
Abstract
Adjuvant glucocorticoid treatment is routinely used in the treatment of ovarian cancer to mitigate the undesirable side effects of chemotherapy, thereby enhancing tolerability to higher cytotoxic drug doses and frequency of treatment cycles. However, in vitro and preclinical in vivo and ex vivo studies indicate that glucocorticoids may spare tumor cells from undergoing cell death through enhanced cell adhesion, promotion of anti-inflammatory signaling, and/or inhibition of apoptotic pathways. The implications of laboratory studies showing potential negative impact on the efficacy of chemotherapy have been long overlooked since clinical investigations have found no apparent survival detriment attributable to adjuvant glucocorticoid use. Importantly, these clinical studies were not randomized and most did not consider glucocorticoid receptor status, a vital determinant of tumor response to glucocorticoid administration. Additionally, the clinically beneficial elements of increased chemotherapy treatment adherence and dosing afforded by adjuvant glucocorticoids may offset and therefore mask their anti-chemotherapy activities. This review summarizes the current evidence on the impact of glucocorticoids in ovarian cancer and discusses the need for further research and development of alternative strategies to ameliorate untoward side effects of chemotherapy.
Similar content being viewed by others


Ovarian cancer, which accounts for approximately 240,000 new cases and 152,000 deaths annually, is the second deadliest gynecologic malignancy worldwide [1]. Current screening and detection methods generally fail to reveal early disease due to non-descript initial symptoms that can include gastric disturbance, abdominal pain, urinary frequency or urgency, changes in bowel habits, and bloating [2]. As a result, only one third of cases are detected at International Federation of Gynecology and Obstetrics (FIGO) stage I or II, where the tumor is confined to the pelvic cavity [3]. When detected at early stages, 5-year survival can approach 90% (for stage IA) due to the efficacy of initial staging surgery and subsequent intravenous chemotherapy, most commonly with platinum-based (i.e., carboplatin) and taxane-based (i.e., paclitaxel) combination therapy.
Most ovarian cancers are detected after the disease has spread beyond the pelvic cavity, which may make surgical cytoreduction challenging. In those cases where surgical outcome is suboptimal, currently defined as tumor remnants > 1 cm in diameter, surgery offers little overall survival benefit [4, 5]. Moreover, while cytoreductive surgery followed by adjuvant chemotherapy often induces remission, the vast majority (> 70%) of patients recur and eventually become resistant to existing chemotherapies [6,7,8]. A key therapeutic goal is thus to optimize chemotherapy efficacy to eliminate residual tumor cells. Toward this goal, glucocorticoids are typically administered to ameliorate the side effects of high-dose chemotherapy that can limit dosing or number of treatment cycles. However, multiple in vitro and preclinical studies indicate glucocorticoid treatment may negatively impact chemotherapeutic efficacy and promote disease progression [9,10,11,12,13,14].
Glucocorticoid Receptor (GR), its Isoforms, and their Differential Impact on Glucocorticoid Response
Glucocorticoids are multifunctional steroid hormones secreted by cells within the zona fasciculata of the adrenal cortex that act to promote tissue and organism homeostasis. Cortisol, the principal active endogenous glucocorticoid in humans, regulates metabolism, antagonizes pro-inflammatory signaling and cell-related immunity, and features prominently in the stress response. Natural glucocorticoids and their synthetic derivatives are commonly used as powerful immunosuppressants for organ transplant recipients and to treat severe autoimmune conditions [15, 16]. Due to their lympholytic activity, they are also used to treat various lymphoid cancers [17].
Glucocorticoids act primarily by binding and activating an intracellular receptor that then translocates to the cell nucleus where it binds as a homo- or heterodimer to specific nucleotide sequences in regulatory regions of target genes to affect their transcription. The activated GR can either enhance or repress gene transcription through multiple mechanisms (reviewed in [18]). Although encoded by a single gene (NR3C1), several functional GR isoforms are produced through alternative RNA splicing and translational start site usage. The differential expression of these isoforms in a cell can determine its sensitivity and response to glucocorticoids, as recently reviewed by Oakley and Cidlowski [19].
The classical effects of glucocorticoids are mediated through the activation and dimerization of the GRα isoform, a 777 amino acid modular protein consisting of a centrally located DNA-binding domain (DBD), an N-terminal transactivation domain that is independent of ligand activation, and a C-terminal ligand-binding domain (LBD) possessing transactivation function requiring conformational changes induced by hormone binding (Fig. 1).

Schematic indicating variations present among the principal identified GR isoforms encoded by the NR3C1 gene. AF1, transactivating function 1; AF2, transactivating function 2; DBD, DNA-binding domain; GCs, glucocorticoids; H, hinge region; LBD, ligand-binding domain
Among the other identified isoforms, the function of GRβ and GRγ are best known and both can act to result in decreased or altered glucocorticoid activity. GRβ is encoded by an alternatively spliced NR3C1 transcript resulting in substitution of 15 non-homologous amino acids for the 50 C-terminal amino acids of GRα, a region of the protein involved in ligand binding [20]. As a consequence, GRβ is unable to bind GR agonists but constitutively localizes to the cell nucleus and blocks GRα activity by acting as a dominant negative inhibitor. Moreover, GRβ affects the expression levels of a subset of genes independent of GRα [21]. Interestingly, this latter activity is blocked by the GR antagonist mifepristone (RU486), which binds both GRα and GRβ [21].
GRγ is also encoded by an alternatively spliced NR3C1 transcript, resulting in insertion of a single arginine addition after amino acid 470, located within the DNA binding domain (Fig. 1). While this addition does not impact the ability of the receptor to bind ligands, it alters the interaction of the receptor with regulatory factors at the site of DNA binding in a context-dependent manner [22]. As a result, GRγ activation results in an overlapping and distinct program of gene transcription activation/repression compared to GRα, thereby altering the cellular response to glucocorticoids. Moreover, since GRγ is less transcriptionally active than GRα, high expression levels of GRγ can result in glucocorticoid insensitivity.
In addition to GRβ and GRγ, which are ubiquitously expressed, two GR isoforms missing portions of their C-terminal ligand-binding domain have been identified in cancer cells that may alter sensitivity to glucocorticoids [23,24,25,26,27]. An additional seven N-terminal truncated GR protein isoforms have been identified that are thought to arise from leaky ribosomal scanning, enabling the use of alternative downstream initiation codons [19, 28, 29]. Altogether, the collective expression of alternative GR isoforms relative to GRα determines cellular sensitivity and the specific cellular response to glucocorticoids. While the vast majority of ovarian tumors express GR, potential differential expression of GR isoforms, which could alter sensitivity and tumor response, has not been determined.
In addition to isoform expression, GR activity is influenced by multiple factors including, but not limited to, co-regulator availability (both co-repressors and co-activators), receptor dimerization with other transcription factors, and post-translational modifications including phosphorylation, sumoylation, ubiquitination, and acetylation (reviewed in [30]). Glucocorticoid activity is also affected by the specific glucocorticoid used, intracellular levels achieved, and their affinity for other steroid hormone receptors. For example, cortisol has high affinity for both GR and mineralocorticoid receptors (MR) and is prevented from activating MR in some tissues, such as distal kidney ductules, through its local conversion to inactive cortisone by 11β-hydroxysteroid dehydrogenase type II (11βHSDII) [31]. Downregulation of 11βHSDII or an increase in 11βHSD type I activity results in increased intracellular cortisol, which can lead to increased GR and MR activation. Dexamethasone (DEX), a clinically valuable potent synthetic glucocorticoid, binds much more avidly to GR than MR, exhibiting a relative binding affinity for MR of 17% as compared to aldosterone [32]. Thus, measurement of GR expression alone, including that of the various GR isoforms, may not fully correlate with glucocorticoid response.
Glucocorticoids Ameliorate Side Effects of Chemotherapy
Frontline chemotherapy regimens for advanced ovarian cancer typically consist of a combination of taxane and platin-based therapy. A common and serious side effect of this regimen is nausea and emesis, which is particularly problematic with cisplatin and to a somewhat lesser extent with carboplatin [33]. Antiemetic agents including serotonin 3 (5-HT3) or neurokinin 1 (NK1) receptor antagonists and glucocorticoids are typically administered to mitigate chemotherapy-induced nausea and vomiting (CINV) [33,34,35]. Glucocorticoids also promote electrolyte balance and stimulate appetite, in part by suppressing edema, pain, and inflammation, in addition to nausea and emesis associated with chemotherapy [34,35,36]. Additionally, they reduce the risk of hypersensitivity reactions to taxanes [37], which can limit patient treatment options.
