talaromyces amestolkiae, T. pinophilus, T. purpurogenus and T. harzianum. The Sørensen’s index of similarity of the two grape cultivars was 0.58. We have identified 45 taxa to species level, although in six cases the identification is only to genus level due to lack of enough molecular data. Taxa were identified using both morphology and molecular techniques. Identified species are listed in Tables 1 and 2 (Supplementary Figs. S1a–d, S2).
Fungal saprophytic diversity and community composition of the two grape cultivars: culture-independent technique
Bioinformatics processing of the sequence data sets
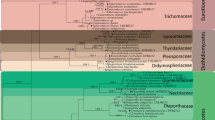
A total of 638,146 quality-filtered fungal ITS reads were obtained after removal of chimeric and the unique tag (3703 sequences) sequences. After normalizing all data sets to a smallest sequence read (45, 246 sequences) and removing all rare taxa, the final analyse data sets contained 226 fungal OTUs. Phylogenetic trees for the top 20 species in different samples of the two cultivars Carbanate Gernischet and Red Globe of Vitis vinifera are given in Fig. 2. With the high number of sequence reads per sample obtained in this study, the sample-based rarefaction curves almost reached saturation for all samples (Fig. 3a). We used the observed OTU richness and Shannon diversity directly for further analyses.

Phylogenetic tree of top 20 species in different samples of the two cultivars Carbanate Gernischet and Red Globe of Vitis vinifera

Rarefaction curves (a), and Venn diagrams show distribution of OTUs across different samples (1–3): (b) in both Carbanate Gernischet (CG) and Red Globe (RG) cultivars, (c) only Carbanate Gernischet and (d) only Red Globe
Fungal saprotrophic OTU diversity and distribution in the two cultivars of Vitis vinifera
Diverse fungi colonized the debris samples derived from Carbanate Gernischet and Red Globe cultivars. Fungal OTU richness was not significantly different between the two V. vinifera cultivars tested in this study, ranging from 122–137 (127.33 ± 4.84 (mean ± SD); Carbanate Gernischet) and 116–141 (126.33 ± 7.54 (mean ± SD); Red Globe) (t = 0.11, P = 0.916). Shannon diversity also showed a similar trend ranging from 1.98–2.35 (2.16 ± 0.11 (mean ± SD); Carbanate Gernischet) and 1.78–1.98 (1.86 ± 0.06 (mean ± SD); Red Globe) (t = 2.41, P = 0.07). In total we detected 226 fungal OTUs with 176 and 189 belonging to the cultivars Carbanate Gernischet and Red Globe, respectively. There were 139 Fungal OTUs shared between the two V. vinifera cultivars and 37 and 50 OTUs were specific to Carbanate Gernischet and Red Globe cultivars. When we took each replicate into account, we detected only moderate proportion of fungal OTUs shared across different replicates (31–44%, 51 OTUs, Fig. 3b). Distributions of fungal OTUs across replicates for the two cultivars and for each specific cultivar are shown in Fig. 3b–d.
Fungal saprophytic community composition: culture-independent technique
The NMDS ordination plot and SIMPER analysis revealed distinct fungal communities in the two cultivars of V. vinifera samples (Table 4 and Supplementary Fig. S3a, b). Overall, fungal community composition of the two cultivars had the overall average dissimilarity of 94.29% (based on Bray–Curtis distance measure) and 30 fungal OTUs mostly responsible for differences in fungal community composition were all together accounting for 94.41% of the overall average dissimilarity (Table 4). The difference in fungal community composition between the two cultivars of V. vinifera was detected across different taxonomic levels (Supplementary Figs. S4, S5).
In Carbanate Gernischet, members of Ascomycota were commonly detected accounting for 77% (46% Sordariomycetes, 19% Eurotiomycetes and 7% Dothideomycetes) of total sequences in this cultivar followed by unidentified phylum (23%; Fungal OTU-7) and Basidiomycota and Zygomycota (less than 0.1%). In Red Globe, almost all sequences were assigned to Ascomycota (97%; 51% Eurotiomycetes, 42% Sordariomycetes, and 3% Dothideomycetes) followed by Basidiomycota (3%; 1% Agaricomycetes and 1% Tremellomycetes) and unidentified phylum (Fungal OTU-7) and Zygomycota were negligible (altogether less than 0.5%). Phylogenetic tree for the abundance at genus level using the top 35 genera detected in the two cultivars are shown in Fig. 4. The difference between the fungal community composition of the two cultivars of V. vinifera were clearly demonstrated at OTU level: Trichothecium roseum OTU-1, Fungal-OTU-7, Aspergillus piperis OTU-5 and Nectriaceae OTU-3 were commonly detected (10–20%) in Carbanate Gernischet, but almost absent in Red Globe (represented by Aspergillus OTU-11, Ilyonectria macrodidyma OTU-4, Aspergillus cibarius OTU-2 and Diaporthaceae OTU-10; 11–19%; Table 4).

Abundance phylogenetic tree at genus level (top 35 genera) in two cultivars [Carbanate Gernischet (CG1-3) and Red Globe (RG1-3)] of Vitis vinifera
Using presence/absence data, we found that for both Vitis vinifera cultivars Ascomycota (Carbanate Gernischet = 151 OTUs and Red Globe = 162 OTUs) was the richest OTU phylum followed by unidentified phylum, Basidiomycota (11–14 OTUs) and Zygomycota (2 OTUs). Patterns of the richest OTU classes and orders were similar for both cultivars: Sordariomycetes (Hypocreales (Carbanate Gernischet = 32 OTUs and Red Globe = 39 OTUs), Sordariales (Carbanate Gernischet = 13 OTUs and Red Globe = 13 OTUs), Microascales (Carbanate Gernischet = 10 OTUs and Red Globe = 11 OTUs)], Eurotiomycetes (Eurotiales, Carbanate Gernischet = 41 OTUs and Red Globe = 38 OTUs) and Dothideomycetes (Pleosporales, Carbanate Gernischet = 7 OTUs and Red Globe = 7 OTUs).
Comparing and matching of traditional and culture-independent approaches
Several commonly detected fungal genera (Aspergillus, Clonostachys and Fusarium) were detected in both approaches. However, there are many highly or frequently detected genera in the mycobiome that were not detected in the traditional method. These include Acrostalagmus, Aureobasidium, Ceratobasidium, Chrysosporium, Ilyonectria, Lasiodiplodia, Microascus, and Trichothecium (Supplementary Table S1). Some frequently isolated fungi, especially the fast growing ones (Rhizopus and Mucor) were not detected in mycobiome analysis.
ITS sequences obtained from both traditional and culture-independent methods were compared using the query and cluster cover. This showed that the saprotrophs detected from the two approaches are consistent in most cases. However, in few cases we found inconsistent identifications which have arisen from the lower level of taxonomic assignment in the culture-independent (amplicon sequencing) as compared with traditional approaches. For example, Diaporthe eres and Alternaria identified via the traditional approach were identified as Diaporthaceae and Pleosporaceae in the culture-independent analyses. A mismatch was also found between Albifimbria viridis and Myrothecium sp., which are classified in the same order (Hypocreales). The sexual morph genus Talaromyces was matched with its potential asexual morph (Penicillium). We found that twelve taxa detected from traditional method form a cluster (91–100% similarity) with 25 fungal OTUs from the culture-independent methods (Table 5). We were able to assign 25 OTUs from NGS: 20 OTUs to genus and 5 OTUs (similarity 99–100%) to species level respectively (Table 5). We removed two OTUs as singletons (Botryosphaeria OTU-178 and Ascomycota OTU-213) as they were detected only once. However, in the direct matching of ITS sequences, these fungal OTUs showed 97 and 100% similarity to Botryospaeria dothidea and Coniella vitis, respectively. The other fungi that we were able to identify to the species level are Aspergillus niger, Clonostachys rosea, Botrytis cinerea, and Albifimbria viridis. Most of the frequently detected genera in the traditional approach (i.e. with relative abundance higher than 5%; Alternaria, Clonostachys, Fusarium) were also detected in culture-independent approach. Rhizopus sp. and Talaromyces sp. were frequently detected in the traditional approach, but exhibited low relative abundances or disappeared in the culture-independent approach.
Fungal functional groups identified using traditional and culture-independent approaches
Among the 45 identified taxa based on traditional method, 17 are well known pathogens on V. vinifera causing severe yield as well as economic loss to viticulture around the world (Table 2). Six species of secondary pathogens of V. vinifera were also identified in this study. Most of the pathogens tend to survive or overwinter on dead plant material as saprotrophs and act as the primary inoculums once the conditions are favourable (Armijo et al. 2016).
In total, 143 fungal OTUs (63% of total fungal OTUs) were successfully assigned for their functions (Supplementary Table S1). We identified six functional groups of fungi associated with dead materials of V. vinifera: saprotrophs, plant pathogens, endophytes, fungal parasites–saprotrophs (mycoparasites–saprotrophs), ectomycorrhizae and animal pathogens. The fungal community was dominated by saprotrophs (102 OTUs) and plant pathogens (22 OTUs), which accounted for 71% and 15% of the function assigned to fungal OTUs in this study. Clonostachys, Lasiodiplodia and Trichothecium, were the most commonly detected plant pathogen genera with relative abundances 1–10%. Botrytis sp., an important fungal pathogen in grape, was also detected with low relative abundance. Endophytes together with endophyte–saprotrophs and endophyte–plant pathogens (9 OTUs) contributed little and most OTUs were detected with low relative abundance, except, Acrostalagmus luteoalbus. All fungal OTUs with their potential functions are listed in Supplementary Table S1.
Checklist of fungi on Vitis
Nine-hundred and six fungal taxa have been reported on Vitis species and are listed in Table 6, although the actual number of fungal taxa associated with this host is likely much higher. It is not possible to reconfirm all previous reports by re-examining collections to confirm their identities. In many cases no fungarium material is linked to the reports, while examining nearly 900 specimens would be an almost impossible task. Even if it was were possible, it would most likely be futile, since molecular data would be needed to establish correct names. This is extremely difficult based on the presently available techniques and not permitted by many fungaria. Most of the 905 taxa reported from Vitis species do not have sequence data. Therefore, recollecting and sequencing these taxa are essential to establish and accurate species list associated with Vitis species.
Discussion
Before the advent of molecular data in taxonomy, studies on the fungi on Vitis were based on traditional methodology and have resulted in hundreds of records of fungi from this host genus (Table 6). Most recent studies have been related to pathogens that affect grape yield and production (Úrbez-Torres et al. 2012, 2013a, b; Dissanayake et al. 2015; Liang et al. 2016; Jayawardena et al. 2015, 2016a; Yan et al. 2015; Chethana et al. 2017) and have resulted in well-resolved taxonomy as they have used molecular data. However, studies on saprobes using molecular data and culture-independent techniques have not been used to identify the fungi on Vitis to date. In this study, we therefore provide the first work comparing saprobes on Vitis sp. using both traditional and culture-independent approaches, with well-resolved taxonomic identifications based on molecular analyses. The taxa derived from both approaches are compared as the same samples were used in the study. We have also established the saprotrophic communities associated with both wine and table grapevine cultivars and demonstrate cultivar specific communities for each grapevine cultivar. A checklist of fungi of Vitis is also provided which is an important resource for viticulture.
Microfungi collected from China, Italy, Russia and Thailand
Sixty-seven saprotrophic taxa from 46 genera were identified in this study (Table 1). Using traditional methodology and analyses of molecular data, we identified two new species, and 41 new host or distribution records for V. vinifera. Taxonomic details, descriptions, photographic plates and phylogenetic analyses are provided in Jayawardena et al. (2018). Some of these genera have a wide distribution. For example, botryosphaerious and Colletotrichum taxa have a wide distribution. These taxa are well-known pathogens and can be spread to other countries undetectable through the exportation of rootstocks. Some genera are only known from one or two countries. This may be due to the lack of data on the fungi associated with this host.
Comparisons of traditional and culture-independent approaches for characterizing the saprotrophic fungal communities associated with two cultivars of Vitis vinifera
Most previous studies on fungi on grapevine have relied on traditional approaches (Table 6). Some recent identification of isolated taxa have incorporated analyses of ITS sequence data (Guo et al. 2003; Promputtha et al. 2007), but this was shown not to be accurate (Ko et al. 2011). The most recent studies on fungal pathogens of grapevine have incorporated multigene analysis to accurately resolve taxa (Dissanayake et al. 2015; Jayawardena et al. 2015; Yan et al. 2015; Chethana et al. 2017).
Most previous studies did not address the total community of fungi on Vitis vinifera. Pancher et al. (2012) carried out an extensive study on endophytes on this host, showing that how various anthropic and nonanthropic factors shape microbial communities. There have been extensive studies on the disease causing agents with more than 150 taxa known to cause various diseases of grapevine. For example, Colletotrichum species cause grape ripe rot of Vitis vinifera worldwide (Jayawardena et al. 2016b). There have however, been no investigation on the saprobes of grapevines using molecular identification and there has been no study using mycobiome analysis to reveal saprotrophic communities. The study of saprotrophs is important, as they not only decay dead leaves and branches, thus beneficial recyclers, but they may also become pathogens when conditions are suitable.
This study therefore fills this void by establishing the saprotrophic fungi on Vitis vinifera using both traditional and culture-independent approaches. In this study we did not obtain similar results from the two methods. In the traditional method, 45 species belonging to 30 genera were identified (Table 2), while in culture-independent method 226 OTUs’ and 72 genera were identified. Even though we isolated directly from the fruiting bodies, some fungi were not able to grow on media. Several single spore isolations were unsuccessful. This may be due to the availability of nutrient content, pH, temperature, and presence of inhibitors and the time of incubation. The number of isolates obtained was less than the actual fungal community and can be misinterpreted (Hugenholtz et al. 1998). These conditions make it difficult to accurately identify and document the vast number of unrecognized taxa (Lücking and Moncada 2017). For example, in this study the total identified taxa from the traditional method were 45. Therefore, to overcome the constraints of traditional methods, culture-independent techniques are proposed as an alternative technique (Hoppe et al. 2016).
The aim of environmental sequence nomenclature is to place names of species of fungi that would otherwise be left undescribed (Lücking and Moncada 2017). These techniques can provide sequence reads almost 1000 times more than the traditional DNA sequencing methods (Lücking and Moncada 2017). Lücking and Moncada (2017) showed that a formally recognized unnamed lichenicolous basidiomycete can be considered as a new genus, with seven new species, although there is no physical type specimens are available. These authors also suggested that this would allow the recognition of thousands of species of voucher less taxa detected through environmental sequencing techniques. However, there are several constrains to NGS methods. DNA may not be recovered from all genotypes and the results of NGS can be biased towards the most abundant organisms at the time of sampling (Ward et al. 1990). The reason for this is that the relative abundance (Fig. 5) of microbial species in a natural habitat is rarely equal. Usually, with a few species being predominant among a larger group of common species makes it difficult to identify the species that are actually present. NGS are mainly based on analysis of ITS regions (Schoch et al. 2012). However, due to the high variability of ITS regions (ITS1 and ITS2), reliable sequence alignments are difficult to obtain for some fungal taxa. Therefore, this method is not reliable for species level identification. The identification levels are usually reported at the genus level or even higher taxonomic levels, such as family or order (Purahong et al. 2018). Another constraint of NGS is that the correspondence of OTU with species can be unreliable. OTUs are defined based on the similarity threshold, usually with a 97% (Sneath and Sokal 1973). However, some species have genes that are 97% similar, which will result in merged OTUs containing multiple species. In the same way, a single species may have paralogs that are < 97% similar, causing the species to be split across two or more species. Some identified clusters, even when a majority, may be false, due to the artifacts including reading errors and chimeras (Sneath and Sokal 1973). Assessing species richness and diversity of a microbial community using culture-independent method (rarefaction curves), suggests that OTUs are observations of organisms with ‘negligible error’. Also, it suggests that the number of reads correlates well with the total number of individuals present in the community. However, if the majority of OTUs are experimental artifacts, the traditional species richness estimations cannot be applied. The measures between sample variations will tend to reflect differences in artifact frequencies rather than biological differences (Sokal and Sneath 1963).

Relative abundance of the top 10% phylum from different samples of the cultivars Carbanate Gernischet and Red Globe of Vitis vinifera
Artifacts can be occurred due to several reasons. PCR amplification steps can be affected by preferentially/differentially that can hinder the detection of some genotypes when analysing bulk DNA extracts from a substrate (Kanagawa 2003). Primer mismatches, a lower rate of primer hybridization, occurrence of heteroduplexes and chimeric amplicons can generate additional signals that do not correspond to the genotypes in the same samples (Suzuki and Giovannoni 1996; Kanagawa 2003). Also, the analysis of fungal rRNA genes limits identification to the genus or family level (Anderson and Cairney 2004).
Dissanayake et al. (2018) in her study using paired-end Illumina sequencing with 55, 822 high quality sequences per endophyte sample (saturated rarefaction curves for all samples) revealed 59 OTUs (the majority containing genera level identification) that were similar to genera revealed by the traditional method (28 species).
Traditional versus culture-independent methods: can matching of these two approaches enable us to identify correct fungal taxonomic information at genus and species levels?
In this study, taxa (OTUs) of Aspergillus, Botrytis, Cladosporium, Clonostachys, Fusarium, Penicillium, Phoma and Talaromyces were identified using both traditional and culture-independent approaches. Some fast growing fungi may be dominant in the culture plates, even though in the natural habitat they may be minorities. Many hyphomycetes tend to grow faster than the other groups of fungi. So, they may suppress the growth of other important, dominant fungi. The fast growing fungi (hyphomycetes) identified in the traditional method such as Mucor and Rhizopus were not recognized in the culture-independent method. The majority of the genera identified in the traditional method are phytopathogens, while in the culture-independent method the majority are saprotrophs. In the traditional method using both morphological and molecular approaches, we were able to identify many taxa to species level, although in six cases the identification is only up to the genus level due to lack of data. We have generated a phytogenetic tree for the genus Colletotrichum using the strains identified in the traditional approach as well as the OTU identified in the culture-independent method. OTU-234, was identified as Glomerellaceae sp. in the culture-independent approach. The blastn result of OTU-234 in NCBI shows 100% similarity to many strains of C. gloeosporioides. Two-hundred and fifteen basepairs of OTU-234 were used in the alignment (Supplementary Fig. S6). In the phylogenetic tree constructed using the ex-type strains of gloeosporioides complex and the truncatum complex, OTU-234 cluster within the truncatum complex, closely to C. curcumae (Supplementary Fig. S7). This example also provides evidence that the NGS sequences is not reliable to identify an organism to the species level. However, in the culture-independent technique we were able to identify 90 out of 226 OTUs to species level, and the rest were identified to genera or family level. These may not be correctly identified as in general NGS fungal taxa identification may be only accurate to the genus level (Purahong et al. 2018), which suggests that the sequence data from the culture-independent approach is inadequate to accurately identify species. The overlap between the two methods in identifying the taxa to the species level is negligible.
Matching between the traditional and culture-independent data allows us to have a better understanding concerning the functional information of the fungal OTUs resulting from culture-independent methods. Next generation sequencing often results in sequences that are associated with taxa, which have not been reported in previous studies (Tejesvi et al. 2010; Ko et al. 2011; Taylor et al. 2016). However, as most of these OTUs are identified to genus or family level, it makes it difficult to relate whether these are actually correctly identified and whether the use of this method is important. In the preparation of the checklist of fungi on Vitis species, the authors had to eliminate most of the taxa that were identified using NGS, as those data can be unreliable. In our study, we compared the sequence similarity between the two cultivars using a 90% similarity of ITS1 sequence data, followed by a manual BLAST based identification of the respective OTUs. We considered the ITS similarity at 98–99% as the same species (Garnica et al. 2016; Jeewon and Hyde 2016). In this criterion, identification of genera can be bias/difficult as some of the data in databases have mistakes or they may be inaccurate. The increase of the ITS similarity to 99–100% can give us better and reliable identification of the species. However, ITS sequence data alone will not be able to identify the complexes genera such as Colletotrichum and Diaporthe to their species level. For a better resolution of these genera protein coding gene regions are required.
Can direct matching between traditional and culture-independent methods help to identify the rare taxa?
Our results show that some singletons, which were usually removed as artifacts or errors of the NGS may actually be real OTUs. In this study, we found two OTUs (Botryospaeria OTU-178 and Ascomycota OTU-213) as singletons and removed them from the analysis. However, with direct matching, we found that these OTUs are Botryospaeria dothidea and Coniella vitis. Therefore, we can assume that not all the singletons are artifacts and matching between traditional and culture-independent methods can help to identify the real rare taxa in the fungal community.
