
Abstract
Neurodegenerative diseases are prominent causes of pain, suffering, and death worldwide. Traditional approaches modelling neurodegenerative diseases are deficient, and therefore, improved strategies that effectively recapitulate the pathophysiological conditions of neurodegenerative diseases are the need of the hour. The generation of human-induced pluripotent stem cells (iPSCs) has transformed our ability to model neurodegenerative diseases in vitro and provide an unlimited source of cells (including desired neuronal cell types) for cell replacement therapy. Recently, CRISPR/Cas9-based genome editing has also been gaining popularity because of the flexibility they provide to generate and ablate disease phenotypes. In addition, the recent advancements in CRISPR/Cas9 technology enables researchers to seamlessly target and introduce precise modifications in the genomic DNA of different human cell lines, including iPSCs. CRISPR-iPSC-based disease modelling, therefore, allows scientists to recapitulate the pathological aspects of most neurodegenerative processes and investigate the role of pathological gene variants in healthy non-patient cell lines. This review outlines how iPSCs, CRISPR/Cas9, and CRISPR-iPSC-based approaches accelerate research on neurodegenerative diseases and take us closer to a cure for neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, Amyotrophic Lateral Sclerosis, and so forth.
Similar content being viewed by others
Introduction
Neurodegenerative diseases/disorders represent one of the major causes of death, disability, and increased social as well as economic burden on affected individuals and their families around the world. In the year 2016, neurological disorders/diseases were the leading cause of disability-adjusted life years and the second leading cause of death globally. Among these, Alzheimer’s disease (AD) and other dementias ranked third for age-standardized disability-adjusted life years, whereas Parkinson’s disease (PD) and motor neuron diseases (MND) ranked 11th and 15th, respectively (Feigin et al. 2019). For decades, attempts to develop effective cures for various neurodegenerative diseases have been ongoing. However, no neurodegenerative disease is curable to date (Durães et al. 2018).
The first step to understand a complex disease process, as is the case with almost all neurodegenerative diseases, is to develop an effective disease model. A disease model is a system displaying the entire set or more frequently a definite subset of the pathological processes underlying an actual disease process. The contribution of animal models, and specifically mouse models, in understanding modern biology is undeniable and irrefutable. A wide variety of different transgenic mice models have been utilized in a diverse range of research applications over the decades. However, they have their drawbacks, especially in modelling neurodegenerative diseases. Mice differ significantly from humans in a variety of therapeutically relevant areas such as anatomical and physiological complexity, life expectancy, neurotransmitter function, anatomical complexity, metabolic rates, and even pharmacological response (Haston and Finkbeiner 2016). Therefore, many scientific investigators and pharmaceutical companies consider datasets obtained from established human cell lines to be more therapeutically translatable as compared to datasets derived from mouse models (Benam et al. 2015; Donowitz et al. 2020).
Traditionally, tissue samples had to be obtained from diseased subjects in order to set up primary cultures and consequently derive the cell line(s) harboring the disease-causing mutation(s) for research applications (Valadez-Barba et al. 2020). However, this approach has several drawbacks. The inherent risk of biopsies combined with the cumbersome and time-consuming procedures/protocols necessary to establish primary cell lines render patient tissue sample acquisition a tedious process (Verma et al. 2020). Post-mortem tissue samples are more likely to represent the terminal stages of a complex disorder/disease and, therefore, may obfuscate critical insights pertaining to early pathological markers, overall disease etiology, and pathophysiological hallmarks exclusive to specific disease stages (Siller et al. 2013). Furthermore, modelling complex polygenetic or multifactorial neurodegenerative disorders like AD, PD, or amyotrophic lateral sclerosis (ALS) requires multiple isogenic cell lines sourced from a diverse patient pool to obtain a comprehensive view of the summative and more importantly combinatorial effects of all the genetic aberrations responsible for the disease phenotype (Poewe et al. 2017; Penney et al. 2020; Trapecar et al. 2021). Finally, modelling a disease using traditional animal cell monocultures may yield inadequate results as the cell growth medium does not resemble the microenvironment of the tissue niche, nor does it account for the various interactions the cell type of interest has with other cellular and non-cellular entities (like the extracellular matrix) in its vicinity (Li et al. 2012; Bolognin et al. 2019; Lam et al. 2019). For example, non-neuronal tissues like astrocytes have been demonstrated to play vital roles in the pathogenesis of complex neurodegenerative processes like AD or PD (Lin et al. 2018; Penney et al. 2020). Therefore, studies aiming to study neurodegenerative disease processes in vitro using only neuronal cells may overlook vital clues pertaining to potential therapeutic targets arising due to the interaction between different cell types or their secreted factors in vivo (Bolognin et al. 2019). All these issues severely delimit attempts to model complex neurodegenerative disease processes. The combination of clustered regularly interspaced short palindromic repeats (CRISPR) and induced pluripotent stem cells (iPSCs) technology can represent a nexus of two promising technologies working in concert to ameliorate and possibly eliminate these challenges.
A Brief Summary of CRISPR and iPSCs
CRISPR
The term CRISPR coined by Jansen and colleagues provided a detailed description of the CRISPR loci derived from in silico analysis of the genomic DNA of several bacterial and archaeal species (Jansen et al. 2002). Later, Barrangou and colleagues reported the first experimental evidence documenting the nucleic acid-mediated adaptive immunity conferred by the CRISPR machinery in a multitude of prokaryotic and archaeal systems (Barrangou et al. 2007). The authors demonstrated the acquisition of short stretches of phage DNA into the bacterial genome when exposed to a bacteriophage infection (Barrangou et al. 2007).
A CRISPR array comprises of short, direct repeats punctuated by stretches of variable DNA sequences, referred to as spacers. When bacteriophage or plasmid DNA is introduced into a bacterial system, they are processed by the Cas1 and Cas2 proteins and integrated as spacers into the CRISPR array. This step is referred to as protospacer acquisition and can be considered analogous to committing a new record in a database. Protospacer acquisition in CRISPR/Cas systems generally requires the presence of a short stretch of nucleotides in the foreign target nucleic acid, referred to as the protospacer adjacent motif (PAM). These spacers function as the basis of the memory required for a targeted host defense response to a subsequent infection. Following the protospacer acquisition, the CRISPR array is transcribed as a precursor transcript (pre-crRNA), which is either bound to a single, multidomain protein named Cas9 or to a multiunit effector complex, forming the crRNA–effector complex. The pre-crRNA is subsequently processed into smaller CRISPR RNAs (crRNAs). On re-exposure to foreign offending nucleic acids, these CRISPR RNAs recognize and guide either the Cas9 protein or the multiunit crRNA–effector complex to cleave and neutralize the infection (Makarova et al. 2015).
The native Cas9 system requires two separate short RNA strands, the mature crRNA and a transactivating crRNA (tracrRNA), to constitute the CRISPR/Cas9–RNA complex capable of generating DNA double-strand breaks (DSBs) at desired target sites. Successful attempts at fusing the tracrRNA and crRNA have enabled the generation of a single chimeric RNA strand called the single guide RNA (sgRNA), which can be effectively packed into any standard genetic payload delivery system. This breakthrough, alongside several others, provides researchers the capability to control the precise site-specific cleavage capabilities of the Cas9 protein and the flexibility conferred by specifically tailored sgRNAs to target nearly any locus in any target organism, including human cell lines and iPSCs. Although several different CRISPR/Cas systems have been repurposed for genome editing over the years, the most widely used CRISPR/Cas system is the Streptococcus pyogenes-derived type II CRISPR/Cas9 system. One of the prominent factors driving this popularity is the simple NGG PAM sequence requirement of the S. pyogenes II CRISPR/Cas9 system. However, this PAM sequence requirement is also one of the major limitations of this system as the Cas9 protein screens and identifies the region immediately downstream of the sgRNA-target DNA duplex in search for an appropriate PAM sequence, failing which its DSB activity is aborted (Makarova et al. 2015; Adli 2018). Therefore, active research aiming to enrich our CRISPR/Cas toolbox is currently underway. Among these, notable mentions include the CRISPR from Prevotella and Francisella (Cpf1) endonuclease protein orthologs derived from Acidaminococcus sp. (AsCpf1) and Lachnospiraceae bacterium (LbCpf1), respectively (Bin Moon et al. 2018; Safari et al. 2019; Alok et al. 2020). Not only do Cpf1 protein naturally require only one sgRNA and recognize a broader range of PAM sequences as compared to the S. pyogenes derived Cas9, but they also generate 5′ overhangs at the cut-site instead of blunt ends like CRISPR/Cas9 (Li et al. 2019).
CRISPR/Cas9 has two catalytic domains (HNH and RuvC), each of which mediates the cleavage of one strand. Together they are responsible for mediating DNA DSBs. A targeted point mutation in either one of the catalytic domains (D10A and H840A for SpCas9) results in the creation of a CRISPR/Cas9 nickase system capable of cleaving any one DNA strand in the target site. Point mutations in both the catalytic domains result in the complete ablation of DNA cleavage activity, resulting in creating a catalytically inactive or “dead” Cas9 (dCas9) complex. Therefore, a dCas9 complex possesses the target sequence specificity of the native CRISPR/Cas9 system but is devoid of the DSB capability. Researchers have exploited this strategy to modulate gene expression patterns in various systems by means of two approaches: CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa). In the CRISPRi approach, scientists fuse the dCas9 protein to a potent repressor complex such as KRuppel-Associated Box (KRAB) and target it to a specific target site, resulting in strong gene repression activity. On the contrary, in the CRISPRa approach, the dCas9 protein is fused to a potent transcriptional activator such as VP64, which consequently upregulates gene expression. The VP64 transcriptional activator is composed of four tandem copies of the transactivation domain (VP16) of the Herpes simplex virus.
To summarize, the CRISPR platform has rapidly evolved with many advancements in the last few years for a wide range of applications, including neurodegenerative disease modelling as is discussed in the following sections.
iPSCs
Stem cells are a critical component of any complex living system which are capable of self-renewal and differentiation into several different terminal cell fates. They are classified as totipotent, pluripotent, multipotent, and unipotent based on the range of terminally differentiated cell lines they can differentiate into. Totipotent cells can differentiate into any cell type affiliated to all three germ layers as well as extra-embryonic tissues like the placenta. Pluripotent cells can differentiate into cell lineages affiliated to all the three germ layers (ectoderm, mesoderm, and endoderm) but not the extra-embryonic tissues. However, both totipotent and pluripotent cells can only be found in the very early stages of embryonic development and hence cannot be utilized due to technical and ethical constraints (Mitalipov and Wolf 2009).
Cell differentiation was traditionally considered to be a unidirectional pathway that commits cells to a particular lineage with a specific set of functions and properties. However, in a culture dish, this idea was established to be incorrect when Shinya Yamanaka and his group were able to successfully reprogram somatic cells, a terminally differentiated cell line, into iPSCs in a culture dish (Takahashi and Yamanaka 2006). The authors utilized a cocktail of four reprogramming factors consisting of Octamer-binding transcription factor (Oct-4), Sex determining region Y-box 2 (Sox2), c-Myc, and Kruppel-like factor 4 (Klf4), which are now referred to as Yamanaka factors, to reprogram mouse and human fibroblasts (Takahashi and Yamanaka 2006; Takahashi et al. 2007). Another group led by James Thomson later generated stable iPSCs using a partially different reprogramming factor cocktail consisting of Oct4, Sox2, Nanog, and Lin28 to reprogram human fibroblasts (Yu et al. 2007). Since then, many genes (Dey et al. 2022) and cell types (Ray et al. 2021; Sundaravadivelu et al. 2021) have been identified that play a crucial role in generating iPSCs efficiently. Various technological advancements have been made with an aim to prospectively generate clinical-grade iPSCs for future biomedical applications (Dey et al. 2017, 2021; Saha et al. 2018a; Haridhasapavalan et al. 2019, 2020; Borgohain et al. 2019). Over the years since the discovery of iPSCs, the use of these cells in disease modelling applications has increased substantially (Young 2012; Singh et al. 2015; Tang et al. 2016; Saha et al. 2018b; Agrawal et al. 2021). Neurodegenerative disease modelling is no exception to this overarching trend, as is discussed in the following sections.
CRISPR and iPSCs in Disease Modelling
CRISPR-iPSC-based disease modelling eliminates the primary barrier surrounding the procurement of diseased tissue samples for traditional disease modelling applications: exposing patients to risky biopsies. This is significant as brain tissue is among the most inaccessible tissue systems in the body and brain biopsies are inherently risky. Cell lines derived from any easily accessible region of a patient’s body can be converted into human iPSCs (Dey et al. 2017, 2021; Haridhasapavalan et al. 2019; Borgohain et al. 2019; Ray et al. 2021; Sundaravadivelu et al. 2021). The iPSCs can subsequently be differentiated into the desired cell type by treating it with an optimized cocktail of growth factors and culture conditions (Young 2012; Hargus et al. 2014; Singh et al. 2015; Tang et al. 2016; Saha et al. 2018b; Agrawal et al. 2021). Using this approach, isogenic cell lines carrying one or multiple disease-relevant mutations can be derived from a single iPSC background, thus significantly eliminating genotypic variability (Beylina et al. 2021).
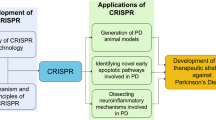
A pool of mutations has been implicated to underlie the pathology of most neurodegenerative disease phenotypes, with varying levels of penetrance. Moreover, multifactorial neurodegenerative diseases like AD, PD, or ALS are significantly modulated by a combination of both genetic and environmental factors (Barber 2012; Giri et al. 2016; Kim et al. 2020b; Tran et al. 2020; Vázquez-Vélez and Zoghbi 2021). Therefore, to model the entire gene pool of mutations responsible for a neurodegenerative disease process, it is necessary to procure cell/tissue samples and generate iPSCs from large cohorts of patients, demonstrating a significant degree of genetic heterogeneity with respect to the disease-causing mutations. Obligatory access to patient cell lines is a persistent limitation of iPSC-based disease models. This limitation is expunged by CRISPR-iPSC-based neurodegenerative disease models. CRISPR/Cas9 allows the seamless targeted editing of genomic DNA of any desired cell line, thus imparting the capability to introduce or correct mutations as desired. Non-patient-derived healthy iPSC lines, as well as patient-derived diseased iPSC lines harboring non-desired mutations, can also be utilized to generate the required isogenic disease models as mutations can easily be introduced or corrected by means of CRISPR/Cas9-mediated genome editing (Chen et al. 2020; Laverde-Paz et al. 2021; Hernández et al. 2021) (Fig. 1). Approaches like CRISPRa or CRISPRi can also be utilized to express or repress the production of specific gene products, respectively (Heman-Ackah et al. 2016; Tian et al. 2019; Inoue 2021). Thus, genome-edited iPSC-based disease models can be utilized to derive actionable insights regarding the mechanistic underpinnings of a disease process and its potential drug targets.

A detailed comparison of traditional, iPSC-based, and CRISPR-iPSC-based disease modelling approaches
In this review, we will discuss some interesting examples of how CRISPR-iPSC-based disease modelling approaches are helping us generate scientific and clinical insights in a vast range of neurodegenerative diseases with unprecedented levels of flexibility, robustness, and reliability. This review also aims to present some of the emerging directions as well as perspectives of neurodegenerative disease research in a seamless narrative.
Alzheimer’s Disease
Disease Pathology and Etiology
According to the National Institutes of Health, AD is the most prevalent neurodegenerative disorder inflicting a significant degree of medical, social, and financial burden on millions of individuals and their families globally (Selkoe et al. 1999). The entire pathological progression of AD can be segmented into three symptomatically characteristic epochs: preclinical AD, mild cognitive impairment due to AD, and dementia due to AD. Preclinical AD is characterized by measurable neurological changes such as abnormal amyloid β (Aβ) levels and decreased glucose metabolism, the early-stage pathological hallmarks of AD. As preclinical AD progresses to mild cognitive impairment due to AD, subtle cognitive and memory changes start to surface, which, although perceptible, does not interfere with an individual’s ability to meet the cognitive rigors of everyday life. Finally, as the disease progresses further, the patient develops Alzheimer’s dementia that is characterized by strong biomarker evidence as well as cognitive and memory decline, which noticeably impairs a person’s ability to function normally in everyday life. AD is, therefore, a progressive condition, the symptomatic manifestations of which worsen with increasing age.
