
Abstract
Disulfide bonds occurred in majority of secreted protein. Formation of correct disulfide bonds are must for achieving native conformation, solubility and activity. Production of recombinant proteins containing disulfide bond for therapeutic, diagnostic and various other purposes is a challenging task of research. Production of such proteins in the reducing cytosolic compartment of E. coli usually ends up in inclusion bodies formation. Refolding of inclusion bodies can be difficult, time and labor consuming and uneconomical. Translocation of these proteins into the oxidative periplasmic compartment provides correct environment to undergo proper disulfide bonds formation and thus achieving native conformation. However, not all proteins can be efficiently translocated to the periplasm with the help of bacterial signal peptides. Therefore, fusion to a small well-folded and stable periplasmic protein is more promising for periplasmic production of disulfide bonded proteins. In the past decades, several full-length proteins or domains were used for enhancing translocation and solubility. Here, protein fusion tags that significantly increase the yields of target proteins in the periplasmic space are reviewed.
Similar content being viewed by others

Introduction
Since the advent of production of recombinant proteins, application of therapeutic and diagnostic proteins as biopharmaceuticals was changed remarkably (Walsh 2014). These proteins are required in huge amount and usually can not be obtained from natural sources due to extremely low availability. Moreover, Genetically engineered proteins with special benefits (e.g. Insulin analogs) are as such molecules which can therefore only be obtained via recombinant technology (Walsh 2000, 2006; Sanchez and Demain 2012). Escherichia coli was the first and still popularly used host for the fast and economical production of recombinant proteins (Vincentelli and Romier 2013; Chance et al. 1981; Choi and Lee 2004; Rosano and Ceccarelli 2014; Lebendiker and Danieli 2014). In-depth knowledge of genetic and biochemical pathways of E. coli and availability of variety of vectors made is an attractive host for such purposes. Although significant improvements have been made at transcription, translation and translocation, still obtaining soluble and bioactive proteins is a major challenge (Pines and Inouye 1999; Baneyx 1999; Rosano and Ceccarelli 2014).
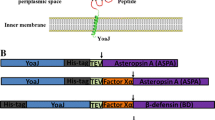
Secreted proteins such as antibodies, enzymes, hormones etc. are used for therapeutic and diagnostic applications. Secreted proteins having two or more cysteines makes disulfide bonds, which is usually vital for structure formation and bioactivity (Creighton 1997b; Creighton et al. 1995; Clarke and Fersht 1993). The cytosol of E. coli is reducing which gives inclusion bodies when such proteins are expressed in the cytosol (Freedman 1989; Hwang et al. 1992; Aslund et al. 1994; Carmel-Harel and Storz 2000; Russel 1995; Messens and Collet 2006). Usually in vitro oxidative refolding is difficult, laborious, time consuming and may be uneconomical depending upon refolding yield (Lilie et al. 1998; Lange and Rudolph 2009; Yamaguchi et al. 2013; Basu et al. 2011). Translocation of these proteins into the E. coli periplasm provides favorable environment for oxidative folding due to the presence of disulfide bond folding and isomerization machinery (Gopal and Kumar 2013; Yoon et al. 2010; Choi and Lee 2004). Moreover, proteases are less abundant in periplasm and also its relatively less crowded than cytosol which reduces the chances of proteolysis and ease in the purification of recombinant proteins (Makrides 1996). To secrete proteins into periplasmic space, a translocation signal sequence must be fused at the N terminus of proteins, but only the fusion of signal sequence is not enough for efficient protein translocation (Fekkes and Driessen 1999; Muller et al. 2001). The sequences on mature protein next to the signal peptidase cut site and other parts of mature protein play an important role in the secretion (Lee et al. 1989; Malik et al. 2006). Under such condition, fusion to a full-length periplasmic protein that is well stable, soluble and properly folded is more promising (Table 1).