For moderate to highly emetic agents such as cisplatin or carboplatin, DEX 12 mg is usually given prior to chemotherapy and daily 8 mg DEX is prescribed for 3 days after completing chemotherapy to prevent acute and delayed CINV [14]. To prevent hypersensitivity reactions, 20 mg of DEX is given before paclitaxel treatment [38]. Since a combination of platinum and paclitaxel is the standard regimen of chemotherapy in ovarian cancer, 8 to 20 mg of DEX is usually given daily for 3 days of the chemotherapy cycle. The incorporation of glucocorticoids into chemotherapy regimens thus increases patient comfort and improves adherence to treatment protocols [39].
Glucocorticoids Promote Tumor Cell Survival
Glucocorticoids induce apoptosis in hematological cell lineages and therefore have proven to be an effective direct anti-neoplastic therapy for cancers derived from these lineages [36]. However, glucocorticoids support the survival of numerous non-hematological cell types, an effect that extends following their malignant transformation. Robust in vitro and ex vivo studies for numerous non-hematological malignancies have identified potential detriments to chemotherapeutic efficacy in patients concurrently treated with glucocorticoids [40,41,42,43,44,45,46,47].
Glucocorticoids inhibit chemotherapy-induced apoptosis in surgically derived tumors from ovary, breast, brain, prostate, liver, pancreas, colon, cervix, bone, skin, and nervous tissue [42]. These effects were observed for diverse chemotherapeutic agents including paclitaxel, cisplatin, 5-fluorouracil, adriamycin, actinomycin D, doxorubicin, and gemcitabine. Since these agents target different cellular functions, the mechanisms underlying this impact of glucocorticoids likely involves a general pro-survival effect such as enhancing cell adhesion, decreasing the likelihood of programmed cell death, or through inducing immunosuppression.
Several studies suggest that glucocorticoid-induced anti-apoptotic gene expression protects ovarian cancer cells from undergoing programmed cell death in the presence of cytotoxic agents [43, 46, 47]. Analogous in vitro, in vivo, and ex vivo findings have been obtained for triple-negative breast cancer (TNBC) [48,49,50,51,52,53,54,55]. Consistent with the breast cancer literature, gene expression studies implicate the anti-inflammatory effects of serum glucocorticoid kinase-1 (SGK1) and dual-specificity phosphatase-1/mitogen-activated protein kinase phosphatase-1 (DUSP1/MKP1) as playing a central role in pro-survival effects on ovarian cancer cells [43, 46]. The evidence for the stark survival benefit experienced by tumor cells across multiple experimental environments is discussed below.
Zhang et al. [47] identified consistent survival effects of DEX in established ovarian cancer cell lines, mouse xenografts, and freshly isolated clinical tumor tissues. Irrespective of experimental modality, treatment with clinically relevant doses of DEX 48 h prior to cisplatin or gemcitabine abrogated the growth inhibitory or apoptotic response of ovarian tumor cells to the chemotherapeutic agent. Remarkably, xenograft tumors in mice treated with DEX and cisplatin grew as fast in vivo as untreated controls, and a similar result was observed for five established cell lines in vitro. Importantly, DEX pretreatment of clinically derived tumor specimens reduced the effect of cisplatin, gemcitabine, and γ-radiation in inducing cell death. The authors conducted preliminary molecular analysis, based on the TNBC literature, and speculated that their findings may be due to the observed DEX-induced increased SGK1 and DUSP1 expression, both of which promote cell survival. However, the precise events underlying glucocorticoid-induced cell resistance to chemotherapeutics are not completely understood. Elucidating the mechanism of this resistance is essential for understanding the impact of glucocorticoid usage in clinical practice and for designing more effective treatment strategies.
Melhem et al. [43] identified a 5- and 10-fold increase in SGK1 and DUSP1 transcript levels, respectively, in GR-positive omental and ovarian tumor samples from ovarian cancer patients as rapidly as 30 min following DEX administration. Concordantly, SKOV3 and HEY8 ovarian cancer cell lines—both GR positive—exhibited similar induction of SGK1 and DUSP1 transcript and protein expression following DEX treatment [43]. Administration of mifepristone reversed this effect, which was taken as confirmation that GR activation was required. Furthermore, cytotoxic paclitaxel treatment efficacy, at doses ranging from 10−8 to 10−6 M, decreased by 40% with 1 h DEX pretreatment, as determined by examining the apoptotic rate in SKOV3 and HEY8 cells [43]. This survival benefit induced by DEX was present up to, and likely beyond, 30 h following the initial treatment with paclitaxel, holding significant implications for the structuring of common chemotherapy regimens. In addition to paclitaxel, DEX inhibited gemcitabine-, γ-radiation-, and cisplatin-induced cell death. Importantly, these effects were observed using clinically relevant doses of DEX and with patient-derived heterogeneous tumor samples, providing the first clinically derived evidence for glucocorticoid-induced survival on ovarian tumors.
More recently, Stringer-Reasor et al. [46] investigated the effect of GR status and glucocorticoid administration in vitro, in vivo, and ex vivo using ovarian cancer cell lines, mouse xenografts, and patient-derived tumors. GR expression was identified as essential for the protective glucocorticoid effect, as cell lines without GR expression did not exhibit a survival benefit due to DEX in the presence of chemotherapy. In GR-positive cell lines, combination carboplatin/gemcitabine therapy was 48% less effective at inducing apoptosis in the presence of DEX as compared to in the absence of DEX. This survival benefit of DEX was abolished by the inclusion of mifepristone or CORT125134, further indicating the effect was mediated by GR activation. CORT125134, also known as relacorilant, is a competitive GR antagonist that, unlike mifepristone, has no affinity for the progesterone receptor or other steroid hormone receptors. In agreement with previous studies, SGK1 and DUSP1 expression was enhanced due to DEX treatment and was proposed as explaining the survival increase [46]. However, glucocorticoids affect a wide array of gene transcripts [56] and may impact both generalized and specific cellular pathways that contribute to altered tumor survival.
Regarding DEX concentrations used for in vitro studies (0.01 or 1.0 μmol/L), commensurate levels are achieved in patients (one dose of 12 mg results in plasma concentrations of between 0.25 and 0.5 μmol/L) [9]; thus, potential detrimental effects of glucocorticoid treatment identified by in vitro studies may have relevance in vivo.
Glucocorticoids Impact Inflammatory Mediators and Immune Function
Tumor necrosis factor alpha (TNFα) is a vital inflammatory cytokine whose expression is significantly enhanced by radiation and by chemotherapy [57,58,59] and is a predictor and effector of tumor cell death [58]. TNFα activates NFκB, a key mediator in inflammatory cell signaling that also acts to increase expression of various cytokines, including TNFα [60, 61]. Glucocorticoids are well-known antagonists of NFκB signaling and, therefore, of TNFα expression and function [57,58,59]. Other cytokines such as interleukin 1 beta (IL-1β), TNF-related apoptosis-inducing ligand (TRAIL), and cluster of differentiation 95-ligand (CD95-L) are similarly inhibited by glucocorticoids following chemotherapy, with authors of numerous studies consequently expressing concern over potential glucocorticoid attenuation of treatment efficacy in several cancers [11, 59, 62].
When pursuing cancer cure, robust host immune surveillance and response is essential for eliminating remnant cancer cells following surgery, radiation, and/or chemotherapy [63]. Initial concerns over glucocorticoid-induced immune suppression were raised over 50 years ago [12, 64]. The effects of prolonged glucocorticoid use in humans may be analogous to results obtained in murine models that show glucocorticoid administration prevents effective elimination of surviving cancer cells by rendering them resistant to cytotoxic treatment and by reducing B- and T-lymphocyte activity and recruitment [65]. Although analogous studies have not been conducted in humans, rapid immune suppression following glucocorticoid treatment is observed [16, 66].
Glucocorticoids Exert Effects on Cell Adhesion, ECM Remodeling, and Metastasis
Part of the resistance to chemotherapy conferred by glucocorticoids may result from their ability to increase cellular adherence to extracellular matrix (ECM) components. While cancer cells are typically able to evade anoikis and proliferate in the absence of attachment, cell–matrix attachment enhances survival of cancer cells in the presence of cytotoxic treatment [67,68,69,70]. Several preclinical studies identifying a glucocorticoid-induced increase in cell survival following radio- and chemotherapy suggest glucocorticoids establish a permissive microenvironment for tumors to attach, migrate, and/or spread by altering expression of adhesion components [67,68,69,70,71,72,73].