AD has two distinct forms: familial early-onset type AD (EOAD) and sporadic late-onset type AD (LOAD). EOAD is characterized by the onset of AD symptoms before the age of 65 years and is responsible for less than 5% of all reported AD cases (Wu et al. 2012). Over 270 highly penetrant mutations in the amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2) genes have been implicated in the pathogenesis of EOAD (Wu et al. 2012; Giri et al. 2016; Oksanen et al. 2017). The presenilin 1 (PSEN1) and presenilin 2 (PSEN2) genes encode components of gamma-secretase, which is the enzyme complex primarily responsible for sequentially cleaving the amyloid precursor protein (APP). Mutations in the PSEN1 and PSEN2 genes have been reported to alter gamma-secretase-mediated APP processivity. This alteration has been hypothesized to result in an increase in the intracellular concentration of the amyloidogenic (compound tending to produce amyloid deposits) Aβ42 peptide, thus promoting EOAD pathogenesis (Carroll and Li 2016; Inoue et al. 2017).
Most individuals who develop AD are aged 65 or older and are affected by LOAD, which is primarily caused by the apolipoprotein E4 (APOE4) variant of the APOE family of fat-binding apolipoproteins (Liu et al. 2013; Oksanen et al. 2017; DeTure and Dickson 2019). APOE2, APOE3, and APOE4 are the three polymorphic APOE alleles observed in humans among which APOE3 has the highest frequency (77.9% of the general population), followed by APOE4 (13.7% of the general population) and, finally, APOE2 (8.4% of the general population) (Lee et al. 2020). Apolipoprotein E (APOE) is an important cholesterol carrier responsible for lipid transportation and injury repair in the brain (Liu et al. 2013). APOE polymorphic alleles are the main genetic factors that are indicative of LOAD risk. Individuals bearing the APOE4 variant of the APOE allele are at an increased risk of developing AD than individuals with the more common APOE3 variant (Liu et al. 2013). This is demonstrated by the fact that the occurrence of the pathological APOE4 allele is significantly enriched in LOAD patients (Lee et al. 2020).
Both EOAD and LOAD pathology are characterized by the formation of intracellular neurofibrillary tangles (NFTs) and extracellular amyloid plaques (APs). NFTs constitute of hyperphosphorylated tau protein, whereas extracellular APs comprise of Aβ peptides. The extracellular Aβ plaques and neurofibrillary tangles tend to aggregate, propagating neuronal dysfunction and cell death, resulting in the pathological hallmarks of the condition (Israel et al. 2012; Wang et al. 2018; Penney et al. 2020; Valadez-Barba et al. 2020). Some of the other pathophysiological indicators of AD include inflammation, blood–brain barrier anomalies, gliosis, altered cellular and endocytic degradation pathways, and elevated DNA damage (Valadez-Barba et al. 2020).
Attempts to identify viable drug targets for AD have been ineffective thus far as no cure or therapeutic strategy for AD exists to date. The overwhelming reliance on mouse disease models to simulate the complex pathology of AD is considered to be one of the key factors behind this failure (Bolognin et al. 2019). This is due to several considerations such as the differences between mouse and human neurobiology, the significantly shorter lifespan of mice, and the significant amino acid composition differences of several protein orthologs implicated in neurodegenerative disease processes (Dawson et al. 2018; Ke et al. 2019; Penney et al. 2020). Furthermore, the lack of viable animal models capable of accurately recapitulating the pathological hallmarks of AD in humans has forced scientists to look for better alternatives such as iPSC-based human disease models (Vethe et al. 2017). To this end, several studies aiming to obtain a deeper understanding of the mechanistic underpinnings underlying AD pathology using the CRISPR-iPSC-based approaches have been reported, some of which are briefly discussed below.
CRISPR-iPSC in Alzheimer’s Disease Modelling and Therapy
The first ever attempt to generate iPSC-based models of EOAD was reported in 2011 after a group successfully generated two EOAD patient fibroblast-derived iPSC lines containing either the PSEN1 mutation or the PSEN2 mutation (Yagi et al. 2011). Both the iPSC lines were subsequently differentiated into neuronal cells, which successfully recapitulated key aspects of EOAD pathology: increased Aβ peptide secretion and an elevated Aβ42 concentration (Yagi et al. 2011). Subsequently, one group reported the generation of iPSC-derived neuronal disease models for LOAD as well as EOAD pathology (Israel et al. 2012). The authors reported several interesting insights pertaining to the complex nature of AD pathology, such as the magnitude of phenotypic variation that can exist between different iPSC-derived neuronal cell lines, thereby demonstrating the need for isogenic iPSC-based disease models (Israel et al. 2012).
In an attempt to better model the biochemical and pathophysiological implications of possessing the APOE4 allele, one group generated iPSCs from patient skin fibroblasts and subsequently used them to produce APOE4-expressing neurons (Wang et al. 2018). The authors utilized a gene-editing approach to generate healthy APOE3 isogenic controls from the diseased APOE4 iPSC lines. Conversion of the disease-causing APOE4 allele to the healthy APOE3 allele eliminated the detrimental effects of APOE4 and consequently rescued the cell from the disease phenotype, thereby implicating the role of the APOE4 allele in the context of LOAD pathology. Furthermore, in this study, the authors also demonstrated the etiology of LOAD pathology, such as an increase in APOE fragmentation, tau phosphorylation, Aβ production, and GABAergic neuron degeneration. Another study utilized CRISPR/Cas9 to correct the PSEN2 mutation in iPSC-derived basal forebrain cholinergic neurons (BFCNs) and reported the reversion of the diseased BFCN phenotype to the healthy BFCN phenotype (Ortiz-Virumbrales et al. 2017).
CRISPR-iPSC-based approaches provide interesting perspectives into how genetic mutations derange the healthy functioning of astrocytes. For example, in order to better understand how astrocytes with a deranged metabolism contribute to the AD pathology, one group generated isogenic diseased and control astrocyte lines from CRISPR/Cas9-edited iPSCs derived from EOAD patients having a mutant PSEN1 gene (Oksanen et al. 2017). The diseased astrocytes demonstrated elevated Aβ production, modified cytokine release, Ca2+ homeostasis dysregulation, increased oxidative stress, depressed lactate secretion, and compromised neuronal function.
Dysregulated expression of a gene product due to copy number aberrations is known to be responsible for autosomal dominant disorders such as AD. A recent study aimed to generate monoallelic, biallelic, and triallelic knockouts of the APP gene in an AD patient-derived triallelic iPSC line, using a paired Cas9 nickase-based strategy (Ye et al. 2021). The genome-edited iPSCs were subsequently differentiated into cortical neurons, demonstrating APP dosage-dependent AD pathogenesis. The isogenic corrected lines (biallelic APP knockouts) also demonstrated a correction of the disease phenotype, thus demonstrating the research and therapeutic potential of gene dosage-manipulation of CRISPR-iPSC-based AD disease models (Ye et al. 2021).
Non-neuronal cells such as fibroblasts have been used to study several aspects of AD pathology over the past two decades (Govoni et al. 1996; Inoue et al. 2017; Iannuzzi et al. 2021; Inoue 2021). Although the premise might sound paradoxical at first glance, fibroblasts are an attractive non-neuronal proxy for modelling some biochemical aspects of AD pathology due to their availability, stability, and robustness. Furthermore, fibroblasts isolated from AD patients have been documented to demonstrate several alterations in their cellular physiology, such as altered ionic permeability, atypical signaling patterns, and increased production of the highly amyloidogenic Aβ42 peptide (Inoue 2021). One group reported the use of a CRISPR/Cas9 synergistic activation mediator-based CRISPRa system to generate a fibroblast model capable of recapitulating aberrant gamma-secretase-mediated APP processivity, which is hypothesized to promote EOAD pathogenesis. In the study, the authors reported several interesting insights about intracellular APP processing and how CRISPRa based approaches can promote the utilization of non-neuronal AD models.
Recent studies have suggested that lipid droplets tend to accumulate in aged microglia, which in turn is associated with a dysfunctional proinflammatory phenotype (Marschallinger et al. 2020; Claes et al. 2021). The TREM2 gene is deeply associated in the regulation of brain lipid biology and is known to encode a microglial lipid-sensor protein. Furthermore, the TREM2 R47H mutation is associated with a significant elevation (up to threefold) in LOAD risk as it is one of the potent microglia-expressed risk factors (Claes et al. 2021). To improve our understanding of microglial lipid accumulation in AD pathogenesis, a group introduced a TREM2 mutation (R47H) in a GFP-expressing iPSC line (AICS-0036 GFP line) using CRISPR/Cas9-based approach (Claes et al. 2021). The genome-edited, mutation-bearing iPSC line as well as its isogenic control was subsequently differentiated into hematopoietic progenitor cells and transplanted into an established chimeric AD mouse model. Interestingly, the iPSC-derived microglia which were xenografted in the chimeric AD mouse model demonstrated reduced in vivo lipid droplet accumulation as well as plaque reactivity, CD9 expression, and plaque-associated APOE secretion (Claes et al. 2021).
Over the recent years, faults in the endolysosomal-autophagy network have been emerging as an important pathological process underlying AD pathology. Therefore, the role of aberrant endolysosomal function in AD development and pathology as well as its underlying genetics is currently an active area of investigation (Hung et al. 2021). The BIN1 (Bridging Integrator 1) gene is involved in the regulation of endosomal dynamics and has been strongly implicated in the development of AD pathology (Lambert et al. 2022). It is also known to demonstrate extensive differential splicing, giving rise to multiple splice variants having different tissue distributions. In order to better understand how different BIN1 isoforms contribute to AD pathogenesis, a group generated isogenic wild type as well as CRISPR/Cas9-mediated BIN1 knockout human iPSC lines, which were subsequently differentiated into neurons and astrocytes via an intermediate neural progenitor stage (Lambert et al. 2022). They reported that overexpression of BIN1 isoform 1 contributed to early-endosome size deregulation which is an early AD pathology marker, thus suggesting that tightly regulated BIN1 isoform 1 expression levels are required for proper neuronal endocytic trafficking (Lambert et al. 2022). The SORL1 gene is responsible for encoding a sorting receptor protein involved in retromer-related endosomal trafficking, thereby regulating the anterograde as well as retrograde movement of APP between the early endosomes and trans-Golgi network (Yin et al. 2015; Knupp et al. 2020). Using a CRISPR-iPSC-based approach, a group generated SORL1-knockout human iPSC-derived neuronal and microglia-like cells to ascertain how SORL1 ablation promotes endosome dysfunction and consequently AD pathogenesis. The authors reported that SORL1 loss results in the induction of early-stage AD cytopathology (endosomal enlargement) in neurons but not the microglia-like cells, suggesting the presence of heterogeneous cell type-specific early AD pathology (Knupp et al. 2020). Using a comparable approach, another group reported the involvement of PSEN1, APP, and SORL1 in a common pathway responsible for the regulation of the endolysosome system, which becomes dysfunctional in AD patients (Hung et al. 2021).
The CLU gene is yet another major genetic AD risk factor which, alongside APOE, regulates cholesterol transport and has a documented role in AD pathogenesis (Yu and Tan 2012; Robbins et al. 2018). Furthermore, Aβ treatment has been previously demonstrated to induce CLU expression in rodent neurons (Killick et al. 2014). To better understand the relationship between the CLU gene and AD, a group generated CRISPR/Cas9-edited, iPSC-derived CLU knockout cortical neurons as well as their isogenic controls and subjected both to Aβ treatment (Robbins et al. 2018). It was observed that the CLU knockout neurons were protected from Aβ-induced phenotypes, while their counterparts underwent dose-dependent degeneration. The findings of the study indicated the role of the CLU gene product in Aβ peptide-mediated AD pathology and established it as a necessary effector of Aβ toxicity (Robbins et al. 2018).
Reports describing CRISPR-iPSC-based three-dimensional organoid culture-based AD disease models have also surfaced in recent years. In one such study, the authors utilized CRISPR/Cas9-mediated genome editing to introduce the faulty APOE4 allele in iPSC-derived neurons, astrocytes, and microglia-like cells cultured in three-dimensional organoid systems (Lin et al. 2018). The results of the study suggested that APOE4 impacts AD pathology through the impairment of astrocyte and microglia-mediated Aβ clearance (Lin et al. 2018).
The SARS-CoV-2 pandemic has promoted the emergence of interesting research perspectives exploring the intersection of virology and CRISPR-iPSC-based AD modelling. One such study tested the neurotropism of SARS-CoV-2 in iPSC models generated using the CRISPR/Cas9 nickase system and reported an increased rate of SARS-CoV-2 infection in neurons and astrocytes containing the APOE4 allele. In the same report, the authors also demonstrated the remarkable effects of remdesivir in treating SARS-CoV-2 infection using CRISPR/Cas9-edited iPSC-derived brain cells, thus providing the medical fraternity with evidence in favor of treating neurological complications in COVID-19 patients using remdesivir (Wang et al. 2021).
Recent developments in iPSCs and CRISPR-iPSC-based disease models offer promising scientific insights in the search for a definitive cure for AD. How these scientific perspectives translate to an effective clinical intervention remains to be seen.
Parkinson’s Disease
Disease Pathology and Etiology
Parkinson’s disease (PD) is the second most prevalent neurodegenerative disease following AD. It is characterized by the progressive loss of dopaminergic neurons in a brain region known as the substantia nigra pars compacta due to the intraneuronal accumulation of misfolded α-synuclein aggregates (Vázquez-Vélez and Zoghbi 2021). The substantia nigra pars compacta is a midbrain structure that houses the dopamine secreting dopaminergic neurons. It plays a critical role in various cognitive activities such as motor behavior, motivation, reward, and working memory (Chinta and Andersen 2005). The death of the dopaminergic neurons due to α-synuclein aggregation disrupts the normal functioning of the basal ganglia, a brain structure responsible for orchestrating and executing movement (Shulman et al. 2011; Vázquez-Vélez and Zoghbi 2021). This ultimately results in the clinical manifestation of PD, which is primarily defined by resting tremors, rigidity, bradykinesia, and postural instability. As the disease progresses, the pathology spreads beyond the confines of the basal ganglia, and more generalized nonmotor symptoms such as dyskinesias (involuntary and erratic body movements), distressing sensory experiences (pain), autonomic dysfunction (urinary incontinence, orthostatic intolerance), and neuropsychiatric manifestations (depression, hallucinations, and dementia) progressively emerge in the patient.
PD is categorized as familial or sporadic based on the family history of the affected individuals. Familial PD patients have a family history of PD symptoms and account for a small fraction (5–15%) of reported cases, whereas spontaneous PD patients develop the disease without any discernable hereditary predisposition (Klein and Westenberger 2012; Tran et al. 2020). The genetic architecture underlying PD pathology is complex and multifactorial. More than 90 variants of genes such as SNCA (Synuclein Alpha), GBA (Glucosylceramidase Beta), LRRK2 (Leucine-Rich Repeat Kinase 2), and MAPT (Microtubule-Associated Protein Tau) have been implicated in PD pathology.
As of now, there exists no pharmaceutical or surgical intervention capable of curing PD. The current lines of pharmaceutical (levodopa) or surgical (deep brain stimulation) interventions provided to PD patients attempt to improve his/her quality of life and decelerate the onset of motor deficits characteristic of the disease process (Kalia and Lang 2015; Poewe et al. 2017; Paff et al. 2020; Sonninen et al. 2020). Therefore, it is imperative that new lines of treatment be designed to bring us closer to a cure to PD.
CRISPR-iPSC in Parkinson’s Disease Modelling and Therapy
One of the first attempts to generate iPSCs from sporadic PD patient-derived fibroblasts was reported in 2009 by Soldner and colleagues (Soldner et al. 2009). The authors of this study successfully differentiated the PD patient-derived iPSCs into dopaminergic neurons, thus validating the viability of iPSC-based approaches to generating PD models using patient-derived fibroblasts (Soldner et al. 2009).