Over two decades of extensive in vivo and in vitro research on protein fusions constructs concluded that fusion tags usually increases the yield and solubility of their fusion partners (Costa et al. 2014; Waugh 2005). Despite all these advancement, still it is difficult to choose the best fusion system for a given protein of interest. In general, selection of fusion tag depends upon the properties of protein of interest itself such as size, stability, and hydrophobicity; the expression site; and the usage of the recombinant protein. After coupling with second protein (fusion tag) the increase in yield and solubility the target proteins varies in each fusions. The detailed mechanism by which fusion proteins improve solubility and yield is not well understood. There is two hypotheses: (a) fusion of a stable or conserved structure to an insoluble recombinant protein may serve to stabilize and promote proper folding of the recombinant protein (Butt et al. 2005) and (b) fusion tags may act as a nucleus of folding “molten globule hypothesis” (Creighton 1997a).
Ideally, an effective periplasmic fusion system should have the following features: (a) efficient translocator; (b) enhance folding and solubility; (c) help in purification; (d) facilitate quantification; (e) minimize proteolysis; (f) no adverse effect on the structure and bioactivity; (g) easy and specific removal of the fusion tag; (h) useful for different classes of proteins and peptides. However, none of the fusion tag is optimal with respect to all of these parameters. Successful examples of each periplasmic fusion proteins are listed in Table 2. In the following sections, merits and demerits of available periplasmic fusion proteins are discussed.
Ecotin
Ecotin (E. coli trypsin inhibitor) is a homodimeric protein which is naturally localized in the periplasmic space (Table 1). The properties of ecotin make it a promising periplasmic fusion tag. It is moderately small in size (16 kDa monomer), extremely stable (tolerates pH 1.0 and 100 °C for 30 min) and contains one disulfide bond in each subunit (Chung et al. 1983). Due to the presence of disulfide bonds, ecotin undergoes a pathway of oxidative folding.
Naturally, ecotin is constitutively expressed (Chung et al. 1983) for the defense of E. coli against trypsin like serine proteases in the digestive tract and neutrophil elastase like serine proteases in the blood. Ecotin had no metabolic role or interaction with other proteins in E. coli (Eggers et al. 2004). The C termini of each monomer in dimeric ecotin protrude in opposite directions (Fig. 1a), which will allow folding of passenger proteins at each end without steric hindrance. Strong affinity of ecotin’s for trypsin like serine protease will facilitate ecotin fusion protein to purify via affinity chromatography. Ecotin’s binding surface has been already randomized (Stoop and Craik 2003) to reduce its affinity to zymogens of serine proteases, which would help to elute ecotin fusion proteins under softer conditions.

Three-dimensional structure of periplasmic fusion proteins. a ecotin (1ECZ), b Maltose binding protein (1DMB), c Z-domain of protein A (1LP1), d ABD-domain of protein G (1EM7), e CBD of endoglucanase (1EXG), f CBD of exoglucanase (modelled 3D structure), g disulfide bond oxidoreductase (1A2J), h Barnase (1RNB)
Moreover, model protein in the ecotin fusion system can be quantatively measured in a very sensitive trypsin inhibition assays (Kang et al. 2005). Even in the cytosol ecotin is stable and active; which makes it suitable candidate to be used as cytoplasmic fusion tag (Kang et al. 2005). Ecotin can also be produced in monomeric native state after removal of the last 10 residues (Pal et al. 1996) Thus, ecotin fusion protein in monomeric state is feasible. Ecotin fusion tag have been used for efficient translocation, solubility enhancement and purification of proteins and peptides (Paal et al. 2009; Malik et al. 2006, 2007).
Maltose-binding protein
Maltose-binding protein (MBP) is cysteine-less relatively large (40.6 kDa) periplasmic protein (Fig. 1b) (Duplay et al. 1984). It is known for its noteworthy solubility enhancement when it is fused at the N terminus of model proteins (Raran-Kurussi et al. 2015; Raran-Kurussi and Waugh 2012; Sachdev and Chirgwin 1998). MBP has been frequently utilized for cytosolic expression but due to its natural periplasmic localization, it is also utilized as periplasmic fusion tag for enhancing secretion, solubility as well as purification of target proteins (Salema and Fernandez 2013; Planson et al. 2003). In certain cases, it was found that MBP attains natively folded state and remains soluble while the passenger proteins could not attained properly folded state and exist as in the state of soluble aggregates (Nallamsetty et al. 2005; Nomine et al. 2001; Sachdev and Chirgwin 1999). The affinity of MBP for maltose is ~1 μM which allowed to purify MBP fusion protein through affinity chromatography (Betton and Hofnung 1996). Moreover, MBP is thermodynamically moderately stable with the T m of 62.8 °C at pH 8.3 (Novokhatny and Ingham 1997) and individual components of MBP fusions are slightly more stable than their counterparts in the fusion protein (Blondel et al. 1996).