DEX was shown to enhance cell binding to fibronectin-coated tissue dishes by up to 200% in a dose-dependent manner across multiple ovarian cancer cell lines [67]. In contrast, cytotoxic cisplatin and/or paclitaxel treatment decreased cellular attachment by 51% and resulted in significant cell death as compared to controls. When simultaneously administered with cisplatin and/or paclitaxel treatment, DEX increased cell survival and adhesion in parallel and in a dose-dependent manner. DEX completely blocked apoptosis induction by cytotoxic treatment [67]. These studies thus contribute to the concerns regarding glucocorticoid usage during radio- and chemotherapy as they support the idea that glucocorticoids may promote treatment failure and disease recurrence.
Mechanistic studies of how glucocorticoids affect cell adhesion have focused largely on integrins, a class of vital cell–cell and cell–ECM binding proteins known to function in cancer progression and initiation (extensively reviewed in [74, 75]). In fact, select integrins have been implicated as therapeutic targets in phase II and III clinical trials. The integrin β1 subfamily, the primary receptor for fibronectin that in turn anchors to fibrillar collagen, consists of heterodimers with variable α subunits. Fibronectin is a putative cancer biomarker with an established role in cell migration; increased fibronectin expression has been implicated in the progression and spread of numerous cancers [76,77,78].
In SKOV-3 and HO-8910 ovarian cancer cells, DEX treatment enhanced β1, α4, and α5 integrin expression by 150–300% and promoted secretion of fibronectin, suggesting a combinatorial increase of both receptor and extracellular ligand for protective ECM–cell bridging [67]. Reducing the binding capacity of β1 integrin to fibronectin using inhibitory antibodies eliminated up to 70% of the survival benefit of DEX during paclitaxel and/or cisplatin therapy [67]. In addition, DEX treatment increased Mucin 1 (Muc1), a large transmembrane glycoprotein that along with similar glycoproteins forms the cancer cell glycocalyx. The glycocalyx can reduce overall integrin binding by decreasing the spatial proximity of the cell membrane to ECM substrates. However, studies have shown that the glycocalyx, and Muc1 in particular, promotes integrin clustering on the cell surface to result in enhanced focal adhesion assembly and increased cell proliferation and survival [79].
The overall impact of glucocorticoid treatment on metastasis is less clear. Tumor metastasis involves acquisition of invasive cell properties characterized by epithelial to mesenchymal cell transition (EMT) and expression of matrix metalloproteinases. In addition, metastasis is enhanced by changes to the tumor microenvironment, largely orchestrated by cancer-associated fibroblasts. Both EMT and stromal fibroblast activation are induced by transforming growth factor (TGF)-β1 signaling [80,81,82,83]. Glucocorticoids both enhance and inhibit TGF-β signaling [80,81,82,83,84], and these signaling pathways synergistically modulate the cell cycle in several cell types [80]. Moreover, TGF-β1 produced pro-survival and adhesion effects both independent of, and synergistic with, DEX in ovarian cancer cells [67]. DEX treatment also enhanced TGF-β type 2 receptor expression in prostate cancer cells [84], a receptor essential for initial binding of TGF-β and activation of type I receptor activity. TGF-β activation of kidney epithelial cell EMT in obstructive neuropathy is mediated by an increase in SGK1, which is associated with increased ECM production and fibrosis [85]. While activation of SGK1 expression could be a point of synergy between GR and TGF-β signaling pathways, DEX has been shown to suppress TGF-β1-induced EMT in mink lung epithelial cells [86] and in A549 lung carcinoma cells [87]. Moreover, DEX alone can promote an epithelial cell phenotype [86, 88]. Thus, an improved understanding of the contextual impact of glucocorticoids on TGFβ signaling in cancer cells and cancer-associated fibroblasts is required.
A recent series of studies provide a mechanism by which glucocorticoids may act beneficially to suppress ovarian cancer metastasis. Lin et al. [89] demonstrated that glucocorticoids increase expression of miRNA-708 in ovarian cancer cell lines, which suppresses migration in vitro and metastasis in vivo. They further showed that miRNA-708 suppresses translation of Rap1B, a member of the Ras-like superfamily involved in regulation of integrin signaling and cell–cell adhesion [90]. In a subsequent study, Lin et al. [91] reported that in an orthotopic mouse model using syngeneic ID8 ovarian cancer cells, DEX treatment inhibited growth of the primary tumor and reduced peritoneal seeding in association with increased miRNA-708 and decreased Rap1B expression. While these studies suggest a potential beneficial impact of adjuvant glucocorticoid therapy, further investigation of the impact of glucocorticoid treatment on ovarian cancer seeding and metastasis using patient-derived xenograft models with known GR isoform expression is required.
Mutations in Breast Cancer, 1 (BRCA1) Affect Glucocorticoid Receptor Activity
A family history of breast or ovarian cancer is the greatest identified risk factor for epithelial ovarian cancer, with most of this heritable risk attributable to germline mutations in the BRCA1 or BRCA2 genes. These mutations confer a lifetime risk of up to 60% for BRCA1 mutation carriers and up to 40% for BRCA2 mutation carriers, compared to 1.4–1.7% for the general population [92,93,94,95]. BRCA1 and BRCA2 mutation carriers are predisposed selectively to the high-grade serous ovarian cancer (HGSOC) histotype, which is the most commonly diagnosed and lethal histotype. The various ovarian cancer histotypes are associated with distinct clinical course, commonly mutated genes, and proposed cell of origin. Numerous studies support the view that the epithelium within the distal fallopian tube is the principal site for the origin of HGSOC [96,97,98]. A current theory is that repetitive exposure of distal fallopian tube epithelial cells to pro-inflammatory signaling molecules present in follicular fluid at the time of ovulation promotes oxidative stress-induced DNA modifications that promote malignant transformation. The well-established anti-inflammatory action of GR signaling has led to investigation of whether cancer-predisposing mutations in BRCA1 associate with impaired GR signaling [99, 100].
Treatment of fallopian tube epithelial cells with DEX blocks TNFα or IL1-β-induced inflammatory signaling [56]. In addition, increased inflammatory signaling is evident in gene expression profiles of fallopian tube epithelia from BRCA1 mutation carriers compared with that from non-mutation carriers who underwent salpingectomy during the post-ovulatory luteal phase [99]. These profiles were consistent with decreased GR activity in tissues from BRCA1 mutation carriers. Moreover, knock-down of BRCA1 protein levels with targeting siRNA in cancer cell lines resulted in decreased GR transactivation whereas expression of exogenous wild-type BRCA1 cDNA increased GR transactivation [99].
GR mRNA and protein levels are positively correlated with BRCA1 expression in patient-derived ovarian cancer specimens [100]. Specifically, BRCA1 repression—either through mutation or promoter hypermethylation—correlates with decreased GR expression. However, adjacent non-cancerous tissue did not exhibit BRCA1 mutation-associated GR inhibition, suggesting that oncogenic signaling pathways combined with reduced functional BRCA1 levels results in decreased GR levels. This is consistent with studies indicating that BRCA1 enhances GR activity and levels in TNBC [101]. Vilasco et al. [101] further showed that in breast cancer cells, BRCA1 potentiates the activation of p38 Mitogen-Activated Protein Kinase (MAPK), which phosphorylates GR at serine-211, which is essential for robust GR signaling. Parallel studies in ovarian cancer cells have not yet been reported.
GR may also regulate BRCA1 expression to impact DNA double-strand break repair. An interesting study by Ritter et al. [102] showed that in the absence of glucocorticoid, GR expression increased functional BRCA1 levels whereas treatment with glucocorticoid decreased BRCA1 protein levels. In the absence of ligand, GR binds to the transactivation subunit, GABPβ, of the ets transcription factor in the promoter region of the BRCA1 gene to stimulate BRCA1 expression. This interaction is disrupted upon glucocorticoid binding to GR, leading to decreased BRCA1 levels. The authors speculate that this mechanism could contribute to an increased risk of breast cancer associated with chronic stress [102, 103]. In this context, glucocorticoid treatment might enhance the efficacy of PARP inhibitors in the treatment of ovarian cancers. DNA homologous repair could be impaired because of glucocorticoid-induced decreased BRCA1 levels. While a decrease in BRCA1 could, in turn, decrease GR expression and glucocorticoid response, this would not be expected to lead to increased BRCA1 since it is the unliganded GR that acts to increase BRCA1 promoter activity. Thus, BRCA1 levels could be decreased by reducing the level of GR binding at the BRCA1 gene promoter by either ligand binding or reducing the level of unliganded GR.