Potential effect of grape cultivars (table grape (Red Globe) and wine grape (Carbanate Gernischet) on fungal saprotrophic community composition and richness
Another aspect of this study was to study whether there is any difference in the fungal communities based on cultivars. In the present study, traditional and culture-independent approaches allows the identification of potential roles of the saprotrophs in the two grapevine cultivars. In this study we identified more than 10 main and important fungal pathogens of grapevine using both methods. With the evaluation of both community composition and community diversity we were able to identify that the fungal communities of the two grape cultivars appear to be different. Alternaria vitis, Albifrimbria viridis, Bipolaris maydis, Botryosphaeria dothidea, Botrytis cinerea, Colletotrichum hebeinse, C. truncatum, C. viniferum, Didymella pomorum, Dothiorella sarmentorum, Epiccocum nigrum, Fusarium sp., Mucor circinelloides, Paraphoma chrysanthemicola, Neopestalotiopsis clavispora, Stagonosporopsis sp.1, Minimedusa sp., Peniophora sp., Penicillium brevicompactum and P. citrinum were recorded only from Red Globe cultivar while Albifimbria verrucaria, Neopestalotiopsis vitis, Pythium amasculinum, Stagonosporopsis sp.2, Trichoderma lixii and Septoriella allojunci were recorded from Carbanate Gernischet cultivar in the traditional method. In the culture-independent approach, Acremonium chrysogenum (OTU-195), Apodus sp. (OTU-235), Ascomycota (OTU-80, 99, 182, 222, 253), Aspergillus sp. (OTU-116, 199), Candida mucifera (OTU-227), Cylindrocarpon sp. (OTU-171), Dactylellina phymatopaga (OTU-58), Davidiella tassiana (OTU-86), Deroxomyces sp. (OTU-225), Fungal (OTU-245, 254), Fusarium cf. dimerum (OTU-162), Helotiales (OTU-91), Hypocreales (OTU-65), Kernia nitida (OTU-184), Kernia pachypleura (OTU-211), Lecanicilium dimorphum (OTU-101), Lentinus squarrosulus (OTU-249), Lophiostoma sp. (OTU-142), Microascales (OTU-138), Metarhizium pinghaense (OTU-210), Myceliophthora fergusii (OTU-198), Myrothecium sp. (OTU-238), Nectriaceae (OTU-97, 194), Papulospora equi (OTU-148), Phialosimplex caninus (OTU-187), Psathyrellaceae (OTU-145), Pseudallescheria angusta (OTU-183), Pyronemataceae (OTU-237), Remersonia sp. (OTU 108), Sordariomycetes (OTU-128), Sordariales (OTU-196, 200, 214), Xylaria sp. (OTU-159) were found only in association with the Red Globe cultivar while Acremonium sp. (OTU-188), Apllosporella yalgorensis (OTU-85), Arachnomyces kanei (OTU-170), Aspergillus melleus (OTU-143), Aspergillus wentii (OTU-175), Cadophora luteo-olivaceae (OTU-146), Ceratobasidiaceae (OTU-219), Chaetomium carinthiacum (OTU-177), Chysisporium lobatum (OTU-150), Cladosporium grevilleae (OTU-121), Dothideomycetes (OTU-140), Eurotiales (OTU-76), Fungal (OTU-82, 104, 165), Gymnascella aurantiaca (OTU-151), Hansfodia sp. (OTU-232), Hypocreales (OTU-49, 92, 202), Lasiophaeriaceae (OTU-189), Leptosphaeria sp. (OTU-205), Magnoporthaceae (OTU- 141, 149), Microascales (OTU-114), Microascus sp. (OTU-185), Microdochium sp. (OTU-225), Nectriaceae (OTU-218), Penicillium ilerdanum (OTU-163), Penicillium neocrassum (OTU-168), Podospora communis (OTU-190), Scopulariopsis sp. (OTU-164), Sordariales (OTU-133), Spiromastix princeps (OTU-139), Thielavia basicola (OTU-155), Trichomaceae (OTU-156, 166) were recorded on from Carbanate Gernischet cultivar.
The difference of the two fungal communities can be due to the geographic variation of the cultivars. This can be a result from the interactions with specific V. vinifera varieties and its soil and climatic conditions (Bokulich et al. 2014). Red Globe cultivar was collected in Beijing, which is a region in North of China and Carbanate Gernischet cultivar was collected from Yunnan which is in the southern part of the country. The difference between the fungal communities in regions may be a function of a neutral process, where these different communities established by chance and lack of species dispersal allows these communities to persist (Martiny et al. 2006). This difference can also be due to the Baas Becking hypothesis, which states that there is no limit to the range of species but that selection sorts these species and defines community composition and diversity in any one area (Hanson et al. 2012). Climate can also co-relates with differences in fungal communities in China, as one moves North up in China, the climate becomes increasingly cold and dry so the pattern of lower fungal species richness in the northern most regions hints that selection might have a role in determining these patterns.
Plant pathogens and endophytes in the saprotrophic fungal community
Species richness and distribution patterns of saprotrophic fungi in a vineyard can provide important insights into the roles of each fungal group for the stability and functioning of its respective ecosystem (Kubartova et al. 2012). However, knowledge of saprotrophic fungi associated with grapevine is very much limited. In this study, we identified 17 primary and six species of secondary pathogens of grapevine as saprobes using the traditional method, while 27 OTUs were identified as both primary and secondary pathogens from dead material of Vitis vinifera in the culture-independent method.
Species of Alternaria are responsible in causing berry rots, raisin molds and rots as well as pedicel and rachis diseases (Barbe and Hewitt 1965; Gonzalez and Tello 2011; Tao et al. 2014, Ariyawansa et al. 2015) and also considered as wound and secondary invaders. Alternaria alternata and A. vitis were isolated in this study. Aspergillus is a causal agent of berry rots as well as a wound and secondary invader (Hewitt 2015). In our study A. aculeatus and A. niger were recorded using the traditional method, while A. aculeatus was also recorded from culture-independent method. Botryosphaerious taxa are well-known to be associated with grapevine canker and die back (Úrbez-Torres et al. 2012, 2013a, b). In our study we identified Botryosphaeria dothidea and Dothiorella sarmentorum as saprotrophs using traditional methodology. Lasiodiplodia was recorded in the culture-independent method.
Botrytis is another genus that we obtained in both traditional and independent approaches. Botrytis cinerea is a pathogen of grapevine causing Botrytis bunch rot and blight all over the world (Fournier et al. 2013; Hyde et al. 2014; Javed et al. 2017). Cladosporium was also recorded in both approaches. Species of this genus cause minor foliage diseases of grapevine, as well as bunch rots (Bensch et al. 2015). Clonostachys is another genus recorded in both approaches. Clonostachys rosea is known to cause root rot of grapevine in Switzerland (Casieri et al. 2009). Colletotrichum hebeiense, C. truncatum and C. viniferum were recorded in the traditional method. Species of this genus cause grape ripe rot affecting the quality and production of grapevine (Yan et al. 2015). Diaporthe eres is another pathogen of grapevine causing die back (Lawrence et al. 2015; Baumgartner et al. 2013; Cinelli et al. 2016; Fischer et al. 2016; Bastide et al. 2017), which was recorded via the traditional methodology as well as via the culture-independent approach.
Species of Fusarium cause wilt disease of grapevine (Castillo-Pando et al. 2001; Gonzalez and Tello 2011). This genus was recorded in both approaches. Neopestalotiopsis vitis recorded from traditional method is a pathogen causing fruit rot, die back and leaf spots of grapevine (Jayawardena et al. 2015, 2016a). Coniella vitis is a pathogen causing white rot of grapes, identified using traditional methods (Chethana et al. 2017). Species of Penicillium are wound and secondary pathogens of grapevines causing bunch rot (Kim et al. 2007). Rhizopus oryzae is another wound and secondary pathogen causing bunch rots of grapevines (Hewitt 2015).
Several genera were identified only in the culture-independent method. Aplosporella is known to cause lesions on grapevine stems in China (Tai 1979). Claviceps is known to be a pathogen on grasses and cereals, but has not been recorded as a pathogen of grapevine (Mey et al. 2002). Therefore, this study provides the first record of this genus on V. vinifera. Cylindrocarpon species are known to cause the black foot disease of grapevine (Abreo et al. 2010, 2012; Mohammadi et al. 2013a, b). Devriesia is a facultative pathogen, but there are no records of this species on V. vinifera (Seifert et al. 2004). Therefore, this study provides the first record of this genus on V. vinifera. Species of Leptosphaeria has been reported as endophytes and saprotrophs of grapevine (Crane and Shearer 1991). However, some species of this genus can be pathogenic to some economically important crops (Fitt et al. 2006). Monographella is a known leaf pathogen on rice, barley, maize and wheat (Daamen et al. 1991; Hock et al. 1992; Tatagiba et al. 2015). However, there are no records of species of this genus associated with grapevine. Therefore, this study provides the first record of Monographella associated with grapevine. Species of Phaeoacremonium are causal agents of Esca disease around the world (Garcia-Benavides et al. 2013). Species of Trichothecium are known to cause berry rot of grapevine, but this is not considered as a major pathogen on grapevine (Oh et al. 2014).
Even though genus Volutella is a facultative pathogen causing leaf spot and cankers (Henricot et al. 2000; Shi and Hsiang 2014), there is no record of this genus occurring on grapevine. Therefore, this study provides the first record of Volutella associated with V. vinifera as a saprotroph.
Among the 45 identified saprotrophic taxa, 17 are well known pathogens of Vitis vinifera causing severe yield as well as economic losses to viticulture worldwide (Table 2). Six secondary pathogens of were also identified in this study. Most of the pathogens tend to survive or overwinter on dead plant material as saprotrophs and act as the primary inoculums once the conditions are favourable (Armijo et al. 2016).
Many studies have shown that most pathogenic fungi can survive unsuitable conditions, such as cold during the winter, by changing their life mode to saprotrophs, and become active pathogens again once the conditions are suitable. Therefore, dead plant materials are the potential primary inocula for plant pathogens in vineyards. In order to avoid this problem, vineyards must be kept clean. If there are any dead grapevines they must be removed and if possible should be burned. This will reduce the pathogenic fungi from year to year.
Checklist of fungi on Vitis
Nine-hundred and five micro- and macro- fungal taxa reported on Vitis species are listed in this study. This is an updated worldwide checklist of fungi on Vitis. These taxa are distributed in 156 families and 343 genera. For each species, family, life mode, diseases caused and the known locality as well as references are provided.
References
Abad ZG, Abad JA, Cacciola SO, Pane A, Faedda R, Moralejo E, Perez-Sierra A, Abad-Campos P, Alvarez-Bernaola LA, Bakonyi J, Jozsa A, Herrero ML, Burgess TI, Cunnington JH, Smith IW, Balci Y, Blomquist C, Henricot B, Denton G, Spies C, McLeod A, Belbahri L, Cooke D, Kageyama K, Uematsu S, Kurbetli I, Degirmenci K (2014) Phytophthora niederhauserii sp. nov., a polyphagous species associated with ornamentals, fruit trees and native plants in 13 countries. Mycologia 106:431–447
Abreo E, Martinez S, Bettucci L, Lupo S (2010) Morphological and molecular characterization of Campylocarpon and Cylindrocarpon spp. associated with black foot disease of grapevines in Uruguay. Australas Plant Pathol 39:446–452
Abreo E, Lupo S, Bettucci L (2012) Fungal community of grapevine trunk diseases: a continuum of symptoms? Sydowia 64:1–12
Agusti-Brisach C, Pérez-Sierra A, García-Figueres F, Montón C, Armengol J (2011) First report of dampingoff caused by Cylindrocarpon pauciseptatum on Pinus radiata in Spain. Plant Dis 95:874
Agusti-Brisach C, Alaniz S, Gramaje D, Perez-Sierra A, Armengol J (2012) First report of Cylindrocladiella parva and Cylindrocladiella peruviana associated with black-foot disease of grapevine in Spain. Plant Dis 96:1381
Agusti-Brisach C, Cabral A, Gonzalez-Dominguez E, Perez-Sierra A, Leon M, Abad-Campos P, Garcia-Jimenez J, Oliveira H, Armengol J (2016) Characterization of Cylindrodendrum, Dactylonectria and Ilyonectria isolates associated with loquat decline in Spain, with description of Cylindrodendrum alicantinum sp. nov. Eur J Plant Pathol 145:103–118
Ahmad S (1969) Fungi of West Pakistan. Biol Soc Pak Monogr 5(Suppl. 1):1–110
Ahmad S (1978) Ascomycetes of Pakistan, Part II. Biol Soc Pak Monogr 8:1–144
Ahmad S, Iqbal SH, Khalid AN (1997) Fungi of Pakistan. Sultan Ahmad Mycological Society of Pakistan, Lahore
Akgul DS, Savas NG, Eskalen A (2014a) First report of wood canker caused by Botryosphaeria dothidea, Diplodia seriata, Neofusicoccum parvum, and Lasiodiplodia theobromae on grapevine in Turkey. Plant Dis 98:568
Akgul DS, Savas NG, Onder S, Ozben S, Kaymak S (2014b) First report of Campylocarpon fasciculare causing black foot disease of grapevine in Turkey. Plant Dis 98:1277
Akgul DS, Gungor Savas N, Teker T, Keykubat B, Mayorquin JS, Eskalen A (2015) Fungal trunk pathogens of Sultana seedless vineyards in Aegean region of Turkey. Phytopathol Mediterr 54:380–393
Alaniz S, Leon M, Vicent A, Garcia-Jimenez J, Abad-Campos P, Armengol J (2007) Characterization of Cylindrocarpon species associated with black foot disease of grapevine in Spain. Plant Dis 91:1187–1193
Alaniz S, Armengol J, Leon M, Garcia-Jimenez J, Abad-Campos P (2009) Analysis of genetic and virulence diversity of Cylindrocarpon liriodendra and C. macrodidymum associated with black foot disease of grapevine. Mycol Res 113:16–23
Alexopoulos CJ (1940) Some fungi from Greece. Mycologia 32:336–358
Alfieri Jr SA, Langdon KR, Wehlburg C, Kimbrough JW (1984) Index of plant diseases in Florida (revised). Florida Department of Agriculture and Consumer Services, Division of Plant Industry Bulletin, vol 11, pp 1–389
Alfieri SA Jr, Langdon KR, Kimbrough JW, El-Gholl NE, Wehlburg C (1994) Diseases and disorders of plants in Florida. Florida Department of Agriculture and Consumer Services, Tallahassee
Al-Saadoon AH, Ameen MKM, Hameed MA, Al-Badran A, Ali Z (2012) First report of grapevine dieback caused by Lasiodiplodia theobromae and Neoscytalidium dimidiatum in Basrah, Southern Iraq. Afr J Biotechnol 11:16165–16171
Alvarez MG (1976) Primer catalogo de enfermedades de plantas Mexicanas. Fitofilo 71:1–169
Alves A, Linaldeddu BT, Deidda A, Scanu B, Phillips AJL (2014) The complex of Diplodia species associated with Fraxinus and some other woody hosts in Italy and Portugal. Fungal Divers 67:143–156
Amrani L, Corio-Costet M-F (2006) A single nucleotide polymorphism in the ß-tubulin gene distinguishing two genotypes of Erysiphe necator expressing different symptoms on grapevine. Plant Pathol 55:505–512
Anderson IC, Cairney JW (2004) Diversity and ecology of soil fungal communities: increased understanding through the application of molecular techniques. Environ microbiol 6:769–779
Ann PJ, Chang T-T, Ko W-H (2002) Phellinus noxius brown root rot of fruit and ornamental trees in Taiwan. Plant Dis 86:820–826
Anonymous (1960) Index of plant diseases in the United States. U.S.D.A. Agricultural Handbook 165:1–531
Anonymous (1979) List of plant diseases in Taiwan. The Plant Protection Society of The Republic of China, Taichung
Ariyawansa HA, Thambugala KM, Manamgoda DS, Jayawardena R, Camporesi E, Boonmee S, Wanasinghe DN, Phookamsak R, Hongsanan S, Singtripop C, Chukeatirote E (2015) Towards a natural classification and backbone tree for Pleosporaceae. Fungal Divers 71:85–139
Armijo G, Schlechter R, Agurto M, Muñoz D, Nuñez C, Arce-Johnson P (2016) Grapevine pathogenic microorganisms: understanding infection strategies and host response scenarios. Front Plant Sci 7:382
Arnold GRW (1986) Lista de Hongos Fitopatogenos de Cuba. Ministerio de Cultura Editorial Cientifico-Tecnica, Habana
Arthur JC (1918) Uredinales of Guatemala based on collections by E.W.D. Holway. I. Introduction. Coleosporiaceae and Uredinaceae. Am J Bot 5:325–336
Arzanlou M, Moshari S, Bakhshi M, Khodaie S (2012) Botryosphaeria dothidea associated with grapevine decline disease in Iran. Australas Plant Dis Notes 7:197–200
Arzanlou M, Narmani A, Moshari S, Khodaei S, Babai-Ahari A (2013) Truncatella angustata associated with grapevine trunk disease in Northern Iran. Arch Phytopathol Pflanzenschutz 46:1168–1181
Arzanlou M, Narmani A, Khodaei S, Moshari S (2014) Pome and stone fruit trees as possible resevoir hosts for Phaeoacremonium spp., the causal agents of grapevine esca disease, in Iran. Arch Phytopathol Pflanzenschutz 47:717–727
Auger J, Esterio M, Ricke G, Perez I (2004a) Black dead arm and basal canker of Vitis vinifera cv. red globe cause by Botryosphaeria obtuse in Chile. Plant Dis 88:1286
Auger J, Esterio M, Perez I, Gubler WD, Eskalen A (2004b) First report of Phaeomoniella chlamydospora on Vitis vinifera and French American hybrids in Chile. Plant Dis 88:1285
Bai J-K (ed) (2000) Flora Fungorum Sinicorum. Sphaeropsidales, Phoma, Phyllosticta, vol 15. Science Press, Beijing
Baker RED, Dale WT (1951) Fungi of Trinidad and Tobago. Mycol Pap 33:1–123
Bakhshi M, Arzanlou M (2017) Multigene phylogeny reveals a new species and novel records and hosts in the genus Ramularia from Iran. Mycol Progr 16:703–712
Bakhshi M, Arzanlou M, Babai-Ahari A (2012) Comprehensive check list of Cercosporoid fungi from Iran. Plant Pathol Quar 2:44–55
Balmas V, Scherm B, Ghignone S, Salem AOM, Cacciola SO, Migheli Q (2005) Characterisation of Phoma tracheiphila by RAPD-PCR, microsatellite-primed PCR and ITS rDNA sequencing and development of specific primers for in planta PCR detection. Eur J Plant Pathol 111:235–247