Staphylococcal protein A
Staphylococcal protein A (SpA) is a surface protein of Gram-positive bacterium Staphylococcus aureus which has strong affinity and high specificity for constant (Fc) part of human immunoglobulins as well as large number of other animals (Eliasson et al. 1988; Cedergren et al. 1993). SpA is a highly soluble 31 kDa protein. Chemically denatured SpA renatures efficiently which assists refolding of the target protein in the SpA fusion system (Samuelsson et al. 1991). SpA is a cysteine-less protein, thus abolishing the chances of interference in disulfide bond formation with fused protein of interest (Kashimura et al. 2013; Uhlen et al. 1984). The gene of SpA is highly repetitive which consists of signal sequence followed by five small highly similar domains (E, D, A, B and C) and C terminal membrane anchoring sequence. The B-domain has been engineered to create smaller variants (7 kDa) of SpA, called as Z-domain (Nilsson et al. 1987). Depending upon localization requirements of the target protein, large number of expression plasmids with or without signal sequences for the production of single Z-domain (7 kDa) or double Z-domains (14 kDa) fusions (Fig. 1c) has been developed (Nilsson et al. 1994, 1996; Hammarberg et al. 1989; Stahl et al. 1989). The fusion protein with Z-domain was more efficiently translocated in comparison to full length SpA proteins (Nilsson et al. 1997).
Streptococcal protein G
Streptococcal protein G (SpG) present on the streptococci surface is a bifunctional receptor and capable of binding with both IgG and serum albumin from different species with different affinities (Nygren et al. 1988). The IgG and albumin binding regions are structurally separated on the SpG. The serum albumin binding region is known as ABD (albumin-binding domain), consists of three binding motifs (each ~5 kDa) (Fig. 1d). Depending upon the localization of the target proteins, ABD with or without signal sequence has been used for expression of fusion protein. Subsequently, fusion proteins were purified via HSA-affinity chromatography in one-step (Hammarberg et al. 1989; Larsson et al. 1996; Stahl et al. 1989).
Cellulose binding domain (CBD)
Nearly 111 residues from endoglucanase (Fig. 1e) and 100 residues from exoglucanase (Fig. 1f) of Cellulomonas fimi, which has high affinity for cellulose, have been used for translocation to periplasmic space and solubility enhancement of target proteins (Gilkes et al. 1988, 1992; Warren et al. 1986; Hwang et al. 2004; Hasenwinkle et al. 1997; Creagh et al. 1996; Ong et al. 1991). The purification of cellulose binding domain fusion protein was achieved via relatively inexpensive ligand matrix (cellulose) (Greenwood et al. 1989, 1992; Ong et al. 1991).
Disulfide bond oxidoreductase
Disulfide bond oxidoreductase (DsbA) is the key enzyme of periplasmic oxidoreductive system (Fig. 1g). It facilitates correct disulfide bond formation via intra- and intermolecular catalysis (Bardwell et al. 1993). In biotechnological applications, target proteins having multiple disulfide bonds (enterokinase catalytic subunit, proinsulin) were fused at the C terminus of DsbA to enhance disulfide bond formation as well as stabilize unfolded target protein via its polypeptide binding site (Collinsracie et al. 1995; Winter et al. 2000). After fusion with DsbA, these proteins were obtained in the well-folded soluble state in the periplasmic space. DsbA is a potent protein thiol oxidase. It has been observed in vitro experiments that DsbA causes non-native disulfide bond formation in proteins having multiple disulfide bonds (Hirudin, BPTI) (Wunderlich and Glockshuber 1993; Zapun and Creighton 1994). Also, in vivo co-expression of DsbA resulted in inclusion bodies formation of IGF-I (Joly et al. 1998).