BRCA1 mutation carriers initially exhibit enhanced responsiveness to platinum-based chemotherapy [104] but develop refractory disease at increased rates and ultimately succumb to their illness [105]. The effect of a BRCA1 mutation may initially inhibit the glucocorticoid-induced ovarian cancer cell survival benefit due to diminished GR signaling, thereby allowing greater initial cytogenicity, but disease recurs due to acquired treatment resistance. Therefore, ovarian cancer cell lines with and without GR isoform expression, and with differential BRCA1 levels may be of use in xenograft or in vitro studies of the interaction between GR isoforms, chemotherapy, and glucocorticoid administration.
Impact of Glucocorticoids on Clinical Outcomes: Need for Further Study
Despite possible detrimental effects of glucocorticoids on the efficacy of chemotherapy at the cellular level, glucocorticoids remain incorporated into standard treatment as an adjunctive medication along with chemotherapy [34, 106]. There is little clinical evidence regarding the impact of glucocorticoid administration and oncological outcomes in ovarian cancer patients. A retrospective study of 245 ovarian cancer patients published in 2004 concluded that there was no evidence glucocorticoid treatment negatively impacted survival [39]. The study included patients who were scheduled to receive at least six courses of systemic chemotherapy, which could include cisplatin, epirubicin, and cyclophosphamide, and compared those who were given glucocorticoids to augment anti-emetic agents (n = 62) to those who did not receive glucocorticoids (n = 183). The primary outcomes assessed in the study were hematologic toxicity and recurrence-free and overall survival. Patients who received glucocorticoid treatment had significantly higher leukocyte counts after chemotherapy compared with patients who did not receive glucocorticoid treatment. Glucocorticoids may prevent chemotherapy-induced leukocytopenia by inhibiting apoptosis of neutrophils [107, 108] making patients less likely to delay or incur dose reduction during an entire treatment course, which are known poor prognostic factors in ovarian cancer [109, 110]. In the study, 65% of patients who had received glucocorticoids completed their chemotherapy as planned, as compared to only 45% of patients who did not receive glucocorticoids. Moreover, 58% of patients who received glucocorticoids had a complete response to their treatment, whereas only 37% of patients who did not receive glucocorticoids had a complete response. Despite these clear beneficial effects of glucocorticoid use, there was no improvement by inclusion of glucocorticoid treatment on either recurrence-free or overall survival, as would be expected. This raises the possibility that detrimental effects of glucocorticoids countering the effects of chemotherapy on tumor cells are offset or masked by beneficial effects on patient well being and treatment compliance.
A recent retrospective study designed to examine the association of GR expression and clinical outcome in ovarian cancer patients has implicated high GR expression as a negative prognostic indicator [111]. GR was shown to be expressed in 65.9% of 341 primary ovarian tumors from patients who received primary cytoreductive surgery and adjuvant chemotherapy. High GR expression, based upon percentage of cells stained for GR immunoreactivity and the intensity of staining, was found to be an independent prognostic factor for decreased recurrence-free survival but not overall survival, adjusting for patient age, grade, stage, and histotype. In contrast, GR expression did not show prognostic value in a study of 85 ovarian cancer patients, with no evidence of poorer survival in a small subset of GR-positive patients, who had received adjuvant glucocorticoid treatment [112]. Neither of these studies included an assessment of adjuvant glucocorticoid treatment. A definitive answer to the question of whether glucocorticoid treatment is without detrimental impact in patients with ovarian carcinoma cannot be provided by a retrospective analysis and would be better addressed in a well-controlled prospective trial that includes tumor GR isoform profiling.
A related question is whether GR activation, in general, promotes ovarian cancer progression. The response of ovarian cancer patients to mifepristone was assessed in a small phase II study of 34 ovarian cancer patients with tumors resistant to cisplatin and paclitaxel. Treatment with mifepristone showed activity with a response rate of 26.5% [113]. While the rationale for the use of mifepristone was as a progesterone receptor antagonist, mifepristone is an equally potent GR antagonist and it is not clear which activity is responsible for the effects observed. Voisin et al. [114] have recently shown that the proliferative effects of 6-oxo-cholestan-3β,5α-diol (OCDO), a tumorigenic metabolite of cholesterol, on breast cancer cells are mediated by GRs. OCDO also activated expression of known glucocorticoid-responsive genes, including SGK1 and DUSP1. Further, they demonstrated that 11βHSDII is the enzyme responsible for OCDO formation whereas 11βHSDI expression inactivates OCDO. Thus, it is possible that inhibiting GR activation in a subset of ovarian cancer patients may impede tumor progression independent of impacting the efficacy of chemotherapeutics.
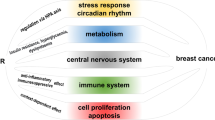
Given the range of GR responses, the use of specifically designed glucocorticoid mimetic agents as adjuvant therapy could be an attractive approach to yield better patient outcomes [115, 116]. Pertaining to solid tumors, it is not presently known how shifting GR target gene expression patterns might affect patient outcomes; however, as we have indicated above, GR may act in both an inhibitory or stimulatory manner in regards to tumor growth and metastasis (summarized in Fig. 2). Selectively limiting the range of target genes affected by GR may prove beneficial for cancer patients. Although selective glucocorticoid receptor agonists and modulators (SEGRAMs) have not been tested clinically, they may present an appealing option for future research and clinical use as an alternative to traditional glucocorticoids.

Summary of the effect of glucocorticoids on pathways involved in cancer cell survival, metastasis, and inflammatory signaling
As already mentioned, DEX in combination with a 5-HT3 and/or NK1 inhibitor is typically recommended as a prophylaxis to prevent acute (defined as the first 24 h after chemotherapy administration) CINV in patients treated with moderately or highly emetogenic chemotherapy. Palonosetron, a second-generation 5-HT3 receptor antagonist with higher affinity and stability than first-generation antagonists, provides protection for both acute and delayed CINV when administered as a single dose. In breast cancer patients given 0.25 mg palonosetron and 8 mg i.v. DEX on day 1 of moderate or highly emetogenic chemotherapy treatment, additional treatment with twice daily 4 mg oral DEX on days 2 and 3 offered no further improvement in preventing CINV as compared to patients receiving placebo on these days [117]. This study indicates that incorporation of second generation 5-HT3 receptor antagonists in CINV prophylaxis regimens may enable the total dose of DEX administered to be safely reduced. Similarly, in patients treated with i.v. DEX + palonosetron + 125 mg oral aprepitant (NK1 receptor antagonist) on day 1 to prevent acute CINV in breast cancer patients treated with a combination of anthracycline and cyclophosphamide, Roila et al. [118] demonstrated that 80 mg aprepitant administered on days 2 and 3 was as effective as twice daily oral DEX for preventing delayed CINV. Of those patients who experienced delayed CINV despite treatment, patients receiving DEX rather than aprepitant reported less severe nausea and fewer emetic episodes; however, this difference was not statistically significant. Although the authors concluded that DEX should be chosen over aprepitant because of its lower cost, the potential benefit of avoiding DEX-induced protection of tumor cells should be considered.
Reduction of DEX treatment for the prevention of hypersensitivity reactions in breast cancer patients receiving paclitaxel has also been investigated. These patients typically receive premedication consisting of DEX and histamine 1 and 2 receptor blockers. Patients susceptible to hypersensitivity reactions tend to exhibit these during their first or second exposure to paclitaxel. Berger et al. [119] have shown that discontinuing hypersensitivity premedications, including DEX, in subsequent paclitaxel treatments in those patients who did not experience a hypersensitivity reaction during their first two exposures did not result in an increased incidence of hypersensitivity reactions. It remains to be determined if reduction of DEX dosing impacts the efficacy of chemotherapy.
Conclusion
Inclusion of glucocorticoids at the time of chemotherapy has clinical benefit associated with its ability to function as an antiemetic and to decrease hypersensitivity responses to cytotoxic agents. Conversely, a preponderance of in vivo, ex vivo, and preclinical animal models point to effects that can undermine the efficacy of cytotoxic chemotherapies (Fig. 2), which could promote residual tumor survival and resistance to chemotherapy. It is also unlikely that all subtypes of ovarian cancer will respond identically to glucocorticoids since they are derived from different cells of origin and not all ovarian cancer tumors express GR. However, studies indicate that most ovarian tumors are GR positive and hence detrimental effects of the glucocorticoid therapy on chemotherapy efficacy could offset the beneficial effects of enabling compliance with prescribed dosing (Fig. 3).