α-synuclein dysfunction is a critical milestone of PD pathology and causes a penetrant, aggressive form of PD accompanied with dementia (Auluck et al. 2010; Stefanis 2012). α-synuclein, one of the three paralogs of the synuclein family, is a 140 amino acid long 14 kDa presynaptic nerve terminal protein that is encoded by the SNCA gene (Fares et al. 2021). It plays critical roles in several neurophysiological processes such as neurotransmitter biosynthesis and release, vesicular trafficking, calcium homeostasis, mitochondrial function, and gene regulation (Auluck et al. 2010; Perez 2020; Singh and Muqit 2020). There is strong evidence suggesting that the aggregation of α-synuclein is promoted by the introduction of point mutations, increases in copy number, and regulatory changes of the SNCA gene at the promoter level (Vázquez-Vélez and Zoghbi 2021). These aggregates are observed in both cell bodies (Lewy bodies) and the processes (Lewy neurites) of neurons principally located in the substantia nigra pars compacta, a key regulatory center of the basal ganglia (Auluck et al. 2010). Therefore, investigations probing the mechanistic basis of neurodegeneration caused by the SNCA gene and its various pathological variants have been an active area of interest in the context of iPSC-based disease modelling (Ryan et al. 2013; Dettmer et al. 2015; Kouroupi et al. 2017; Ke et al. 2019). For example, one group reported a successful attempt at developing a PD model capable of recapitulating the pathological hallmarks of dysfunctional α-synuclein overexpression propagated neurodegeneration (Devine et al. 2011). The authors used midbrain dopaminergic neurons generated from iPSCs obtained from a spontaneous PD patient possessing three copies of the SNCA gene (Devine et al. 2011). Another study investigating the deleterious effects of SNCA triplications using iPSC-based disease models described how α-synuclein overexpression impairs the differentiation of neuronal progenitor cells from iPSCs (Oliveira et al. 2015). Similarly, iPSC-based PD models harboring other gene mutations responsible for promoting PD pathology such as LRRK2, PINK1, PARK2, GBA1, GBA, and parkin have also been developed and elaborately explored by several groups over the past decade (Nguyen et al. 2011; Jiang et al. 2012; Lin et al. 2016; Ke et al. 2019; Hu et al. 2020).
In 2016, using a CRISPR-iPSC-based indel approach, Soldner and colleagues deleted and replaced a region of the intron-4 enhancer element of the SNCA gene containing PD-associated single nucleotide polymorphisms (SNPs) with different allelic variants (Soldner et al. 2016). In doing so, they generated a collection of isogenic iPSC-derived neuronal cell lines representing an allelic series of the SNP of interest. On interrogating the entire set of allelic variants for PD pathology-specific markers, the authors identified a risk variant in a noncoding distal enhancer element of the SNCA gene responsible for upregulating α-synuclein expression and consequently promoting the pathological descent demonstrated by PD patients.
Besides the range of issues pertaining to heterogeneous genetic backgrounds, researchers working towards the generation of genome-edited iPSC-based disease models for polygenic diseases such as PD face another pertinent challenge. This challenge is the painstaking and time-consuming process of screening genome-edited cell lines to separate the desired clones containing the on-target gene knock-ins from the aberrant clones plagued by unwanted mutations such as random integrations, on-target indels, and second-allele indels. To streamline this challenging ordeal, an automated high-throughput, fluorescent marker-based, genome-edited iPSC phenotype screening workflow was demonstrated by Arias-Fuenzalida and colleagues. The authors utilized donor vectors to deliver fluorescent protein tags alongside the desired SNCA mutation-bearing sequences inside iPSC-derived neuroepithelial stem cells, an early neurodevelopment disease model (Arias-Fuenzalida et al. 2017). Following CRISPR/Cas9-mediated integration at the appropriate locus, the desired clones could be seamlessly separated from the aberrant clones using fluorescence-activated cell sorting (FACS) (Arias-Fuenzalida et al. 2017). A recent study reported another innovative approach to FACS-based enrichment of dopaminergic neurons (Überbacher 2019). The group performed a study that generated novel CRISPR/Cas9-edited iPSC-derived dopaminergic neuronal lines endogenously expressing fluorescent tyrosine hydroxylase-enhanced green fluorescent protein reporter (Überbacher 2019). Tyrosine hydroxylase is the rate-limiting enzyme of the dopamine biosynthetic pathway and is commonly employed as a dopaminergic neuron-specific marker protein (Haavik and Toska 1998; White and Thomas 2012). Since a typical iPSC differentiation workflow yields a heterogeneous population of dopaminergic neurons contaminated with other unsuccessfully differentiated cell types, the reporter-based approach demonstrated significant improvements in the identification and sorting of dopaminergic neurons as compared to conventional manual approaches (Überbacher 2019).
Our understanding of the mechanistic underpinnings of α-synuclein aggregation, kinetics, as well as its associated pathology has improved considerably over the past few years due to the concerted efforts of many different groups employing a diverse range of chemical, biological, and biophysical approaches (Wördehoff et al. 2015; Fauerbach and Jovin 2018; de Oliveira and Silva 2019; Ruggeri et al. 2020; Srivastava et al. 2020). In one such approach, a group seeded exogenous fibrillar α-synuclein in neuroblastoma cells (SH-SY5Y) and iPSC-derived neurons, neither of which possessed PD-pathology promoting genetic mutations (Gao et al. 2019). Using CRISPR/Cas9, the neuroblastoma cells (SH-SY5Y) were genome-edited to obtain isogenic α-synuclein knockout and α-synuclein expressing control cell lines. The authors monitored inclusion body formation and investigated the temporal behavior of autophagy/lysosomal proteins in the exogenous α-synuclein fibril-treated neurons. They reported evidence suggesting a connection between the formation of cytoplasmic α-synuclein inclusion bodies and changes in the autophagy/lysosome markers. Furthermore, they also discussed the viability of autophagy-promoting interventions such as 5-AMP-activated protein kinase activation as potential therapeutic routes for managing PD (Gao et al. 2019).
LRRK2 gene mutations are responsible for causing both sporadic and familial PD (Sánchez‐Danés et al. 2012; Qing et al. 2017). After the ground-breaking discovery of iPSCs by Yamanaka group (Takahashi and Yamanaka 2006), several reports describing different aspects of PD pathology in LRRK2 mutant iPSC-based disease models began to surface (Nguyen et al. 2011; Sánchez‐Danés et al. 2012). However, as Qing and colleagues observe, many of the iPSC-based contemporary studies ignored the non-trivial differences between the genetic backgrounds of the patient and non-isogenic control cell lines (Qing et al. 2017). The authors articulated the possibility that these differences could potentially mask critical insights pertaining to PD pathogenesis and even obscure promising novel drug targets (Qing et al. 2017). The generated LRRK2 mutation-bearing dopaminergic neurons demonstrated decreased neurite length (< 2000 μm) and branching. Furthermore, the neurons with longer neurite lengths (> 2000 μm) were positive for Serine-129 phosphorylated α-synuclein, thereby implying the involvement of this post translational modification in the formation and maintenance of long neurites (Qing et al. 2017).
Dopaminergic neurons are not the only CRISPR/Cas9-edited iPSC-derived products under active investigation due to their role in PD. Several genes with established causative roles in the pathogenesis of PD are also responsible for mediating critical processes in astrocytes (Sonninen et al. 2020). Therefore, several groups have explored the roles of non-neuronal cells such as astrocytes in PD pathology using CRISPR-iPSC-based astrocyte disease models (Booth et al. 2017; Santos et al. 2017; Ke et al. 2019). Using CRISPR/Cas9, one group FLAG-tagged the endogenous SNCA locus of PD-affected as well as healthy iPSC lines and subsequently differentiated them into astrocytes to monitor intracellular α-synuclein accumulation (di Domenico et al. 2019). They also used CRISPR/Cas9 to correct a particular pathological point mutation (G2019S) in the LRRK2 gene of a mutant iPSC line to generate isogenic diseased as well as disease phenotype-rescued astrocytes. Using astrocyte-dopaminergic neuron co-culture models, the group demonstrated that the astrocytes in PD patients exhibit severely compromised lysosomal α-synuclein degradation, thus resulting in progressive neurodegeneration. They also discovered that diseased astrocytes accumulate and transfer α-synuclein to neighboring neurons, thus significantly contributing to the growing body of research exploring the role astrocytes play in PD pathology (di Domenico et al. 2019). Another group corroborated these findings using a similar approach involving isogenic iPSC-derived, CRISPR/Cas9-edited diseased and disease phenotype-rescued astrocytes (Sonninen et al. 2020). They reported that LRRK2 (G2019S) and GBA (N370S) mutant astrocytes demonstrate upregulated α-synuclein expression, elevated astrocytic Ca2+ levels, increased inflammatory reactivity, compromised mitochondrial DNA maintenance, and metabolic homeostasis perturbation such as altered polyamine metabolism (Sonninen et al. 2020).
The GBA gene is responsible for encoding the lysosomal hydrolase glucocerebrosidase, an enzyme which hydrolyses the sphingolipid waste product glucosylceramide into ceramide and is a documented risk factor for PD (Sidransky and Lopez 2012; Mullin et al. 2019). The GBA gene is particularly interesting in the context of CRISPR/Cas9-based genome editing as it shares a 96% sequence homology with a pseudogene GBAPI (Hanss et al. 2020). The presence of pseudogenes having a high degree of sequence similarity with the target gene complicates genome editing attempts. This is due to the higher risk of off-target effects and accidental pseudogene modification, resulting in a potentially pathological imbalance in the gene/pseudogene transcript ratio. Hanss and colleagues described a detailed CRISPR-Cas9-based approach involving a screening strategy capable of gene vs. pseudogene discrimination, thereby enabling the insertion and correction of point mutations in the GBA gene without any concomitant alterations in GBAPI (Hanss et al. 2020).
Genes involved in the pathology of neurodegenerative diseases such as AD and ALS have been reported to be associated with PD-associated genes. For example, the LRP10 (Lipoprotein Receptor-Related Protein 10) gene has been recently reported to interact with the AD susceptibility gene SORL1 in a study involving both iPSC-derived cell lines (astrocytes and neurons) and CRISPR/Cas9-edited LRP10 knockout cell lines (HEK293T and HuTu 80) but not a combination of the two (Grochowska et al. 2021). The study nevertheless provided interesting insights pertaining to the intertwined nature of the genetic architecture of different neuropathological conditions. In another interesting study, Harjuhaahto and colleagues demonstrated the viability of iPSC-derived motor neurons in which either the CHCHD2 gene or CHCHD10 gene had been knocked out using a CRISPR/Cas9-mediated genome editing approach (Harjuhaahto et al. 2020). This is an interesting finding because dominant CHCHD10 mutations are known to promote a range of pathological motor neuron conditions, whereas dominant CHCHD2 mutations are known to cause PD (Shi et al. 2016, p. 2; Harjuhaahto et al. 2020). The authors of the study noted that although both the CHCHD2 and CHCHD10 knockout iPSC lines were viable and pluripotent, compromised mitochondrial respiration (increased proton leakage and respiratory rate) was observed in both knockout iPSC and iPSC-derived neuronal cell lines (Harjuhaahto et al. 2020). Interestingly, reciprocal compensatory increases in CHCHD2/CHCHD10 levels were observed in either of the knockout cell lines. Furthermore, both the knockout motor neuron cell lines demonstrated significantly overlapping transcriptome profiles when compared to isogenic controls. These findings are indicative of the fact that a CHCHD2-CHCHD10 complex is required for efficient mitochondrial respiration, a fact corroborated by other studies (Burstein et al. 2018; Harjuhaahto et al. 2020; Xiao et al. 2020). This study bears significance in both the context of PD and ALS, discussed later in the review.
Knockout studies conducted over the past few years have yielded a lot of information pertaining to the effects of several new genes implicated in PD pathology. In one such study, a group knocked out three different early-onset autosomal recessive PD-associated genes (PRKN, DJ-1, and ATP13A2) in three separate iPSC lines, all of which were subsequently differentiated into midbrain dopaminergic neurons (Ahfeldt et al. 2020). DJ-1 is known to have various functions such as transcriptional, chaperone, protease, and mitochondrial regulation as well as antioxidative stress reaction (Ariga et al. 2013). ATP13A2 is a lysosomal P5-type transport ATPase which is known to be involved in transmembrane cationic transport (Estrada-Cuzcano et al. 2017; Spataro et al. 2019). PRKN is a cytosolic ubiquitin E3 ligase which ubiquitinates a number of cytosolic and outer mitochondrial membrane protein targets (Seirafi et al. 2015). Using the knockout cell lines and their corresponding isogenic controls, proteomics as well as a transcriptomics datasets were generated by the authors for deeper evaluation of the molecular insights (Ahfeldt et al. 2020). Although all the three knockout cell lines experienced elevated oxidative stress levels as well as dysregulated lysosomal and mitochondrial function, only the PRKN knockout line demonstrated an increase in death rate. Furthermore, the authors uncovered evidence suggesting the presence of shared as well as distinct dysregulated oxidative stress and autophagy-lysosomal pathways among the three knockout cell lines. This study highlights the need for further studies studying individual as well as the combinatorial effects of different genes on PD pathology (Ahfeldt et al. 2020). In that regard, a very recent lab resource paper reported the generation of CRISPR/Cas9-edited homozygous PRKN, PINK1 and double PINK1/PRKN knockout iPSC lines which can be differentiated into neuronal and glial cells for studying PD and related neuropathological conditions (Chen et al. 2022). The PINK1 and PRKN genes encode proteins involved in mitophagy-mediated dysfunctional mitochondria degradation, and mutations in these genes are among the most common causes of early-onset familial PD (Bradshaw et al. 2021). VPS13C is another notable PD risk factor belonging to a gene family (the VPS13 gene family) responsible for encoding lipid transfer proteins which localize in several distinct contact sites located in-between membranous proteins. Using a CRISPR-iPSC-based approach, a group generated VPS13C knockout iPSC-derived neuronal cell lines in which they studied knockout-mediated lysosomal function alteration. Combining these insights with that derived from VPS13C knockout HeLa cells and VPS13C−/− mice, the group demonstrated the perturbation of lysosomal lipid homeostasis in VPS13C-deficient cells, thereby suggesting the involvement of VPS13C in PD pathogenesis-relevant pathways (Hancock-Cerutti et al. 2022).
Simple two-dimensional cell culture-based disease models fail to recreate these complex physiological conditions, whereas three-dimensional cell culture-based disease models recreate in vivo conditions and consequently recapitulate disease processes much more effectively. To demonstrate this, Bolognin and colleagues developed a CRISPR-iPSC-based microfluidic PD disease model that explicitly illustrated the superiority of three-dimensional cell culture-based disease models over their traditional two-dimensional counterparts (Bolognin et al. 2019). The authors used both healthy and LRRK2 (G2019S) mutation-bearing iPSC lines to generate diseased and disease-corrected isogenic pairs using a footprint-free piggyBac vector system-potentiated CRISPR/Cas9-based genome editing approach. The genome-edited iPSCs were subsequently differentiated into neuroepithelial stem cells, wherein the authors observed notable differences in the neuronal differentiation outcomes between the parallelly maintained two-dimensional and three-dimensional cultures. Exciting developments and advancements have been made in CRISPR-iPSC-based PD modelling. How these developments ultimately lead us to a cure for this complex yet debilitating malady remains to be seen.
Huntington’s Disease
Disease Pathology and Etiology
Huntington’s disease (HD) is an adult-onset, hereditary, monogenic, autosomal dominant, and fully penetrant neurodegenerative condition. It is characterized by protein misfolding and aggregation in the brain, resulting in progressive psychiatric disruption, cognitive decline, and loss of motor coordination (Schulte and Littleton 2011; Ross and Tabrizi 2011; McColgan and Tabrizi 2018). The first clinical manifestations of HD’s motor, cognitive, and behavioral characteristics arise at a mean age of 35 (Roos 2010). The average life expectancy for HD is approximately two decades from the age of symptomatic onset (Roos 2010; Schulte and Littleton 2011).