Barnase
Barnase is an enzymatically inactive variant (H102A) of extracellular RNAse from Bacillus amyloliquefaciens (Fig. 1h). It is monomeric, cysteine-less protein of relatively small size. For biotechnological applications, enzymatically inactive variant of RNAse was used as a fusion protein to enhance the secretion of cysteine-knot peptides in the periplasmic space. It was found that majority of the cysteine-knot peptides were in the native state when fused with barnase (Schmoldt et al. 2005). Moreover, the Barnase fusion protein could be purified via immobilized barstar (Barnase inhibitor) in single step (Schmoldt et al. 2005).
Conclusion
Every protein is unique and due to their different applications such as academic research, diagnostic or therapeutic usage, the quantity and purity level vary. Therefore, no single fusion tag will address every requirement. Fusion tags are helpful in enhancing their solubility and stability. Protein fusion tag with μM-nM ligand affinity generally results in 90–99 % purity after affinity chromatography. Removal of protein fusion tag and producing recombinant protein with authentic N terminal adds another layer of complexity. When considering which protein fusion to use, important queries should keep in mind such as: nature of protein itself, how much protein required, application of protein, is fusion tag removal necessary or not, how much additional residues could be tolerated at N terminal? To remove most part of the fusion protein, highly specific protease cleavage site (TEV protease, thrombin, enterokinase, etc.) could be placed in the linker region between fusion tag and model protein. Also, non-specific proteases such as trypsin could be used to generate authentic N terminus as demonstrated in the case of Ecotin-proinsulin fusion protein (Malik et al. 2007). If authentic N terminus is must for the application, ubiquitin fusion technology could be used as successfully demonstrated in ecotin–ubiquitin–peptide fusion system (Paal et al. 2009).
References
Aslund F, Ehn B, Miranda-Vizuete A, Pueyo C, Holmgren A (1994) Two additional glutaredoxins exist in Escherichia coli: glutaredoxin 3 is a hydrogen donor for ribonucleotide reductase in a thioredoxin/glutaredoxin 1 double mutant. Proc Natl Acad Sci USA 91(21):9813–9817
Baneyx F (1999) Recombinant protein expression in Escherichia coli. Curr Opin Biotechnol 10(5):411–421
Bardwell JC, Lee JO, Jander G, Martin N, Belin D, Beckwith J (1993) A pathway for disulfide bond formation in vivo. Proc Natl Acad Sci USA 90(3):1038–1042
Basu A, Li X, Leong SS (2011) Refolding of proteins from inclusion bodies: rational design and recipes. Appl Microbiol Biotechnol 92(2):241–251. doi:10.1007/s00253-011-3513-y
Betton JM, Hofnung M (1996) Folding of a mutant maltose-binding protein of Escherichia coli which forms inclusion bodies. J Biol Chem 271(14):8046–8052
Blondel A, Nageotte R, Bedouelle H (1996) Destabilizing interactions between the partners of a bifunctional fusion protein. Protein Eng 9(2):231–238
Butt TR, Edavettal SC, Hall JP, Mattern MR (2005) SUMO fusion technology for difficult-to-express proteins. Protein Expr Purif 43(1):1–9
Carmel-Harel O, Storz G (2000) Roles of the glutathione- and thioredoxin-dependent reduction systems in the Escherichia coli and saccharomyces cerevisiae responses to oxidative stress. Annu Rev Microbiol 54:439–461
Cedergren L, Andersson R, Jansson B, Uhlen M, Nilsson B (1993) Mutational analysis of the interaction between staphylococcal protein-a and human Igg(1). Protein Eng 6(4):441–448
Chance RE, Kroeff EP, Hoffmann JA, Frank BH (1981) Chemical, physical, and biologic properties of biosynthetic human insulin. Diabetes Care 4(2):147–154