Baloyi MA, Halleen F, Mostert L, Eskalen A (2013) First report of Togninia minima perithecia on esca- and Petri-diseased grapevines in South Africa. Plant Dis 97:1247
Barbe GD, Hewitt WB (1965) Principal fungus in summer bunch rot of grapes. Phytopathology 55:815
Baroncelli R, Sreenivasaprasad S, Lane CR, Thon MR, Sukno SA (2014) First report of Colletotrichum acutatum sensu lato (Colletotrichum godetiae) causing anthracnose on grapevine (Vitis vinifera) in the United Kingdom. New Dis Rep 29:26
Barr ME (1990) Some dictyosporous genera and species of Pleosporales in North America. N Y Bot Gard 62:1–92
Barr ME (1992) Notes on the Lophiostomataceae (Pleosporales). Mycotaxon 45:191–221
Baskarathevan J, Jaspers MV, Jones EE, Ridgway HJ (2008) Distribution of Botryosphaeria species causing grapevine dieback and decline in New Zealand vineyards. NZ Plant Protect 61:392
Baskarathevan J, Jaspers MV, Ridgway HJ, Eirian JE (2012) Incidence and distribution of Botryosphaeriaceous species in New Zealand vineyards. Eur J Plant Pathol 132:549–560
Bastide F, Serandat I, Gombert J, Laurent E, Morel E, Kolopp J, Guillermin PL, Hamon B, Simoneau P, Berruyer R, Poupard P (2017) Characterization of fungal pathogens (Diaporthe angelicae and D. eres) responsible for umbel browning and stem necrosis on carrot in France. Plant Pathol 66:239–253
Batista AC, Ciferri R (1962) The Chaetothyriales. Sydowia 3:1–129
Batra LR (1973) Nematosporaceae (Hemiascomycetidae): taxonomy, pathogenicity, distribution, and vector relationships. U.S.D.A. Agric Res Serv Tech Bull 1469:1–71
Baumgartner K, Fujiyoshia PT, Travadon R, Castlebury LA, Wilcox WF, Rolshausen PE (2013) Characterization of species of Diaporthe from wood cankers of grape in eastern North American vineyards. Plant Dis 97:912–920
Beale RE, Pitt D (1990) Biological and integrated control of Fusarium basal rot of Narcissus using Minimedusa polyspora and other micro-organisms. Plant Pathol 39:477–488
Bechet M, Sapta-Forda A (1981) Investigation on the growth, development and control of Alternaria vitis Cav., the pathogen of alternariosis of grapevine. Romania Univ’ersitatea Babes-Bloya, pp 147–156
Begon M, Harper JL, Townsend CR (1993) Ecology: Individuals, Population and Communities, 3rd edn. Blackwell Science, Boston
Begoude AD, Slippers B, Wingfeld MJ, Roux J (2010) Botryosphaeriaceae associated with Terminalia catappa in Cameroon, South Africa and Madagascar. Mycol Progr 9:101–123
Benavides PG, Zamorano PM, Ocete CA, Maistrello L, Ocete R (2013) Biodiversity of pathogenic wood fungi isolated from Xylotrechus arvicola (Olivier) galleries in vine shoots. OENO One 47:73–81
Bendezu-Euribe MV, Alvarez LA (2012) The perfect stage of powdery mildew of grapevine caused by Erysiphe necator found in Peru. Plant Dis 96:768
Benjamin CR, Slot A (1969) Fungi of Haiti. Sydowia 23:125–163
Bensch K, Groenewald JZ, Braun U, Dijksterhuis J, de Jesús Y-MM, Crous PW (2015) Common but different: the expanding realm of Cladosporium. Stud Mycol 82:23–74
Berkeley MJ (1872) Notices of North American fungi. Grevillea 1:33–39
Berndt R (2004) A checklist of Costa Rican rust fungi. In: Agerer R, Piepenbring M, Blanz P (eds) Frontiers in Basidiomycete Mycology. IHW-Verlag, Berlin
Berraf-Tebbal A, Bouznad Z, Santos JM, Coelho MA, Peros J-P, Phillips AJL (2011) Phaeoacremonium species associated with Eutypa dieback and esca of grapevines in Algeria. Phytopathol Mediterr 50:S86–S97
Berraf-Tebbal A, Guereiro MA, Phillips AJL (2014) Phylogeny of Neofusicoccum species associated with grapevine trunk disease in Algeria, with decription of Neofusicoccum algeriense sp. nov. Phytopathol Mediterr 53:416–427
Berthier J (1976) Monographie des Typhula, Pistillaria et des genres voisins. Bull Soc Linn Lyon 45:1–213
Bezerra JL, De Lima TEF (2012) Phaeotrichoconis crotalariae, endophytic on Vitis labrusca, in Brazil. Mycotaxon 120:291–294
Billones RG, Ridgway HJ, Jones EE, Jaspers MV (2010) First report of Neofusicoccum macroclavatum as a canker pathogen of grapevine in New Zealand. Plant Dis 94:1504
Blair JE, Coffey MD, Park SY, Geiser DM, Kang S (2008) A multi-locus phylogeny for Phytophthora utilizing markers derived from complete genome sequences. Fungal Genet Biol 45:266–277
Bobev S (2009) Reference guide for the diseases of cultivated plants. http://nt.ars-grin.gov/fungaldatabases
Boesewinkel HJ (1982) A list of 142 new plant disease recordings from New Zealand and short notes on three diseases. Australas Plant Pathol 11:40–43
Boidin J, Lanquetin P (1987) Le genre Scytinostroma Donk (basidiomycetes, Lachnocladiaceae). Biblioth Mycol 114:1–130
Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG (2013) Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat Methods 10:57–59
Bokulich NA, Thorngate JH, Richardson PM, Millis DA (2014) Microbial biogeography of wine grapes is conditioned by cultivar, vintage and climate. Proc Natl Acad Sci 111:E139–E148
Bolay A (2005) Powdery mildews of Switzerland (Erysiphaceae). Cryptogr Helv 20:1–176
Booth C (1966) The genus Cylindrocarpon. Mycol Pap 104:1–56
Brasier CM, Sanchez-Hernandez E, Kirk SA (2003) Phytophthora inundata sp. nov., a part heterothallic pathogen of trees and shrubs in wet or flooded soils. Mycol Res 107:477–484
Braun U (1998) A monograph of Cercosporella, Ramularia and allied genera (Phytopathogenic Hyphomycetes), vol 2. IHW-Verlag, Eching, p 337
Braun U, Crous PW, Dugan F, Groenewald JE, De Hoog GS (2003) Phylogeny and taxonomy of Cladosporium-like hyphomycetes, including Davidiella gen. nov., the teleomorph of Cladosporium s. str. Mycol Prog 2:3–18
Brenckle JF (1918) North Dakota fungi—II. Mycologia 10:199–221
Briceno EX, Latorre BA (2007) Outbreaks of Cladosporium rot associated with delayed harvest wine grapes in Chile. Plant Dis 91:1060
Briceno EX, Latorre BA (2008) Characterization of Cladosporium rot in grapevines, a problem of growing importance in Chile. Plant Dis 92:1635–1642
Bucher VVC, Hyde KD, Pointing SB, Reddy CA (2004) Production of wood decay enzymes, mass loss and lignin solubilization in wood by marine ascomycetes and their anamorphs. Fungal Divers 15:1–14
Burdsall HH Jr (1976) A new species of Laeticorticium (Aphyllophorales, Corticiaceae) from the southern Appalachians. J Elisha Mitchell Sci Soc 91:243–245
Burdsall HH Jr (1985) A contribution to the taxonomy of the genus Phanerochaete. Mycol Mem 10:1–165
Burgess TI, Barber PA, Hardy J (2005) Bothryosphaeria spp. associated with Eucalyptus in Western Australia including description of Fusicoccum macroclavatum sp. nov. Australas Plant Pathol 34:557–567
Buritica P (1999) La familia Phakopsoraceae en el neotropico III, generos: Batistopsora y Phakopsora. Rev Acad Colomb Cienc 23:271–305
Buritica P, Pardo Cardona VM (1996) Flora Uredineana Colombiana. Rev Acad Colomb Cienc 20:183–236
Cai L, Udayanga D, Manamgoda DS, Maharachchikumbura SSN, McKenzie EHC, Guo LD, Liu XZ, Bahkali A, Hyde KD (2011) The need to carry out re-inventory of plant pathogenic fungi. Trop Plant Pathol 36:205–213
Campbell WA, Miller JH, Thompson GE (1950) Notes on some wood-decaying fungi of Georgia. Plant Dis Rep 34:128–134
Candolfi-Arballo O, Valenzuela-Solano C, Gubler WD, Hernández-Martínez R (2010) Botryosphaeriaceae species associated with grapevine decline in Mexico. Phytopathol Mediterr 49:105–106
Cannon PF, Hawksworth DL, Sherwood-Pike MA (1985) The British ascomycotina: An annotated checklist. Commonwealth Mycological Institute, Kew, Surrey
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Carlucci A, Frisullo S (2009) First report of Diplodia corticola on grapevine in Italy. J Plant Pathol 91:231–240
Carlucci A, Cibelli F, Lops F, Raimondo ML (2015) Characterization of Botryosphaeriaceae species as causal agents of trunk disease on Grapevines. Plant Dis 99:1678–1688
Carlucci A, Lops F, Mostert L, Halleen F, Raimondo ML (2017) Occurrence fungi causing black foot on young grapevines and nursery rootstock plants in Italy. Phytopathol Mediterr 56:10–39
Carmaran CC, Novas MV (2003) A review of spegazzini taxa of Periconia and Sporocybe after over 115 years. Fungal Divers 14:67–76
Carraro L, Maifreni M, Bartolomeoli I, Martino ME, Novelli E, Frigo F, Marino M, Cardazzo B (2011) Comparison of culture-dependent and -independent methods for bacterial community monitoring during Montasio cheese manufacturing. Res Microbiol 162:231–239
Carter MV (1991) The status of Eutypa lata as a pathogen. Phytopathol Pap 32:1–59
Cash EK (1952) A record of the fungi named by J.B. Ellis (Part 1). U.S.D.A. Spec Publ 2:1–165
Cash EK (1953) A record of the fungi named by J.B. Ellis (Part 2). U.S.D.A. Spec Publ 2:167–345
Cash EK (1954) A record of the fungi named by J.B. Ellis (Part 3). U.S.D.A. Spec Publ 2:347–518
Casieri L, Hofstetter V, Viret O, Gindro K (2009) Fungal communities living in the wood of different cultivars of young Vitis vinifera plants. Phytopathol Mediterr 48:73–83
Castellani E, Ciferri R (1937) Prodromus Mycoflorae Africae Orientalis Italicae. Istituto Agricolo Coloniale Italiano, Firenze
Castillo-Pando M, Somers A, Green CD, Priest M, Sriskathades M (2001) Fungi associated with dieback of Semillon grapevines in the Hunter Valley of New South Wales. Australas Plant Pathol 30:59–63
Cavara F (1888) Intorno al disseccamento dei grappoli della vite. Atti dell’Istituto Botanico della Università e Laboratorio Crittogamico di Pavia 1:293–324
Cavara F (1889) I Funghi Parassiti delle Piante Coltivate ed Utili Essicati, Delineati e Descriti: 21
Chandra S (1974) Some new leaf-spot diseases from Allahabad (India). Nova Hedwig 47:35–102
Chardon CE, Toro RA (1930) Mycological explorations of Colombia. J Dept Agric Porto Rico 14:195–369
Chatasiri S, Ono Y (2008) Phylogeny and taxonomy of the Asian grapevine leaf rust fungus, Phakopsora euvitis, and its allies (Uredinales). Mycoscience 49:66–74
Chebil S, Fersi R, Yakoub A, Chenenaoui S, Chattaoui M, Melki I, Zemni H, Rhouma A, Durante G, Zacchi E, Mliki A (2014) First report of Botryosphaeria dothidea, Diplodia seriata, and Neofusicoccum luteum associated with canker and dieback of grapevines in Tunisia. Plant Dis 98:420
Checa J (2004) Dictyosporic Dothideales. Flora Mycol Iber 6:1–162
Chen Q, Jiang JR, Zhang GZ, Cai L, Crous PW (2015) Resolving the Phoma enigma. Stud Mycol 82:137–217
Chethana KWT, Zhou Y, Zhang W, Liu M, Xing QK, Li XH, Yan JY (2017) Coniella vitis sp. nov. is the common pathogen of white rot in Chinese vineyards. Plant Dis 101:2123–2136
Chiarappa L (1959) Wood decay of the grapevine and its relationship with black measles disease. Phytopathology 49:510–519
Chicau G, Aboim-Inglez M, Cabral S, Cabral JPS (2000) Phaeoacremonium chlamydosporum and Phaeoacremonium angustius associated with esca and grapevine decline in Vinho Verde grapevines in northwest Portugal. Phytopathol Mediterr 39:80–86
Cho WD, Shin HD (eds) (2004) List of plant diseases in Korea, 4th edn. Korean Society of Plant Pathology, Suwon
Chomnunti P, Hongsanan S, Aguirre-Hudson B, Tian Q, Peršoh D, Dhami MK, Alias AS, Xu J-C, Liu X-Z, Stadler M, Hyde KD (2014) The sooty moulds. Fungal Divers 66:1–36
Choueiri E, Jreijiri F, Chlela P, Louvet G, Lecomte P (2006) Occurrence of grapevine decline and first report of black dead arm associated with Botryosphaeria obtusa in Lebanon. Plant Dis 90:115
Choueiri E, Jreijiri F, El Amil R, Chlela P, Bugaret Y, Liminana JM, Mayet V, Lecomte P (2009) First report of black foot disease associated with Cylindrocarpon sp. in Lebanon. J Plant Pathol 91:231–240
Chupp C (1953) Monograph of the fungus genus Cercospora. Author, Ithaca
Ciferri R (1929) Micoflora Domingensis. Lista de los hongos hasta la fecha indicados en Santo Domingo. Publ Estac Estac Agron Moca Ser B Bot 14:1–260
Ciferri R (1961) Mycoflora Domingensis Integrata. Quaderno 19:1539
Cinelli T, Mondello V, Marchi G, Burruano S, Alves A, Mugnai L (2016) First report of Diaporthe eres associated with cane blight of grapevine (Vitis vinifera) in Italy. Plant Dis 100:532
Cloete M, Fischer M, Mostert L, Halleen F (2014) A novel Fomitiporia species associated with esca on grapevine in South Africa. Mycol Prog 13:303–311
Cloete M, Fischer M, Mostert L, Halleen F (2015) Hymenochaetales associated with esca-related wood rots on grapevine with a special emphasis on the status of esca in South African vineyards. Phytopathol Mediterr 54:299–312
Cloete M, Fischer M, Du Plessis IL, Mostert L, Halleen F (2016) A new species of Phellinus sensu stricto associated with esca on grapevine in South Africa. Mycol Prog 15:25
Comont G, Mayet V, Corio-Costet MF (2016) First report of Lasiodiplodia viticola, Spencermartinsia viticola and Diplodia intermedia associated with Vitis vinifera grapevine decline in French vineyards. Plant Dis 100:2328
Cook RP, Dubé AJ (1989) Host–pathogen index of plant diseases in South Australia. South Australian Department of Agriculture, Adelaide
Cooke MC (1878) Ravenel’s American fungi. Grevillea 6:129–146
Cooke WB (1983) The 1979 Oklahoma foray. Mycologia 75:752–755
Correia KC, Camara MPS, Barbosa MAG, Sales R, Agusti-Brisach C, Gramaje D, Leon M, Garcia-Jimenez J, Abad-Campos P, Armengol J, Michereff SJ (2013) Fungal trunk pathogens associated with table grape decline in Northeastern Brazil. Phytopathol Mediterr 52:380–387
Correia KC, Silva MA, Netto MSB, Vieira WAS, Camara MPS, Michereff SJ (2016a) First report of grapevine dieback caused by Neoscytalidium hyalinum in Brazil. Plant Dis 100:213
Correia KC, Silva MA, de Morais Jr MA, Armengol J, Phillips AJL, Camara MPS, Michereff SJ (2016b) Phylogeny, distribution and pathogenicity of Lasiodiplodia species associated with dieback of table grape in the main Brazilian exporting region. Plant Pathol 65:92–103
Costa MEA, Camara MS (1952) Species Aliquae Mycologicae Lusitaniae. Portugaliae Acta Biol Ser A 3:294–307
Coutinho IBL, Freire FCO, Lima CS, Lima JS, Goncalves FJT, Machado AR, Silva AMS, Cardoso JE (2017) Diversity of genus Lasiodiplodia associated with perennial tropical fruit plants in northeastern Brazil. Plant Pathol 66:90–104
Crane JL, Shearer CA (1991) A nomenclator of Leptosphaeria V. Cesati & G. DeNotaris. Ill Nat Hist Surv Biol Notes 34:1–355
Crous PW, Braun U (2003) Mycosphaerella and its anamorphs: 1. Names published in Cercospora and Passalora. Centraalbureau voor Schimmelcultures, Utrecht
Crous PW, Gams W (2000) Phaeomoniella chlamydospora gen. et comb. nov., a causal organism of Petri grapevine decline and esca. Phytopathol Mediterr 39:112–118
Crous PW, Braun U, Wingfield MJ, Wood AR, Shin HD, Summerell BA, Alfenas AC, Cumagun CJR, Groenewald JZ (2009) Phylogeny and taxonomy of obscure genera of microfungi. Persoonia 22:139–161
Crous PW, Wingfield MJ, Guarro J, Hernandez-Restrepo M, Sutton DA, Acharya K, Barber PA, Boekhout T, Dimitrov RA, Duenas M, Dutta AK, Gene J, Gouliamova DE, Groenewald M, Lombard L, Morozova OV, Sarkar J, Smith MTh, Stchigel AM, Wiederhold NP, Alexandrova AV, Antelmi I, Armengol J, Barnes I, Cano-Lira JF, Castaneda-Ruiz RF, Contu M, PrR Courtecuisse, da Silveira AL, Decock CA, de Goes A, Edathodu J, Ercole E (2015) Fungal Planet description sheets: 320–370. Persoonia 34:167–266
Crous PW, Wingfield MJ, Burgess TI, St J Hardy GE, Barber PA, Alvarado P, Barnes CW, Buchanan PK, Heykoop M, Moreno G, Thangavel R, van der Spuy S, Barili A, Barrett S, Cacciola SO, Cano-Lira JF, Crane C, Decock C, Gilbertoni TB, Guarro J, Guevara-Suarez M, Hubka V, Kolarik M, Lira CRS, Ordonez ME, Padamsee M, Ryvarden L, Soares AM, Stchigel AM, Sutton DA, Vizzini A, Weir BS, Acharya K, Aloi F (2017) Fungal Planet description sheets: 558–624. Persoonia 38:240–384
Cruywagen EM, Slippers B, Roux J, Wingfield MJ (2017) Phylogenetic species recognition and hybridisation in Lasiodiplodia: a case study on species from baobabs. Fungal Biol 121:420–436
Cunningham GH (1965) Polyporaceae of New Zealand. Bull N Z Dept Ind Res 164:1–304
Cunnington J (2003) Pathogenic fungi on introduced plants in Victoria. A host list and literature guide for their identification. Department of Primary Industries, Victoria
Cunnington JH, Priest MJ, Powney RA, Cother NJ (2007) Diversity of Botryosphaeria species on horticultural plants in Victoria and New South Wales. Australas Plant Pathol 36:157–159
da Silva MA, Correia KC, Barbosa MAG, Camara MPS, Gramaje D, Michereff SJ (2017) Characterization of Phaeoacremonium isolates associated with petri disease of table grape in Northeastern Brazil, with description of Phaeoacremonium nordesticola sp. nov. Eur J Plant Pathol 149:695–709
Daamen RA, Langerak CJ, Stol W (1991) Surveys of cereal diseases and pests in the Netherlands 3. Monographella nivalis and Fusarium spp. in winter wheat fields and seed lots. Neth J Plant Pathol 97:105–114
Damm U, Crous PW, Fourie PH (2007) Botryosphaeriaceae as potential pathogens of Prunus species in South Africa, with descriptions of Diplodia africana and Lasiodiplodia plurivora sp. nov. Mycologia 99:664–680
Damm U, Woudenberg JHC, Cannon PF, Crous PW (2009) Colletotrichum species with curved conidia from herbaceous hosts. Fungal Divers 39:45–87
Damm U, Cannon PF, Woudenberg JHC, Crous PW (2012) The Colletotrichum acutatum species complex. Stud Mycol 73:37–113
Daranagama DA, Camporesi E, Jeewon R, Liu XZ, Stadler M, Lumyong S, Hyde KD (2016) Taxonomic rearrangement of Anthostomella (Xylariaceae) based on a multigene phylogeny and morphology. Cryptogam Mycol 37:509–538
de Sousa Dias MR, Lucas MT (1972) Fungi Lusitaniae XXIII. Agron. Lusit 33:173–184
de Sousa Dias MR, Lucas MT, Lopes MC (1987) Fungi Lusitaniae XXX. Agron Lusit 42:179–188
de Wet J, Slippers B, Preisig O, Wingfield BD, Tsopelas P, Wingfield MJ (2009) Molecular and morphological characterization of Dothiorella casuarinae sp. nov. and other Botryosphaeriaceae with diplodia-like conidia. Mycologia 101:503–511