HD is caused by a CAG triplet repeat expansion mutation in a gene located on the short arm of chromosome 4p16.3 called the Huntingtin (HTT) gene (Roos 2010; Ross and Tabrizi 2011). The HTT gene is an essential gene that is most strongly expressed in the brain, and surprisingly the testes, but is expressed at lower levels throughout the body. It is known to interact with over 200 different proteins and plays a vital role in critical cellular processes such as axonal trafficking, transcriptional control, and apoptotic regulation (Schulte and Littleton 2011). The number of CAG repeats present in the mutant HTT gene of symptomatic HD patients is generally observed to lie between 36 and 100 and in some cases even over 100 (Langbehn et al. 2010; Roos 2010; Schulte and Littleton 2011). Healthy individuals have less than 27 CAG repeats, whereas reduced HD penetrance is observed in individuals having less than 40 repeats. However, full HD penetrance is observed for individuals having 40 or more CAG repeats (McColgan and Tabrizi 2018). The number of the CAG triplet repeats present in the mutant HTT gene is inversely correlated with the age of onset (Langbehn et al. 2010).
The expanded CAG repeat in the mutant HTT gene encodes an expanded polyglutamine stretch in the Huntingtin (HTT) protein (Ross and Tabrizi 2011). This polyglutamine expansion in the mutant HTT (mHTT) protein results in the presentation of a toxic gain-of-function phenotype. The polyglutamine stretch causes the mHTT protein to misfold, resulting in intracellular aggregates within the brain cells. The rate of protein aggregation is proportional to the magnitude of the polyglutamine expansion in the mHTT protein (Schulte and Littleton 2011). This is consistent with the inverse correlation between the age of symptomatic onset in HD patients and the number of CAG repeats in their respective mutant Huntingtin genes, as previously discussed. These protein aggregates tend to overload the cell’s ubiquitin-proteasomal degradation system, thus resulting in a progressive deterioration of the affected cell’s homeostatic integrity (Schulte and Littleton 2011). Currently, there are no effective lines of treatment that prevents the onset or progression of HD. The presently available therapeutic regimes can only provide symptomatic relief to HD patients.
CRISPR-iPSC in Huntington’s Disease Modelling and Therapy
The limited clinical translatability of the insights derived from animal HD models is one of the notable reasons behind the failure of the research community to develop a cure. Moreover, most mouse HD models have a far higher number of CAG repeats in comparison to their adult HD patient counterparts, thus further complicating disease modelling efforts (Csobonyeiova et al. 2020). Therefore, it is not surprising that iPSC-based and CRISPR-iPSC-based HD models are experiencing substantial growth in research and clinical interest in recent years.
Some of the earliest reports of iPSC-based HD models go back as early as 2010 (Zhang et al. 2010). One such study aimed to better understand HD disease pathology by generating striatal neurons from an HD patient-derived iPSC line having 72 CAG repeats. This study is particularly interesting in the context of striatal neurons because the mHTT protein causes transcriptional and transport inhibition of brain-derived neurotrophic factor, a factor critical for striatal neuron maintenance. The authors of the study reported several interesting findings, such as the stability of CAG stretches over multiple passages in HD patient-derived iPSCs, and growth factor-deprivation promoted elevation in caspase activity (Zhang et al. 2010). In another interesting study, a group packaged a healthy HTT gene flanked by homologous arms in a bacterial artificial chromosome and delivered it inside an HD patient fibroblast-derived iPSC line via nucleofection. The iPSC-derived neural stem cells were subsequently transplanted in the striatum of an HD mouse model’s brain, which successfully underwent differentiation in vivo into neuronal and glial cells (An et al. 2012).
As the interest surrounding CRISPR, iPSCs, and their application in disease modelling applications developed, CRISPR-iPSC-based HD modelling approaches started gaining significant momentum. Very recently, a group recently reported a CAG repeat tract contraction strategy using an engineered Cas9 nuclease variant recognizing the NGN PAM sequence instead of the NGG PAM sequence (recognized by the wild-type variant of the Cas9 protein), thus enabling more precise targeting of the faulty HTT gene (Oura et al. 2021). In 2014, a group reported an attempt to generate isogenic iPSC lines containing 97 CAG repeats in the HTT gene using a CRISPR/Cas9-based knock-in strategy. They successfully demonstrated the viability of utilizing CRISPR-assisted homologous recombination in CRISPR-iPSC-based HD disease modelling applications (An et al. 2014). Another group reported a dual CRISPR/Cas9 guide RNA-based approach to selectively knock-out the mutant HTT gene in a fibroblast-derived iPSC line and an iPSC-derived neural progenitor cell line by exclusively targeting mutation-specific PAM sites (Shin et al. 2016). Since HD patients homozygous for the mutant allele are very rare and one functional copy of the HTT gene is sufficient to maintain cellular integrity, this proof-of-principle study demonstrated a potential CRISPR-iPSC-based route to model and eventually cure HD by completely ablating mHTT expression inside a cell (Shin et al. 2016). The authors also discussed the translatability of their approach to a wide variety of other devastating dominant gain-of-function disorders such as AD, PD, and ALS (Shin et al. 2016).
The existence of non-trivial genetic differences between different iPSC lines and their therapeutic and clinical implications is well reported and discussed in the literature (Kajiwara et al. 2012; Wang et al. 2020). These differences can arise due to different factors ranging from genetic heterogeneities in the donor cell lines to qualitative and quantitative differences in the differentiation protocols utilized to derive different iPSCs (Kajiwara et al. 2012). This problem is compounded for CRISPR-iPSC-based systems due to the additional involvement of the CRISPR/Cas9-specific molecular machinery such as expression vector cassettes and transfection agents. These differences in genetic backgrounds among different iPSC lines are important considerations in disease modelling applications. Genome-wide expression analysis has revealed higher numbers of gene expression differences between iPSC-based HD disease models and their non-isogenic controls compared to isogenic controls, thus illustrating how heterogeneous genetic backgrounds can generate false insights about the disease transcriptome (Xu et al. 2017). To ameliorate this pertinent issue, fully removable selection cassettes such as the piggyBac transposon as well as selection cassette-free approaches to deliver the CRISPR/Cas9 payload into iPSCs have been explored by different groups (Yusa 2013; Xu et al. 2017; Malankhanova et al. 2020). Furthermore, isogenic HD disease models are being established by different groups using CRISPR-iPSC-based approaches, thus enriching the repertoire of tools currently available to effectively model HD. For example, a group established several homozygous HEK293T cell lines containing different numbers of CAG repeats generated using several different genome editing strategies involving both the wild-type CRISPR/Cas9 system and the CRISPR/Cas9 nickase system (Dabrowska et al. 2020). They also established a mutation corrected and HTT knock-out variant of an established HD patient-derived iPSC line (ND42222) to serve as effective isogenic controls in future proteome as well as transcriptome profiling studies carried using neuronal cell lines derived from the same iPSC line (Dabrowska et al. 2020).
A recent study aiming to develop and validate a model for the purpose of estimating the diagnosed prevalence of HD propounded that HD prevalence is increasing with time (Crowell et al. 2021). Therefore, it is essential that we develop better therapeutic approaches to manage and eventually cure HD. The CRISPR-iPSC-based disease models discussed provide some promising perspectives towards that common goal.
Amyotrophic Lateral Sclerosis
Disease Pathology and Etiology
ALS, also referred to as von Gehrig’s disease, is a multisystem neurodegenerative disorder (Mejzini et al. 2019). ALS is primarily characterized by upper and lower motor neuron degeneration. The motor neuron degradation eventually leads to progressive muscular atrophy, weakness, and voluntary muscle paralysis resulting in the complete inability to initiate or control voluntary movements. Similar to AD, PD, and HD, no clinical intervention to effectively cure ALS is available to date (Mejzini et al. 2019). All lines of current ALS treatment are purely focused on delaying disease progression and providing symptomatic relief (van Es et al. 2017; Chen 2020).
ALS is a complex polygenetic multifactorial disorder with a combination of genetic, environmental, and age-related factors considered to be responsible for the symptomatic onset of the disease. ALS can be categorized as familial ALS or sporadic ALS, depending on whether the patient had any prior family history or not. Although over 90% of reported ALS cases are sporadic in nature, the remaining 10% reports of familial ALS almost always demonstrate an autosomal dominant pattern of inheritance. Many potentially causative or disease-modifying genes have been identified to potentiate the pathological hallmarks of ALS by genome-wide association studies, with varying levels of characterization and confidence (Mejzini et al. 2019). However, the identities of the most penetrant gene mutations such as the SOD1 (superoxide dismutase 1), FUS (fused in sarcoma), TARDBP (TAR DNA-binding protein 43), TBK1 (TANK-binding kinase 1), and C9ORF72 (chromosome 9 open reading frame 72) genes are well characterized (Nguyen et al. 2018; Burk and Pasterkamp 2019; Mejzini et al. 2019; Masrori and Van Damme 2020; Amado and Davidson 2021).
CRISPR-iPSC in ALS Modelling and Therapy
Since the first SOD1 mutant mouse disease models were developed in 1994, several animal models such as Caenorhabditis elegans, Drosophila melanogaster, Danio rerio, and even higher mammal models such as pigs and primates have been used by different research groups. However, clinical translatability of the animal model-derived insights to human trials has always remained dismal, as evidenced by the fact that no cure for ALS exists to date (Mejzini et al. 2019; Liguori et al. 2021; Braems et al. 2021). iPSC-based ALS disease models soon came into prominence given the advantages they presented over conventional animal-based disease models, as already discussed in the context of other neurodegenerative diseases.
Using iPSC-derived motor neuronal cell lines carrying mutations in the SOD1, TDP-43, or C9ORF72 gene, one group demonstrated the influence of the cell’s genetic background in determining the levels of intracellular protein aggregation (Seminary et al. 2018). They also established that protein aggregation itself is insufficient to trigger the cellular heat shock response (Seminary et al. 2018). Since then, a variety of genes with differing levels of penetrance are responsible for ALS. Similar attempts to recapitulate the pathological hallmarks of different mutations such as the FUS mutation or C9ORF72 mutation using iPSC-derived motor neuron cell lines sourced from patients can be found in the literature (Bilican et al. 2012; Sareen et al. 2013; Japtok et al. 2015). The effects of ALS causing mutations on iPSC-derived non-neuronal cell lines such as astrocytes have also been reported. In one such report, the authors demonstrated that C9ORF72 mutation-bearing astrocytes secrete soluble neurotoxic factors, which aggravate oxidative stress in healthy motor neurons (Birger et al. 2019).
CRISPR-iPSC-based disease models and studies leveraging the combined potential of iPSCs and CRISPR/Cas9-based genome editing have been reported. One such CRISPR-iPSC-based study attempted to rescue iPSC-derived motor neuron cells from the deleterious effects of the C9ORF72 mutation (Krishnan et al. 2020). The C9ORF72 mutation occurs due to a hexanucleotide repeat expansion (GGGGCC) in the first intron of the C9ORF72 gene, which is suggested to result in the formation of abnormal mRNA transcripts responsible for causing a range of downstream issues. Some of these issues include mRNA misprocessing and faulty transcription, protein sequestration and aggregation, and the translation of abnormal protein fragments that form neuronal inclusions (Mejzini et al. 2019). In the study, the authors carried out a CRISPR/Cas9-based minimal promoter knockout of the first exon of C9ORF72 in an iPSC line containing around a thousand copies of the hexanucleotide repeat in the first intronic region located downstream of the first exon (Krishnan et al. 2020). The motor neuron cells generated thereafter from the edited iPSC lines demonstrated reduced neurodegenerative phenotypes, suggesting the application of CRISPR/Cas9-based promoter region targeting as a viable and potentially usable therapeutic approach to correct toxic gene product-triggered disease phenotypes (Krishnan et al. 2020).
Another CRISPR-iPSC-based study published used CRISPR/Cas9-corrected iPSC-derived motor neuron lines originally sourced from familial ALS patients positive for SOD1 and FUS mutations (Wang et al. 2017). In this study, familial ALS patient-derived fibroblasts bearing a SOD1 mutation (A272C) as well as a FUS mutation (G1566A) were each utilized to establish a single mutation-bearing iPSC line. Following this, mutation-corrected isogenic controls of the two mutant iPSC lines were generated using CRISPR/Cas9-based genome editing. Finally, the mutation-bearing iPSCs as well as their corresponding isogenic controls were differentiated into motor neurons. The authors further proceeded to perform genome-wide RNA-seq analysis of the iPSC-derived motor neuron cell line derived from the SOD1 mutant and its corresponding isogenic control iPSC line (Wang et al. 2017). A total of 899 aberrant transcripts were uncovered including transcripts to genes involved in critical processes such as signal transduction, extracellular matrix organization, homeostatic equilibrium maintenance, and neurogenesis implicating that a few different pathways may be involved in SOD1-associated early ALS pathogenesis (Wang et al. 2017). Apart from identifying potential early pathological markers of motor neuron decay due to SOD1 mutation, the authors also demonstrated the viability of using integration-free single-stranded oligodeoxynucleotide templates instead of the more conventionally used integrating plasmid-based constructs to deliver the corrected template strand to the Cas9 protein in vivo (Wang et al. 2017).
The combination of whole-genome sequencing-based gene variant discovery and CRISPR-iPSC-based disease modelling techniques has accelerated the discovery of rare yet highly penetrant ALS-causing novel gene variants which only certain ethnic groups are predisposed to (Yun et al. 2020). A sizeable fraction of the published studies have reported the utilization of CRISPR/Cas9-corrected patient-derived iPSC lines to generate isogenic disease models. As previously discussed, the versatility of CRISPR-iPSC-based disease modelling applications renders access to patient-derived cell lines an optional requirement. One such attempt has been recently reported wherein a group generated CRISPR-iPSC-based isogenic ALS disease models harboring a SOD1 missense mutation (G93A) by utilizing a CRISPR/Cas9-based targeted knock-in approach on healthy iPSC lines. The genome-edited iPSCs were subsequently differentiated into motor neurons, demonstrating the pathological signatures of intracellular misfolded SOD1 aggregate accumulation, such as reduced axonal length and branching, presynaptic and postsynaptic abnormalities, and anomalous electrical activity (Kim et al. 2020a). In another study, a group generated CRISPR/Cas9-mediated SQSTM1 knockout iPSC-derived cortical neurons and assessed its impact on neuronal physiology (Poon et al. 2021). SQSTM1 is a multifunctional protein involved in a wide range of cellular processes such as oxidative stress response, metabolic reprogramming, and autophagy. The authors demonstrated that the SQSTM1 knockout iPSC-derived cortical neurons demonstrated alterations in mitochondrial gene expression patterns and functionality, autophagy flux, and mitochondrial spare respiratory capacity. Based on their findings, the authors suggested the role of SQSTM1 to be related to mitochondrial function (Poon et al. 2021).
The neuromuscular junction (NMJ) is a very specialized structure where a motor neuron nerve terminal forms a synaptic junction with a muscle fiber. It is responsible for converting neuronal electrical signals into muscular contraction-initiating electrical activity. The NMJ also happens to be one of the best investigated and disease-prone synaptic structures of the nervous system. In fact, NMJ dismantling has been observed in ALS patients (Rodríguez Cruz et al. 2020). There is a growing body of evidence that indicates the need to consider muscle fibers alongside motor neurons as well as their functional interface, i.e., the NMJ, as important players in ALS initiation, progression, and therapy (Cappello and Francolini 2017; Jongh et al. 2021). However, the particulars of NMJ degeneration in ALS, such as its temporal relationship with motor neuron death or the exact series of events culminating in the disassembly of the NMJ circuitry, are poorly understood. It is essential that human NMJ disease models are utilized in functional and mechanistic studies as rodent NMJs are significantly different from human NMJs in terms of size, complexity, fragmentation, and temporal stability (Stoklund Dittlau et al. 2021). To that end, several iPSC-derived three-dimensional cell culture, organoids, and microfluidics-based ALS disease models aimed to model the NMJ have been reported (Cappello and Francolini 2017; Osaki et al. 2018; Faustino Martins et al. 2020). In one such study, a group utilized CRISPR/Cas9 to introduce point mutations in SOD1(G85R), TARDBP (G298S), and PFN1 (G118V), thereby establishing three mutant iPSC lines (Pereira et al. 2021), which were subsequently used to derive organoid cultures. The authors observed some distinct disease phenotypes such as innervated NMJs in the SOD1 and PFN1 mutants and a decrease in innervated NMJ area in the TARDBP mutant, suggesting the need to identify the mechanistic links between specific genes, their mutations, and aberrant NMJ physiology in the context of ALS (Pereira et al. 2021). Another notable mention among such reports is a recent attempt by a group to co-culture isogenic diseased (FUS mutant) and CRISPR/Cas9-corrected iPSC-derived motor neurons and human primary mesoangioblast-derived myotubes in a compartmentalized microfluidic device (Stoklund Dittlau et al. 2021). Mesoangioblasts are vessel-associated mesenchymal stem cells that are isolated from adult skeletal muscle tissue (Yucel and Blau 2019). The authors reported several interesting insights pertaining to the impact of the FUS mutation on NMJ formation as well as potential therapeutic strategies to ameliorate the detrimental effects of the mutation. They also demonstrated the spontaneous formation of NMJs between the motor neurons and myotubes under the influence of the chemotactic and volumetric gradients created between the two compartments of the microfluidic device (Stoklund Dittlau et al. 2021).