Choi JH, Lee SY (2004) Secretory and extracellular production of recombinant proteins using Escherichia coli. Appl Microbiol Biotechnol 64(5):625–635. doi:10.1007/s00253-004-1559-9
Chung CH, Ives HE, Almeda S, Goldberg AL (1983) Purification from Escherichia coli of a periplasmic protein that is a potent inhibitor of pancreatic proteases. J Biol Chem 258(18):1032–1038
Clarke J, Fersht AR (1993) Engineered disulfide bonds as probes of the folding pathway of Barnase—increasing the stability of proteins against the rate of denaturation. Biochemistry 32(16):4322–4329
Collinsracie LA, Mccolgan JM, Grant KL, Diblasiosmith EA, Mccoy JM, Lavallie ER (1995) Production of recombinant bovine enterokinase catalytic subunit in Escherichia coli using the novel secretory fusion partner Dsba. Bio-Technol 13(9):982–987
Costa S, Almeida A, Castro A, Domingues L (2014) Fusion tags for protein solubility, purification and immunogenicity in Escherichia coli: the novel Fh8 system. Front Microbiol 5:63. doi:10.3389/fmicb.2014.00063
Creagh AL, Ong E, Jervis E, Kilburn DG, Haynes CA (1996) Binding of the cellulose-binding domain of exoglucanase Cex from Cellulomonas fimi to insoluble microcrystalline cellulose is entropically driven. Proc Natl Acad Sci USA 93(22):12229–12234
Creighton TE (1997a) How important is the molten globule for correct protein folding? Trends Biochem Sci 22(1):6–10
Creighton TE (1997b) Protein folding coupled to disulphide bond formation. Biol Chem 378(8):731–744
Creighton TE, Zapun A, Darby NJ (1995) Mechanisms and catalysts of disulfide bond formation in proteins. Trends Biotechnol 13(1):18–23
Duplay P, Bedouelle H, Fowler A, Zabin I, Saurin W, Hofnung M (1984) Sequences of the male gene and of its product, the maltose-binding protein of Escherichia-Coli-K12. J Biol Chem 259(16):606–613
Eggers CT, Murray LA, Delmar VA, Day AG, Craik CS (2004) The periplasmic serine protease inhibitor ecotin protects bacteria against neutrophil elastase. Biochem J 379:107–118
Eliasson M, Olsson A, Palmcrantz E, Wiberg K, Inganas M, Guss B, Lindberg M, Uhlen M (1988) Chimeric IgG-binding receptors engineered from staphylococcal protein A and streptococcal protein G. J Biol Chem 263(9):4323–4327
Engel H, Mottl H, Keck W (1992) A modified vector for the controlled high-level overproduction of staphylococcal protein A fusion proteins in the periplasm of Escherichia coli. Protein Expr Purif 3(2):108–113
Fekkes P, Driessen AJ (1999) Protein targeting to the bacterial cytoplasmic membrane. Microbiol Mol Biol Rev 63(1):161–173
Freedman RB (1989) Protein disulfide isomerase: multiple roles in the modification of nascent secretory proteins. Cell 57(7):1069–1072
Gilkes NR, Warren RA, Miller RC Jr, Kilburn DG (1988) Precise excision of the cellulose binding domains from two Cellulomonas fimi cellulases by a homologous protease and the effect on catalysis. J Biol Chem 263(21):10401–10407
Gilkes NR, Jervis E, Henrissat B, Tekant B, Miller RC Jr, Warren RA, Kilburn DG (1992) The adsorption of a bacterial cellulase and its two isolated domains to crystalline cellulose. J Biol Chem 267(10):6743–6749
Gopal GJ, Kumar A (2013) Strategies for the production of recombinant protein in Escherichia coli. Protein J 32(6):419–425. doi:10.1007/s10930-013-9502-5
Greenwood JM, Gilkes NR, Kilburn DG, Miller RC, Warren RAJ (1989) Fusion to an endoglucanase allows alkaline-phosphatase to bind to cellulose. FEBS Lett 244(1):127–131
Greenwood JM, Ong E, Gilkes NR, Warren RAJ, Miller RC, Kilburn DG (1992) Cellulose-binding domains—potential for purification of complex proteins. Protein Eng 5(4):361–365