Deighton FC (1976) Studies on Cercospora and allied genera. VI. Pseudocercospora Speg., Pantospora Cif. and Cercoseptoria Petr. Mycol Pap 140:1–168
Deighton J (2016) Fungi in ecosystem processes, vol 31. CRC Press, Boca Raton
Denaree JB, Runner GA (1942) Control of grape diseases and insects in Eastern United States. Diseases principally of Amaerican bunch grapes. US Dept Agr Farm Bull 1893:1–28
Dennis RWG (1964) Remarks on the genus Hymenoscyphus S.F. Gray, with observations on sundry species referred by Saccardo and others to the genera Helotium Pezizella or Phialea. Persoonia 3:29–80
Dennis RWG (1970) Kew Bulletin Additional Series III. Fungus flora of Venezuela and adjacent countries. Verlag von J Cramer, Weinheim
Diaz GA, Latorre BA (2014) Infection caused by Phaeomoniella chlamydospora associated with esca-like symptoms in grapevine in Chile. Plant Dis 98:351–360
Diaz GA, Prehn D, Latorre BA (2011) First report of Cryptovalsa ampelina and Eutypella leprosa associated with grapevine trunk diseases in Chile. Plant Dis 95:490
Diaz GA, Elfar K, Latorre BA (2012) First report of Seimatosporium botan associated with trunk disease of grapevine (Vitis vinifera) in Chile. Plant Dis 96:1696
Diaz G, Auger J, Besoain X, Bordeu E, Latorre BA (2013) Prevalence and pathogenicity of fungi associated with grapevine trunk diseases in Chilean vineyards. Cien Inv Agr 40:327–339
Dingley JM, Fullerton RA, McKenzie EHC (1981) Survey of agricultural pests and diseases. Technical Report, vol 2. Records of fungi, bacteria, algae, and angiosperms pathogenic on Plants in Cook Islands, Fiji, Kiribati, Niue, Tonga, Tuvalu, and Western Samoa. F.A.O., Rome
Dissanayake AJ, Zhang W, Mei L, Chukeatirote E, Yan JY, Li XH, Hyde KD (2015) Lasiodiplodia pseudotheobromae causes pedicel and peduncle discolouration of grapes in China. Australas Plant Pathol 10:21
Dissanayake AJ, Purahong W, Wubet T, Hyde KD, Wei Z, Xu HY, Zhang GJ, Yuan FC, Liu M, Xing Q, Li XH, Yan JY (2018) Direct comparison of culture dependent and cultire independent molecular approaches reveal the diversity of fungal endophytic communities in stems of grapevine (Vitis vinifera). Fungal Divers (Under review)
Dohadwala MM, Vita JA (2009) Grapes and Cardiovascular Disease. J Nutr 139:1788S–1793S
Doidge EM (1950) The South African fungi and lichens to the end of (1945). Bothalia 5:1–1094
Doilom MW, Shuttleworth LA, Roux J, Chukeatirote E, Hyde KD (2015) Botryosphaeriaceae associated with Tectona grandis (teak) in northern Thailand. Phytotaxa 233:1–26
Du Z, Fan X-L, Hyde KD, Yang Q, Liang Y-M, Tian C-M (2016) Phylogeny and morphology reveal two new species of Diaporthe form Betula spp. in China. Phytotaxa 269:090–102
Duarte S, Pascoal C, Cássio F, Bärlocher F (2006) Aquatic hyphomycete diversity and identity affect leaf litter decomposition in microcosms. Oecologia 147:658–666
Dudka IO, Heluta VP, Tykhonenko YY, Andrianova TV, Hayova VP, Prydiuk MP, Dzhagan VV, Isikov VP (2004) Fungi of the Crimean Peninsula. M.G. Kholodny Institute of Botany, National Academy of Sciences of Ukraine, Kiev
Dugan FM, Schubert K, Braun U (2004) Check-list of Cladosporium names. Schlechtendalia 11:1–103
Dupont J, Laloui W, Roquebert MF (1998) Partial ribosomal DNA sequences show an important divergence between Phaeoacremonium species isolated from Vitis vinifera. Mycol Res 102:631–637
Dupont J, Laloui W, Magnin S, Larignon P, Roquebert M-F (2000) Phaeoacremonium viticola, a new species associated with Esca disease of grapevine in France. Mycologia 92:499–504
Dupont J, Magnin S, Cesari C, Gatica M (2002) ITS and β-tubulin markers help delineate Phaeoacremonium species, and the occurrence of P. parasiticum in grapevine disease in Argentina. Mycol Res 106:1143–1150
Ebbels DL, Allen DJ (1979) A supplementary and annotated list of plant diseases, pathogens and associated fungi in Tanzania. Phytopathol Pap 22:1–89
Edgar RC (2004) Muscle: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113
Edgar RC (2013) UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 10:996–998
Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R (2011) UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200
El-Buni AM, Rattan SS (1981) Check list of Libyan Fungi. Al Faateh University, Fac Sci, Dept. Bot, Tripoli
Elenkin AA (1909) Title unknown. Bolezni Rast 4:68
Ellis JB, Everhart BM (1904) New species of fungi from various localities. J Mycol 10:167–170
Epstein L (2008) Botryosphaeria-related dieback and control investigated in noncoastal California grapevines. Calif Agric 62:161–166
Erwin DC, Ribeiro OK (1996) Phytophthora diseases worldwide. APS Press, St. Paul
Esfandiari E, Petrak F (1950) Pilze aus Iran. Sydowia 4:11–38
Eskalen A, Rooney-Latham S, Gubler WD (2005) First report of perithecia of Phaeoacremonium viticola on grapevine (Vitis vinifera) and ash tree (Fraxinus latifolia) in California. Plant Dis 89:686–686
Essakhi S, Mugnai L, Crous PW, Groenewald JZ, Surico G (2008) Molecular and phenotypic characterisation of novel Phaeoacremonium species isolated from esca diseased grapevines. Persoonia 21:119–134
Fadrosh DW, Ma B, Gajer P, Sengamalay N, Ott S, Brotman RM, Ravel J (2014) An improved dual-indexing approach for multiplexed 16S rRNA gene sequencing on the Illumina MiSeq platform. Microbiome 2:6
Faedda R, Agosteo GE, Schena L, Mosca S, Frisullo S, di San Magnano, Lio G, Cacciola SO (2011) Colletotrichum clavatum sp. nov. identified as the causal agent of olive anthracnose in Italy. Phytopathol Mediterr 50:283–302
Farr ML (1973) An annotated list of Spegazzini’s fungus taxa, Vol. 1. Biblioth Mycol 35:1–823
Farr DF, Castlebury LA, Rossman AY, Erincik O (2001) Greeneria uvicola, cause of bitter rot of grapes, belongs in the Diaporthales. Sydowia 53:185–199
Farr DF, Elliott M, Rossman AY, Edmonds RL (2005) Fusicoccum arbuti sp. nov. causing cankers on Pacific madrone in western North America with notes on Fusicoccum dimidiatum, the correct name for Scytalidium dimidiatum and Nattrassia mangiferae. Mycologia 97:730–741
Fautrey F, Lambotte JBE (1896) Espèces nouvelles ou rares de la Côte d’Or. Revue Mycologique Toulouse. 18:68–71
Ferrada EE, Diaz GA, Zoffoli JP, Latorre BA (2014) First report of blossom blight caused by Sclerotinia sclerotiorum on Japanese plum, nectarine, and sweet cherry orchards in Chile. Plant Dis 98:695
Fischer M, Edwards J, Cunnington JH, Pascoe IG (2005) Basidiomycetous pathogens on grapevine: a new species from Australia—Fomitiporia australiensis. Mycotaxon 92:85–96
Fischer M, Schneider P, Kraus C, Molnar M, Dubois C, D’Aguiar D, Haag N (2016) Grapevine trunk disease in German viticulture: occurence of lesser known fungi and first report of Phaeoacremonium viticola and P. fraxinopennsylvanicum. Vitis 55:145–156
Fitt BD, Brun H, Barbetti MJ, Rimmer SR (2006) World-wide importance of phoma stem canker (Leptosphaeria maculans and L. biglobosa) on oilseed rape (Brassica napus). In: Fitt BDL, Evans N, Howlett BJ, Cooke BM (eds) Sustainable strategies for managing Brassica napus (oilseed rape) resistance to Leptosphaeria maculans (phoma stem canker). Springer, Dordrecht, pp 3–15
Foister CE (1961) The economic plant diseases of Scotland. Techn Bull Dept Agric Fish Scotland 1:1–210
Forster H, Coffey MD (1993) Molecular taxonomy of Phytophthora megasperma based on mitochondrial and nuclear DNA polymorphisms. Mycol Res 97:1101–1112
Fotouhifar K-B, Hedjaroude G-A, Leuchtmann A (2010) ITS rDNA phylogeny of Iranian strains of Cytospora and associated teleomorphs. Mycologia 102:1369–1382
Fourie PH, Halleen F (2002) Investigation on the occurence of Phaeomoniella chlamydospora in canes of rootstock mother vines. Australas Plant Pathol 31:425–426
Fournier E, Gladieux P, Giraud T (2013) The ‘Dr Jekyll and Mr Hyde fungus’: noble rot versus gray mold symptoms of Botrytis cinerea on grapes. Evol Applic 6:960–969
French AM (1987) California plant disease host index. Part 1: fruit and nuts. California Department of Food Agriculture, Sacramento
French AM (1989) California plant disease host index. California Department of Food Agriculture, Sacramento
Gadgil PD (2005) Fungi on trees and shrubs in New Zealand. In: Fungi of New Zealand, vol 4. Fungal Diversity Press, Hong Kong
Gao Z-M, Li Y-M, Li X-L, Zhang Z-H, Yiny J-F (2009) Comparison of the biological characteristics of Botrytis cinerea isolates from different hosts. Mycosystema 28:370–377
Gaponenko NI (1965) Survey of the fungi of Bukhara. Akad Sa Uzbekh
García B, López L, Velázquez M, Pérez-Moreno J (1998) Mycorrhizal and saprotrophic macrofungi in declining sacred-fir stands. Micol Neotrop Apl 11:9–21
Garcia-Benavides P, Martin Zamorano P, Ocete Perez CA, Maistrello L, Ocete Rubio R (2013) Biodiversity of pathogenic wood fungi isolated from Xylotrechus arvicola (Olivier) galleries in vine shoots. J Int Sci Vigne Vin 47:73–81
Garcia-Blazquez G, Constantinescu O, Telleria MT, Martin MP (2006) Preliminary checklist of Albuginales and Peronosporales (Chromista) reported from the Iberian Peninsula and Balearic Islands. Mycotaxon 98:185–188
Garnica S, Riess K, Schön ME, Oberwinkler F, Setaro SD (2016) Divergence times and phylogenetic patterns of Sebacinales, a highly diverse and widespread fungal lineage. PLoS ONE 11:e0149531
Gatica M, Cesari C, Magnin S, Dupont J (2001) Phaeoacremonium species and Phaeomoniella chlamydospora in vines showing “hoja de malvon” and young vine decline symptoms in Argentina. Phytopathol Mediterr 40:S317–S324
Ge Q, Chen Y, Xu T (2009) Flora Fungorum Sinicorum. Pestalotiopsis, vol 38. Science Press, Beijing, p 235
Georghiou GP, Papadopoulos C (1957) A second list of Cyprus fungi. Government of Cyprus, Department of Agriculture, Nicosia
Gerlach W, Ershad D (1970) Beitrag zur Kenntnis der Fusarium und Cylindrocarpon-Arten in Iran. Nova Hedwig 20:725–784
Giatgong P (1980) Host index of plant diseases in Thailand, 2nd edn. Mycology Branch, Plant Pathology and Microbiology Division, Department of Agriculture and Cooperatives, Bangkok
Gilbertson RL, Martin KJ, Lindsey JP (1974) Annotated check list and host index for Arizona wood-rotting fungi. Univ Arizona Agric Exp Sta Techn Bull 209:1–48
Gilman JC (1932) First supplementary list of parasitic fungi from Iowa. Iowa State Coll J Sci 6:357–365
Gilman JC, Archer WA (1929) The Fungi of Iowa Parasitic on Plants. Iowa State Coll J Sci 3:299–507
Gimenez-Jaime A, Aroca A, Raposo R, Garcia-Jimenez J, Armengol J (2006) Occurrence of fungal pathogens associated with grapevine nurseries and the decline of young vines in Spain. J Phytopathol 154:598–602
Gomes RR, Glienke C, Videira SIR, Lombard L, Groenewald JZ, Crous PW (2013) Diaporthe: a genus of endophytic, saprobic and plant pathogenic fungi. Persoonia 31:1–41
Gonzalez CD, Chaverri P (2017) Corinectria, a new genus to accommodate Neonectria fuckeliana and C. constricta sp. nov. from Pinus radiata in Chile. Mycol Prog 16(11–12):1015–1027
Gonzalez Fragoso R (1916) Bosquejo de una Florula Hispalense de Micromicetos. Trab Mus Nac Ci Nat Ser Bot 10:1–221
Gonzalez Fragoso R (1917) Fungi novi vel minus cognitarum horti botanici matritense. Trab Mus Nac Ci Nat Ser Bot 12:1–99
Gonzalez Fragoso R (1921) Algunos Demaciaceos de la Flora espanola. Bol Real Soc Esp Hist Nat 21:93–99
Gonzalez V, Tello ML (2011) The endophytic mycota associated with Vitis vinifera in central Spain. Fungal Diver 47:29–42
Goos RD (2010) The Mycota of Rhode Island: a checklist of the fungi recorded in Rhode Island (including lichens and myxomycetes). R I Nat Hist Surv 4:222
Gorter GJMA (1977) Index of plant pathogens and the diseases they cause in cultivated plants in South Africa. Republic South Africa Dept Agric Techn Serv. Pl Protect Res Inst Sci Bull 392:1–177
Graham AB, Johnston PR, Weir BS (2009) Three new Phaeoacremonium species on grapevines in New Zealand. Australas Plant Pathol 38:505–513
Gramaje D, Alaniz S, Perez Sierra A, Abad-Campos P, Garcia-Jimenez J, Armengol J (2008) First report of Phaeoacremonium scolyti causing petri disease of grapevine in spain. Plant Dis 92:836
Gramaje D, Armengol J, Mohammadi H, Banihashemi Z, Mostert L (2009) Novel Phaeoacremonium species associated with petri disease and esca of grapevine in Iran and Spain. Mycologia 101:920–929
Gramaje D, Aguilar MI, Armengol J (2011) First report of Phaeoacremonium krajdenii causing petri disease of grapevine in Spain. Plant Dis 95:615
Grand LF (ed) (1985) North Carolina Plant Disease Index. North Carolina Agric Res Serv Techn Bull 240:1–157
Grasso S (1984) Infezioni di Fusarium oxysporum e di Cylindrocarpon destructans associate a una moria di giovani piante di vite in Sicilia. Inf Fitopatol 1:59–63
Greene HC (1955) Notes on Wisconsin parasitic fungi. XXI. Trans. Wisconsin Acad Sci 44:29–43
Greene HC (1956) Notes on Wisconsin parasitic fungi. XXII. Trans. Wisconsin Acad Sci 45:177–191
Greene HC (1963) Notes on Wisconsin parasitic fungi. XXIX. Trans Wisconsin Acad Sci 52:229–253
Greene HC (1966) Notes on Wisconsin parasitic fungi. XXXII. Trans Wisconsin Acad Sci 55:147–166
Greuter W, Poelt J, Raimondo FM (1991) A checklist of Sicillian fungi. Bocconea 2:222
Guba EF (1961) Monograph of Monochaetia and Pestalotia. Harvard University Press, Cambridge
Guerber JC, Liu B, Correll JC, Johnston PR (2003) Characterization of diversity in Colletotrichum acutatum sensu lato by sequence analysis of two gene introns, mtDNA and intron RFLPs, and mating compatibility. Mycologia 95:872–895
Guo Y-L, Liu X-J (eds) (2003) Flora Fungorum Sinicorum. Mycovellosiella, Passalora, Phaeoramularia, vol. 20 Science Press, Beijing
Guo LD, Huang GR, Wang Y, He WH, Zheng WH, Hyde KD (2003) Molecular identification of white morphotype strains of endophytic fungi from Pinus tabulaeformis. Mycol Res 107:680–688
Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, Methé B (2011) Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 21:494–504
Halleen F, Crous PW, Petrini O (2003) Fungi associated with healthy grapevine cuttings in nurseries, with special reference to pathogens involved in the decline of young vines. Australas Plant Pathol 32:47–52
Hammer Ø, Harper DAT, Ryan PD (2001) PAST: paleontological statistics software package for education and data analysis. Palaeontol Electron 4:1–9
Hanlin RT (1963) A revision of the Ascomycetes of Georgia. Georgia Agric Exp Sta Mimeo Ser ns 175:1–65
Hanlin RT (1966) Host index to the Basidiomycetes of Georgia. Georgia Agric Exp Sta Mimeo Ser ns 260:1–30
Hansford CG (1947) New or interesting tropical fungi—I. Proc Linn Soc London 158:28–50
Hanson CA, Fuhrman JA, Horner-Devine MC, Martiny JBH (2012) Beyond biogeographic patterns:processes shaping the microbial landscape. Nat RevMicrobiol 10:497–506
Harvey JM (1955) A method of forecasting decay in California storage grapes. Phytopathology 45:229–232
Henricot B, Pérez Sierra A, Prior C (2000) A new blight disease on Buxus in the UK caused by the fungus Cylindrocladium. Plant Pathol 49:805–805
Hernandez JC (2004) Bases Corologicas de Flora Micologica Iberica. Adiciones y numeros 2179–2238. Cuad Trab Fl Micol Iber 20:1–94
Hewitt WB (1951) Grape dead-arm control. Plant Dis Rep 35:142–143
Hewitt WB (2015) Miscellaneous berry rots and raisin molds. In: Gubler WD, Uyemoto JK (eds) Compendium of grape diseases, disorders and pests (Wilcox WF. American phytopathological society, USA, pp 22–24
Hirayama K, Tanaka K (2011) Taxonomic revision of Lophiostoma and Lophiotrema based on reevaluation of morphological characters and molecular analyses. Mycoscience 52:401–412
Hirooka Y, Rossman AY, Zhuang W-Y, Salgado-Salazar C, Chaverri P (2013) Species delimitation for Neonectria coccinea group including the causal agents of beech bark disease in Asia, Europe, and North America. Mycosystema 32:485–517
Hock J, Dittrich U, Renfro BL, Kranz J (1992) Sequential development of pathogens in the maize tarspot disease complex. Mycopathologia 117:157–161
Hofstetter V, Buyck B, Croll D, Viret O, Couloux A, Gindro K (2012) What if esca disease of grapevine were not a fungal disease? Fungal Divers 54:51–67
Holevas CD, Chitzanidis A, Pappas AC, Tzamos EC, Elena K, Psallidas PG, Alivizatos AS, Panagopoulos CG, Kyriakopoulou PE, Bem FP, Lazscaris DN (2000) Disease agents of cultivated plants observed in Greece from 1981 to 1990. Benaki Phytopathol Inst Kiphissia Athens 19:1–96
Hoppe B, Purahong W, Wubet T, Kahl T, Bauhus J, Arnstadt T, Hofrichter M, Buscot F, Krüger D (2016) Linking molecular deadwood-inhabiting fungal diversity and community dynamics to ecosystem functions and processes in Central European forests. Fungal Divers 77:367–379
Hrustic J, Delibasic G, Stankovic I, Grahovac M, Krstic B, Bulajic A, Tanovic B (2015) Monilinia spp. causing brown rot of stone fruit in Serbia. Plant Dis 99:709–717
Hsieh W-H, Goh T-K (1990) Cercospora and similar fungi from Taiwan. Maw Chang Book, Taipei
Huang F, Udayanga D, Wang X, Hou X, Mei X, Fu Y, Hyde KD, Li H (2015) Endophytic Diaporthe associated with Citrus: a phylogenetic reassessment with seven new species from China. Fungal Biol 119:331–347
Hugenholtz P, Goebel BM, Pace NR (1998) Impact of culture-independent studies on the 23 emerging phylogenetic view of bacterial diversity. J Bacteriol 180:4765–4774
Hyde KD, Bussaban B, Paulus B, Crous PW, Lee S, Mckenzie EH, Photita W, Lumyong S (2007) Diversity of saprobic microfungi. Biodivers Conserv 16:7–35
Hyde Kevin D, Abd-Elsalam Kamel, Cai Lei (2010) Morphology: still essential in a molecular world. Mycotaxon 114:439–451