Among the four neurodegenerative diseases discussed, ALS has the most rapid progression. Therefore, it is imperative that more efforts be concentrated to establish a cure for this debilitating disease. CRISPR-iPSC-based disease modelling approaches, as summarized above, are a positive stride in this front.
Neurodegenerative Diseases: The Big Picture
The deranged genetic and biochemical signatures underlying the pathological and clinical manifestations of a neurodegenerative disease process are highly complex and heterogeneous. However, the overarching pattern of neurodegenerative progression with respect to the pathological markers, molecular players, and disease markers bear interesting congruencies as well as overlaps across most neurodegenerative diseases (Cornblath et al. 2020). For example, TREM2, a microglial receptor encoding gene responsible for stimulating phagocytosis and suppressing proinflammatory cytokine production, has been associated with AD, FTD, PD, and ALS (Lee et al. 2020).
It is a well-established fact that pathological protein aggregation is among the primary drivers of any neurodegenerative disease process. There are, however, multiple concurrently acting mechanisms responsible for causing protein aggregation-mediated cellular dysfunction and degeneration (Davis et al. 2018; Cornblath et al. 2020). For example, the presence of Aβ, tau protein, α-synuclein, and TDP-43 protein aggregates have been reported in a variety of post-mortem brain tissue samples sourced from patients suffering from some form of neurodegeneration. This is a surprising finding as Aβ and tau protein aggregation is a hallmark of AD, α-synuclein is primarily implicated in PD, and TDP-43 mutations are best characterized in connection to ALS and frontotemporal dementia (FTD) (Strong 2012; Cornblath et al. 2020). Interestingly, the notable overlaps in the clinical, genetic, and pathological signatures of FTD and ALS have propagated the interpretation that these two diseases lie on a single spectrum (Ling et al. 2013; Ghosh and Lippa 2015). There is clinical evidence suggesting that a sparse subset of HD patients are susceptible to developing some clinical manifestations of ALS because mHTT may predispose motor neurons to TDP-43 associated pathology and promote the manifestation of some atypical pathological features (Tada et al. 2012). It has even been suggested that tau protein and α-synuclein together constitute a neurodegeneration-promoting deleterious positive feedback loop in which they promote the fibrillization and solubility of each other (Moussaud et al. 2014). Furthermore, in vitro and animal model-based studies suggest that these dysfunctional aggregate-forming proteins mutually interact among themselves, causing unique, concomitant patterns of neurological dysfunction and heterogeneous clinical manifestations of the disease markers (Cornblath et al. 2020). A thorough understanding of the underlying patterns and similarities between all the neurodegenerative disease phenotypes is therefore essential to design holistic, robust, and clinically translatable disease models leveraging the capabilities of the CRISPR/Cas9-mediated genome editing and iPSCs.
The four neurodegenerative diseases and disease modelling approaches covered in depth in the above sections are representative of the overarching trends, patterns, strategies, and emerging approaches in the domain of neurodegenerative disease modelling and research. In the following sections, four distinct groups of neurodegenerative diseases are discussed to further delineate the mechanistically as well as pathologically interrelated and interconnected nature of various neurodegenerative diseases.
Polyglutamine Disorders
HD belongs to a group of disorders known as polyglutamine disorders, all of which are characterized by an expansion of the glutamine-encoding CAG codon in different gene loci (Nóbrega and Pereira de Almeida 2018). In HD, the polyglutamine tract expansion occurs in the HTT gene, which eventually triggers the pathological hallmarks of the disease, as already discussed. Aside from HD, there are eight other polyglutamine disorders that are currently known to exist, which includes spinal and bulbar muscular atrophy (SBMA), dentatorubral-pallidoluysian atrophy, and six variants of spinocerebellar ataxia (SCA) (Banno et al. 2009). Since there are several fundamental similarities between these diseases, many of the disease modelling strategies and developments discussed for HD extend to other polyglutamine disorders as well.
Spinocerebellar ataxias (SCAs) are a collection of neurodegenerative diseases characterized by a progressive loss of balance, coordination, and speech slurring. Over 40 genetically distinct subtypes of SCA have been identified, which include both non-repeat mutations and repeat mutations (Klockgether et al. 2019). Therefore, only a subset of SCAs can be categorized as polyglutamine disorders. Several CRISPR-iPSC-based protocols for the generation of various SCA disease models such as SCA2 (CAG expansion) and SCA12 (CAG/CTG expansion) have surfaced over the years (Marthaler et al. 2016; Feng et al. 2021). However, to date, only one CRISPR-iPSC-based study involving polyglutamine SCAs has been reported for spinocerebellar ataxia type 3 (SCA3) (Ouyang et al. 2018). SCA3 is characterized by a polyglutamine tract expansion in the human ATXN3 gene, which is an important component of the ubiquitin–proteasome protein degradation system (Zeng et al. 2018). In the study, SCA3 patient-derived iPSCs were subjected to a CRISPR/Cas9-mediated knockout of the expanded stretch of CAG repeats in exon 10 of the mutant ATXN3 gene. The gene-corrected iPSCs retained pluripotent status and were subsequently differentiated into neural stem cells and, finally, neural cells. The gene-corrected ATXN3 gene was observed to be able to express functional Ataxin-3 protein, capable of binding ubiquitin (Ouyang et al. 2018).
Spinal and bulbar muscular atrophy (SBMA), also referred to as Kennedy’s disease, is a late-onset, X-linked, neuromuscular condition characterized by progressive lower motor neuron degeneration, primary myopathy, weakness, muscular atrophy, and complex multi-organ involvement resulting in non-neurological features like gynecomastia, testicular atrophy, reduced fertility, and erectile dysfunction (Grunseich et al. 2014; Querin et al. 2018). It is caused by an abnormal polyglutamine tract expansion in the AR (androgen receptor) gene located on the X chromosome. AR requires coregulatory proteins to affect a multitude of downstream signaling pathways responsible for critical cellular functions like chromatin accessibility modulation, transcriptional machinery recruitment, and transactivation enhancement. However, mutant polyglutamine stretch-bearing AR receptors have been observed to cause transcriptional dysregulation resulting in the clinical hallmarks of SBMA. A group recently explored the possibility of using naturally occurring truncated AR isoforms lacking the expanded polyglutamine stretch as a genomic regulatory switch in androgen-responsive tissues (Lim et al. 2021). In the study, the authors identified the most abundantly expressed AR isoform from a pool of nine candidate AR isoforms using RNA-seq data obtained from dihydrotestosterone-treated iPSC-derived motor neurons as well as other cadaver-derived neuronal tissue samples. They also utilized CRISPR/Cas9-mediated genome editing to insert an in-frame FLAG tag 5′ of the isoform encoding gene for immunofluorescence-based validation studies. This study represents a deviation from the general trend of CRISPR-iPSC-based neurodegenerative disease modelling studies, which typically involve studying and contrasting the characteristics of isogenic iPSC-derived, CRISPR/Cas9-edited diseased and disease corrected cell lines for a particular disease phenotype (Lim et al. 2021).
Motor Neuron Disease
Although the terms ALS and motor neuron disease (MND) are often used interchangeably, MND encompasses an entire family of clinical phenotypes characterized by progressive and selective motor neuron degeneration. Among MNDs, ALS is most prevalent in adults, whereas spinal muscular atrophy (SMA) is most prevalent in children (Strong 2012; Bowerman et al. 2018; Mejzini et al. 2019). Although there are some notable differences between the pathophysiological intricacies and progression patterns of ALS and SMA, animal model-based studies have suggested several overarching similarities such as selective motor neuron vulnerability, anomalous motor neuron excitability, and functional collapse of neuromuscular junctions due to motor neuron damage (Comley et al. 2016; Bowerman et al. 2018). Deeper molecular links between ALS and SMA involving the RNAP II/U1 snRNP machinery have also been reported (Chi et al. 2018). Furthermore, SMA is a monogenic disease, whereby aberrations or deletions of just one gene, the SMN1 (survival motor neuron 1) gene, are responsible for over 98% of reported cases (Li et al. 2020). Therefore, SMA is much simpler to model compared to ALS, which is known to be precipitated by a plethora of genetic and environmental factors. Interestingly the SMN1 gene has a paralog, SMN2 (survival motor neuron 2), which is present in all SMA patients. However, the SMN2 gene is unable to compensate for the aberrant SMN1 because it possesses a translationally silent C-to-T transition in its seventh exon, which flips an exonic splicing enhancer into an exonic splicing silencer. Thus, the ensuing alternative splicing causes over 90% of the translated protein to be truncated and hence non-functional (Li et al. 2020). One group reported a CRISPR/Cpf1-based genome editing strategy to convert the offending T in the seventh exon back to a C, thus making the edited SMN2 gene transcriptionally equivalent to the SMN1 gene. The gene correction was carried out in a patient-derived iPSC cell line, which was subsequently differentiated into motor neurons demonstrating significantly rescued SMN protein expression. This was also the first report wherein a group demonstrated efficient CRISPR/Cpf1 homology-directed repair-mediated genome editing in human cells (Zhou et al. 2018). A study published later reported the proof-of-principle of a novel therapeutic strategy to correct SMA involving the utilization of CRISPR/Cas9-mediated splicing-regulatory element disruption of SMA patient-derived iPSCs (Li et al. 2020). The splicing-corrected iPSCs were differentiated into spinal motor neurons, demonstrating appreciable disease phenotype decreases. This was showed by restored SMN protein expression and decreased apoptosis as compared to the unedited iPSC-derived motor neurons functioning as the negative controls (Li et al. 2020).
Neuronal Ceroid Lipofuscinoses
Neuronal ceroid lipofuscinoses (NCL), also referred to as Batten disease, are a collection of neurodegenerative lysosomal storage disorders characterized by an overaccumulation of lipofuscin, a fluorescent mixture of partially digested proteins and lipids, that progressively builds up with age in the lysosomes of various cell types, including neurons (Adler et al. 2015; Tang et al. 2021). Most neurodegenerative diseases tend to be present in adults and develop with advancing senility. However, an exception to this trend is juvenile neuronal ceroid lipofuscinosis (JNCL), the most common form of NCL. Batten disease typically presents between 4 and 7 years of age and is characterized by blindness followed by severe CNS deterioration resulting in epilepsy, motor deficits, cognitive decline, and ultimately death, all within the third decade of life (Gomez-Giro et al. 2019). The vision loss characteristic of JNCL is due to the degeneration of the light-sensing photoreceptor cells present in the outer neural retina, the exact mechanism of which is not well characterized (Gomez-Giro et al. 2019; Tang et al. 2021).
JNCL is caused by mutations in the CLN3 gene. Although the exact function of the CLN3 gene is still not fully understood, it has been observed to be active in the lysosomes and endosomes of a cell, suggesting that it plays a crucial role in interconnecting cytoskeletal dynamics to endocytic membrane trafficking (Mirza et al. 2019). A total of 78 different CLN3 mutations have been reported as per the NCL Mutation and Patient Database (https://www.ucl.ac.uk/ncl/CLN3mutationtable.htm) (Gomez-Giro et al. 2019). Since JNCL is a pediatric neurodegenerative disease, patient tissue samples are particularly difficult to source. Therefore, the requirement for standardized non-patient JNCL disease models is particularly high. A very recent iPSC-based study using patient iPSC-derived retinal pigment epithelium (RPE) disease models has reported the importance of CNL3 gene in photoreceptor cell integrity via photoreceptor outer segment phagocytosis (Tang et al. 2021). A group reported the generation of a JNCL disease model harboring one such pathologic variant (CLN3Q352X) from healthy iPSC lines using CRISPR/Cas9-mediated genome editing (Gomez-Giro et al. 2019). Using the genome-edited iPSC disease models, the authors were able to recapitulate JNCL hallmarks in iPSC-derived endothelial cells. Half of the cerebral organoids generated from the genome-edited iPSCs aborted growth and development before reaching the differentiation endpoint. The organoids which managed to reach the differentiation endpoint demonstrated several pathological markers relevant to the early stages of the disease process. The authors also documented the manifestation of early pathological markers associated with synaptic formation and neurotransmission (Gomez-Giro et al. 2019). Another group reported a successful attempt at correcting a very common 1.02 kb deletion spanning exons 7 and 8 of the CLN3 gene, using a CRISPR/Cas9-based HDR approach in iPSCs derived from homozygous as well as compound heterozygous patients. The attempt was successful, as exemplified by the presence of mutation corrected CLN3 and wildtype CLN3 transcript expression in both the genome-edited iPSC lines (Burnight et al. 2018).
Frontotemporal Dementia
Frontotemporal dementia (FTD) is another example of a group of heterogeneous disorders typically characterized by cognitive impairment impacting the frontal and temporal lobes of the brain, resulting in progressive brain atrophy. Other motor disorders such as supranuclear palsy, parkinsonian disorders, corticobasal syndrome, and MNDs share a number of clinically overlapping features with FTD (Lines et al. 2020). The gene mutations underlying FTD have significant overlaps with the mutations responsible for causing other neurodegenerative conditions as well (Abramzon et al. 2020). For example, over 40 mutations in the MAPT gene, one of the genes implicated in PD pathology, have been linked to FTD (Espay and Litvan 2011; Lines et al. 2020). FTD shares conspicuous similarities with other prominent neurodegenerative conditions as well, making it an interesting yet challenging disorder to treat. To that end, various iPSC and CRISPR-iPSC-based disease models have been developed by different groups over the years.
ALS and FTD are considered two ends of a single spectrum disorder where pure FTD and pure ALS represent two distinct polarities of an otherwise indiscrete continuum (Ling et al. 2013; Ugbode and West 2021). Therefore, CRISPR-iPSC-based disease models aiming to model the deleterious effects of the C9ORF72 mutation in the context of ALS also bear relevance for FTD. A hexanucleotide (GGGGCC) repeat expansion in the first intron of the C9ORF72 gene is a well-established cause for ALS (Braems et al. 2020). The mutation results in the expression of glycine-arginine and proline-arginine dipeptide repeat proteins, which tend to bind to a large number of intracellular proteins, thereby promoting aggregate formation and consequently DNA damage (Babu et al. 2021). A study aiming to better understand the mechanism of dipeptide repeat protein-mediated DNA damage using a CRISPR/iPSC-based C9ORF72 disease model revealed that dipeptide repeat proteins such as proline-arginine repeat proteins compromise the proper functioning DNA double-strand repair pathways by inhibiting the action of key proteins such as nucleophosmin (Andrade et al. 2020).
Mutations in the CHMP2B gene, responsible for encoding the charged multivesicular body protein 2B, have been identified across the entire FTD-ALS spectrum and are responsible for causing FTD-3 (FTD linked to chromosome 3) (Zhang et al. 2017; Ugbode and West 2021). Mutations in the CHMP2B gene, responsible for encoding the charged multivesicular body protein 2B, have been identified across the entire FTD-ALS spectrum. This protein is a core component of the ESCRT (endosomal sorting complexes required for transport) machinery and therefore plays a critical role in orchestrating a cell’s topologically unique membrane budding and scission events (Schmidt and Teis 2012; Ugbode and West 2021). It is in fact a component of the ESCRT-III complex and is therefore directly involved in cell surface receptor maintenance (Zhang et al. 2017). Since mutations of the CHMP2B gene are rare, a deep understanding of the cellular and molecular events underlying FTD-3 is non-existent (Zhang et al. 2017; Ugbode and West 2021). To better understand the same, a group used a CRISPR-iPSC-based approach in which the global gene expression profile of iPSC-derived FTD-3 neurons was compared to their CRISPR/Cas9-edited disease phenotype rescued isogenic controls (Zhang et al. 2017). The diseased neurons exhibited atypical endosomes as well as several mitochondrial defects such as faulty cristae formation, mitochondrial respiration deficiencies, and elevated reactive oxygen species concentration. Furthermore, they observed evidence indicative of perturbed iron homeostasis in the diseased neurons. The authors were able to successfully recapitulate and ablate the disease phenotype-specific histopathological markers in the diseased and disease-rescued cell lines, respectively, thus establishing the validity of the disease model. In this study, not only did the authors develop a CRISPR-iPSC-based disease model capable of recapitulating the disease phenotype of this rare disorder, but they also established a new tool to model intracellular iron accumulation in the context of neurodegenerative diseases (Zhang et al. 2017).