Collective evidence indicates that adjuvant therapy with currently used GR agonists have both a beneficial and deleterious impact on concurrent chemotherapy treatment. These opposing activities may balance each other to mask an impact on overall or progression-free survival, resulting in clinical studies concluding that adjuvant glucocorticoids do not negatively impact the patient
The key question of whether glucocorticoid therapy has demonstrable negative impact on disease-free or overall survival of ovarian cancer patients remains. Currently, only one retrospective study has addressed this question. While this study indicates there is no impact of DEX on survival, this is surprising given the significant impact of DEX on adherence to scheduled chemotherapy. A carefully designed prospective study controlling for histotype, stage, and chemotherapy schedule completion, and considering GR isoform expression is required to address this question. Such studies are difficult to conduct given the recognized beneficial effects of glucocorticoids. As further SEGRAMs are developed and tested preclinically, they should be incorporated into clinical trials as substitutes for currently used GR agonists in chemotherapy regimens.
References
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F (2015) Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer 136(5):E359–E386. https://doi.org/10.1002/ijc.29210
Devlin SM, Diehr PH, Andersen MR, Goff BA, Tyree PT, Lafferty WE (2010) Identification of ovarian cancer symptoms in health insurance claims data. J Women's Health (Larchmt) 19(3):381–389. https://doi.org/10.1089/jwh.2009.1550
Maringe C, Walters S, Butler J, Coleman MP, Hacker N, Hanna L, Mosgaard BJ, Nordin A, Rosen B, Engholm G, Gjerstorff ML, Hatcher J, Johannesen TB, McGahan CE, Meechan D, Middleton R, Tracey E, Turner D, Richards MA, Rachet B, ICBP Module 1 Working Group (2012) Stage at diagnosis and ovarian cancer survival: evidence from the International Cancer Benchmarking Partnership. Gynecol Oncol 127(1):75–82. https://doi.org/10.1016/j.ygyno.2012.06.033
Chang SJ, Bristow RE, Chi DS, Cliby WA (2015) Role of aggressive surgical cytoreduction in advanced ovarian cancer. J Gynecol Oncol 26(4):336–342. https://doi.org/10.3802/jgo.2015.26.4.336
Elattar A, Bryant A, Winter-Roach BA, Hatem M, Naik R. (2011) Optimal primary surgical treatment for advanced epithelial ovarian cancer. Cochrane Database Syst Rev CD007565
Agarwal R, Kaye SB (2003) Ovarian cancer: strategies for overcoming resistance to chemotherapy. Nat Rev Cancer 3(7):502–516. https://doi.org/10.1038/nrc1123
Rubin SC, Randall TC, Armstrong KA, Chi DS, Hoskins WJ (1999) Ten-year follow-up of ovarian cancer patients after second-look laparotomy with negative findings. Obstet Gynecol 93(1):21–24
Winter WE 3rd, Maxwell GL, Tian C, Sundborg MJ, Rose GS, Rose PG et al (2008) Tumor residual after surgical cytoreduction in prediction of clinical outcome in stage IV epithelial ovarian cancer: a gynecologic oncology group study. J Clin Oncol 26:83–89
Rutz HP (2002) Effects of corticosteroid use on treatment of solid tumours. Lancet 360(9349):1969–1970. https://doi.org/10.1016/S0140-6736(02)11922-2
Rutz HP, Herr I (2004) Interference of glucocorticoids with apoptosis signaling and host–tumor interactions. Cancer Biol Ther 3:315–318
Herr I, Ucur E, Herzer K, Okouoyo S, Ridder R, Krammer PH, von Knebel Doeberitz M, Debatin KM (2003) Glucocorticoid cotreatment induces apoptosis resistance toward cancer therapy in carcinomas. Cancer Res 63(12):3112–3120
Sherlock P, Hartmann H (1962) Adrenal steroids and the pattern of metastasis of breast cancer. JAMA 181(4):313–317. https://doi.org/10.1001/jama.1962.03050300033007
Iversen HG, Hjort GH (1958) The influence of corticoid steroids on the frequency of spleen metastasis in patients with breast cancer. Acta Pathologica Microbiologica Scandinavica 44:205–212
Sui M, Chen F, Chen Z, Fan W (2006) Glucocorticoids interfere with therapeutic efficacy of paclitaxel against human breast and ovarian xenograft tumors. Int J Cancer 119(3):712–717. https://doi.org/10.1002/ijc.21743
Flammer JR, Rogatsky I (2011) Minireview: glucocorticoids in autoimmunity: unexpected targets and mechanisms. Mol Endocrinol 25(7):1075–1086. https://doi.org/10.1210/me.2011-0068
Coutinho AE, Chapman KE (2011) The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol Cell Endocrinol 335(1):2–13. https://doi.org/10.1016/j.mce.2010.04.005
Smith LK, Cidlowski JA (2010) Glucocorticoid-induced apoptosis of healthy and malignant lymphocytes. Prog Brain Res 182:1–30. https://doi.org/10.1016/S0079-6123(10)82001-1
Ratman D, Vanden Berghe W, Dejager L, Libert C, Tavernier J, Beck IM, de Bosscher K (2013) How glucocorticoid receptors modulate the activity of other transcription factors: a scope beyond tethering. Mol Cell Endocrinol 380(1-2):41–54. https://doi.org/10.1016/j.mce.2012.12.014
Oakley RH, Cidlowski JA (2013) The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132(5):1033–1044. https://doi.org/10.1016/j.jaci.2013.09.007
Oakley RH, Sar M, Cidlowski JA (1996) The human glucocorticoid receptor beta isoform. Expression, biochemical properties, and putative function. J Biol Chem 271(16):9550–9559. https://doi.org/10.1074/jbc.271.16.9550
Kino T, Manoli I, Kelkar S, Wang Y, YA S, Chrousos GP (2009) Glucocorticoid receptor (GR) beta has intrinsic, GRalpha-independent transcriptional activity. Biochem Biophys Res Commun 381(4):671–675. https://doi.org/10.1016/j.bbrc.2009.02.110
Thomas-Chollier M, Watson LC, Cooper SB, Pufall MA, Liu JS, Borzym K, Vingron M, Yamamoto KR, Meijsing SH (2013) A naturally occurring insertion of a single amino acid rewires transcriptional regulation by glucocorticoid receptor isoforms. Proc Natl Acad Sci U S A 110(44):17826–17831. https://doi.org/10.1073/pnas.1316235110
Moalli PA, Pillay S, Krett NL, Rosen ST (1993) Alternatively spliced glucocorticoid receptor messenger RNAs in glucocorticoid-resistant human multiple myeloma cells. Cancer Res 53(17):3877–3879
de Lange P, Segeren CM, Koper JW, Wiemer E, Sonneveld P, Brinkmann AO, White A, Brogan IJ, de Jong FH, Lamberts SW (2001) Expression in hematological malignancies of a glucocorticoid receptor splice variant that augments glucocorticoid receptor-mediated effects in transfected cells. Cancer Res 61(10):3937–3941
Krett NL, Pillay S, Moalli PA, Greipp PR, Rosen ST (1995) A variant glucocorticoid receptor messenger RNA is expressed in multiple myeloma patients. Cancer Res 55(13):2727–2729
Gaitan D, DeBold CR, Turney MK, Zhou P, Orth DN, Kovacs WJ (1995) Glucocorticoid receptor structure and function in an adrenocorticotropin-secreting small cell lung cancer. Mol Endocrinol 9(9):1193–1201. https://doi.org/10.1210/mend.9.9.7491111
Oakley RH, Cidlowski JA (2011) Cellular processing of the glucocorticoid receptor gene and protein: new mechanisms for generating tissue-specific actions of glucocorticoids. J Biol Chem 286(5):3177–3184. https://doi.org/10.1074/jbc.R110.179325
NZ L, Cidlowski JA (2005) Translational regulatory mechanisms generate N-terminal glucocorticoid receptor isoforms with unique transcriptional target genes. Mol Cell 18:331–342