Hyde KD, Nilsson RH, Alias SA, Ariyawansa HA, Blair JE, Cai L, de Cock AWAM, Dissanayake AJ, Glockling SL, Goonasekara ID, Gorczak M, Hahn M, Jayawardena RS, van Kan JAL, Laurence MH, Lévesque CA, Li XH, Liu JK, Maharachchikumbura SSN, Manamgoda DS, Martin FN, McKenzie EHC, McTaggart AR, Mortimer PE, Nair PVR, Pawłowska J, Rintoul TL, Shivas RG, Spies CFJ, Summerell BA, Taylor PWJ, Terhem RB, Udayanga D, Vaghefi N, Walther G, Wilk M, Wrzosek M, Xu JC, Yan JY, Zhou N (2014) One stop shop: backbones trees for important phytopathogenic genera: I. Fungal Divers 67:21–125
Hyde KD, Hongsanan S, Jeewon R, Bhat DJ, McKenzie EHC, Jones EBG, Phookamsak R, Ariyawansa HA, Boonmee S, Zhao Q, Abdel-Aziz FA, Abdel-Wahab MA, Banmai S, Chomnunti P, Cui BK, Daranagama DA, Das K, Dayarathne MC, de Silva NI, Dissanayake AJ, Doilom M, Ekanayaka AH, Gibertoni TB, Góes-Neto A, Huang SK, Jayasiri SC, Jayawardena RS, Konta S, Lee HB, Li WJ, Lin CG, Liu JK, Lu YZ, Luo ZL, Manawasinghe IS, Manimohan P, Mapook A, Niskanen T, Norphanphoun C, Papizadeh M, Perera RH, Phukhamsakda C, Richter C, de Santiago ALCMA, Drechsler-Santos ER, Senanayake IC, Tanaka K, Tennakoon TMDS, Thambugala KM, Tian Q, Tibpromma S, Thongbai B, Vizzini A, Wanasinghe DN, Wijayawardene NN, Wu HX, Yang J, Zeng XY, Zhang H, Zhang JF, Bulgakov TS, Camporesi E, Bahkali AH, Amoozegar AM, Araujo-Neta LS, Ammirati JF, Baghela A, Bhatt RP, Bojantchev S, Buyck B, da Silva GA, de Lima CLF, de Oliveira RJV, de Souza CAF, Dai YC, Dima B, Duong TT, Ercole E, Mafalda-Freire F, Ghosh A, Hashimoto A, Kamolhan S, Kang JC, Karunarathna SC, Kirk PM, Kytövuori I, Lantieri A, Liimatainen K, Liu ZY, Liu XZ, Lücking R, Medardi G, Mortimer PE, Nguyen TTT, Promputtha I, Raj KNA, Reck MA, Lumyong S, Shahzadeh-Fazeli SA, Stadler M, Soudi MR, Su HY, Takahashi T, Tangthirasunun N, Uniyal P, Wang Y, Wen TC, Xu JC, Zhang ZK, Zhao YC, Zhou JZ, Zhu L (2016) Fungal diversity notes 367–490: taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers 80:1–270
Hyde KD, Norphanphoun C, Abreu VP, Bazzicalupo A, Chethana KT, Clericuzio M, Dayarathne MC, Dissanayake AJ, Ekanayaka AH, He MQ, Hongsanan S, Huang SK, Jayasiri SC, Jayawardena RS, Karunarathna A, Konta S, Kušan I, Lee H, Li J, Lin CG, Liu NG, Lu YZ, Luo ZL, Manawasinghe IS, Mapook A, Perera RH, Phookamsak R, Phukhamsakda C, Siedlecki I, Soares AM, Tennakoon D, Tian Q, Tibpromma S, Wanasinghe DN, Xiao YP, Yang J, Zeng XY, Abdel-Aziz FA, Li WJ, Senanayake IC, Shang QJ, Daranagama DA, de Silva NI, Thambugala KM, Abdel-Wahab MA, Bahkali AH, Berbee ML, Boonmee S, Bhat DJ, Bulgakov TS, Buyck B, Camporesi E, Castañeda-Ruiz RF, Chomnunti P, Doilom M, Dovana F, Gibertoni TB, Jadan M, Jeewon R, Jones EBG, Kang JC, Karunarathna SC, Lim YW, Liu JK, Liu ZY, Plautz HL Jr, Lumyong S, Maharachchikumbura SSN, Matočec N, McKenzie EHC, MeŠić A, Miller D, Pawlowska J, Pereira OL, Promputtha I, Romero AI, Ryvarden L, Su HY, Suetrong S, Tkalčec Z, Vizzini A, Wen TC, Wisitrassameewong K, Wrzosek M, Xu JC, Zhao Q, Zhao RL, Mortimer P (2017) Fungal diversity notes 603–708: taxonomic and phylogenetic notes on genera and species. Fungal Divers 87:1–235
Index Fungorum (2018) http://www.indexfungorum.org/names/names.asp
Jackson HS (1931) The rusts of South America based on the Holway collections—V. Mycologia 23:463–503
Jarvis WR, Traquair JA (1984) Bunch rot of grapes caused by Aspergillus aculeatus. Plant Dis 68:718–719
Javed S, Javaid A, Anwar W, Majeed RA, Akhtar R, Naqvi SF (2017) First Report of Botrytis Bunch Rot of Grapes Caused by Botrytis cinerea in Pakistan. Plant Dis 101:1036
Jayawardena RS, Zhang W, Liu M, Maharachchikumbura SSN, Zhou Y, Huang J-B, Nilthong S, Wang Z-Y, Li XH, Yan JY, Hyde KD (2015) Identification and characterization of Pestalotiopsis-like fungi related to grapevine diseases in China. Fungal Biol 119:348–361
Jayawardena RS, Liu M, Maharachchikumbura SS, Zhang W, Xing Q, Hyde KD, Nilthong S, Li X, Yan J (2016a) Neopestalotiopsis vitis sp. nov. causing grapevine leaf spot in China. Phytotaxa 258:63–74
Jayawardena RS, Hyde KD, Damm U, Cai L, Liu M, Li XH, Zhang W, Zhao WS, Yan JY (2016b) Notes on currently accepted species of Colletotrichum. Mycosphere 7:1192–1260
Jayawardena RS, Hyde KD, Chethana KWT, Daranagama DA, Dissanayake AJ, Goonasekara ID, Manawasinghe IS, Mapook A, Jayasiri SC, Karunarathne A, Li C-G, Phukhamsakda C, Senanayake IC, Wanasinghe DN, Camporesi E, Bulgakov TS, Li XH, Liu M, Zhang W, Yan JY (2018) Mycosphere Notes 102–168: saprotrophic fungi on Vitis in China, Italy, Russia and Thailand. Mycosphere 9:1–114
Jeewon R, Hyde KD (2016) Establishing species boundaries and new taxa among fungi: recommendations to resolve taxonomic ambiguities. Mycosphere 7:1669–1677
Jenkins AE, Bitancourt AA (1940–1963) Myrangiales Selecti Exsiccati. Nos. 1–550. Exsiccati set
Jones DR, Baker RH (2007) Introductions of non-native plant pathogens into Great Britain, (1970–2004). Plant Pathol 56:891–910
Ju Y-M, Rogers JD (1995) Pareutypella gen. nov. for two long-ostiolate pyrenomycetes from Taiwan. Mycologia 87:891–895
Ju Y-M, Rogers JD (1999) The Xylariaceae of Taiwan (excluding Anthostomella). Mycotaxon 73:343–440
Kakalikova L, Jankura E, Srobarova A (2009) First report of Alternaria bunch rot of grapevines in Slovakia. Australas Plant Pathol 4:68–69
Kamal (2010) Cercosporoid fungi of India. Bishen Singh Mahendra Pal Singh, Dehra Dun, p 351
Kanagawa T (2003) Bias and artifacts in multitemplate polymerase chain reaction (PCR). J Biosci Bioengin 96:317–323
Kanematsu S, Minaka N, Kobayashi T, Kudo A, Ohtsu Y (2000) Molecular phylogenetic analysis of Ribosomal DNA internal transcribed spacer regions and comparison of fertility in Phomopsis isolates from fruit trees. J Gen Plant Pathol 66:191–201
Kantschaveli LA (1928) New species of fungi from Georgia. Bolezni Rastenij 17:81–94
Karimi MR, Mahmoodi B, Kazemiyan M (2001) First report of esca of grapevine in Iran. Phytopathol Mediterr 40:S481
Kepley JB, Reeves FB, Jacobi WR, Adams GC (2015) Species associated with Cytospora canker on Populus tremuloides. Mycotaxon 130:783–805
Kern FD, Thurston HW Jr, Whetzel HH (1934) Uredinales. Monogr Univ Puerto Rico B 2:262–303
Khan S, Kamal M (1974) Additions to the parasitic fungi of West Pakistan—III. Mycopathol Mycol Appl 52:29–43
Kim J-D, Shin HD (1998) Taxonomic studies on Cercospora and allied genera in Korea (I). Korean J Mycol 26:327–341
Kim WK, Sang HK, Woo SK, Park MS, Paul NC, Yu SH (2007) Six species of Penicillium associated with blue mold of grape. Mycobiology 35:180–185
Ko TWK, Stephenson SL, Bahkali AH, Hyde KD (2011) From morphology to molecular biology: can we use sequence data to identify fungal endophytes? Fungal Divers 50:113
Kobayashi T (2007) Index of fungi inhabiting woody plants in Japan. Host, distribution and literature. Zenkoku-Noson-Kyoiku Kyokai Publishing, Tokyo
Koike ST, Bettiga LJ, Nguyen TT, Gubler WD (2016) First report of Cylindrocladiella lageniformis and C. peruviana as grapevine pathogens in California. Plant Dis 100:1783–1784
Kõljalg U, Nilsson RH, Abarenkov K, Tedersoo L, Taylor AF, Bahram M, Bates ST, Bruns TD, Bengtsson-Palme J, Callaghan TM, Douglas B (2013) Towards a unified paradigm for sequence-based identification of fungi. Mol Ecol 22:5271–5277
Konstantinia-Sulidu A (1939) Parasitische Pilze Mazedoniens. Hedwig 78:284–298
Korf RP, Pearson RC, Zhuang WY, Dubos B (1986) Pseudopezicula (Helotiales, Peziculoideae), a new discomycete genus for pathogens causing an angular leaf scorch of grapes (“Rotbrenner”). Mycotaxon 26:457–471
Koshkelova EN, Frolov IP (1973) Microflora of Kopet-Dag lowland and Central Karakum (micromycetes). Ylym, Ashkhabad
Kotlaba F (1997) Some uncommon or rare polypores (Polyporales s.l.) collected on uncommon hosts. Czech Mycol 50:133–142
Kozich JJ, Westcott SL, Baxter NT, Highlander SK (2013) Schloss PD: development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl Environ Microbiol 79:5112–5120
Kubatova A, Kolarik M, Pazoutova S (2004) Phaeoacremonium rubrigenum-Hyphomycete associated with bark beetles found in Czechia. Folia Microbiol 49:99–104
Kubartova A, Ottosson E, Dahlberg A, Stenlid J (2012) Patterns of fungal communities among and within decaying logs, revealed by 454sequencing. Mol Ecol 21:4514–4532
Kummuang N, Smith BJ, Diehl SV, Graves CH Jr (1996) Muscadine grape berry rot diseases in Mississippi: disease identification and incidence. Plant Dis 80:238–243
Kunin V, Engelbrektson A, Ochman H, Hugenholtz P (2010) Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12:118–123
Kuntze O (1898) Revisio generum plantarum: vascularium omnium atque cellularium multarum secundum leges nomeclaturae internationales cum enumeratione plantarum exoticarum in itinere mundi collectarum. A. Felix 3:1–576
Kuo K-C, Leu L-S (1998) Phomopsis vitimegaspora: a new pathogenic Phomopsis from vines. Mycotaxon 66:497–499
Lane C, Kirk P (2012) Introduction to fungal plant pathogens. CAB International, Cambridge, pp 1–6
Langrell SRH, Morel O, Robin C (2011) Touchdown nested multiplex PCR detection of Phytophthora cinnamomi and P. cambivora from French and English chestnut grove soils. Fungal Biol 115:672–682
Lardner R, Stummer BE, Sosnowski MR, Scott ES (2005) Molecular identification and detection of Eutypa lata in grapevine. Mycol Res 109:799–808
Larignon P, Dubos B (1997) Fungi associated with esca disease in grapevine. Eur J Plant Pathol 103:147–157
Larignon P, Dubos B (2001) The villainy of black dead arm. Wines Vines 82:86–89
Larsen MJ, Nakasone KK (1984) Additional new taxa of Laeticorticium (Aphyllophorales, Corticiaceae). Mycologia 76:528–532
Larter LNH, Martyn EB (1943) A preliminary list of plant diseases in Jamaica. Mycol Pap 8:1–16
Latorre BA, Guerrero MJ (2001) First report of shoot blight of grapevine caused by Sclerotinia sclerotiorum in Chile. Plant Dis 85:1122
Lawrence DP, Travadon R, Baumgartner K (2015) Diversity of Diaporthe species associated with wood cankers of fruit and nut crops in northern California. Mycologia 107:926–940
Lawrence DP, Travadon R, Pouzoulet J, Rolshausen PE, Wilcox WF, Baumgartner K (2017a) Characterization of Cytospora isolates from wood cankers of declining grapevine in North America, with the descriptions of two new Cytospora species. Plant Pathol 66:713–725
Lawrence DP, Peduto Hand F, Gubler WD, Trouillas FP (2017b) Botryosphaeriaceae species associated with dieback and canker disease of bay laurel in northern California with the description of Dothiorella californica sp. nov. Fungal Biol 121:347–360
Lee Y-H, Cho W-D, Kim WK, Jin K-S, Lee E-J (1991) Report on host-unrecorded diseases identified from economical crops in Korea. Res Rep Rural Developm Admin 33:15–19
Li W, Liu J, Bhat DJ, Camporesi E, Xu J, Hyde KD (2014) Introducing the novel species, Dothiorella symphoricarposicola, snowberry in Italy. Cryptog Mycol 35:257–270
Li GJ, Hyde KD, Zhao RN, Hongsanan S, Abdel-Aziz FA, AbdelWahab MA, Alvarado P, Alves-Silva G, Ammirati JF, Ariyawansa HA, Baghela A, Bahkali AH, Beug M, Bhat DJ, Bojantchev D, Boonpratuang T, Bulgakov TS, Camporesi E, Boro MC, Ceska O, Chakraborty D, Chen JJ, Chethana KWT, Chomnunti P, Consiglio G, Cui BK, Dai DQ, Dai YC, Daranagama DA, Das K, Dayarathne MC, Crop ED, De Oliveira RJV, de Souza CAF, de Souza JI, Dentinger BTM, Dissanayake AJ, Doilom M, Drechsler-Santos ER, Ghobad-Nejhad M, Gilmore SP, Góes-Neto A, Gorczak M, Haitjema GH, Hapuarachchi KK, Hashimoto A, He MQ, Henske JK, Hirayama K, Iribarren MJ, Jayasiri SC, Jayawardena RS, Jeon SJ, Jerónimo GH, Jesus AL, Jones EBG, Kang JC, Karunarathna SC, Kirk PM, Konta S, Kuhnert E, Langer E, Lee HS, Lee HB, Li WJ, Li XH, Liimatainen K, Lima DX, Lin CG, Liu JK, Liu XZ, Liu ZY, Luangsa-ard JJ, Lücking R, Lumbsch HT, Lumyong S, Leaño EM, Marano AV, Matsumura M, McKenzie EHC, Mongkolsamrit S, Mortimer PE, Nguyen TTT, Niskanen T, Norphanphoun C, O’Malley MA, Parnmen S, Pawłowska J, Perera RH, Phookamsak R, Phukhamsakda C, Pires-Zottarelli CLA, Raspe´ O, Reck MA, Rocha SCO, de Santiago ALCMA, Senanayake IC, Setti L, Shang QJ, Singh SK, Sir EB, Solomon KV, Song J, Srikitikulchai P, Stadler M, Suetrong S, Takahashi H, Takahashi T, Tanaka K, Tang LP, Thambugala KM, Thanakitpipattana D, Theodorou MK, Thongbai B, Thummarukcharoen T, Tian Q, Tibpromma S, Verbeken A, Vizzini A, Vlasa´k J, Voigt K, Wanasinghe DN, Wang Y, Weerakoon G, Wen HA, Wen TC, Wijayawardene NN, Wongkanoun S, Wrzosek M, Xiao YP, Xu JC, Yan JY, Yang J, Yang SD, Hu Y, Zhang JF, Zhao J, Zhou LW, Persˇoh D, Phillips AJL, Maharachchikumbura SSN (2016) Fungal divers notes 253–366: taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers 78:1–237
Li WJ, Bhat DJ, Camporesi E, Tian Q, Wijayawardene NN, Dai DQ, Phookamsak R, Chomnunti P, Bahkali AH, Hyde KD (2015) New asexual morph taxa in Phaeosphaeriaceae. Mycosphere 6:681–708
Liang C, Jayawardena RS, Zhang W, Wang X, Liu M, Liu L, Zang C, Xu X, Hyde KD, Yan J, Li X, Zhao K (2016) Identification and characterization of Pseudocercospora species causing grapevine leaf spot in China. J Phytopathol 164:75–85
Linaldeddu BT, Deidda A, Scanu B, Franceschini A, Serra S, Berraf-Tebbal A, Zouaoui Boutiti M, Ben Jamaa ML, Philips AJL (2015) Diversity of Botryosphaeriaceae species associated with grapevine and other woody hosts in Italy, Algeria and Tunisia, with descriptions of Lasiodiplodia exigua and Lasiodiplodia mediterranea sp. nov. Fungal Divers 71:201–214
Linaldeddu BT, Deidda A, Scanu B, Franceschini A, Alves A, Abdollahzadeh J, Phillips AJL (2016) Phylogeny, morphology and pathogenicity of Botryosphaeriaceae, Diatrypaceae and Gnomoniaceae associated with branch diseases of hazelnut in Sardinia (Italy). Eur J Plant Pathol 146:259–279
Litzenberger SC, Farr ML, Lip HT (1962) A preliminary list of Cambodian plant diseases. Div Agric Nat Res, USAID Minist Agric Phnom-Penh, Cambodia
Liu PSW (1977) A supplement to a host list of plant diseases in Sabah, Malaysia. Phytopathol Pap 21:1–49
Liu X-J, Guo Y-L (eds) (1998) Flora Fungorum Sinicorum. Pseudocercospora, vol 9. Science Press, Beijing
Liu JK, Hyde KD, Jones EBG, Ariyawansa HA, Bhat DJ, Boonmee S, Maharachchikumbura S, McKenzie EHC, Phookamsak R, Phukhamsakda C, Shenoy BD, Abdel-Wahab MA, Buyck B, Chen J, Chethana KWT, Singtripop C, Dai DQ, Dai YC, Daranagama DA, Dissanayake AJ, Doliom M, D’souza MJ, Fan XL, Goonasekara ID, Hirayama K, Hongsanan S, Jayasiri SC, Jayawardena RS, Karunarathna SC, Li WJ, Mapook A, Norphanphoun C, Pang KL, Perera RH, Peršoh D, Pinruan U, Senanayake IC, Somrithipol S, Suetrong S, Tanaka K, Thambugala KM, Tian Q, Tibpromma S, Udayanga D, Wijayawardena NN, Wanasinghe D, Wisitrassameewong K, Abdel-Aziz FA, Adamčík S, Bahkali AH, Boonyuen N, Bulgakov T, Callac P, Chomnunti P, Greiner K, Hashimoto A, Hofstetter V, Kang JC, Lewis D, Li XH, Liu XX, Liu ZY, Matumura M, Mortimer PE, Rambold G, Randrianjohany E, Sato G, Sri-Indrasutdhi V, Tian CM, Verbeken A, von Brackel W, Wang Y, Wen TC, Xu JC, Yan JY, Zhao RL, Camporesi E (2015) Fungal diversity notes 1–110: taxonomic and phylogenetic contributions to fungal species. Fungal Divers 72:1–197
Liu QL, Li GQ, Li JQ, Chen SF (2016a) Botrytis eucalypti, a novel species isolated from diseased Eucalyptus seedlings in South China. Mycol Progr 15:1057–1079
Liu M, Zhang W, Zhou Y, Liu Y, Yan JY, Li XH, Jayawardena RS, Hyde KD (2016b) First report of twig anthracnose on grapevine caused by Colletotrichum nymphaeae in China. Plant Dis 100:2530
Lombard L, Houbraken J, Decock C, Samson RA, Meijer M, Réblová M, Groenewald JZ, Crous PW (2016) Generic hyper-diversity in Stachybotriaceae. Persoonia 36:156
Longland JM, Sutton TB (2008) Factors affecting the infection of fruit of Vitis vinifera by the bitter rot pathogen Greeneria uvicola. Phytopathology 98:580–584
Lorsuwan C, Tontyaporn S, Visarathanonth N, Manoch L, Kakishima M (1984) Materials for the rust flora in Thailand I. Trans Mycol Soc Jpn 25:57–65
Lotz-Winter H, Hofmann T, Kirschner R, Kursawe M, Trampe T, Piepenbring M (2011) Fungi in the Botanical Garden of the University of Frankfurt. Z Mykol 77:89–122
Lu B, Hyde KD, Ho WH, Tsui KM, Taylor JE, Wong KM, Yanna, Zhou D (2000) Checklist of Hong Kong Fungi. Fungal Diversity Press, Hong Kong
Lubbe CM, Denman S, Cannon PF, Groenewald JZ, Lamprecht SC, Crous PW (2004) Characterization of Colletotrichum species associated with diseases of Proteaceae. Mycologia 96:1268–1279
Lücking R, Moncada B (2017) Dismantling Marchandiomphalina into Agonimia (Verrucariaceae) and Lawreymyces gen. nov. (Corticiaceae): setting a precedent to the formal recognition of thousands of voucherless fungi based on type sequences. Fungal Divers 84:119–138
Luongo L, Santori A, Riccioni L, Belisario A (2011) Phomopsis sp. associated with post-harvest fruit rot of kiwifruit in Italy. J Plant Pathol 93:205–209
Luque J, Martos S, Phillips AJL (2005) Botryosphaeria viticola sp. nov. on grapevines: a new species with a Dothiorella anamorph. Mycologia 97:1111–1121
Luque J, Martos S, Aroca A, Raposo R, Garcia-Figueres F (2009) Symptoms and fungi associated with declining mature grapevine plants in northeast Spain. J Plant Pathol 91:381–390
Magoč T, Salzberg SL (2011) FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27:2957–2963
Magyar D, Shoemaker RA, Bobvos J, Crous PW, Groenewald JZ (2011) Pyrigemmula, a novel hyphomycete genus on grapevine and tree bark. Mycol Progr 10:307–314
Maharachchikumbura SSN, Guo LD, Chukeatirote E, Bahkali AH, Hyde KD (2011) Pestalotiopsis—morpholgy, phylogeny, biochemsitry and diversity. Fungal Divers 50:167–187
Maharachchikumbura SSN, Crous PW, Groenewald JZ, Xu J, Hyde KD (2014) Pestalotiopsis revisited. Stud Mycol 79:121–186
Maharachchikumbura SN, Hyde KD, Jones EBG, McKenzie EHC, Huang SK, Abdel-Wahab MA, Daranagama DA, Dayarathne M, D’souza MJ, Goonasekara ID, Hongsanan S, Jayawardena RS, Kirk PM, Konta S, Liu JK, Liu Z-Y, Norphanphoun C, Pang KL, Perera RH, Senanayake IC, Shang Q, Shenoy BD, Xiao Y, Bahkali AH, Kang J, Somrothipol S, Suetrong S, Wen T, Xu J (2015) Towards a natural classification and backbone tree for Sordariomycetes. Fungal Divers 72:199