Hammarberg B, Nygren PA, Holmgren E, Elmblad A, Tally M, Hellman U, Moks T, Uhlen M (1989) Dual affinity fusion approach and its use to express recombinant human insulin-like growth factor-Ii. Proc Natl Acad Sci USA 86(12):4367–4371
Hasenwinkle D, Jervis E, Kops O, Liu C, Lesnicki G, Haynes CA, Kilburn DG (1997) Very high-level production and export in Escherichia coli of a cellulose binding domain for use in a generic secretion-affinity fusion system. Biotechnol Bioeng 55(6):854–863
Hayhurst A (2000) Improved expression characteristics of single-chain Fv fragments when fused downstream of the Escherichia coli maltose-binding protein or upstream of a single immunoglobulin-constant domain. Protein Expr Purif 18(1):1–10. doi:10.1006/prep.1999.1164
Honjo E, Watanabe K (1999) Expression of mature pokeweed antiviral protein with or without C-terminal extrapeptide in Escherichia coli as a fusion with maltose-binding protein. Biosci Biotechnol Biochem 63(7):1291–1294. doi:10.1271/bbb.63.1291
Hwang C, Sinskey AJ, Lodish HF (1992) Oxidized redox state of glutathione in the endoplasmic reticulum. Science 257(5076):1496–1502
Hwang S, Ahn J, Lee S, Lee TG, Haam S, Lee K, Ahn IS, Jung JK (2004) Evaluation of cellulose-binding domain fused to a lipase for the lipase immobilization. Biotechnol Lett 26(7):603–605
Joly JC, Leung WS, Swartz JR (1998) Overexpression of Escherichia coli oxidoreductases increases recombinant insulin-like growth factor-I accumulation. Proc Natl Acad Sci USA 95(6):2773–2777
Kang SH, Park JJ, Chung SS, Bang OS, Chung CH (2005) Strategies for assaying deubiquitinating enzymes. Method Enzymol 398:500–508
Kashimura A, Okawa K, Ishikawa K, Kida Y, Iwabuchi K, Matsushima Y, Sakaguchi M, Sugahara Y, Oyama F (2013) Protein A-mouse acidic mammalian chitinase-V5-His expressed in periplasmic space of Escherichia coli possesses chitinase functions comparable to CHO-expressed protein. PLoS One 8(11):e78669. doi:10.1371/journal.pone.0078669
Lange C, Rudolph R (2009) Suppression of protein aggregation by l-arginine. Curr Pharm Biotechnol 10(4):408–414
Larsson M, Brundell E, Nordfors L, Hoog C, Uhlen M, Stahl S (1996) A general bacterial expression system for functional analysis of cDNA-encoded proteins. Protein Expr Purif 7(4):447–457
Lebendiker M, Danieli T (2014) Production of prone-to-aggregate proteins. FEBS Lett 588(2):236–246. doi:10.1016/j.febslet.2013.10.044
Lee C, Li P, Inouye H, Brickman ER, Beckwith J (1989) Genetic-studies on the inability of beta-galactosidase to be translocated across the Escherichia coli cytoplasmic membrane. J Bacteriol 171(9):4609–4616
Lilie H, Schwarz E, Rudolph R (1998) Advances in refolding of proteins produced in E. coli. Curr Opin Biotechnol 9(5):497–501
Makrides SC (1996) Strategies for achieving high-level expression of genes in Escherichia coli. Microbiol Rev 60(3):512
Malik A, Rudolph R, Sohling B (2006) A novel fusion protein system for the production of native human pepsinogen in the bacterial periplasm. Protein Expr Purif 47(2):662–671
Malik A, Jenzsch M, Lubbert A, Rudolph R, Sohling B (2007) Periplasmic production of native human proinsulin as a fusion to E. coli ecotin. Protein Expr Purif 55(1):100–111. doi:10.1016/j.pep.2007.04.006
Messens J, Collet JF (2006) Pathways of disulfide bond formation in Escherichia coli. Int J Biochem Cell Biol 38(7):1050–1062
Muller M, Koch HG, Beck K, Schaefer U (2001) Protein traffic in bacteria: multiple routes from the ribosome to and across the membrane. Prog Nucleic Acid Res Mol Biol 66(66):107–157