Ramamoorthy S, Cidlowski JA (2013) Exploring the molecular mechanisms of glucocorticoid receptor action from sensitivity to resistance. Endocr Dev 24:41–56. https://doi.org/10.1159/000342502
Weikum ER, Knuesel MT, Ortlund EA, Yamamoto KR (2017) Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat Rev Mol Cell Biol 18(3):159–174. https://doi.org/10.1038/nrm.2016.152
Krozowski Z, Li KX, Koyama K, Smith RE, Obeyesekere VR, Stein-Oakley A et al (1999) The type I and type II 11beta-hydroxysteroid dehydrogenase enzymes. J Steroid Biochem Mol Biol 69(1-6):391–401. https://doi.org/10.1016/S0960-0760(99)00074-6
Martin PM, Rolland PH, Raynaud JP (1981) Macromolecular binding of dexamethasone as evidence for the presence of mineralocorticoid receptor in human breast cancer. Cancer Res 41(3):1222–1226
Jordan K, Sippel C, Schmoll HJ (2007) Guidelines for antiemetic treatment of chemotherapy-induced nausea and vomiting: past, present, and future recommendations. Oncologist 12(9):1143–1150. https://doi.org/10.1634/theoncologist.12-9-1143
Italian Group for Antiemetic R (1995) Dexamethasone, granisetron, or both for the prevention of nausea and vomiting during chemotherapy for cancer. N Engl J Med 332:1–5
Italian Group for Antiemetic R (2000) Dexamethasone alone or in combination with ondansetron for the prevention of delayed nausea and vomiting induced by chemotherapy. N Engl J Med 342:1554–1559
Pufall MA (2015) Glucocorticoids and cancer. Adv Exp Med Biol 872:315–333. https://doi.org/10.1007/978-1-4939-2895-8_14
Boulanger J, Boursiquot JN, Cournoyer G, Lemieux J, Masse MS, Almanric K, Guay MP, Comité de l’évolution des pratiques en oncologie (2014) Management of hypersensitivity to platinum- and taxane-based chemotherapy: cepo review and clinical recommendations. Curr Oncol 21(4):e630–e641. https://doi.org/10.3747/co.21.1966
Schwartz JR (2012) Dexamethasone premedication for prophylaxis of taxane toxicities: can the doses be reduced when paclitaxel or docetaxel are given weekly? J Oncol Pharm Pract 18(2):250–256. https://doi.org/10.1177/1078155211409473
Munstedt K, Borces D, Bohlmann MK, Zygmunt M, von Georgi R (2004) Glucocorticoid administration in antiemetic therapy: is it safe? Cancer 101(7):1696–1702. https://doi.org/10.1002/cncr.20534
Bruera E, Roca E, Cedaro L, Carraro S, Chacon R (1985) Action of oral methylprednisolone in terminal cancer patients: a prospective randomized double-blind study. Cancer Treat Rep 69(7-8):751–754
De Camp G (1961) Corticosteroid therapy in bronchial carcinoma and other malignant tumors in the thorax. Munch Med Wochenschr [trans] 103:2026–2030
Keith BD (2008) Systematic review of the clinical effect of glucocorticoids on nonhematologic malignancy. BMC Cancer 8(1):84. https://doi.org/10.1186/1471-2407-8-84
Melhem A, Yamada SD, Fleming GF, Delgado B, Brickley DR, Wu W, Kocherginsky M, Conzen SD (2009) Administration of glucocorticoids to ovarian cancer patients is associated with expression of the anti-apoptotic genes SGK1 and MKP1/DUSP1 in ovarian tissues. Clin Cancer Res 15(9):3196–3204. https://doi.org/10.1158/1078-0432.CCR-08-2131
Plengvanit U, Viranuvatti V (1964) Treatment of primary carcinoma of the liver with nitrogen mustard and prednisolone: report of 51 cases. Am J Gastroenterol 42:521–528
Runnebaum IB, Bruning A (2005) Glucocorticoids inhibit cell death in ovarian cancer and up-regulate caspase inhibitor cIAP2. Clin Cancer Res 11(17):6325–6332. https://doi.org/10.1158/1078-0432.CCR-05-0182
Stringer-Reasor EM, Baker GM, Skor MN, Kocherginsky M, Lengyel E, Fleming GF, Conzen SD (2015) Glucocorticoid receptor activation inhibits chemotherapy-induced cell death in high-grade serous ovarian carcinoma. Gynecol Oncol 138(3):656–662. https://doi.org/10.1016/j.ygyno.2015.06.033
Zhang C, Marme A, Wenger T, Gutwein P, Edler L, Rittgen W et al (2006) Glucocorticoid-mediated inhibition of chemotherapy in ovarian carcinomas. Int J Oncol 28(2):551–558
Agyeman AS, Jun WJ, Proia DA, Kim CR, Skor MN, Kocherginsky M, Conzen SD (2016) Hsp90 inhibition results in glucocorticoid receptor degradation in association with increased sensitivity to paclitaxel in triple-negative breast cancer. Horm Cancer 7(2):114–126. https://doi.org/10.1007/s12672-016-0251-8
Pang D, Kocherginsky M, Krausz T, Kim SY, Conzen SD (2006) Dexamethasone decreases xenograft response to paclitaxel through inhibition of tumor cell apoptosis. Cancer Biol Ther 5(8):933–940. https://doi.org/10.4161/cbt.5.8.2875
Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD (2001) Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem 276(20):16649–16654. https://doi.org/10.1074/jbc.M010842200
Wu W, Pew T, Zou M, Pang D, Conzen SD (2005) Glucocorticoid receptor-induced MAPK phosphatase-1 (MPK-1) expression inhibits paclitaxel-associated MAPK activation and contributes to breast cancer cell survival. J Biol Chem 280(6):4117–4124. https://doi.org/10.1074/jbc.M411200200
Skor MN, Wonder EL, Kocherginsky M, Goyal A, Hall BA, Cai Y, Conzen SD (2013) Glucocorticoid receptor antagonism as a novel therapy for triple-negative breast cancer. Clin Cancer Res 19(22):6163–6172. https://doi.org/10.1158/1078-0432.CCR-12-3826
Kim MJ, Chae JS, Kim KJ, Hwang SG, Yoon KW, Kim EK, Yun HJ, Cho JH, Kim J, Kim BW, Kim H, Kang SS, Lang F, Cho SG, Choi EJ (2007) Negative regulation of SEK1 signaling by serum- and glucocorticoid-inducible protein kinase 1. EMBO J 26(13):3075–3085. https://doi.org/10.1038/sj.emboj.7601755
Chen Z, Lan X, Wu D, Sunkel B, Ye Z, Huang J, Liu Z, Clinton SK, Jin VX, Wang Q (2015) Ligand-dependent genomic function of glucocorticoid receptor in triple-negative breast cancer. Nat Commun 6:8323. https://doi.org/10.1038/ncomms9323
Pan D, Kocherginsky M, Conzen SD (2011) Activation of the glucocorticoid receptor is associated with poor prognosis in estrogen receptor-negative breast cancer. Cancer Res 71(20):6360–6370. https://doi.org/10.1158/0008-5472.CAN-11-0362
Backman S, Kollara A, Haw R, Stein L, Brown TJ (2014) Glucocorticoid-induced reversal of interleukin-1beta-stimulated inflammatory gene expression in human oviductal cells. PLoS One 9(5):e97997. https://doi.org/10.1371/journal.pone.0097997
Neta R (1997) Modulation of radiation damage by cytokines. Stem Cells 15(Suppl 2):87–94
Di Maggio FM, Minafra L, Forte GI, Cammarata FP, Lio D, Messa C et al (2015) Portrait of inflammatory response to ionizing radiation treatment. J Inflamm (Lond) 12(1):14. https://doi.org/10.1186/s12950-015-0058-3
Voloshin T, Alishekevitz D, Kaneti L, Miller V, Isakov E, Kaplanov I, Voronov E, Fremder E, Benhar M, Machluf M, Apte RN, Shaked Y (2015) Blocking IL1beta pathway following paclitaxel chemotherapy slightly inhibits primary tumor growth but promotes spontaneous metastasis. Mol Cancer Ther 14(6):1385–1394. https://doi.org/10.1158/1535-7163.MCT-14-0969
Collart MA, Baeuerle P, Vassalli P (1990) Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol 10(4):1498–1506. https://doi.org/10.1128/MCB.10.4.1498
Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV (1990) Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med 171(1):35–47. https://doi.org/10.1084/jem.171.1.35