Maharachchikumbura SSN, Larigon P, Hyde KD, Al-Sadi AM, Liu Z (2016) Characteriztion of Neopestalotiopsis, Pestalotiopsis and Truncatella species associated with grapevine trunk disease in France. Phytopathol Mediterr 55:380–390
Mahdian S, Zafari D (2017) First report of table grape blue mold caused by Penicillium sumatrense in Iran. Plant Dis 101:244
Makovetz S (1933) Contributions to the mycoflora of the Ukraine. Bull Jard Bot Kieff 16:71–87
Maluta D-R, Larignon P (1991) Pied-noir: mieux vaut prévenir. Viticulture 11:71–72
Manamgoda DS, Rossman AY, Castlebury LA, Crous PW, Madrid H, Chukeatirote E, Hyde KD (2014) The genus Bipolaris. Stud Mycol 79:221–288
Maneval WE (1937) A list of the Missouri Fungi; with special reference to plant pathogens and wood-destroying species. Univ Missouri Stud Sci Ser 12:1–150
Marais PG (1979) Fungi associated with root rot in vineyards in the Western Cape. Phytophylactica 11:65–68
Marais PG (1980) Fungi associated with decline and death of nursery grapevines in the Western Cape. Phytophylactica 12:9–13
Marchesi JR, Ravel J (2015) The vocabulary of microbiome research: a proposal. Microbiome 3:31
Marin-Felix Y, Groenewald JZ, Cai L, Chen Q, Marincowitz S, Barnes I, Bensch K, Braun U, Camporesi E, Damm U, de Beer ZW, Dissanayake A, Edwards J, Giraldo A, Hernández-Restrepo M, Hyde KD, Jayawardena RS, Lombard L, Luangsa-ard J, McTaggart AR, Rossman AY, Sandoval-Denis M, Shen M, Shivas RG, Tan YP, van der Linde EJ, Wingfield MJ, Wood AR, Zhang JQ, Zhang Y, Crous PW (2017) Genera of phytopathogenic fungi: GOPHY 1. Stud Mycol 86:99–216
Martin MT, Cobos R (2007) Identification of fungi associated with grapevine decline in Castilla y Leon (Spain). Phytopathol Mediterr 46:18–25
Martin MT, Martin L, de-Francisco MT, Cobos R (2009) First report of Lasiodiplodia theobromae and Cryptovalsa ampelina associated with grapevine decline from Castilla y Leon, Spain. Plant Dis 93:545
Martin MT, Martin L, Cuesta MJ, Garcia-Benavides P (2011a) First report of Cylindrocarpon pauciseptatum associated with grapevine decline from Castilla y Leon, Spain. Plant Dis 95:361
Martin MT, Martin L, Cuesta MJ (2011b) First report of Neofusicoccum mediterraneum and N. austral causing decay in Vitis vinifera in Castilla y Leon, Spain. Plant Dis 95:876
Martin FN, Blair JE, Coffey MD (2014) A combined mitochondrial and nuclear multilocus phylogeny of the genus Phytophthora. Fungal Genet Biol 66:19–32
Martiny JBH, Bohannan BJM, Brown JH, Colwell RK, Fuhrman JA, Green JL, Horner-Devine MC, Kane M, Krumins JA, Kuske CR, Morin PJ, Naeem S, Ovreås L, Reysenbach AL, Smith VH, Staley JT (2006) Microbial biogeography: putting microorganisms on the map. Nat Rev Microbiol 4:102–112
Mathur RS (1979) The Coelomycetes of India. Bishen Singh Mahendra Pal Singh, Delhi
Mayorquin JS, Wang DH, Twizeyimana M, Eskalen A (2016) Identification, distribution, and pathogenicity of Diatrypaceae and Botryosphaeriaceae associated with citrus branch canker in the Southern California desert. Plant Dis 100:2402–2413
McDonald V, Eskalen A (2011) Botrysphaeriaceae species associated with avacado branch cankers in California. Plant Dis 95:1465–1473
McKirdy SJ, Riley IT, Cameron IJ (1999) First report of grapevine downy mildew (Plasmopara viticola) in commercial viticulture in Western Australia. Plant Dis 83:301
McLeod A, Botha WJ, Meitz JC, Spies CFJ, Tewoldemedhin YT, Mostert L (2009) Morphological and phylogenetic analyses of Pythium species in South Africa. Mycol Res 113:933–951
Melnik VA, Popushoi IS (1992) Imperfect fungi on species of trees and shrubs (U.S.S.R.). Unknown journal or publisher
Mena-Portales J, Delgado-Rodriguez G, Heredia-Abarca G (2000) New combinations for species of Sporidesmium s.l. Bol Soc Micol Madrid 25:265–269
Mendes MAS, da Silva VL, Dianese JC, Ferreira M, Santos CD, Gomes NE, Urben A, Castro C (1998) Fungos em Plantas no Brasil. Embrapa-SPI/Embrapa-Cenargen, Brasilia
Mey G, Oeser B, Lebrun MH, Tudzynski P (2002) The biotrophic, non-appressorium-forming grass pathogen Claviceps purpurea needs a Fus3/Pmk1 homologous mitogen-activated protein kinase for colonization of rye ovarian tissue. Mol Plant Microbe Interact 15:303–312
Michailides TJ, Peacock W, Christensen P, Morgan DP, Felts D (2002) First report of Aspergillus vine canker of table grapes caused by Aspergillus niger. Plant Dis 86:75
Milholland RD (1994) Macrophoma rot. In: Pearson RC, Goheen AC (eds) Compendium of grape diseases. APS Press, St Paul
Miller JW (1991) Bureau of Plant Pathology. Tri-ol Tech Rep Div Pl Ind Florida 30:2–4
Mills SD, Foerster H, Coffey MD (1991) Taxonomic structure of Phytophthora cryptogea and P. drechsleri based on isozyme and mitochondrial DNA analyses. Mycol Res 95:31–48
Mirzaei S, Nahvi MJ, Khaledi E, Abdollahzadeh J, Amini J, Abrinbana M (2015) Molecular and morphological characterization of Endoconidioma populi from Kurdistan province, Iran. Mycol Iran 2:127–133
Mitchell JI, Zuccaro A (2006) Sequences, the environment and fungi. Mycologist 20:62–74
Mohammadi H, Banihashemi Z (2012) First report of Phaeoacremonium inflatipes and Phaeoacremonium mortoniae associated with grapevine Petri disease in Iran. J Agric Sci Technol 14:1405–1414
Mohammadi H, Banihashemi Z, Gramaje D, Armengol J (2013a) Fungal pathogens associated with grapevine trunk diseases in Iran. J Agric Sci Technol 15:137–150
Mohammadi H, Gramaje D, Banihashemi Z, Armengol J (2013b) Characterization of Diplodia seriata and Neofusicoccum parvum associated with grapevine decline in Iran. J Agric Sci Technol 15:603–616
Moller WJ, Kasimatis AN, Kissler JJ (1974) A dying arm disease of grape in California. Plant Dis Rep 58:869–871
Mondello V, Lo Piccolo S, Conigliaro G, Alfonzo A, Torta L, Burruano S (2013) First report of Neofusicoccum vitifusiforme and presence of the Botryosphaeriaceae species associated with Botryosphaeria dieback of grapevine in Sicily (Italy). Phytopathol Mediterr 52:388–396
Montagne JFC (1856) Sylloge generum specierumque plantarum cryptogamarum, vol 1. J. B. Bailliere, Paris, pp. 1–498
Moralejo E, Clemente A, Descals E, Belbahri L, Calmin G, Lefort F, Spies CFJ, McLeod A (2008) Pythium recalcitrans sp. nov. revealed by multigene phylogenetic analysis. Mycologia 100:310–319
Morgan DP, Michailides TJ (2004) First report of melting decay of ‘Red Globe’ grapes in California. Plant Dis 88:1047
Mostert L, Crous PW, Kang JC, Phillips AJL (2001) Species of Phomopsis and a Libertella sp. occurring on grapevines with specific reference to South Africa: morphological, cultural, molecular and pathological characterization. Mycologia 93:146–167
Mostert L, Halleen F, Creaser ML, Crous PW (2004) Cryptovalsa ampelina, a forgotten shoot and cane pathogen of grapevines. Australas Plant Pathol 33:295–299
Mostert L, Halleen F, Fourie P, Crous PW (2006) A review of Phaeoacremonium species involved in Petri disease and esca of grapevines. Phytopathol Mediterr 45:12–29
Mouchacca J (2009) Novel fungal taxa from the arid Middle East introduced prior to the year 1940. II. Anamorphic fungi: hypomycetes. Cryptog Mycol 30:199–222
Mugnai L, Graniti A, Surico G (1999) Esca (black measles) and brown wood-streaking: two old and elusive diseases of grapevines. Plant Dis 83:404–418
Mujica F, Oehrens B (1967) Segunda addenda a flora fungosa Chilena. Boletin Tecnico 27:1–78
Mujica F, Vergara C (1945) Flora fungosa Chilena. Indice preliminar de los huespedes de los hongos chilenos y sus referencias bibliograficas. Imprenta Stanley
Mulenko W, Majewski T, Ruszkiewicz-Michalska M (2008) A preliminary checklist of micromycetes in Poland, vol 9. W. Szafer Institute of Botany, Polish Academy of Sciences, p 752
Mundkur BB (1943) Indian species of Phakopsora and Bubakia. Mycologia 35:538–545
Mundkur BB, Thirumalachar MJ (1946) Revisions of and additions to Indian fungi. I. Mycol Pap 16:1–27
Muthumary J, Qaiser AS, Sutton BC (1986) Reassessment of Monochaetia terminaliae. Trans Br Mycol Soc 87:103–108
Nag Raj TR (1988) Redisposals and redescriptions in the Monochaetia-Seiridium, Pestalotia-Pestalotiopsis complexes. VIII. On the status of Monochaetia camelliae, M. osyridella and Pestalotia gaurae. Mycotaxon 32:121–132
Nag Raj TR (1993) Coelomycetous anamorphs with appendage-bearing conidia. Mycologue Publications, Waterloo, pp 1–1101
Nag Raj TR, Kendrick B (1975) A monograph of Chalara and allied genera. Wilfrid Laurier University Press, Ontario
Nagorny P (1930) Die pilsflora der Kaukasischen Weinreve. Trav Jard Bot Tiflis Ser II 5:1–201
Nakagiri A, Tadayoshi ITO, Hatamoto M (1994) Brown zonate spot of grape caused by Briosia ampelophaga. Jpn J Phytopathol 60:608–612
Nakaune R, Tatsuki M, Matsumoto H, Ikoma Y (2016) First report of a new postharvest disease of grape caused by Cadophora luteo-olivacea. J Gen Plant Pathol 82:116–119
Nasyrov O (1964) Some species of fungi Imperfecti, new for Turkmenistan in the valley of the middle Ama-Darya. Akad Nauk Turkmen SSR Iav Ser Biol Nauk 3:1–116
Nattrass RM (1961) Host lists of Kenya fungi and bacteria. Mycol Pap 81:1–46
Navarrete F, Abreo E, Bettucci L, Martinez S, Lupo S (2009) First report of Greeneria uvicola as cause of grapevine dead-arm dieback in Uruguay. Australas Plant Dis Notes 4:117–119
Netto MSB, Assuncao IP, Lima GSA, Marques MW, Lima WG, Monteiro JHA, Michereff SJ, Phillips AJL, Camara MPS, de Queiroz Balbino V (2014) Species of Lasiodiplodia associated with papaya stem-end rot in Brazil. Fungal Divers 67:127–141
Netto MSB, Lima WG, Correia KC, Da Silva CFB, Thon M, Martins RB, Miller RNG, Michereff SJ, Camara MPS (2017) Analysis of phylogeny, distribution, and pathogenicity of Botryosphaeriaceae species associated with gummosis of Anacardium in Brazil, with a new species of Lasiodiplodia. Fungal Biol 121:437–451
Nguyen NH, Song Z, Bates ST, Branco S, Tedersoo L, Menke J, Schilling JS, Kennedy PG (2015) FUNGuild: an open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol 20:241–248
Niessl G (1871) Beiträge zur Kenntniss der Pilze. Beschreibung neuer und wenig bekannter Pilze. Verhandlungen des Naturforschenden Vereines in Brünn 10:153–217
Nogueira AF Jr, Fischer IH, Braganca CAD, Massola NS Jr, Amorim L (2016) Identification of Botryosphaeriaceae species that cause stylar-end rot of guavas and characterisation of the disease monocycle. Eur J Plant Pathol 144:271–287
Nomura Y, Takamatsu S, Fujioka K (2003) Teleomorph of Erysiphe necator var. necator on Vitis vinifera and Ampelopsis brevipedunculata var. heterophylla (Vitaceae) newly found in Japan. Mycoscience 44:157–158
Norse D (1974) Plant diseases in Barbados. Phytopathol Pap 18:1–38
O’Donnell K, Cigelnik E, Nirenberg HI (1998) Molecular systematics and phylogeography of the Gibberella fujikuroi species complex. Mycologia 90:465–493
Oh S-Y, Nam K-W, Yoon D-H (2014) Identification of Acremonium acutatum and Trichothecium roseum isolated from grape with white stain symptom in Korea. Mycobiology 42:269–273
Ono Y (2000) Taxonomy of the Phakopsora ampelopsidis species complex on vitaceous hosts in Asia includes a new species, P. euvitis. Mycologia 92:154–173
Ono Y (2016) Mixed infections of grapevine leaf rusts Phakopsora meliosmae-myrianthae and P. montana in Japan. J Gen Plant Pathol 82:149–153
Orieux L, Felix S (1968) List of plant diseases in Mauritius. Phytopathol Pap 7:1–48
Oudemans P, Coffey MD (1991) Isozyme comparison within and among worldwide sources of three morphologically distinct species of Phytophthora. Mycol Res 95:19–30
Ozben S, Demirci F, Degirmenci K, Uzunok S (2012) First report of Cylindrocarpon macrodidymum associated with black foot diseases of Grapevine in Turkey. Plant Dis 96:762
Padwick GW (1946) Notes on Indian fungi. IV. Mycol Pap 17:1–12
Pan FY, Huang Y, Lin L, Zhou YM, Wei RF, Guo WF, Yin L, Lu J (2016) First report of Colletotrichum capsici causing grape ripe rot in Guangxi, China. Plant Dis 100:2531
Pancher M, Ceol M, Corneo PE, Longa CMO, Yousaf S, Pertot I, Campisano A (2012) Fungal endophytic communities in grapevines (Vitis vinifera L.) respond to crop management. Appl Environ Microbiol 78:4308–4317
Pande A (2008) Ascomycetes of Peninsular India. Scientific Publishers (India), Jodhpur
Pantidou ME (1973) Fungus–host index for Greece. Benaki Phytopathological Institute, Kiphissia, Athens
Paolinelli-Alfonso M, Villalobos-Escobedo JM, Rolshausen P, Herrera-Estrella A, Galindo-Sánchez C, López-Hernández JF, Hernandez-Martinez R (2016) Global transcriptional analysis suggests Lasiodiplodia theobromae pathogenicity factors involved in modulation of grapevine defensive response. BMC Genomics 17:615
Pardo Cardona VM (1998) Distribucion de las especies colombianas de Uredinales segun los grupos taxonomicos de sus hospederos. Rev Fac Nal Agr Medellin 51:285–319
Park M-J, Choi Y-J, Hong S-B, Shin H-D (2010) Genetic variability and mycohost association of Ampelomyces quisqualis isolates inferred from phylogenetic analyses of ITS rDNA and actin gene sequences. Fungal Biol 114:235–247
Parkinson LE, Shivas RG, Dann EK (2017) Pathogenicity of Nectriaceous fungi on avocado in Australia. Phytopathology 107:1479–1485
Parris GK (1959) A revised host index of Mississippi plant diseases. Miss State Univ Bot Dept Misc Publ 1:1–146
Pascoe I, Cottral E (2000) Developments in grapevine trunk diseases research in Australia. Phytopathol Mediterr 39:68–75
Passerini (1872) Erb Critt Ital Ser 2:595
Patil MS, Mahamulkar SH (1999) Studies on Meliolaceae of India IV. Indian Phytopathol 52:245–253
Paul YS, Thakur VK (2006) Indian Erysiphaceae. Scientific Publishers (India), Jodhpur
Pavlic-Zupanc D, Piskur B, Slippers B, Wingfield MJ, Jurc D (2015) Molecular and morphological characterization of Dothiorella species associated with dieback of Ostrya carpinifolia in Slovenia and Italy. Phytopathol Mediterr 54:222–231
Pearson RC, Smith FD, Dubos B (1988) Angular leaf scorch, a new disease of grapevines in North America caused by Pseudopezicula tetraspora. Plant Dis 72:796–800
Peng L-J, Sun T, Yang Y-L, Cai L, Hyde KD, Bahkali AH, Liu Z-Y (2013) Colletotrichum species on grape in Guizhou and Yunnan provinces, China. Mycoscience 54:29–41
Pennycook SR (1989) Plant diseases recorded in New Zealand, vols 1–3. Plant Diseases Division, DSIR, Auckland
Peregrine WTH, Ahmad KB (1982) Brunei: a first annotated list of plant diseases and associated organisms. Phytopathol Pap 27:1–87
Peregrine WTH, Siddiqi MA (1972) A revised and annotated list of plant diseases in Malawi. Phytopathol Pap 16:1–51
Péros JP, Jamaux-Despréaux I, Berger G, Gerba D (1999) The potential importance of diversity in Eutypa lata and co-colonising fungi in explaining variation in development of grapevine dieback. Mycol Res 103:1385–1390
Peter G, Tornai-Lehoczki J, Suzuki M, Dlauchy D (2005) Metschnikowia viticola sp. nov., a new yeast species from grape. Antonie van Leeuwenhoek Suppl 87:155–160
Petit E, Gubler WD (2005) Characterization of Cylindrocarpon species, the cause of black foot disease of grapevine in California. Plant Dis 89:1051–1059
Petit E, Gubler WD (2007) First report of Cylindrocarpon liriodendri causing black foot disease of grapevine in California. Plant Dis 91:1060
Petit E, Barriault E, Baumgartner K, Wilcox WF, Rolshausen PE (2011) Cylindrocarpon species associated with black-foot of grapevine in northeastern United States and southeastern Canada. Am J Enol Vitic 62:177–183
Petrak F (1953) Ein Beitrag zur Pilzflora Floridas. Sydowia 7:103–116
Petrak F, Sydow H (1927) Die Gattungen der Pyrenomyzeten, Sphaeropsideen und Melanconieen. 1. Die phaeosporen Sphaeropsideen und die Gattung Macrophoma. Repert Spec Nov Regni Veg 42:1–551
Petrini LE (1992) Rosellinia species of the temperate zones. Sydowia 44:169–281
Petrini LE (2013) Rosellinia—a world monograph. Biblioth Mycol 205:410
Pettersson OV, Leong SL, Lantz H, Rice T, Dijksterhuis J, Houbraken J, Samson RA, Schnurer J (2011) Phylogeny and intraspecific variation of the extreme xerophile, Xeromyces bisporus. Fungal Biol 115:1100–1111
Phengsintham P, Braun U, McKenzie EHC, Chukeatirote E, Cai L, Hyde KD (2013) Monograph of Cercosporoid fungi from Thailand. Plant Pathol Q 3:67–138
Phillips AJL (1998) Botryosphaeria dothidea and other fungi associated with excoriose and dieback of grapevines in Portugal. J Phytopathol 146:327–332
Phillips AJL (1999) The relationship between Diaporthe perjuncta and Phomopsis viticola on grapevines. Mycologia 91:1001–1007
Phillips AJL (2000) Excoriose, cane blight and related diseases of grapevines: a taxonomic review of the pathogens. Phytopathol Mediterr 39:341–356
Phillips AJL, Lucas MT (1997) The taxonomic status of Macrophoma flaccid and Macrophoma reniformis and their relationship to Botryosphaeria dothidea. Sydowia 49:150–159
Phillips AJL, Fonseca F, Povoa V, Castilho R, Nolasco G (2002) A reassessement of the anamorphic fungus Fusicoccum luteum and description of its teleomorph Botryosphaeria lutea sp. nov. Sydowia 54:59–77
Phillips A, Alves A, Correia A, Luque J (2005) Two new species of Botryosphaeria with brown, 1-septate ascospores and Dothiorella anamorphs. Mycologia 97:513–529
Pintos Varela C, Redondo Fernandez V, Aguin Casal O, Ferreiroa Martinez V, Mansilla Vazquez JP (2016) First report of Pleurostoma richardsiae causing grapevine trunk disease in Spain. Plant Dis 100:2168
Pintos VC, Redondo Fernandez V, Aguin Casal O, Mansilla Vazuez JP (2011) First report of cankers and dieback caused by Neofusicoccum mediterraneum and Diplodia corticola on Grapevine in Spain. Plant Dis 95:1315
Piqueras CM, Herrera D, Latorre BA (2014) First report of high boscalid resistance in Botrytis cinerea associated with the H272L mutation in grapevine in Chile. Plant Dis 98:1441
Pirnia M, Zare R, Zamanizadeh HR, Khodaparast A (2012) New records of cercosporoid hyphomycetes from Iran. Mycotaxon 120:157–169