Nallamsetty S, Austin BP, Penrose KJ, Waugh DS (2005) Gateway vectors for the production of combinatorially-tagged His(6)-MBP fusion proteins in the cytoplasm and periplasm of Escherichia coli. Protein Sci 14(12):2964–2971
Nilsson B, Moks T, Jansson B, Abrahmsen L, Elmblad A, Holmgren E, Henrichson C, Jones TA, Uhlen M (1987) A synthetic Igg-binding domain based on staphylococcal protein-A. Protein Eng 1(2):107–113
Nilsson J, Nilsson P, Williams Y, Pettersson L, Uhlen M, Nygren PA (1994) Competitive elution of protein-a fusion proteins allows specific recovery under mild conditions. Eur J Biochem 224(1):103–108
Nilsson J, Jonasson P, Samuelsson E, Stahl S, Uhlen M (1996) Integrated production of human insulin and its C-peptide. J Biotechnol 48(3):241–250
Nilsson J, Stahl S, Lundeberg J, Uhlen M, Nygren PA (1997) Affinity fusion strategies for detection, purification, and immobilization of recombinant proteins. Protein Expr Purif 11(1):1–16
Nomine Y, Ristriani T, Laurent C, Lefevre JF, Weiss E, Trave G (2001) Formation of soluble inclusion bodies by HPV E6 oncoprotein fused to maltose-binding protein. Protein Expr Purif 23(1):22–32
Novokhatny V, Ingham K (1997) Thermodynamics of maltose binding protein unfolding. Protein Sci 6(1):141–146
Nygren PA, Eliasson M, Abrahmsen L, Uhlen M, Palmcrantz E (1988) Analysis and use of the serum albumin binding domains of streptococcal protein G. J Mol Recognit 1(2):69–74
Ong E, Gilkes NR, Miller RC, Warren RAJ, Kilburn DG (1991) Enzyme immobilization using a cellulose-binding domain—properties of a beta-glucosidase fusion protein. Enzyme Microb Technol 13(1):59–65
Paal M, Heel T, Schneider R, Auer B (2009) A novel Ecotin-Ubiquitin-Tag (ECUT) for efficient, soluble peptide production in the periplasm of Escherichia coli. Microb Cell Fact 8:7. doi:10.1186/1475-2859-8-7
Pal G, Szilagyi L, Graf L (1996) Stable monomeric form of an originally dimeric serine proteinase inhibitor, ecotin, was constructed via site directed mutagenesis. FEBS Lett 385(3):165–170
Pines O, Inouye M (1999) Expression and secretion of proteins in E. coli. Mol Biotechnol 12(1):25–34
Planson AG, Guijarro JI, Goldberg ME, Chaffotte AF (2003) Assistance of maltose binding protein to the in vivo folding of the disulfide-rich C-terminal fragment from Plasmodium falciparum merozoite surface protein 1 expressed in Escherichia coli. Biochemistry 42(45):13202–13211
Raran-Kurussi S, Waugh DS (2012) The ability to enhance the solubility of its fusion partners is an intrinsic property of maltose-binding protein but their folding is either spontaneous or chaperone-mediated. PLoS One 7(11):e49589. doi:10.1371/journal.pone.0049589
Raran-Kurussi S, Keefe K, Waugh DS (2015) Positional effects of fusion partners on the yield and solubility of MBP fusion proteins. Protein Expr Purif 110:159–164. doi:10.1016/j.pep.2015.03.004
Rosano GL, Ceccarelli EA (2014) Recombinant protein expression in Escherichia coli: advances and challenges. Front Microbiol 5:172. doi:10.3389/fmicb.2014.00172
Russel M (1995) Thioredoxin genetics. Methods Enzymol 252:264–274
Sachdev D, Chirgwin JM (1998) Order of fusions between bacterial and mammalian proteins can determine solubility in Escherichia coli. Biochem Biophys Res Commun 244(3):933–937
Sachdev D, Chirgwin JM (1999) Properties of soluble fusions between mammalian aspartic proteinases and bacterial maltose-binding protein. J Protein Chem 18(1):127–136
Salema V, Fernandez LA (2013) High yield purification of nanobodies from the periplasm of E. coli as fusions with the maltose binding protein. Protein Expr Purif 91(1):42–48. doi:10.1016/j.pep.2013.07.001