Hallahan DE, Virudachalam S, Kuchibhotla J, Kufe DW, Weichselbaum RR (1994) Membrane-derived second messenger regulates x-ray-mediated tumor necrosis factor alpha gene induction. Proc Natl Acad Sci U S A 91(11):4897–4901. https://doi.org/10.1073/pnas.91.11.4897
Ochsenbein AF, Sierro S, Odermatt B, Pericin M, Karrer U, Hermans J, Hemmi S, Hengartner H, Zinkernagel RM (2001) Roles of tumour localization, second signals and cross priming in cytotoxic T-cell induction. Nature 411(6841):1058–1064. https://doi.org/10.1038/35082583
Haid M (1981) Steroid antiemesis may be harmful. N Engl J Med 304:1237
Jusko WJ (1995) Pharmacokinetics and receptor-mediated pharmacodynamics of corticosteroids. Toxicology 102(1-2):189–196. https://doi.org/10.1016/0300-483X(95)03047-J
Meier CA (1996) Mechanisms of immunosuppression by glucocorticoids. Eur J Endocrinol 134(1):50. https://doi.org/10.1530/eje.0.1340050
Chen YX, Wang Y, CC F, Diao F, Song LN, Li ZB et al (2010) Dexamethasone enhances cell resistance to chemotherapy by increasing adhesion to extracellular matrix in human ovarian cancer cells. Endocr Relat Cancer 17(1):39–50. https://doi.org/10.1677/ERC-08-0296
Liotta LA, Kohn EC (2001) The microenvironment of the tumour–host interface. Nature 411(6835):375–379. https://doi.org/10.1038/35077241
Moran TJ, Gray S, Mikosz CA, Conzen SD (2000) The glucocorticoid receptor mediates a survival signal in human mammary epithelial cells. Cancer Res 60(4):867–872
Herr I, Pfitzenmaier J (2006) Glucocorticoid use in prostate cancer and other solid tumours: implications for effectiveness of cytotoxic treatment and metastases. Lancet Oncol 7(5):425–430. https://doi.org/10.1016/S1470-2045(06)70694-5
Hidalgo AA, Montecinos VP, Paredes R, Godoy AS, McNerney EM, Tovar H, Pantoja D, Johnson C, Trump D, Onate SA (2011) Biochemical characterization of nuclear receptors for vitamin D3 and glucocorticoids in prostate stroma cell microenvironment. Biochem Biophys Res Commun 412(1):13–19. https://doi.org/10.1016/j.bbrc.2011.06.181
Volden PA, Conzen SD (2013) The influence of glucocorticoid signaling on tumor progression. Brain Behav Immun 30(Suppl):S26–S31. https://doi.org/10.1016/j.bbi.2012.10.022
Sundahl N, Clarisse D, Bracke M, Offner F, Berghe WV, Beck IM (2016) Selective glucocorticoid receptor-activating adjuvant therapy in cancer treatments. Oncoscience 3(7-8):188–202. https://doi.org/10.18632/oncoscience.315
Desgrosellier JS, Cheresh DA (2010) Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 10(1):9–22. https://doi.org/10.1038/nrc2748
Kobayashi M, Sawada K, Kimura T. (2017) Potential of integrin inhibitors for treating ovarian cancer: a literature review. Cancers (Basel) 9
Mierke CT, Frey B, Fellner M, Herrmann M, Fabry B (2011) Integrin alpha5beta1 facilitates cancer cell invasion through enhanced contractile forces. J Cell Sci 124(3):369–383. https://doi.org/10.1242/jcs.071985
Lou X, Han X, Jin C, Tian W, Yu W, Ding D, Cheng L, Huang B, Jiang H, Lin B (2013) SOX2 targets fibronectin 1 to promote cell migration and invasion in ovarian cancer: new molecular leads for therapeutic intervention. OMICS 17(10):510–518. https://doi.org/10.1089/omi.2013.0058
Knowles LM, Gurski LA, Engel C, Gnarra JR, Maranchie JK, Pilch J (2013) Integrin alphavbeta3 and fibronectin upregulate slug in cancer cells to promote clot invasion and metastasis. Cancer Res 73(20):6175–6184. https://doi.org/10.1158/0008-5472.CAN-13-0602
Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, Hudak JE, Lakins JN, Wijekoon AC, Cassereau L, Rubashkin MG, Magbanua MJ, Thorn KS, Davidson MW, Rugo HS, Park JW, Hammer DA, Giannone G, Bertozzi CR, Weaver VM (2014) The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 511(7509):319–325. https://doi.org/10.1038/nature13535
Kanatani Y, Kasukabe T, Okabe-Kado J, Hayashi S, Yamamoto-Yamaguchi Y, Motoyoshi K, Nagata N, Honma Y (1996) Transforming growth factor beta and dexamethasone cooperatively enhance c-jun gene expression and inhibit the growth of human monocytoid leukemia cells. Cell Growth Differ 7(2):187–196
Takuma A, Kaneda T, Sato T, Ninomiya S, Kumegawa M, Hakeda Y (2003) Dexamethasone enhances osteoclast formation synergistically with transforming growth factor-beta by stimulating the priming of osteoclast progenitors for differentiation into osteoclasts. J Biol Chem 278(45):44667–44674. https://doi.org/10.1074/jbc.M300213200
Ranganathan P, Agrawal A, Bhushan R, Chavalmane AK, Kalathur RK, Takahashi T et al (2007) Expression profiling of genes regulated by TGF-beta: differential regulation in normal and tumour cells. BMC Genomics 8(1):98. https://doi.org/10.1186/1471-2164-8-98
Salem S, Harris T, Mok JS, Li MY, Keenan CR, Schuliga MJ et al (2012) Transforming growth factor-beta impairs glucocorticoid activity in the A549 lung adenocarcinoma cell line. Br J Pharmacol 166(7):2036–2048. https://doi.org/10.1111/j.1476-5381.2012.01885.x
Li Z, Chen Y, Cao D, Wang Y, Chen G, Zhang S, Lu J (2006) Glucocorticoid up-regulates transforming growth factor-beta (TGF-beta) type II receptor and enhances TGF-beta signaling in human prostate cancer PC-3 cells. Endocrinology 147(11):5259–5267. https://doi.org/10.1210/en.2006-0540
Cheng J, Truong LD, Wu X, Kuhl D, Lang F, Du J (2010) Serum- and glucocorticoid-regulated kinase 1 is upregulated following unilateral ureteral obstruction causing epithelial-mesenchymal transition. Kidney Int 78(7):668–678. https://doi.org/10.1038/ki.2010.214
Zhang L, Lei W, Wang X, Tang Y, Song J (2010) Glucocorticoid induces mesenchymal-to-epithelial transition and inhibits TGF-beta1-induced epithelial-to-mesenchymal transition and cell migration. FEBS Lett 584(22):4646–4654. https://doi.org/10.1016/j.febslet.2010.10.038
Yang HW, Lee SA, Shin JM, Park IH, Lee HM (2017) Glucocorticoids ameliorate TGF-beta1-mediated epithelial-to-mesenchymal transition of airway epithelium through MAPK and snail/slug signaling pathways. Sci Rep 7(1):3486. https://doi.org/10.1038/s41598-017-02358-z
Godoy P, Lakkapamu S, Schug M, Bauer A, Stewart JD, Bedawi E, Hammad S, Amin J, Marchan R, Schormann W, Maccoux L, von Recklinghausen I, Reif R, Hengstler JG (2010) Dexamethasone-dependent versus -independent markers of epithelial to mesenchymal transition in primary hepatocytes. Biol Chem 391(1):73–83. https://doi.org/10.1515/BC.2010.010
Lin KT, Yeh YM, Chuang CM, Yang SY, Chang JW, Sun SP, Wang YS, Chao KC, Wang LH (2015) Glucocorticoids mediate induction of microRNA-708 to suppress ovarian cancer metastasis through targeting Rap1B. Nat Commun 6:5917. https://doi.org/10.1038/ncomms6917
Bos JL, de Rooij J, Reedquist KA (2001) Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol 2(5):369–377. https://doi.org/10.1038/35073073
Lin KT, Sun SP, JI W, Wang LH (2017) Low-dose glucocorticoids suppresses ovarian tumor growth and metastasis in an immunocompetent syngeneic mouse model. PLoS One 12(6):e0178937. https://doi.org/10.1371/journal.pone.0178937
King MC, Marks JH, Mandell JB, New York Breast Cancer Study G (2003) Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302:643–646
Antoniou A, Pharoah PD, Narod S, Risch HA, Eyfjord JE, Hopper JL et al (2003) Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet 72(5):1117–1130. https://doi.org/10.1086/375033