Mutations in the PGRN gene results in the haploinsufficiency of progranulin which accounts for up to 10% of all documented FTD cases (Lines et al. 2020). In fact, the disease phenotype for PGRN mutations manifests in a dose-dependent manner. Patients heterozygous for PGRN mutations experience decreased progranulin expression and are consequently affected by familial FTD. However, patients homozygous for the mutant PGRN gene experience a complete cessation of progranulin expression and consequently experience symptoms corresponding to that of the lysosomal storage disorder, NCL (Smith et al. 2012). One group developed patient-sourced PGRN mutation-bearing iPSC-derived neuronal models to study the effects of progranulin haploinsufficiency in FTD patients (Almeida et al. 2012). Using the model, the authors recapitulated several key markers indicative of the disease phenotype, such as the elevation of cytoplasmic TDP-43 levels, a finding in agreement with past reports (Lines et al. 2020). To better understand how the dose-dependent nature of PGRN mutations contribute to this duality of disease outcome, another group generated FTD patient-sourced PGRN mutation-bearing iPSC-derived human cortical neurons. They also generated a mutation-corrected isogenic control line using CRISPR/Cas9-based genome editing. The authors observed that the PGRN mutant neurons exhibited histopathological changes common to both FTD and NCL pathology such as lipofuscin accumulation, enlarged electron-dense vesicles, and granular osmiophilic deposits. Furthermore, they uncovered evidence suggesting that progranulin and its cleaved product, granulin E, are responsible for the activation of cathepsin D. Cathepsin D is a lysosomal protease capable of degrading a wide range of long-lived substrates, including disease-associated proteins such as α-synuclein, APP, and tau protein. Some variants of the cathepsin D-encoding gene, CTSD, have been linked to increased risk of AD, PD, and NCL (Valdez et al. 2017; Bunk et al. 2021). Therefore, the authors hypothesized that the dose-dependent, PGRN loss-propagated decline in cathepsin D activity compromises normal lysosomal function, a key factor underlying both FTD and NCL pathology (Valdez et al. 2017).
CRISPR-iPSC-based disease modelling approaches are also deepening our understanding of the mechanistic underpinnings of FTD and propelling us closer to a viable medical intervention for this yet incurable disease (Zhang et al. 2017; Jiang et al. 2018; Hofmann et al. 2019; Lines et al. 2020).
Conclusions and Future Perspectives
The immense potential of CRISPR/Cas9-mediated genome editing, iPSCs, and the combination of the two in the context of neurodegenerative disease modelling and therapeutics is undisputed. iPSC-based neurodegenerative disease models have experienced a significant increase in adoption over the past half-decade due to the advantages and convenience it offers over other more traditional disease modelling approaches, as already discussed. With the accumulation of research and the consequent maturation of this domain, the quality of disease models generated by the scientific community at large is improving impressively. For example, when iPSC-based disease modelling was at its nascency, insights derived from diseased iPSCs were often compared to non-isogenic control iPSCs derived from healthy family and/or gender-matched control subjects (Doss and Sachinidis 2019). This soon raised concerns regarding genetic background heterogeneity and its resulting confounding effects on the obtained datasets, prompting a methodological improvement in study design. As a result, most recent studies report the use of isogenic controls in the generated datasets and reported insights.
There are still several challenges in the road ahead. Prolonged in vitro culture can result in iPSCs developing passage-induced mutations by accumulating genetic aberrations such as chromosomal anomalies, genetic instability, copy number variations, and heterozygosity loss. Moreover, pre-existing random mutations extant in a small fraction of the parental somatic cells can get expanded because of the clonal expansion step present in any iPSC generation workflow. There have also been reports of genetic mutations and genomic instability arising in iPSCs as an unintended consequence of the reprogramming process, which raises concerns regarding their tumorigenicity (Yoshihara et al. 2017; Doss and Sachinidis 2019). The immense heterogeneity across different iPSC lines as well as their derivatives in terms of parameters such as differentiation potential, residual epigenetic memory, immunogenicity, tumorigenicity, parental cell line health, lineage commitment preference, batch to batch experimental variability, reprogramming protocol used, differentiation efficiency, and co-occurring contaminating cell populations bring into question valid safety concerns regarding their therapeutic application in a clinical setting (Yoshihara et al. 2017; Paik et al. 2018; Attwood and Edel 2019; Doss and Sachinidis 2019; Huang et al. 2019). Also, the technical and practical limitations in current high-throughput sequencing methodologies may obfuscate oncogenic aberrations in generated iPSC lines (Attwood and Edel 2019).
Like the current distributed consensus with respect to the therapeutic viability of iPSCs, concerns surrounding the safety and efficacy of the CRISPR/Cas9 genome editing platform have also surfaced over the past several years. Concerns regarding potential off-target edits caused by the CRISPR/Cas9 system have been voiced by multiple groups (Zhang et al. 2015; Modell et al. 2022). Other concerns such as in vivo payload delivery, immunogenicity, tissue system targeting, and unwanted genetic mosaicism (due to concurrently occurring cell division and genome editing) also remain prominent matters of contention in the scientific community (Mehta and Merkel 2020; Nishizono et al. 2020; Gohil et al. 2021; Modell et al. 2022).
Attempts to resolve these pertinent issues surrounding both iPSCs and CRISPR/Cas9 technology are currently under active exploration. For example, highly precise surface-marker and fluorescence-based cell sorting approaches to yield pure populations of iPSCs and iPSC-derivatives have been reported by several groups (Lehnen et al. 2017; Arias-Fuenzalida et al. 2017; Schmitt et al. 2017; Hoshina et al. 2018; Patterson et al. 2020; Sriram et al. 2021). Several recent reports have also described promising approaches to improving the efficiency and reliability of the CRISPR/Cas9 system while minimizing the occurrences of unwanted off-target edits. For example, very recently, a group developed a synthetic light-sensitive sgRNA system that fragments on light irradiation, thereby offering scientists more granular spatio-temporal control of the entire genome editing process (Carlson-Stevermer et al. 2020). Another group recently reported an innovative competitive inhibition-based method of shielding off-target sites from the Cas9 endonuclease by co-delivering off-target site-directed short guide RNAs alongside the on-target guide RNA (Coelho et al. 2020). Highly precise single base editor CRISPR/Cas9 systems, an area of active research, now enable the seamless execution of targeted nucleotide edits without the need to introduce double-stranded DNA breaks at the target site. This development considerably improves the efficiency and simplicity of introducing as well as correcting SNPs for disease modelling and therapeutic applications (Cheng et al. 2019; Chen et al. 2021; Li et al. 2021).
Over the past few years, iPSCs have slowly started entering the realm of clinical application. In August 2018, the Kyoto University Hospital conducted the world’s first clinical trial to treat Parkinson’s disease patients using human iPSC-derived dopaminergic progenitor cells. Dopaminergic progenitor cells were derived from clinical-grade iPSC cell lines, subjected to a battery of quality checks and ultimately transplanted into the bilateral putamen of PD patients (Takahashi 2020). On January 14, 2022, the Keio University announced the successful transplantation of human iPSC-derived neural progenitor cells into the participant of a clinical study aiming to treat subacute spinal cord injury (https://www.keio.ac.jp/en/press-releases/2022/Jan/14/49-92092/) (Nagoshi et al. 2020; Sugai et al. 2021). In 2020, a study published a systematic multi-database analysis of iPSC-based clinical trials featuring a total of 131 studies being conducted (Deinsberger et al. 2020). More are expected to start in the coming years. However, clinical trials featuring genome-edited iPSCs are yet to see the light of day. As the protocols for the generation of CRISPR/Cas 9-edited iPSCs are standardized and the current technical issues are resolved, it is reasonable to assume that with time, CRISPR-iPSC-based therapeutic protocols will enter mainstream clinical application.
References
Abramzon YA, Fratta P, Traynor BJ, Chia R (2020) The overlapping genetics of amyotrophic lateral sclerosis and frontotemporal dementia. Front Neurosci 14:42. https://doi.org/10.3389/fnins.2020.00042
Adler L, Boyer NP, Chen C et al (2015) The 11-cis retinal origins of lipofuscin in the retina. In: Progress in Molecular Biology and Translational Science. Elsevier, pp e1–e12
Adli M (2018) The CRISPR tool kit for genome editing and beyond. Nat Commun 9:1911. https://doi.org/10.1038/s41467-018-04252-2
Agrawal A, Narayan G, Gogoi R, Thummer RP (2021) Recent advances in the generation of β-cells from induced pluripotent stem cells as a potential cure for diabetes mellitus. In: Turksen K (ed) Cell Biology and Translational Medicine, vol 14. Stem Cells in Lineage Specific Differentiation and Disease. Springer International Publishing, Cham, pp 1–27
Ahfeldt T, Ordureau A, Bell C et al (2020) Pathogenic pathways in early-onset autosomal recessive Parkinson’s disease discovered using isogenic human dopaminergic neurons. Stem Cell Reports 14:75–90. https://doi.org/10.1016/j.stemcr.2019.12.005
Almeida S, Zhang Z, Coppola G et al (2012) Induced pluripotent stem cell models of progranulin-deficient frontotemporal dementia uncover specific reversible neuronal defects. Cell Rep 2:789–798. https://doi.org/10.1016/j.celrep.2012.09.007
Alok A, Sandhya D, Jogam P et al (2020) The rise of the CRISPR/Cpf1 system for efficient genome editing in plants. Front Plant Sci 11:264. https://doi.org/10.3389/fpls.2020.00264
Amado DA, Davidson BL (2021) Gene therapy for ALS: a review. Mol Ther S1525001621001957. https://doi.org/10.1016/j.ymthe.2021.04.008
An MC, O’Brien RN, Zhang N et al (2014) Polyglutamine disease modeling: epitope based screen for homologous recombination using CRISPR/Cas9 system. PLoS Currents 6. https://doi.org/10.1371/currents.hd.0242d2e7ad72225efa72f6964589369a
An MC, Zhang N, Scott G et al (2012) Genetic correction of Huntington’s disease phenotypes in induced pluripotent stem cells. Cell Stem Cell 11:253–263. https://doi.org/10.1016/j.stem.2012.04.026
Andrade NS, Ramic M, Esanov R et al (2020) Dipeptide repeat proteins inhibit homology-directed DNA double strand break repair in C9ORF72 ALS/FTD. Mol Neurodegeneration 15:13. https://doi.org/10.1186/s13024-020-00365-9
Arias-Fuenzalida J, Jarazo J, Qing X et al (2017) FACS-assisted CRISPR-Cas9 genome editing facilitates Parkinson’s disease modeling. Stem Cell Reports 9:1423–1431. https://doi.org/10.1016/j.stemcr.2017.08.026
Ariga H, Takahashi-Niki K, Kato I et al (2013) Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid Med Cell Longev 2013:e683920. https://doi.org/10.1155/2013/683920
Attwood S, Edel M (2019) iPS-Cell Technology and the problem of genetic instability—can it ever be safe for clinical use? JCM 8:288. https://doi.org/10.3390/jcm8030288
Auluck PK, Caraveo G, Lindquist S (2010) α-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu Rev Cell Dev Biol 26:211–233. https://doi.org/10.1146/annurev.cellbio.042308.113313
Babu M, Favretto F, de Opakua AI et al (2021) Proline/arginine dipeptide repeat polymers derail protein folding in amyotrophic lateral sclerosis. Nat Commun 12:3396. https://doi.org/10.1038/s41467-021-23691-y
Banno H, Katsuno M, Suzuki K et al (2009) Neuropathology and therapeutic intervention in spinal and bulbar muscular atrophy. IJMS 10:1000–1012. https://doi.org/10.3390/ijms10031000
Barber RC (2012) The genetics of Alzheimer’s disease. Scientifica 2012:1–14. https://doi.org/10.6064/2012/246210
Barrangou R, Fremaux C, Deveau H et al (2007) CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. https://doi.org/10.1126/science.1138140
Benam KH, Dauth S, Hassell B et al (2015) Engineered in vitro disease models. Annu Rev Pathol Mech Dis 10:195–262. https://doi.org/10.1146/annurev-pathol-012414-040418
Beylina A, Langston RG, Rosen D et al (2021) Generation of fourteen isogenic cell lines for Parkinson’s disease-associated leucine-rich repeat kinase (LRRK2). Stem Cell Research 53:102354. https://doi.org/10.1016/j.scr.2021.102354
Bilican B, Serio A, Barmada SJ et al (2012) Mutant induced pluripotent stem cell lines recapitulate aspects of TDP-43 proteinopathies and reveal cell-specific vulnerability. Proc Natl Acad Sci 109:5803–5808. https://doi.org/10.1073/pnas.1202922109
Bin Moon S, Lee JM, Kang JG et al (2018) Highly efficient genome editing by CRISPR-Cpf1 using CRISPR RNA with a uridinylate-rich 3′-overhang. Nat Commun 9:3651. https://doi.org/10.1038/s41467-018-06129-w
Birger A, Ben-Dor I, Ottolenghi M et al (2019) Human iPSC-derived astrocytes from ALS patients with mutated C9ORF72 show increased oxidative stress and neurotoxicity. EBioMedicine 50:274–289. https://doi.org/10.1016/j.ebiom.2019.11.026
Bolognin S, Fossépré M, Qing X et al (2019) 3D cultures of Parkinson’s disease-specific dopaminergic neurons for high content phenotyping and drug testing. Adv Sci 6:1800927. https://doi.org/10.1002/advs.201800927
Booth HDE, Hirst WD, Wade-Martins R (2017) The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci 40:358–370. https://doi.org/10.1016/j.tins.2017.04.001
Borgohain MP, Haridhasapavalan KK, Dey C et al (2019) An insight into DNA-free reprogramming approaches to generate integration-free induced pluripotent stem cells for prospective biomedical applications. Stem Cell Rev Rep 15:286–313. https://doi.org/10.1007/s12015-018-9861-6
Bowerman M, Murray LM, Scamps F et al (2018) Pathogenic commonalities between spinal muscular atrophy and amyotrophic lateral sclerosis: converging roads to therapeutic development. Eur J Med Genet 61:685–698. https://doi.org/10.1016/j.ejmg.2017.12.001