Samuelsson E, Wadensten H, Hartmanis M, Moks T, Uhlen M (1991) Facilitated in vitro refolding of human recombinant insulin-like growth factor I using a solubilizing fusion partner. Biotechnology (N Y) 9(4):363–366
Sanchez S, Demain A (2012) Special issue on the production of recombinant proteins. Biotechnol Adv 30(5):1100–1101. doi:10.1016/j.biotechadv.2011.12.004
Schmoldt HU, Wentzel A, Becker S, Kolmar H (2005) A fusion protein system for the recombinant production of short disulfide bond rich cystine knot peptides using barnase as a purification handle. Protein Expr Purif 39(1):82–89
Stahl S, Sjolander A, Nygren PA, Berzins K, Perlmann P, Uhlen M (1989) A dual expression system for the generation, analysis and purification of antibodies to a repeated sequence of the Plasmodium-falciparum antigen Pf155/Resa. J Immunol Methods 124(1):43–52
Stoop AA, Craik CS (2003) Engineering of a macromolecular scaffold to develop specific protease inhibitors. Nat Biotechnol 21(9):1063–1068
Tait AR, Straus SK (2011) Overexpression and purification of U24 from human herpesvirus type-6 in E. coli: unconventional use of oxidizing environments with a maltose binding protein-hexahistine dual tag to enhance membrane protein yield. Microb Cell Fact 10:51. doi:10.1186/1475-2859-10-51
Uhlen M, Guss B, Nilsson B, Gotz F, Lindberg M (1984) Expression of the gene encoding protein A in Staphylococcus aureus and coagulase-negative staphylococci. J Bacteriol 159(2):713–719
Vincentelli R, Romier C (2013) Expression in Escherichia coli: becoming faster and more complex. Curr Opin Struct Biol 23(3):326–334. doi:10.1016/j.sbi.2013.01.006
Walsh G (2000) Biopharmaceutical benchmarks. Nat Biotechnol 18(8):831–833. doi:10.1038/78720
Walsh G (2006) Biopharmaceutical benchmarks 2006. Nat Biotechnol 24(7):769–776. doi:10.1038/nbt0706-769
Walsh G (2014) Biopharmaceutical benchmarks 2014. Nat Biotechnol 32(10):992–1000. doi:10.1038/nbt.3040
Warren RA, Beck CF, Gilkes NR, Kilburn DG, Langsford ML, Miller RC Jr, O’Neill GP, Scheufens M, Wong WK (1986) Sequence conservation and region shuffling in an endoglucanase and an exoglucanase from Cellulomonas fimi. Proteins 1(4):335–341
Waugh DS (2005) Making the most of affinity tags. Trends Biotechnol 23(6):316–320
Winter J, Neubauer P, Glockshuber R, Rudolph R (2000) Increased production of human proinsulin in the periplasmic space of Escherichia coli by fusion to DsbA. J Biotechnol 84(2):175–185
Wunderlich M, Glockshuber R (1993) In vivo control of redox potential during protein-folding catalyzed by bacterial protein disulfide-isomerase (Dsba). J Biol Chem 268(33):24547–24550
Yamaguchi S, Yamamoto E, Mannen T, Nagamune T (2013) Protein refolding using chemical refolding additives. Biotechnol J 8(1):17–31. doi:10.1002/biot.201200025
Yoon SH, Kim SK, Kim JF (2010) Secretory production of recombinant proteins in Escherichia coli. Recent Pat Biotechnol 4(1):23–29
Zapun A, Creighton TE (1994) Effects of Dsba on the disulfide folding of bovine pancreatic trypsin-inhibitor and alpha-lactalbumin. Biochemistry 33(17):5202–5211
Acknowledgments
This study was funded by the National Plan for Science, Technology and Innovation (MAARIFAH), King Abdulaziz city for Science and Technology, Kingdom of Saudi Arabia, Award Number (12-MED-2932).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
I confirm that there is no known conflict of interest associated with this review article.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Malik, A. Protein fusion tags for efficient expression and purification of recombinant proteins in the periplasmic space of E. coli . 3 Biotech 6, 44 (2016). https://doi.org/10.1007/s13205-016-0397-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-016-0397-7