Mavaddat N, Peock S, Frost D, Ellis S, Platte R, Fineberg E, Evans DG, Izatt L, Eeles RA, Adlard J, Davidson R, Eccles D, Cole T, Cook J, Brewer C, Tischkowitz M, Douglas F, Hodgson S, Walker L, Porteous ME, Morrison PJ, Side LE, Kennedy MJ, Houghton C, Donaldson A, Rogers MT, Dorkins H, Miedzybrodzka Z, Gregory H, Eason J, Barwell J, McCann E, Murray A, Antoniou AC, Easton DF, EMBRACE (2013) Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J Natl Cancer Inst 105(11):812–822. https://doi.org/10.1093/jnci/djt095
Chen S, Parmigiani G (2007) Meta-analysis of BRCA1 and BRCA2 penetrance. J Clin Oncol 25(11):1329–1333. https://doi.org/10.1200/JCO.2006.09.1066
Kurman RJ, Shih Ie M (2010) The origin and pathogenesis of epithelial ovarian cancer: a proposed unifying theory. Am J Surg Pathol 34(3):433–443. https://doi.org/10.1097/PAS.0b013e3181cf3d79
Crum CP, Drapkin R, Miron A, Ince TA, Muto M, Kindelberger DW, Lee Y (2007) The distal fallopian tube: a new model for pelvic serous carcinogenesis. Curr Opin Obstet Gynecol 19(1):3–9. https://doi.org/10.1097/GCO.0b013e328011a21f
Tone AA, Salvador S, Finlayson SJ, Tinker AV, Kwon JS, Lee CH, Cohen T, Ehlen T, Lee M, Carey MS, Heywood M, Pike J, Hoskins PJ, Stuart GC, Swenerton KD, Huntsman DG, Gilks CB, Miller DM, McAlpine JN (2012) The role of the fallopian tube in ovarian cancer. Clin Adv Hematol Oncol 10(5):296–306
Tone AA, Virtanen C, Shaw P, Brown TJ (2012) Prolonged postovulatory proinflammatory signaling in the fallopian tube epithelium may be mediated through a BRCA1/DAB2 axis. Clin Cancer Res 18(16):4334–4344. https://doi.org/10.1158/1078-0432.CCR-12-0199
Fang YY, Li D, Cao C, Li CY, Li TT (2014) Glucocorticoid receptor repression mediated by BRCA1 inactivation in ovarian cancer. BMC Cancer 14(1):188. https://doi.org/10.1186/1471-2407-14-188
Vilasco M, Communal L, Hugon-Rodin J, Penault-Llorca F, Mourra N, Wu Z et al (2013) Loss of glucocorticoid receptor activation is a hallmark of BRCA1-mutated breast tissue. Breast Cancer Res Treat 142(2):283–296. https://doi.org/10.1007/s10549-013-2722-8
Ritter HD, Antonova L, Mueller CR (2012) The unliganded glucocorticoid receptor positively regulates the tumor suppressor gene BRCA1 through GABP beta. Mol Cancer Res 10(4):558–569. https://doi.org/10.1158/1541-7786.MCR-11-0423-T
Antonova L, Aronson K, Mueller CR (2011) Stress and breast cancer: from epidemiology to molecular biology. Breast Cancer Res 13(2):208. https://doi.org/10.1186/bcr2836
Quinn JE, Carser JE, James CR, Kennedy RD, Harkin DP (2009) BRCA1 and implications for response to chemotherapy in ovarian cancer. Gynecol Oncol 113(1):134–142. https://doi.org/10.1016/j.ygyno.2008.12.015
Kennedy RD, Quinn JE, Mullan PB, Johnston PG, Harkin DP (2004) The role of BRCA1 in the cellular response to chemotherapy. J Natl Cancer Inst 96(22):1659–1668. https://doi.org/10.1093/jnci/djh312
Markman M, Kennedy A, Webster K, Kulp B, Peterson G, Belinson J (2000) Paclitaxel-associated hypersensitivity reactions: experience of the gynecologic oncology program of the Cleveland Clinic Cancer Center. J Clin Oncol 18(1):102–105. https://doi.org/10.1200/JCO.2000.18.1.102
Nakagawa M, Terashima T, D'Yachkova Y, Bondy GP, Hogg JC, van Eeden SF (1998) Glucocorticoid-induced granulocytosis: contribution of marrow release and demargination of intravascular granulocytes. Circulation 98(21):2307–2313. https://doi.org/10.1161/01.CIR.98.21.2307
Liles WC, Dale DC, Klebanoff SJ (1995) Glucocorticoids inhibit apoptosis of human neutrophils. Blood 86(8):3181–3188
Fauci JM, Whitworth JM, Schneider KE, Subramaniam A, Zhang B, Frederick PJ, Kilgore LC, Straughn JM Jr (2011) Prognostic significance of the relative dose intensity of chemotherapy in primary treatment of epithelial ovarian cancer. Gynecol Oncol 122(3):532–535. https://doi.org/10.1016/j.ygyno.2011.05.023
Hanna RK, Poniewierski MS, Laskey RA, Lopez MA, Shafer A, Van Le L, Crawford J, Dale DC, Gehrig PA, Secord AA, Havrilesky LJ, Lyman GH (2013) Predictors of reduced relative dose intensity and its relationship to mortality in women receiving multi-agent chemotherapy for epithelial ovarian cancer. Gynecol Oncol 129(1):74–80. https://doi.org/10.1016/j.ygyno.2012.12.017
Veneris JT, Darcy KM, Mhawech-Fauceglia P, Tian C, Lengyel E, Lastra RR, Pejovic T, Conzen SD, Fleming GF (2017) High glucocorticoid receptor expression predicts short progression-free survival in ovarian cancer. Gynecol Oncol 146(1):153–160. https://doi.org/10.1016/j.ygyno.2017.04.012
Woenckhaus J, Franke FE, Hackethal A, Von Georgi R, Munstedt K (2006) Glucocorticosteroid receptors in ovarian carcinomas. Oncol Rep 15(5):1137–1140
Rocereto TF, Saul HM, Aikins JA Jr, Paulson J (2000) Phase II study of mifepristone (RU486) in refractory ovarian cancer. Gynecol Oncol 77(3):429–432. https://doi.org/10.1006/gyno.2000.5789
Voisin M, de Medina P, Mallinger A, Dalenc F, Huc-Claustre E, Leignadier J, Serhan N, Soules R, Ségala G, Mougel A, Noguer E, Mhamdi L, Bacquié E, Iuliano L, Zerbinati C, Lacroix-Triki M, Chaltiel L, Filleron T, Cavaillès V, Al Saati T, Rochaix P, Duprez-Paumier R, Franchet C, Ligat L, Lopez F, Record M, Poirot M, Silvente-Poirot S (2017) Identification of a tumor-promoter cholesterol metabolite in human breast cancers acting through the glucocorticoid receptor. Proc Natl Acad Sci U S A 114(44):E9346–E9E55. https://doi.org/10.1073/pnas.1707965114
Schacke H, Berger M, Rehwinkel H, Asadullah K (2007) Selective glucocorticoid receptor agonists (SEGRAs): novel ligands with an improved therapeutic index. Mol Cell Endocrinol 275(1-2):109–117. https://doi.org/10.1016/j.mce.2007.05.014
Sundahl N, Bridelance J, Libert C, De Bosscher K, Beck IM (2015) Selective glucocorticoid receptor modulation: new directions with non-steroidal scaffolds. Pharmacol Ther 152:28–41. https://doi.org/10.1016/j.pharmthera.2015.05.001
Aapro M, Fabi A, Nole F, Medici M, Steger G, Bachmann C, Roncoroni S, Roila F (2010) Double-blind, randomised, controlled study of the efficacy and tolerability of palonosetron plus dexamethasone for 1 day with or without dexamethasone on days 2 and 3 in the prevention of nausea and vomiting induced by moderately emetogenic chemotherapy. Ann Oncol 21(5):1083–1088. https://doi.org/10.1093/annonc/mdp584
Roila F, Ruggeri B, Ballatori E, Del Favero A, Tonato M (2014) Aprepitant versus dexamethasone for preventing chemotherapy-induced delayed emesis in patients with breast cancer: a randomized double-blind study. J Clin Oncol 32(2):101–106. https://doi.org/10.1200/JCO.2013.51.4547
Berger MJ, Vargo C, Vincent M, Shaver K, Phillips G, Layman R, Macrae E, Mrozek E, Ramaswamy B, Wesolowski R, Shapiro CL, Lustberg MB (2015) Stopping paclitaxel premedication after two doses in patients not experiencing a previous infusion hypersensitivity reaction. Support Care Cancer 23(7):2019–2024. https://doi.org/10.1007/s00520-014-2556-x
Acknowledgements
Supported by Canadian Institutes of Health Research grants MOP142364 and MOP106679.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Djedovic, V., Lee, YY., Kollara, A. et al. The Two Faces of Adjuvant Glucocorticoid Treatment in Ovarian Cancer. HORM CANC 9, 95–107 (2018). https://doi.org/10.1007/s12672-017-0319-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12672-017-0319-0