Pitt WM, Huang R, Steel CC, Savocchia S (2010) Identification, distribution and current taxonomy of Botryosphaeriaceae species associated with grapevine decline in New South Wales and South Australia. Aust J Grape Wine Res 16:258–271
Pitt WM, Úrbez-Torres JR, Trouillas FP (2013) Dothiorella vidmadera, a novel species from grapevines in Australia and notes on Spencermartinsia. Fungal Divers 61:209–219
Pitt WM, Úrbez-Torres JR, Trouillas FP (2015) Dothiorella and Spencermartinsia, new species and records from grapevines in Australia. Australas Plant Pathol 44:43–56
Pollack FG (1987) An annotated compilation of Cercospora names. Mycol Mem 12:1–212
Pons N, Sutton BC (1988) Cercospora and similar fungi on yams (Dioscorea species). Mycol Pap 160:1–78
Pota S, Chatasiri S, Unartngam J, Yamaoka Y, Hosaka K, Ono Y (2015) Taxonomic identity of a Phakopsora fungus causing the grapevine leaf rust disease in Southeast Asia and Australasia. Mycoscience 56:198–204
Preston DA (1945) Host index of Oklahoma plant diseases. Oklahoma Agric Coll Agric Exp Sta Techn Bull T 21:1–168
Priest MJ (2006) Fungi of Australia: Septoria. ABRS/CSIRO Publishing, Canberra/Melbourne
Prodorutti D, De Luca F, Michelon L, Pertot I (2009) Susceptibility to Armillaria mellea root rot in grapevine rootstocks commonly grafted onto Teroldego Rotaliano. Phytopathol Mediterr 48:285–290
Promputtha I, Lumyong S, Dhanasekaran V, McKenzie EHC, Hyde KD, Jeewon R (2007) A phylogenetic evaluation of whether endophytes become saprotrophs at host senescence. Microb Ecol 53:579–590
Promputtha I, Hyde KD, McKenzie EH, Peberdy JF, Lumyong S (2010) Can leaf degrading enzymes provide evidence that endophytic fungi becoming saprobes? Fungal Divers 41:89–99
Purahong W, Wubet T, Krüger D, Buscot F (2018) Molecular evidence strongly supports deadwood-inhabiting fungi exhibiting unexpected tree species preferences in temperate forests. ISME J 12:article 289
Qiu Y, Steel CC, Ash GJ, Savocchia S (2011) Survey of Botryosphaeriaceae associated with grapevine decline in the Hunter Valley and Mudgee grape growing regions of New South Wales. Australas Plant Pathol 40:1–11
Raabe RD (1966) Check list of plant diseases previously unreported in Hawaii. Plant Dis Rep 50:411–414
Raabe RD, Conners IL, Martinez AP (1981) Checklist of plant diseases in Hawaii. College of Tropical Agriculture and Human Resources, University of Hawaii. Information Text Series No. 22. Hawaii Inst Trop Agric Human Resources
Radulescu E, Negru A, Docea E (1973) Septoriozele din Romania. Ed Acad Rep Soc Romania Bucarest
Ragunathan AN, Ramakrishnan K (1973) Rust fungi of Madras State. VII. Kuehneola, Hamaspora, Phragmotelium, Phragmidium, Diorchidium, Ravenelia, Hapalophragmium and Nyssopsora. Mysore J Agric Sci 7:73–86
Raimondo ML, Lops F, Carlucci A (2014) Phaeoacremonium italicum sp. nov., associated with esca of grapevine in southern Italy. Mycologia 106:1119–1126
Rajak RC, Pandey AK (1985) Fungi from Jabalpur-II. Indian J Mycol Plant Pathol 15:186–194
Rajchenberg M, Robledo G (2013) Pathogenic polypores in Argentina. For Pathol 43:171–184
Rappaz F (1987) Taxonomy and nomenclature of the octosporous Diatrypaceae. Mycol Helv 2:285–648
Rastogi G, Sani RK (2011) Molecular techniques to assess microbial community structure, function, and dynamics in the environment. In: Microbes and microbial technology, Springer, New York, pp29–57
Reddy MS, Reddy KRC (1983) Greeneria fruit rot—an endemic disease of grape in India. Indian Phytopathol 36:110–114
Rego C, Oliveira H, Carvalho A, Phillips A (2000) Involvement of Phaeoacremonium spp. and Cylindrocarpon destructans with grapevine decline in Portugal. Phytopathol Mediterr 39:76–79
Riley EA (1960) A revised list of plant diseases in Tanganyika Territory. Mycol Pap 75:1–42
Rolshausen PE, Trouillas F, Gubler WD (2004) Identification of Eutypa lata by PCR-RFLP. Plant Dis 88:925–929
Rolshausen PE, Akgul DS, Perez R, Eskalen A, Gispert C (2013) First report of wood canker caused by Neoscytalidium dimidiatum on grapevine in California. Plant Dis 97:1511
Rolshausen PE, Baumgartner K, Travadon R, Pouzoulet J, Fujiyoshi P, Wilcox WF (2014) Identification of Eutypa spp. causing Eutypa dieback of grapvine in Eastern North America. Plant Dis 98:483–491
Rooney-Latham S, Janousek CN, Eskalen A, Gubler WD (2008) First report of Aspergillus carbonarius causing Sour rot of table grapes (Vitis vinifera) in California. Plant Dis 92:651
Roux J, Wingfield MJ, Morris MJ (1997) Botryosphaeria dothidea, a pathogen of Acacia mearnsii in South Africa. S Afr J Sci 93:xii
Rusanov VA, Bulgakov TS (2008) Powdery mildew fungi of Rostov region. Mikol Fitopatol 42:314–322
Ruszkiewicz-Michalska M, Michalski M (2005) Phytopathogenic micromycetes in central Poland. I. Peronosporales and Erysiphales. Acta Mycol 40:223–250
Saccardo PA (1878) Fungi Veneti novi vel critici vel mycologiae Venetae addendi. Ser VII Michelia 1:133–221
Saccardo P, Traverso G (1903) Contribuzione alla flora micologica della Sardegna. Ann Myc 1:427–444
Saito S, Margosan D, Michailides TJ, Xiao CL (2016) Botrytis californica, a new cryptic species in the B. cinerea species complex causing gray mold in blueberries and table grapes. Mycologia 108:330–343
Sakalidis ML, Slippers B, Wingfield BD, St J Hardy GE, Burgess TI (2013) The challenge of understanding the origin, pathways and extent of fungal invasions: global populations of the Neofusicoccum parvum—N. ribis species complex. Div Distrib 19:873–883
Salazar-Yepes M, Buritica P, Cadena-Gomez G (2002) Implicaciones de los estudios sobre biodiversidad de los Uredinales (Royas) en la region cafetera colombiana. Cenicafe 53:219–238
Sami S, Mohammadi H, Heydarnejad J (2014) Phaeoacremonium species associated with necrotic wood of pome fruit trees in Iran. J Plant Pathol 96:487–495
Sanoamuang N, Jitjak W, Rodtong S, Whalley AJS (2013) Gelatinomyces siamensis gen. sp. nov. (Ascomycota, Leotiomycetes, incertae sedis) on bamboo in Thailand. IMA Fungus 4:71–87
Santos C, Fragoeiro S, Valentim H, Phillips A (2006) Phenotypic characterisation of Phaeoacremonium and Phaeomoniella strains isolated from grapevines: enzyme production and virulence of extra-cellular filtrate on grapevine calluses. Sci Horti 107:123–130
Santos JM, Correia VG, Phillips AJL (2010) Primers for mating-type diagnosis in Diaporthe and Phomopsis: their use in teleomorph induction in vitro and biological species definition. Fungal Biol 114:255–270
Santos R, Blume E, Muniz M, Harakawa R, Garrido R, Rego C (2014) Characterization of Campylocarpon pseudofasciculare associated with black foot of grapevine in southern Brazil. Phytopathol Mediterr 53:06–415
Sarbhoy AK, Agarwal DK (1990) Descriptions of tropical plant pathogenic fungi. Set 1 to 10. Malhotra Publ. House, New Delhi
Sarbhoy AK, Lal G, Varshney JL (1971) Fungi of India (1967–71). Navyug Traders, New Delhi
Savas NG, Akgul DS, Albaz EA (2015) First report of Ilyonectria liriodendra associated with black foot disease of grapevine in Turkey. Plant Dis 99:1855
Savulescu T, Rayss T (1935) The species of Cercospora parasitic on Vine leaves in Palestine. Rev Pathol Veg 22:3
Sawada K (1943) Descriptive catalogue of the Formosan fungi. Part IX. Rep Dept Agric Govt Res Inst Formosa 86:1–178
Sawada K (1959) Descriptive catalogue of Taiwan (Formosan) fungi. XI. Spec Publ Coll Agric Natl Taiwan Univ 8:1–268
Sawant IS, Narkar SP, Shetty DS, Upadhyay A, Sawant SD (2012) First report of Colletotrichum capsici causing anthracnose on grapes in Maharashtra, India. New Dis Rep 25:2
Scheck HJ, Vasquez SJ, Gubler WD (1998a) First report of black-foot disease, caused by Cylindroncarpon obtusisporum, of grapevine in California. Plant Dis 82:448
Scheck HJ, Vasquez SJ, Fogle D, Gubler WD (1998b) Grape growers report losses to black-foot and grapevine decline. Calif Agric 52:19–23
Scheper RWA, Crane DC, Whisson DL, Scott ES (2000) The Diaporthe teleomorph of Phomopsis taxon 1 on grapevine. Mycol Res 104:226–231
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Chen W (2012) Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci USA 109:6241–6246
Schubert K, Braun U (2005) Taxonomic revision of the genus Cladosporium s.l. 4. Species reallocated to Asperisporium, Dischloridium, Fusicladium, Passalora, Pseudoasperisporum and Stenella. Fungal Divers 20:187–208
Schulzer S (1870) Mykologische Beiträge. Verhandlungen der Zoologisch-Botanischen Gesellschaft Wien 20:635–658
Seifert KA (1985) A monograph of Stilbella and some allied Hyphomycetes. Stud Mycol 27:1–234
Seifert KA, Axelrood PE (1998) Cylindrocarpon destructans var. destructans. Can J Plant Pathol 20:115–117
Seifert KA, McMullen CR, Yee D, Reeleder RD, Dobinson KF (2003) Molecular differentiation and detection of ginseng-adapted isolates of the root rot fungus Cylindrocarpon destructans. Phytopathology 93:1533–1542
Seifert KA, Nickerson NL, Corlett M, Jackson ED, Louis-Seize G, Davies RJ (2004) Devriesia, a new hyphomycete genus to accommodate heat-resistant, Cladosporium-like fungi. Can J Bot 82:914–926
Senanayake IC, Maharachchikumbura SS, Hyde KD, Bhat JD, Jones EG, McKenzie EH, Dai DQ, Daranagama DA, Dayarathne MC, Goonasekara ID, Konta S, Li WJ, Shang QJ, Stadler M, Wijayawardene NN, Xiao YP, Norphanphoun C, Li Q, Liu XZ, Bahkali AH, Kang JC, Wang Y, Wen TC, Wendt L, Xu JC, Camporesi E (2015) Towards unraveling relationships in Xylariomycetidae (Sordariomycetes). Fungal Divers 73:73–144
Sergeeva V, Priest M, Nair NG (2005) Preliminary studies on Pestalotiopsis species from southern hemisphere conifers in Australasia and South Africa. Australas Plant Pathol 34:255–258
Serra R, Peterson SW (2007) Penicillium astrolabium and Penicillium neocrassum, two new species isolated from grapes and their phylogenetic placement in the P. olsonii and P. brevicompactum clade. Mycologia 99:78–87
Sessa L, Abreo E, Bettucci L, Lupo S (2016) Botryosphaeriaceae species associated with wood diseases of stone and pome fruits trees: symptoms and virulence across different hosts in Uruguay. Eur J Plant Pathol 146:519–530
Setati ME, Jacobson D, Bauer FF (2015) Sequence-based analysis of the Vitis vinifera L. cv Cabernet Sauvignon grape must mycobiome in three South African vineyards employing distinct agronomic systems. Front microbial 6:article1358
Sharma HC, Agarwal DK (1997) A new anthracnose diseae of grape vine (Vitis vinifera) in India. Indian Phytopathol 50:150
Shaw CG (1973) Host fungus index for the Pacific Northwest—I. Hosts. Wash State Univ Agric Exp Sta Bull 765:1–121
Shi F, Hsiang T (2014) First Report of Pseudonectria buxi causing Volutella Blight on Boxwood (Buxus sp.) in Beijing, China. Plant Dis 98:1282–1282
Shirouzu T, Harada Y (2004) Notes on species of Helminthosporium and its allied genera in Japan. Mycoscience 45:17–23
Shivas RG (1989) Fungal and bacterial diseases of plants in Western Australia. J R Soc West Australia 72:1–62
Shivas RG, Tan YP, Edwards J, Dinh Q, Maxwell A, Andjic V, Liberato JR, Anderson C, Beasley DR, Bransgrove K, Coates LM, Cowan K, Daniel R, Dean JR, Lomavatu MF, Mercado-Escueta D, Mitchell RW, Thangavel R, Tran-Nguyen LTT, Weir BS (2016) Colletotrichum species in Australia. Australas Plant Pathol 45:447–464
Shoemaker RA (1992) Catalogue of Pleospora names and types 1. Alphabetically in hard copy and diskette 2. Sortable Data Base on Diskette (available from the author or herb. BPI). Unknown journal or publisher
Simmonds JH (1966) Host index of plant diseases in Queensland. Queensland Department of Primary Industries, Brisbane
Simonyan SA (1981) Mycoflora of Botanical Gardens and Arboreta in Armenia. Hayka
Slippers B, Smit WA, Crous PW, Coutinho TA, Wingfield BD, Wingfield MJ (2007) Taxonomy, phylogeny and identification of Botryosphaeriaceae associated with pome and stone fruit trees in South Africa and other regions of the world. Plant Pathol 56:128–139
Smetham GM, Ades PK, Peros J-P, Ford R (2010) Genetic structure of the grapevine fungal pathogen Phaeomoniella chlamydospora in southeastern Australia and southern France. Plant Pathol 59:736–744
Sneath PH, Sokal RR (1973) Numerical taxonomy. The principles and practice of numerical classification. Freeman WH, San Francisco
Sogonov MV, Castlebury LA, Rossman AY, Mejia LC, White JF (2008) Leaf-inhabiting genera of the Gnomoniaceae, Diaporthales. Stud Mycol 62:1–79
Soliman EA, Abdel-Azeem AM, Salem FM, Nafady NA, Mehesien MT, Ahmed AIS, Ibrahem ME, Abdel-Azeem MA, Hassan SS (2016) A taxonomic analysis of fungi collected and described from Egypt up to (1931). Stud Fungi 1:11–33
Sokal RR, Sneath PH (1963) Principles of numerical taxonomy. San Francisco, p 359
Sosnowski MR, Lardner R, WicksTJ Scott ES (2007) The influence of grapevine cultivar and isolate of Eutypa lata on wood and foliar symptoms. Plant Dis 91:924–931
Sousa da Câmara M (1950) Catalogus Fungorum Juresi (Serra do Gerês) ad Mycofloram Lusitanicam. Agron Lusit 12:89–122
Spegazzini (1878) Riv Viticolt Enolog Conegliano 2:727
Stevenson JA (1975) Fungi of Puerto Rico and the American Virgin Islands. Contr Reed Herb 23:743
Stevenson JA, Wellman FL (1944) A preliminary account of the plant diseases of El Salvador. J Wash Acad Sci 34:259–268
Stewart EJ (2012) Growing unculturable bacteria. J Bacteriol 194:4151–4160
Stoykov DY (2012) Diaporthales. Fungi Bulga 8:319
Stoykow DY, Denchev CM (2006) Current knowledge of Diaporthales (Ascomycota) in Bulgaria. Mycol Balcan 3:179–185
Sultan A, Johnston PR, Park D, Robertson AW (2011) Two new pathogenic ascomycetes in Guignardia and Rosenscheldiella on New Zealand’s pygmy mistletoes (Korthalsella: Viscaceae). Stud Mycol 68:237–247
Sumstine DR (1949) The Albert Commons collection of fungi in the herbarium of the Academy of Natural Sciences in Philadelphia. Mycologia 41:11–23
Sutton BC (1969) Type studies of Coniella, Anthasthoopa, and Cyclodomella. Can J Bot 47:603–608
Sutton BC (1975) Coelomycetes. V. Coryneum. Mycol Pap 138:1–224
Sutton BC (1980) The Coelomycetes. Fungi Imperfecti with Pycnidia, Acervuli and Stromata. Commonwealth Mycological Institute, Kew, Surrey
Suzuki MT, Giovannoni SJ (1996) Bias caused by template annealing in the amplification of mixtures of 16S rRNA genes by PCR. Appl Environ Microbiol 62:625–630
Swett CL, Bourret T, Gubler WD (2016) Characterizing the brown spot pathosystem in late-harvest table grapes (Vitis vinifera L.) in the California Central Valley. Plant Dis 100:2204–2210
Tai FL (1979) Sylloge Fungorum Sinicorum. Sci Press, Acad Sin Peking, pp1527
Tanaka K, Harada Y (2003) Pleosporales in Japan (2): the genus Lophiotrema. Mycoscience 44:115–121
Tao WC, Zhang W, Yan JY, Hyde KD, McKenzie EH, Li XH, Wang Y (2014) A new Alternaria species from grapevine in China. Mycol Prog 13:999
Tassi F (1902) I generi Phyllosticta Pers., Phoma Fr., Macrophoma (Sacc.) Berl. & Voglino e i loro generi analoghi, giusta la legge d’analogia. Bollettino del Laboratorio de Orto Botanico Reale Universita Siena 5:1–76
Tatagiba SD, DaMatta FM, Rodrigues FÁ (2015) Leaf gas exchange and chlorophyll a fluorescence imaging of rice leaves infected with Monographella albescens. Phytopathology 105:180–188
Taylor A, St J Hardy GE, Wood P, Burgess T (2005) Identification and pathogenicity of Botryosphaeria species associated with grapevine decline in Western Australia. Australas Plant Pathol 34:187–195
Taylor DL, Walters WA, Lennon NJ, Bochicchio J, Krohn A, Caporaso JG, Pennanen T (2016) Accurate estimation of fungal diversity and abundance through improved lineage-specific primers optimized for Illumina amplicon sequencing. Appl Environ Microbiol 82:7217–7226
Tejesvi MV, Ruotsalainen AL, Markkola AM, Pirttilä AM (2010) Root endophytes along a primary succession gradient in northern Finland. Fungal Divers 41:125–134
Teng SC (1996) Fungi of China. Mycotaxon Ltd, Ithaca
Teodoro NG (1937) An Enumeration of Philippine Fungi. Tech Bull Dept Agric Comm Manila 4:1–585
Terral JF, Tabard E, Bouby L, Ivorra S, Pastor T, Figueiral I, Picq S, Chevance JB, Jung C, Fabre L, Tardy C (2009) Evolution and history of grapevine (Vitis vinifera) under domestication: new morphometric perspectives to understand seed domestication syndrome and reveal origins of ancient European cultivars. Ann Bot 105:443–455
Thaung MM (1984) Some fungi of Cercospora complex from Burma. Mycotaxon 19:425–452
Thaung MM (2008a) Pathologic and taxonomic analysis of leaf spot and tar spot diseases in a tropical dry to wet monsoon ecosystem of lowland Burma. Australas Plant Pathol 37:180–197
Thaung MM (2008b) Report: some noteworthy fungi of Burma. Australas Mycol 27:111–116
Thaung MM (2008c) Biodiversity survey of coelomycetes in Burma. Australas Mycol 27:74–110
Tibpromma S, Hyde KD, Jeewon R, Maharachchikumbura SSN, Liu JK, Bhat DJ, Jones EBG, McKenzie EHC, Camporesi E, Bulgakov TS, Doilom M, ALCMA Santiago, Das K, Manimohan P, Gibertoni TB, Lim YW, Ekanayaka AH, Thongbai B, Lee HB, Yang JB, Kirk PM, Sysouphanthong P, Singh SK, Boonmee S, Dong W, Raj KNA, Latha KPD, Phookamsak R, Phukhamsakda C, Konta S, Jayasiri SC, Norphanphoun C, Tennakoon DS, Li J, Dayarathne MC, Perera RH, Xiao Y, Wanasinghe DN, Senanayake IC, Goonasekara ID, de Silva NI, Mapook A, Jayawardena RS, Dissanayake AJ, Manawasinghe IS, Chethana KWT, Luo ZL, Hapuarachchi KK, Baghela A, Soares AM, Vizzini A, Meiras-Ottoni A, Mešić A, Dutta AK, de Souza CAF, Richter C, Lin CG, Chakrabarty D, Daranagama DA, Lima DX, Chakraborty D, Ercole E, Wu F, Simonini G, Vasquez G, da Silva GA, Plautz Jr. HL, Ariyawansa HA, Lee H, Kušan I, Song J, Sun J, Karmakar J, Hu K, Semwal KC, Thambugala KM, Voigt K, Acharya K, Rajeshkumar KC, Ryvarden L, Jadan M, Hosen MI, Mikšík M, Samarakoon MC, Wijayawardene NN, Kim NM, Matočec N, Singh PN, Tian Q, Bhatt RP, de Oliveira RJV, Tulloss RE, Aamir S, Kaewchai S, Marathe SD, Khan S, Hongsanan S, Adhikari S, Mehmood T, Bandyopadhyay TK, Svetasheva TY, Nguyen TTT, Antonín V, Li WJ, Wang Y, Indoliya Y, Tkalčec Z, Elgorban AM, Bahkali AH, Tang AMC, Su HY, Zhang H, Promputtha I, Luangsa-ard J, Xu J, Yan JY, Ji-Chuan Y, Stadler M, Mortimer PE, Chomnunti P, Zhao Q, Phillips AJL, Nontachaiyapoom S, Wen TC, Karunarathna SC (2017) Fungal diversity notes 491–602: taxonomic and phylogenetic contributions to fungal taxa. Fungal Divers 83:1–261