Bradshaw AV, Campbell P, Schapira AHV et al (2021) The PINK1—Parkin mitophagy signalling pathway is not functional in peripheral blood mononuclear cells. PLoS ONE 16:e0259903. https://doi.org/10.1371/journal.pone.0259903
Braems E, Swinnen B, Van Den Bosch L (2020) C9orf72 loss-of-function: a trivial, stand-alone or additive mechanism in C9 ALS/FTD? Acta Neuropathol 140:625–643. https://doi.org/10.1007/s00401-020-02214-x
Braems E, Tziortzouda P, Van Den Bosch L (2021) Exploring the alternative: fish, flies and worms as preclinical models for ALS. Neurosci Lett 759:136041. https://doi.org/10.1016/j.neulet.2021.136041
Bunk J, Prieto Huarcaya S, Drobny A et al (2021) Cathepsin D variants associated with neurodegenerative diseases show dysregulated functionality and modified α-synuclein degradation properties. Front Cell Dev Biol 9:581805. https://doi.org/10.3389/fcell.2021.581805
Burk K, Pasterkamp RJ (2019) Disrupted neuronal trafficking in amyotrophic lateral sclerosis. Acta Neuropathol 137:859–877. https://doi.org/10.1007/s00401-019-01964-7
Burnight ER, Bohrer LR, Giacalone JC et al (2018) CRISPR-Cas9-mediated correction of the 1.02 kb common deletion in CLN3 in induced pluripotent stem cells from patients with batten disease. CRISPR J 1:75–87. https://doi.org/10.1089/crispr.2017.0015
Burstein SR, Valsecchi F, Kawamata H et al (2018) In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum Mol Genet 27:160–177. https://doi.org/10.1093/hmg/ddx397
Cappello V, Francolini M (2017) Neuromuscular junction dismantling in amyotrophic lateral sclerosis. IJMS 18:2092. https://doi.org/10.3390/ijms18102092
Carlson-Stevermer J, Kelso R, Kadina A et al (2020) CRISPRoff enables spatio-temporal control of CRISPR editing. Nat Commun 11:5041. https://doi.org/10.1038/s41467-020-18853-3
Carroll CM, Li Y-M (2016) Physiological and pathological roles of the γ-secretase complex. Brain Res Bull 126:199–206. https://doi.org/10.1016/j.brainresbull.2016.04.019
Chen CX-Q, You Z, Abdian N et al (2022) Generation of homozygous PRKN, PINK1 and double PINK1/PRKN knockout cell lines from healthy induced pluripotent stem cells using CRISPR/Cas9 editing. Stem Cell Res 62:102806. https://doi.org/10.1016/j.scr.2022.102806
Chen JJ (2020) Overview of current and emerging therapies for amyotrophic lateral sclerosis. Am J Manag Care 26:S191–S197. https://doi.org/10.37765/ajmc.2020.88483
Chen L, Park JE, Paa P et al (2021) Programmable C: G to G: C genome editing with CRISPR-Cas9-directed base excision repair proteins. Nat Commun 12:1384. https://doi.org/10.1038/s41467-021-21559-9
Chen S, Luo Z, Ward C et al (2020) Generation of two LRRK2 homozygous knockout human induced pluripotent stem cell lines using CRISPR/Cas9. Stem Cell Res 45:101804. https://doi.org/10.1016/j.scr.2020.101804
Cheng T-L, Li S, Yuan B et al (2019) Expanding C-T base editing toolkit with diversified cytidine deaminases. Nat Commun 10:3612. https://doi.org/10.1038/s41467-019-11562-6
Chi B, O’Connell JD, Iocolano AD et al (2018) The neurodegenerative diseases ALS and SMA are linked at the molecular level via the ASC-1 complex. Nucleic Acids Res 46:11939–11951. https://doi.org/10.1093/nar/gky1093
Chinta SJ, Andersen JK (2005) Dopaminergic neurons. Int J Biochem Cell Biol 37:942–946. https://doi.org/10.1016/j.biocel.2004.09.009
Claes C, Danhash EP, Hasselmann J et al (2021) Plaque-associated human microglia accumulate lipid droplets in a chimeric model of Alzheimer’s disease. Mol Neurodegener 16:50. https://doi.org/10.1186/s13024-021-00473-0
Coelho MA, De Braekeleer E, Firth M et al (2020) CRISPR GUARD protects off-target sites from Cas9 nuclease activity using short guide RNAs. Nat Commun 11:4132. https://doi.org/10.1038/s41467-020-17952-5
Comley LH, Nijssen J, Frost-Nylen J, Hedlund E (2016) Cross-disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology: selective Vulnerability in ALS and SMA. J Comp Neurol 524:1424–1442. https://doi.org/10.1002/cne.23917
Cornblath EJ, Robinson JL, Irwin DJ et al (2020) Defining and predicting transdiagnostic categories of neurodegenerative disease. Nat Biomed Eng 4:787–800. https://doi.org/10.1038/s41551-020-0593-y
Crowell V, Houghton R, Tomar A et al (2021) Modeling manifest Huntington’s disease prevalence using diagnosed incidence and survival time. Neuroepidemiology 55:361–368. https://doi.org/10.1159/000516767
Csobonyeiova M, Polak S, Danisovic L (2020) Recent overview of the use of iPSCs Huntington’s disease modeling and therapy. IJMS 21:2239. https://doi.org/10.3390/ijms21062239
Dabrowska M, Ciolak A, Kozlowska E et al (2020) Generation of new isogenic models of Huntington’s disease using CRISPR-Cas9 technology. IJMS 21:1854. https://doi.org/10.3390/ijms21051854
Davis AA, Leyns CEG, Holtzman DM (2018) Intercellular spread of protein aggregates in neurodegenerative disease. Annu Rev Cell Dev Biol 34:545–568. https://doi.org/10.1146/annurev-cellbio-100617-062636
Dawson TM, Golde TE, Lagier-Tourenne C (2018) Animal models of neurodegenerative diseases. Nat Neurosci 21:1370–1379. https://doi.org/10.1038/s41593-018-0236-8
de Oliveira GAP, Silva JL (2019) Alpha-synuclein stepwise aggregation reveals features of an early onset mutation in Parkinson’s disease. Commun Biol 2:374. https://doi.org/10.1038/s42003-019-0598-9
Deinsberger J, Reisinger D, Weber B (2020) Global trends in clinical trials involving pluripotent stem cells: a systematic multi-database analysis. npj Regen Med 5:15. https://doi.org/10.1038/s41536-020-00100-4
Dettmer U, Newman AJ, Soldner F et al (2015) Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat Commun 6:7314. https://doi.org/10.1038/ncomms8314
DeTure MA, Dickson DW (2019) The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegeneration 14:32. https://doi.org/10.1186/s13024-019-0333-5
Devine MJ, Ryten M, Vodicka P et al (2011) Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat Commun 2:440. https://doi.org/10.1038/ncomms1453
Dey C, Narayan G, Krishna Kumar H et al (2017) Cell-penetrating peptides as a tool to deliver biologically active recombinant proteins to generate transgene-free induced pluripotent stem cells. Stud Stem Cells Res Ther 3:006–015. https://doi.org/10.17352/sscrt.000011
Dey C, Raina K, Haridhasapavalan KK et al (2021) Chapter 9 - An overview of reprogramming approaches to derive integration-free induced pluripotent stem cells for prospective biomedical applications. In: Birbrair A (ed) Recent Advances in iPSC Technology. Academic Press, pp 231–287
Dey C, Raina K, Thool M et al (2022) Chapter 2 - Auxiliary pluripotency-associated genes and their contributions in the generation of induced pluripotent stem cells. In: Birbrair A (ed) Molecular Players in iPSC Technology. Academic Press, pp 29–94
di Domenico A, Carola G, Calatayud C et al (2019) Patient-specific iPSC-derived astrocytes contribute to non-cell-autonomous neurodegeneration in Parkinson’s disease. Stem Cell Rep 12:213–229. https://doi.org/10.1016/j.stemcr.2018.12.011
Donowitz M, Turner JR, Verkman AS, Zachos NC (2020) Current and potential future applications of human stem cell models in drug development. J Clin Investig 130:3342–3344. https://doi.org/10.1172/JCI138645
Doss MX, Sachinidis A (2019) Current challenges of iPSC-based disease modeling and therapeutic implications. Cells 8:403. https://doi.org/10.3390/cells8050403
Durães F, Pinto M, Sousa E (2018) Old drugs as new treatments for neurodegenerative diseases. Pharmaceuticals 11:44. https://doi.org/10.3390/ph11020044
Espay AJ, Litvan I (2011) Parkinsonism and frontotemporal dementia: the clinical overlap. J Mol Neurosci 45:343–349. https://doi.org/10.1007/s12031-011-9632-1
Estrada-Cuzcano A, Martin S, Chamova T et al (2017) Loss-of-function mutations in the ATP13A2/PARK9 gene cause complicated hereditary spastic paraplegia (SPG78). Brain 140:287–305. https://doi.org/10.1093/brain/aww307
Fares MB, Jagannath S, Lashuel HA (2021) Reverse engineering Lewy bodies: how far have we come and how far can we go? Nat Rev Neurosci 22:111–131. https://doi.org/10.1038/s41583-020-00416-6
Fauerbach JA, Jovin TM (2018) Pre-aggregation kinetics and intermediates of α-synuclein monitored by the ESIPT probe 7MFE. Eur Biophys J 47:345–362. https://doi.org/10.1007/s00249-017-1272-0
Faustino Martins J-M, Fischer C, Urzi A et al (2020) Self-organizing 3D human trunk neuromuscular organoids. Cell Stem Cell 26:172-186.e6. https://doi.org/10.1016/j.stem.2019.12.007
Feigin VL, Nichols E, Alam T et al (2019) Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18:459–480. https://doi.org/10.1016/S1474-4422(18)30499-X
Feng H, Li Q, Margolis RL, Li PP (2021) Generation of a human induced pluripotent stem cell line JHUi003-A with homozygous mutation for spinocerebellar ataxia type 12 using genome editing. Stem Cell Res 53:102346. https://doi.org/10.1016/j.scr.2021.102346
Gao J, Perera G, Bhadbhade M et al (2019) Autophagy activation promotes clearance of α-synuclein inclusions in fibril-seeded human neural cells. J Biol Chem 294:14241–14256. https://doi.org/10.1074/jbc.RA119.008733
Ghosh S, Lippa CF (2015) Clinical subtypes of frontotemporal dementia. Am J Alzheimers Dis Other Demen 30:653–661. https://doi.org/10.1177/1533317513494442
Giri M, Lü Y, Zhang M (2016) Genes associated with Alzheimer’s disease: an overview and current status. CIA 665. https://doi.org/10.2147/CIA.S105769
Gohil N, Bhattacharjee G, Lam NL et al (2021) CRISPR-Cas systems: challenges and future prospects. In: Progress in Molecular Biology and Translational Science. Elsevier, pp 141–151
Gomez-Giro G, Arias-Fuenzalida J, Jarazo J et al (2019) Synapse alterations precede neuronal damage and storage pathology in a human cerebral organoid model of CLN3-juvenile neuronal ceroid lipofuscinosis. Acta Neuropathol Commun 7:222. https://doi.org/10.1186/s40478-019-0871-7
Govoni S, Gasparini L, Racchi M, Trabucchi M (1996) Peripheral cells as an investigational tool for Alzheimer’s disease. Life Sci 59:461–468. https://doi.org/10.1016/0024-3205(96)00325-6
Grochowska MM, Carreras Mascaro A, Boumeester V et al (2021) LRP10 interacts with SORL1 in the intracellular vesicle trafficking pathway in non-neuronal brain cells and localises to Lewy bodies in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol 142:117–137. https://doi.org/10.1007/s00401-021-02313-3
Grunseich C, Rinaldi C, Fischbeck K (2014) Spinal and bulbar muscular atrophy: pathogenesis and clinical management. Oral Dis 20:6–9. https://doi.org/10.1111/odi.12121
Haavik J, Toska K (1998) Tyrosine hydroxylase and Parkinson’s disease. Mol Neurobiol 16:285–309. https://doi.org/10.1007/BF02741387
Hancock-Cerutti W, Wu Z, Xu P et al (2022) ER-lysosome lipid transfer protein VPS13C/PARK23 prevents aberrant mtDNA-dependent STING signaling. J Cell Biol 221:e202106046. https://doi.org/10.1083/jcb.202106046
Hanss Z, Boussaad I, Jarazo J et al (2020) Quality control strategy for CRISPR-Cas9-based gene editing complicated by a pseudogene. Front Genet 10. https://doi.org/10.3389/fgene.2019.01297
Hargus G, Ehrlich M, Hallmann A-L, Kuhlmann T (2014) Human stem cell models of neurodegeneration: a novel approach to study mechanisms of disease development. Acta Neuropathol 127:151–173. https://doi.org/10.1007/s00401-013-1222-6
Haridhasapavalan KK, Borgohain MP, Dey C et al (2019) An insight into non-integrative gene delivery approaches to generate transgene-free induced pluripotent stem cells. Gene 686:146–159. https://doi.org/10.1016/j.gene.2018.11.069
Haridhasapavalan KK, Raina K, Dey C et al (2020) An insight into reprogramming barriers to iPSC generation. Stem Cell Rev Rep 16:56–81. https://doi.org/10.1007/s12015-019-09931-1
Harjuhaahto S, Rasila TS, Molchanova SM et al (2020) ALS and Parkinson’s disease genes CHCHD10 and CHCHD2 modify synaptic transcriptomes in human iPSC-derived motor neurons. Neurobiol Dis 141:104940. https://doi.org/10.1016/j.nbd.2020.104940
Haston KM, Finkbeiner S (2016) Clinical trials in a dish: the potential of pluripotent stem cells to develop therapies for neurodegenerative diseases. Annu Rev Pharmacol Toxicol 56:489–510. https://doi.org/10.1146/annurev-pharmtox-010715-103548
Heman-Ackah SM, Bassett AR, Wood MJA (2016) Precision modulation of neurodegenerative disease-related gene expression in human iPSC-derived neurons. Sci Rep 6:28420. https://doi.org/10.1038/srep28420
Hernández D, Morgan Schlicht S, Daniszewski M et al (2021) Generation of a gene-corrected human isogenic iPSC line from an Alzheimer’s disease iPSC line carrying the London mutation in APP (V717I). Stem Cell Res 53:102373. https://doi.org/10.1016/j.scr.2021.102373
Hofmann JW, Seeley WW, Huang EJ (2019) RNA binding proteins and the pathogenesis of frontotemporal lobar degeneration. Annu Rev Pathol Mech Dis 14:469–495. https://doi.org/10.1146/annurev-pathmechdis-012418-012955
Hoshina A, Kawamoto T, Sueta S-I et al (2018) Development of new method to enrich human iPSC-derived renal progenitors using cell surface markers. Sci Rep 8:6375. https://doi.org/10.1038/s41598-018-24714-3
Hu X, Mao C, Fan L et al (2020) Modeling Parkinson’s disease using induced pluripotent stem cells. Stem Cells Int 2020:1–15. https://doi.org/10.1155/2020/1061470
Huang C-Y, Liu C-L, Ting C-Y et al (2019) Human iPSC banking: barriers and opportunities. J Biomed Sci 26:87. https://doi.org/10.1186/s12929-019-0578-x
Hung C, Tuck E, Stubbs V et al (2021) SORL1 deficiency in human excitatory neurons causes APP-dependent defects in the endolysosome-autophagy network. Cell Rep 35:109259. https://doi.org/10.1016/j.celrep.2021.109259
Iannuzzi F, Frisardi V, Annunziato L, Matrone C (2021) Might fibroblasts from patients with Alzheimer’s disease reflect the brain pathology? A Focus on the Increased Phosphorylation of Amyloid Precursor Protein Tyr682 Residue. Brain Sci 11:103. https://doi.org/10.3390/brainsci11010103
Inoue K (2021) CRISPR-activated patient fibroblasts for modeling of familial Alzheimer’s disease. Neurosci Res S0168010221000717. https://doi.org/10.1016/j.neures.2021.03.008
Inoue K, Oliveira LMA, Abeliovich A (2017) CRISPR transcriptional activation analysis unmasks an occult γ-secretase processivity defect in familial Alzheimer’s disease skin fibroblasts. Cell Rep 21:1727–1736. https://doi.org/10.1016/j.celrep.2017.10.075
Israel MA, Yuan SH, Bardy C et al (2012) Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482:216–220. https://doi.org/10.1038/nature10821
Jansen R, Embden JDV, Gaastra W, Schouls LM (2002) Identification of genes that are associated with DNA repeats in prokaryotes. Mol Microbiol 43:1565–1575. https://doi.org/10.1046/j.1365-2958.2002.02839.x