Tiffany LH, Gilman JC (1965) Iowa Ascomycetes IV. Diatrypaceae. Iowa State Coll J Sci 40:121–161
Torrejón M (2013) Fungi of Cyprus: new data on micro-and macrofungi. Acta Mycol 48:207–218
Trakunyingcharoen T, Lombard L, Groenewald JZ, Cheewangkoon R, Toanun C, Alfenas AC, Crous PW (2014) Mycoparasitic species of Sphaerellopsis, and allied lichenicolous and other genera. IMA Fungus 5:391–414
Trakunyingcharoen T, Lombard L, Groenewald JZ, Cheewangkoon R, To-anun C, Crous PW (2015) Caulicolous Botryosphaeriales from Thailand. Persoonia 34:87–99
Travadon R, Baumgartner K (2015) Molecular polymorphism and phenotypic diversity in the eutypa dieback pathogen Eutypa lata. Phytopathology 105:255–264
Travadon R, Lawrence PD, Rooney-Latham S, Gubler WD, Wilcox WF, Rolshausen PE, Baumgartner K (2015) Cadophora species associated with wood-decay of grapevine in North America. Fungal Biol 119:53–66
Trouillas F, Gubler WD (2004) Identification and characterization of Eutypa leptoplaca, a new pathogen of grapevine in Northern California. Mycol Res 108:1195–1204
Trouillas FP, Gubler WD (2010) Pathogenicity of Diatrypaceae species in grapevines in California. Plant Dis 94:867–872
Trouillas FP, Úrbez-Torres JR, Gubler WD (2010) Diversity of diatrypaceous fungi associated with grapevine canker diseases in California. Mycologia 102:319–336
Trouillas FP, Pitt WM, Sosnowski MR, Huang R, Peduto F, Loschiavo A, Savocchia S, Scott ES (2011) Taxonomy and DNA phylogeny of Diatrypaceae associated with Vitis vinifera and other woody plants in Australia. Fungal Divers 49:203–223
Tsui CKM, Hyde KD, Hodgkiss IJ (2001) Colonization patterns of wood-inhabiting fungi on baits in Hong Kong rivers, with reference to the effects of organic pollution. Antonie Van Leeuwenhoek J Microb 79:33–38
Tubaki K (1958) Studies on the Japanese Hyphomycetes. V J Hattori Bot Lab 20:142–244
Udayanga D, Liu X, McKenzie EHC, Chukeatirote E, Bahkali AHA, Hyde KD (2011) The genus Phomopsis: biology, applications, species concepts and names of common phytopathogens. Fungal Divers 50:189–225
Udayanga D, Liu X-Z, Crous PW, McKenzie EHC, Chukeatirote E, Hyde KD (2012) A multi-locus phylogenetic evaluation of Diaporthe (Phomopsis). Fungal Divers 56:157–171
Udayanga D, Castlebury LA, Rossman AY, Chukeatirote E, Hyde KD (2014a) Insights into the genus Diaporthe: phylogenetic species delimitation in the D. eres species complex. Fungal Divers 67:203–229
Udayanga D, Castlebury LA, Rossman AY, Hyde KD (2014b) Species limits in Diaporthe: molecular re-assessment of D. citri, D. cytosporella, D. foeniculina and D. rudis. Persoonia 32:83–101
Uecker FA, Kuo K-C (1992) A new Phomopsis with long paraphyses. Mycotaxon 44:425–433
Unamuno PLM (1941) Enumeracion y distribucion geografica de los ascomicetos de la Peninsula Iberica y de las Islas Baleares. Mem Real Acad Ci Exact Madrid 8:1–403
Underhill DM, Iliev ID (2014) The mycobiota: interactions between commensal fungi and the host immune system. Nat Rev Immunol 14:405–416
Úrbez-Torres JR (2011) The status of Botryosphaeriaceae species infecting grapevines. Phytopathol Mediterr 50:5–45
Úrbez-Torres JR, Gubler WD (2009) Pathogenicity of Botryosphaeriaceae species isolated from grapevine cankers in California. Plant Dis 93:584–592
Úrbez-Torres JR, Leavitt GM, Voegel TM, Gubler WD (2006) Identification and distribution of Botryosphaeria spp. associated with grapevine cankers in California. Plant Dis 90:1490–1503
Úrbez-Torres JR, Gubler WD, Luque J (2007) First report of Botryosphaeria iberica and B. viticola associated with grapevine decline in California. Plant Dis 91:772
Úrbez-Torres JR, Battany M, Bettiga LJ, Gispert C, McGourty G, Roncoroni J, Smith RJ, Verdegaal P, Gubler WD (2010a) Botryosphaeriaceae species spore-trapping studies in California vineyards. Plant Dis 94:717–724
Úrbez-Torres JR, Peduto F, Gubler WD (2010b) First report of grapevine cankers caused by Lasiodiplodia crassispora and Neofusicoccum mediterraneum in California. Plant Dis 94:785
Úrbez-Torres JR, Peduto F, Rooney-Latham S, Gubler WD (2010c) First report of Diplodia corticola causing grapevine (Vitis vinifera) cankers and trunk cankers and dieback of canyon live oak (Quercus chrysolepis) in California. Plant Dis 94:785
Úrbez-Torres JR, Peduto F, Striegler RK, Urrea-Romero KE, Rupe JC, Cartwright RD, Gubler WD (2012) Characterization of fungal pathogens associated with grapevine trunk diseases in Arkansas and Missouri. Fungal Divers 52:169–189
Úrbez-Torres JR, Peduto F, Vossen PM, Krueger WH, Gubler WD (2013a) Olive twig and branch dieback: etiology, incidence, and distribution in California. Plant Dis 97:231–244
Úrbez-Torres JR, Peduto F, Smith RJ, Gubler WD (2013b) Phomopsis dieback: a grapevine trunk disease caused by Phomopsis viticola in California. Plant Dis 97:1571–1579
Úrbez-Torres JR, Haag P, Bowen P, O’Gorman DT (2014) Grapevine trunk diseases in British Columbia: incidence and characterization of the fungal pathogens associated with black foot disease of Grapevine. Plant Dis 98:456–468
Úrbez-Torres JR, Adams P, Kamas J, Gubler WD (2009) Identification, incidence, and pathogenicity of fungal species associated with grapevine dieback in Texas. Am J Enol Vitic 60:497–507
Urquhart EJ, Sun LJ, Punja ZK (1997) Identification of species of Tilletiopsis using random amplified polymorphic DNA analysis. Can J Plant Pathol 19:380–389
Urtiaga R (1986) Indice de enfermedades en plantas de Venezuela y Cuba. Impresos en Impresos Nuevo Siglo. S.R.L., Barquisimeto, Venezuela
Uzuhashi S, Tojo M, Kakishima M (2010) Phylogeny of the genus Pythium and description of new genera. Mycoscience 51:337–365
Váczy KZ (2017) First report of Seimatosporium vitis associated with grapevine trunk disease symptoms in Hungary. Plant Dis 101:253
Vail ME, Gubler WD, Adaskaveg JE (1995) First report of Pleurotus ostreatus causing a wood rot of grapevines in California vineyards. Plant Dis 79:1187
Valencia D, Torres C, Camps R, López E, Celis-Diez JL, Besoain X (2015) Dissemination of Botryosphaeriaceae conidia in vineyards in the semiarid Mediterranean climate of the Valparaíso Region of Chile. Phytopathol Mediterr 54:394
Van Coller GJ, Denman S, Groenewald JZ, Lamprecht SC, Crous PW (2005) Characterisation and pathogenicity of Cylindrocladiella spp. associated with root and cutting rot symptoms of grapevines in nurseries. Australas Plant Pathol 34:489–498
Van Niekerk JM, Crous PW, Groenewald JZ, Fourie PH, Halleen F (2004a) DNA phylogeny, morphology and pathogenicity of Botryosphaeria species on grapevines. Mycologia 96:781–798
Van Niekerk JM, Groenewald JZ, Verkley GJM, Fourie PH, Wingfeld MJ, Crous PW (2004b) Systematic reappraisal of Coniella and Pilidiella, with specific reference to species ocurring on Eucalyptus and Vitis in South Africa. Mycol Res 108:283–303
Van Niekerk JM, Groenewald JZ, Farr DF, Fourie PH, Halleen F, Crous PW (2005) Reassessment of Phomopsis species on grapevines. Australas Plant Pathol 34:27–39
Van Niekerk JM, Fourie PH, Halleen F, Crous PW (2006) Botryosphaeria spp. as grapevine trunk disease pathogens. Phytopathol Mediterr 45:S43–S54
Van Niekerk JM, Bester W, Halleen F, Crous PW, Fourie PH (2010) First report of Lasiodiplodia crassispora as a pathogen of grapevine trunks in South Africa. Plant Dis 94:1063
Vandenkoornhuyse P, Quaiser A, Duhamel M, Le Van A, Dufresne A (2015) The importance of the microbiome of the plant holobiont. New Phytol 206:1196–1206
Vanev SG, Sameva EF, Bakalova GG (1997) Order Sphaeropsidales. Fungi Bulga 3:1–335
Vasilyeva LN (1990) New pyrenomycetous species from Kunashir. Mikol Fitopatol 24:207–210
Vasilyeva L, Li Y, Stephenson S (2009) Some pyrenomycetous fungi new to China. Mycotaxon 109:415–428
Vasilyeva L, Chernyshev A, Stephenson SL (2010) Pyrenomycetes of the Russian Far East 4: Family Nitschkiaceae (Coronophorales, Ascomycota). Mycologia 102:233–247
Vasudeva RS (1963) Indian Cercosporae. Indian Council of Agricultural Research, New Delhi
Verkley GJM, Dukik K, Renfurm R, Goker M, Stielow JB (2014) Novel genera and species of coniothyrium-like fungi in Montagnulaceae (Ascomycota). Persoonia 32:25–51
Victor D, Crous PW, Janse BJH, Van Zyl WH, Wingfield MJ, Alfenas AC (1998) Systematic appraisal of species complexes within Cylindrocladiella. Mycol Res 102:273–279
Vitale A, Castello I, Polizzi G (2008) First report of Aspergillus vine canker on table grapes caused by Aspergillus niger in Europe. Plant Dis 92:1471
Voglmayr H, Riethmuller A, Goker M, Weiss MJ, Oberwinkler F (2004) Phylogenetic relationships of Plasmopara, Bremia and other genera of downy mildew pathogens with pyriform haustoria based on Bayesian analysis of partial LSU rDNA sequence data. Mycol Res 108:1011–1024
Vohník M, Burdíková Z, Vyhnal A, Koukol O (2011) Interactions between testate amoebae and saprotrophic microfungi in a Scots pine litter microcosm. Microb Ecol 61:660–668
von Arx JA, Mueller E (1954) Die Gattungen der amerosporen Pyrenomyceten. Beitr Kryptogamenfl Schweiz 11:1–434
Voytyuk SO, Heluta VP, Wasser SP, Nevo E, Takamatsu S, Volz PA (2009) Biodiversity of the Powdery Mildew Fungi (Erysiphales, Ascomycota) of Israel: biodiversity of cyanoprocaryotes, algae and fungi of Israel, vol 7. Koeltz Scientific Books, Königstein
Walker A-S, Gautier A, Confais J, Martinho D, Viaud M, Le Pecheur P, Dupont J, Fournier E (2011) Botrytis pseudocinerea, a new cryptic species causing gray mold in French vineyards in sympatry with Botrytis cinerea. Phytopathology 101:1433–1445
Wang HK, Hyde KD, Soytong K, Lin FC (2008) Fungal diversity on fallen leaves of Ficus in northern Thailand. J Zhejiang Univ Sci B 9:835–841
Wang Y, Wang CW, Gao L (2015) First report of Fusarium proliferatum causing fruit rot on grape (Vitis vinifera) in China. Plant Dis 99:1180
Wangikar PD, Raut JG, Gopalkrishna N (1969) Drying of grape vines caused by Hendersonula toruloidea. Indian Phytopathol 22:403–404
Ward DM, Weller R, Bateson MM (1990) 16S rRNA sequences reveal numerous uncultured microorganisms in a natural community. Nature 345:63–65
Watson AJ (1971) Foreign bacterial and fungus diseases of food, forage, and fiber crops. U.S.D.A. Agricultural Research Service, Washington DC
Weber RW, Stenger E, Meffert A, Matthias HAHN (2004) Brefeldin A production by Phoma medicaginis in dead pre-colonized plant tissue: a strategy for habitat conquest? Mycol Res 108:662–671
Weir BS, Johnston PR, Damm U (2012) The Colletotrichum gloeosporioides species complex. Stud Mycol 73:115–180
White TJ, Bruns T, Lee S, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR protocols: a guide to methods and applications. Academic, New York, pp 315–322
White C-L, Halleen F, Fischer M, Mostert L (2011) Characterisation of the fungi associated with esca diseased grapevines in South Africa. Phytopathol Mediterr 50:204–223
Whitelaw-Weckert MA, Nair NG, Lamont R, Alonso M, Priest MJ, Huang R (2007) Root infection of Vitis vinifera by Cylindrocarpon liriodendra in Australia. Australas Plant Pathol 36:403–406
Whitelaw-Weckert MA, Rahman L, Appleby LM, Hall A, Clark AC, Waite H, Hardie WJ (2013) Co-infection by Botryosphaeriaceae and Ilyonectria spp. fungi during propagation causes decline of young grafted grapevines. Pl Pathol 62:1226–1237
Whiteside JO (1966) A revised list of plant diseases in Rhodesia. Kirkia 5:87–196
Whiting E, Cunha MG, Gubler WD (2005) Phaeomoniella chlamydospora and Phaeoacremonium species distinguished through cultural characters and ribosomal DNA sequence analysis. Mycotaxon 92:351–360
Wicht B, Petrini O, Jermini M, Gessler C, Broggini GAL (2012) Molecular, proteomic and morphological characterization of the ascomycete Guignardia bidwellii, agent of grape black rot: a polyphasic approach to fungal identification. Mycologia 104:1036–1045
Wiehe PO (1948) The plant diseases and fungi recorded from Mauritius. Mycol Pap 24:1–39
Wijayawardene NN, Hyde KD, Rajeshkumar KC, Hawksworth DL, Madrid H, Kirk PM, Braun U, Singh RV, Crous PW, Kukwa M, Lücking R, Kurtzman CP, Yurkov A, Haelewaters D, Aptroot A, Lumbsch HT, Timdal E, Ertz D, Etayo J, Phillips AJL, Groenewald JZ, Papizadeh M, Selbmann L, Dayarathne MC, Weerakoon G, Jones EBG, Suetrong S, Tian Q, Castañeda-Ruiz RF, Bahkali AH, Pang K-L, Tanaka K, Dai DQ, Sakayaroj J, Hujslová M, Lombard L, Shenoy BD, Suija A, Maharachchikumbura SSN, Thambugala KM, Wanasinghe DN, Sharma BO, Gaikwad S, Pandit G, Zucconi L, Onofri S, Egidi E, Raja HA, Kodsueb R, Cáceres MES, Pérez-Ortega S, Fiuza PO, Monteiro JS, Vasilyeva LN, Shivas RG, Prieto M, Wedin M, Olariaga I, Lateef AA, Agrawal Y, Fazeli SAS, Amoozegar MA, Zhao GZ, Pfliegler WP, Sharma G, Oset M, Abdel-Wahab MA, Takamatsu S, Bensch K, de Silva NI, De Kesel A, Karunarathna A, Boonmee S, Pfister DH, Lu Y-Z, Luo Z-L, Boonyuen N, Daranagama DA, Senanayake IC, Jayasiri SC, Samarakoon MC, Zeng X-Y, Doilom M, Quijada L, Rampadarath S, Heredia G, Dissanayake AJ, Jayawardena RS, Perera RH, Tang L-Z, Phukhamsakda C, Hernández-Restrepo M, Ma X, Tibpromma S, Gusmao LFP, Weerahewa D, Karunarathna SC (2017) Notes for genera: Ascomycota. Fungal Divers 86:1–594
Wijayawardene NN, Hyde KD, Divakar PK, Rajeshkumar KC, Weerahewa D, Delgado G, Wang Y, Fu L (2018) Notes for genera update—Ascomycota: 6616–6821. Mycosphere 9:115–140
Witbooi WR, Taylor MA, Fourie JF (2000) In vitro studies on the effect of harvest maturity, relative humidity, wetness period and cold-storage temperature, on the infection potential of Aspergillus niger and Rhizopus stolonifer on Thompson Seedless table grapes. Decid Fruit Grower 50:S1–S14
Wolf FA, Garren KH, Miller JK (1938) Fungi of the Duke Forest and their relation to forest pathology. Bull School For Duke Univ 2:1–122
Wong MKM, Hyde KD (2001) Diversity of fungi on six species of Gramineae and one species of Cyperaceae in Hong Kong. Mycol Res 105:1485–1491
Wu DD, Fu G, Hu FY, Ye YF, Mou HF, Qin LL, Jiang N (2015) First report of Neofusicoccum parvum causing panicle blight and leaf spot on Vitis heyneana in China. Plant Dis 99:417
Yan J-Y, Peng Y-L, Xie Y, Li X-H, Yao S-W, Tang M-L, Wang Z-Y (2011a) First report of grapevine trunk disease caused by Botryosphaeria obtusa in China. Plant Dis 95:616
Yan J-Y, Li X-H, Kong FF, Wang Z-Y, Gong L-Z, He H-P (2011b) Occurrence of grapevine trunk disease caused by Botryosphaeria rhodina in China. Plant Dis 95:219
Yan J, Xie Y, Yao S-W, Wang Z, Li X (2012) Characterization of Botryosphaeria dothidea, the casual agent of grapevine canker in China. Australas Plant Pathol 41:351–357
Yan J-Y, Jayawardena MMRS, Goonasekara ID, Wang Y, Zhang W, Liu M, Huang J-B, Wang Z-Y, Shang J-J, Peng Y-L, Bahkali A, Hyde KD, Li X-H (2015) Diverse species of Colletotrichum associated with grapevine anthracnose in China. Fungal Divers 71:233–246
Yang T, Groenewald JZ, Cheewangkoon R, Jami F, Abdollahzadeh J, Lombard L, Crous PW (2017) Families, genera, and species of Botryosphaeriales. Fungal Biol 121:322–346
Yanna Ho WHH, Hyde KD (2002) Fungal succession on fronds of Phoenix hanceana in Hong Kong. Fungal Divers 10:185–211
Yilmaz N, Visagie CM, Houbraken J, Frisvad JC, Samson RA (2014) Polyphasic taxonomy of the genus Talaromyces. Stud in Mycol 78:175–341
Ying-Ren L (2012) Flora Fungorum Sinicorum: Rhytismatales, vol 40. Science Press, Beijing
Zapparata A, Da Lio D, Sarrocco S, Vannacci G, Baroncelli R (2017) First report of Colletotrichum godetiae causing grape (Vitis vinifera) berry rot in Italy. Plant Dis 101:1051–1052
Zervakis G, Dimou D, Balis C (1998) A check-list of the Greek macrofungi including hosts and biogeographic distribution: I. Basidiomycotina. Mycotaxon 66:273–336
Zhang TY (2003) Flora Fungorum Sinicorum. Alternaria, vol 16. Science Press, Beijing
Zhang L, Zhang GL, Qian X, Li GY (2009) First report of verticillium wilt of grapevine (Vitis vinifera) caused by Verticillium dahlia in China. Plant Dis 93:841
Zheng RY, Liu XY (2005) Actinomucor elegans var. meitauzae, the correct name for A. taiwanensis and Mucor meitauzae (Mucorales, Zygomycota). Nova Hedwig 80:419–432
Zhou YJ, Zhang J, Wang XD, Yang L, Jiang DH, Li GQ, Hsiang T, Zhuang WY (2014) Morphological and phylogenetic identification of Botrytis sinoviticola, a novel cryptic species causing gray mold disease of table grapes (Vitis vinifera) in China. Mycologia 106:43–56
Zhou N, Chen Q, Carroll G, Zhang N, Shivas RG,Cai L (2015) Polyphasic characterization of four new plant pathogenic Phyllosticta species from China, Japan, and the United States. Fung Biol 119:433–446
Zhuang W-Y (ed) (2001) Higher fungi of Tropical China. Mycotaxon Ltd, Ithaca
Zhuang W-Y (ed) (2005) Fungi of northwestern China. Mycotaxon Ltd, Ithaca
Živkoviċ S, Vasiċ T, Anđelković S, Jevremoviċ D, Trkulja V (2012) Identification and characterization of Eutypa lata on grapevine in Serbia. Plant Dis 96:913–913
Zubkova RD (1971) Genus novum Saccharomycetacearum e Kazachstania. Botanicheskie Materialy Gerbariga Instituta Botaniki Akademii Nauk Kazakh. SSR 7:53–56
Acknowledgements
This work was financially supported by Beijing Talent Programm for Dr. Jiye Yan, CARS-29 and JNKYT201605.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jayawardena, R.S., Purahong, W., Zhang, W. et al. Biodiversity of fungi on Vitis vinifera L. revealed by traditional and high-resolution culture-independent approaches. Fungal Diversity 90, 1–84 (2018). https://doi.org/10.1007/s13225-018-0398-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-018-0398-4