Japtok J, Lojewski X, Naumann M et al (2015) Stepwise acquirement of hallmark neuropathology in FUS-ALS iPSC models depends on mutation type and neuronal aging. Neurobiol Dis 82:420–429. https://doi.org/10.1016/j.nbd.2015.07.017
Jiang H, Ren Y, Yuen EY et al (2012) Parkin controls dopamine utilization in human midbrain dopaminergic neurons derived from induced pluripotent stem cells. Nat Commun 3:668. https://doi.org/10.1038/ncomms1669
Jiang S, Wen N, Li Z et al (2018) Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl Psychiatry 8:265. https://doi.org/10.1038/s41398-018-0319-z
Jongh R, Spijkers XM, Pasteuning-Vuhman S et al (2021) Neuromuscular junction-on-a-chip: ALS disease modeling and read-out development in microfluidic devices. J Neurochem 157:393–412. https://doi.org/10.1111/jnc.15289
Kajiwara M, Aoi T, Okita K et al (2012) Donor-dependent variations in hepatic differentiation from human-induced pluripotent stem cells. Proc Natl Acad Sci 109:12538–12543. https://doi.org/10.1073/pnas.1209979109
Kalia LV, Lang AE (2015) Parkinson’s disease. Lancet 386:896–912. https://doi.org/10.1016/S0140-6736(14)61393-3
Ke M, Chong C-M, Su H (2019) Using induced pluripotent stem cells for modeling Parkinson’s disease. WJSC 11:634–649. https://doi.org/10.4252/wjsc.v11.i9.634
Killick R, Ribe EM, Al-Shawi R et al (2014) Clusterin regulates β-amyloid toxicity via Dickkopf-1-driven induction of the wnt–PCP–JNK pathway. Mol Psychiatry 19:88–98. https://doi.org/10.1038/mp.2012.163
Kim BW, Ryu J, Jeong YE et al (2020a) Human motor neurons with SOD1-G93A mutation generated from CRISPR/Cas9 gene-edited iPSCs develop pathological features of amyotrophic lateral sclerosis. Front Cell Neurosci 14:604171. https://doi.org/10.3389/fncel.2020.604171
Kim G, Gautier O, Tassoni-Tsuchida E et al (2020b) ALS genetics: gains, losses, and implications for future therapies. Neuron 108:822–842. https://doi.org/10.1016/j.neuron.2020.08.022
Klein C, Westenberger A (2012) Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2:a008888–a008888. https://doi.org/10.1101/cshperspect.a008888
Klockgether T, Mariotti C, Paulson HL (2019) Spinocerebellar ataxia. Nat Rev Dis Primers 5:24. https://doi.org/10.1038/s41572-019-0074-3
Knupp A, Mishra S, Martinez R et al (2020) Depletion of the AD risk gene SORL1 selectively impairs neuronal endosomal traffic independent of amyloidogenic app processing. Cell Rep 31:107719. https://doi.org/10.1016/j.celrep.2020.107719
Kouroupi G, Taoufik E, Vlachos IS et al (2017) Defective synaptic connectivity and axonal neuropathology in a human iPSC-based model of familial Parkinson’s disease. Proc Natl Acad Sci USA 114:E3679–E3688. https://doi.org/10.1073/pnas.1617259114
Krishnan G, Zhang Y, Gu Y et al (2020) CRISPR deletion of the C9ORF72 promoter in ALS/FTD patient motor neurons abolishes production of dipeptide repeat proteins and rescues neurodegeneration. Acta Neuropathol 140:81–84. https://doi.org/10.1007/s00401-020-02154-6
Lam D, Enright HA, Cadena J et al (2019) Tissue-specific extracellular matrix accelerates the formation of neural networks and communities in a neuron-glia co-culture on a multi-electrode array. Sci Rep 9:4159. https://doi.org/10.1038/s41598-019-40128-1
Lambert E, Saha O, Soares Landeira B et al (2022) The Alzheimer susceptibility gene BIN1 induces isoform-dependent neurotoxicity through early endosome defects. Acta Neuropathol Commun 10:4. https://doi.org/10.1186/s40478-021-01285-5
Langbehn DR, Hayden MR, Paulsen JS, and the PREDICT-HD Investigators of the Huntington Study Group (2010) CAG-repeat length and the age of onset in Huntington disease (HD): a review and validation study of statistical approaches. Am J Med Genet 153B:397–408. https://doi.org/10.1002/ajmg.b.30992
Laverde-Paz MJ, Nuytemans K, Wang L et al (2021) Derivation of stem cell line UMi028-A-2 containing a CRISPR/Cas9 induced Alzheimer’s disease risk variant p.S1038C in the TTC3 gene. Stem Cell Res 52:102258. https://doi.org/10.1016/j.scr.2021.102258
Lee C, Willerth SM, Nygaard HB (2020) The use of patient-derived induced pluripotent stem cells for Alzheimer’s disease modeling. Prog Neurobiol 192:101804. https://doi.org/10.1016/j.pneurobio.2020.101804
Lehnen D, Barral S, Cardoso T et al (2017) IAP-based cell sorting results in homogeneous transplantable dopaminergic precursor cells derived from human pluripotent stem cells. Stem Cell Rep 9:1207–1220. https://doi.org/10.1016/j.stemcr.2017.08.016
Li J, Yu W, Huang S et al (2021) Structure-guided engineering of adenine base editor with minimized RNA off-targeting activity. Nat Commun 12:2287. https://doi.org/10.1038/s41467-021-22519-z
Li J-J, Lin X, Tang C et al (2020) Disruption of splicing-regulatory elements using CRISPR/Cas9 to rescue spinal muscular atrophy in human iPSCs and mice. Natl Sci Rev 7:92–101. https://doi.org/10.1093/nsr/nwz131
Li T, Zhu L, Xiao B et al (2019) CRISPR-Cpf1-mediated genome editing and gene regulation in human cells. Biotechnol Adv 37:21–27. https://doi.org/10.1016/j.biotechadv.2018.10.013
Li JX, Valadez AV, Zuo P, Nie Z (2012) Microfluidic 3D cell culture: potential application for tissue-based bioassays. Bioanalysis 4:1509–1525. https://doi.org/10.4155/bio.12.133
Liguori F, Amadio S, Volonté C (2021) Fly for ALS: Drosophila modeling on the route to amyotrophic lateral sclerosis modifiers. Cell Mol Life Sci. https://doi.org/10.1007/s00018-021-03905-8
Lim WF, Forouhan M, Roberts TC et al (2021) Gene therapy with AR isoform 2 rescues spinal and bulbar muscular atrophy phenotype by modulating AR transcriptional activity. Sci Adv 7:eabi6896. https://doi.org/10.1126/sciadv.abi6896
Lin L, Göke J, Cukuroglu E et al (2016) Molecular features underlying neurodegeneration identified through in vitro modeling of genetically diverse Parkinson’s disease patients. Cell Rep 15:2411–2426. https://doi.org/10.1016/j.celrep.2016.05.022
Lin Y-T, Seo J, Gao F et al (2018) APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98:1141-1154.e7. https://doi.org/10.1016/j.neuron.2018.05.008
Lines G, Casey JM, Preza E, Wray S (2020) Modelling frontotemporal dementia using patient-derived induced pluripotent stem cells. Mol Cell Neurosci 109:103553. https://doi.org/10.1016/j.mcn.2020.103553
Ling S-C, Polymenidou M, Cleveland DW (2013) Converging mechanisms in ALS and FTD: disrupted RNA and protein homeostasis. Neuron 79:416–438. https://doi.org/10.1016/j.neuron.2013.07.033
Liu C-C, Kanekiyo T, Xu H, Bu G (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol 9:106–118. https://doi.org/10.1038/nrneurol.2012.263
Makarova KS, Wolf YI, Alkhnbashi OS et al (2015) An updated evolutionary classification of CRISPR–Cas systems. Nat Rev Microbiol 13:722–736. https://doi.org/10.1038/nrmicro3569
Malankhanova T, Suldina L, Grigor’eva E et al (2020) A human induced pluripotent stem cell-derived isogenic model of Huntington’s disease based on neuronal cells has several relevant phenotypic abnormalities. JPM 10:215. https://doi.org/10.3390/jpm10040215
Marschallinger J, Iram T, Zardeneta M et al (2020) Lipid-droplet-accumulating microglia represent a dysfunctional and proinflammatory state in the aging brain. Nat Neurosci 23:194–208. https://doi.org/10.1038/s41593-019-0566-1
Marthaler AG, Schmid B, Tubsuwan A et al (2016) Generation of an isogenic, gene-corrected control cell line of the spinocerebellar ataxia type 2 patient-derived iPSC line H271. Stem Cell Res 16:180–183. https://doi.org/10.1016/j.scr.2015.12.028
Masrori P, Van Damme P (2020) Amyotrophic lateral sclerosis: a clinical review. Eur J Neurol 27:1918–1929. https://doi.org/10.1111/ene.14393
McColgan P, Tabrizi SJ (2018) Huntington’s disease: a clinical review. Eur J Neurol 25:24–34. https://doi.org/10.1111/ene.13413
Mehta A, Merkel OM (2020) Immunogenicity of Cas9 protein. J Pharm Sci 109:62–67. https://doi.org/10.1016/j.xphs.2019.10.003
Mejzini R, Flynn LL, Pitout IL et al (2019) ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci 13:1310. https://doi.org/10.3389/fnins.2019.01310
Mirza M, Vainshtein A, DiRonza A et al (2019) The CLN3 gene and protein: what we know. Mol Genet Genomic Med 7. https://doi.org/10.1002/mgg3.859
Mitalipov S, Wolf D (2009) Totipotency, pluripotency and nuclear reprogramming. In: Martin U (ed) Engineering of Stem Cells. Springer, Berlin Heidelberg, Berlin, Heidelberg, pp 185–199
Modell AE, Lim D, Nguyen TM et al (2022) CRISPR-based therapeutics: current challenges and future applications. Trends Pharmacol Sci 43:151–161. https://doi.org/10.1016/j.tips.2021.10.012
Moussaud S, Jones DR, Moussaud-Lamodière EL et al (2014) Alpha-synuclein and tau: teammates in neurodegeneration? Mol Neurodegeneration 9:43. https://doi.org/10.1186/1750-1326-9-43
Mullin S, Hughes D, Mehta A, Schapira AHV (2019) Neurological effects of glucocerebrosidase gene mutations. Eur J Neurol 26:388-e29. https://doi.org/10.1111/ene.13837
Nagoshi N, Okano H, Nakamura M (2020) Regenerative therapy for spinal cord injury using iPSC technology. Inflamm Regener 40:40. https://doi.org/10.1186/s41232-020-00149-0
Nguyen HN, Byers B, Cord B et al (2011) LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell 8:267–280. https://doi.org/10.1016/j.stem.2011.01.013
Nguyen HP, Van Broeckhoven C, van der Zee J (2018) ALS genes in the genomic era and their implications for FTD. Trends Genet 34:404–423. https://doi.org/10.1016/j.tig.2018.03.001
Nishizono H, Yasuda R, Laviv T (2020) Methodologies and challenges for CRISPR/Cas9 mediated genome editing of the mammalian brain. Front Genome Ed 2:602970. https://doi.org/10.3389/fgeed.2020.602970
Nóbrega C, Pereira de Almeida L (eds) (2018) Polyglutamine disorders. Springer International Publishing, Cham
Oksanen M, Petersen AJ, Naumenko N et al (2017) PSEN1 mutant iPSC-derived model reveals severe astrocyte pathology in Alzheimer’s disease. Stem Cell Rep 9:1885–1897. https://doi.org/10.1016/j.stemcr.2017.10.016
Oliveira LMA, Falomir-Lockhart LJ, Botelho MG et al (2015) Elevated α-synuclein caused by SNCA gene triplication impairs neuronal differentiation and maturation in Parkinson’s patient-derived induced pluripotent stem cells. Cell Death Dis 6:e1994–e1994. https://doi.org/10.1038/cddis.2015.318
Ortiz-Virumbrales M, Moreno CL, Kruglikov I et al (2017) CRISPR/Cas9-correctable mutation-related molecular and physiological phenotypes in iPSC-derived Alzheimer’s PSEN2 N141I neurons. Acta Neuropathol Commun 5:77. https://doi.org/10.1186/s40478-017-0475-z
Osaki T, Uzel SGM, Kamm RD (2018) Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci Adv 4:eaat5847. https://doi.org/10.1126/sciadv.aat5847
Oura S, Noda T, Morimura N et al (2021) Precise CAG repeat contraction in a Huntington’s disease mouse model is enabled by gene editing with SpCas9-NG. Commun Biol 4:771. https://doi.org/10.1038/s42003-021-02304-w
Ouyang S, Xie Y, Xiong Z et al (2018) CRISPR/Cas9-targeted deletion of polyglutamine in spinocerebellar ataxia type 3-derived induced pluripotent stem cells. Stem Cells Dev 27:756–770. https://doi.org/10.1089/scd.2017.0209
Paff M, Loh A, Sarica C et al (2020) Update on current technologies for deep brain stimulation in Parkinson’s disease. JMD 13:185–198. https://doi.org/10.14802/jmd.20052
Paik DT, Tian L, Lee J et al (2018) Large-scale single-cell RNA-Seq reveals molecular signatures of heterogeneous populations of human induced pluripotent stem cell-derived endothelial cells. Circ Res 123:443–450. https://doi.org/10.1161/CIRCRESAHA.118.312913
Patterson K, Linask KL, Beers J, Zou J (2020) Generation of two tdTomato reporter induced pluripotent stem cell lines (NHLBIi003-A-1 and NHLBIi003-A-2) by AAVS1 safe harbor gene-editing. Stem Cell Research 42:101673. https://doi.org/10.1016/j.scr.2019.101673
Penney J, Ralvenius WT, Tsai L-H (2020) Modeling Alzheimer’s disease with iPSC-derived brain cells. Mol Psychiatry 25:148–167. https://doi.org/10.1038/s41380-019-0468-3
Pereira JD, DuBreuil DM, Devlin A-C et al (2021) Human sensorimotor organoids derived from healthy and amyotrophic lateral sclerosis stem cells form neuromuscular junctions. Nat Commun 12:4744. https://doi.org/10.1038/s41467-021-24776-4
Perez RG (2020) Editorial: The protein alpha-synuclein: its normal role (in neurons) and its role in disease. Front Neurosci 14:116. https://doi.org/10.3389/fnins.2020.00116
Poewe W, Seppi K, Tanner CM et al (2017) Parkinson disease. Nat Rev Dis Primers 3:17013. https://doi.org/10.1038/nrdp.2017.13
Poon A, Saini H, Sethi S et al (2021) The role of SQSTM1 (p62) in mitochondrial function and clearance in human cortical neurons. Stem Cell Rep 16:1276–1289. https://doi.org/10.1016/j.stemcr.2021.03.030
Qing X, Walter J, Jarazo J et al (2017) CRISPR/Cas9 and piggyBac-mediated footprint-free LRRK2-G2019S knock-in reveals neuronal complexity phenotypes and α-Synuclein modulation in dopaminergic neurons. Stem Cell Res 24:44–50. https://doi.org/10.1016/j.scr.2017.08.013
Querin G, Bede P, Marchand-Pauvert V, Pradat P-F (2018) Biomarkers of spinal and bulbar muscle atrophy (SBMA): a comprehensive review. Front Neurol 9:844. https://doi.org/10.3389/fneur.2018.00844
Ray A, Joshi JM, Sundaravadivelu PK et al (2021) An overview on promising somatic cell sources utilized for the efficient generation of induced pluripotent stem cells. Stem Cell Rev Rep. https://doi.org/10.1007/s12015-021-10200-3
Robbins JP, Perfect L, Ribe EM et al (2018) Clusterin is required for β-amyloid toxicity in human iPSC-derived neurons. Front Neurosci 12. https://doi.org/10.3389/fnins.2018.00504
Rodríguez Cruz PM, Cossins J, Beeson D, Vincent A (2020) The neuromuscular junction in health and disease: molecular mechanisms governing synaptic formation and homeostasis. Front Mol Neurosci 13:610964. https://doi.org/10.3389/fnmol.2020.610964
Roos RA (2010) Huntington’s disease: a clinical review. Orphanet J Rare Dis 5:40. https://doi.org/10.1186/1750-1172-5-40
Ross CA, Tabrizi SJ (2011) Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10:83–98. https://doi.org/10.1016/S1474-4422(10)70245-3
Ruggeri FS, Flagmeier P, Kumita JR et al (2020) The influence of pathogenic mutations in α-synuclein on biophysical and structural characteristics of amyloid fibrils. ACS Nano 14:5213–5222. https://doi.org/10.1021/acsnano.9b09676
Ryan SD, Dolatabadi N, Chan SF et al (2013) Isogenic human iPSC Parkinson’s model shows nitrosative stress-induced dysfunction in MEF2-PGC1α transcription. Cell 155:1351–1364. https://doi.org/10.1016/j.cell.2013.11.009
Safari F, Zare K, Negahdaripour M et al (2019) CRISPR Cpf1 proteins: structure, function and implications for genome editing. Cell Biosci 9:36. https://doi.org/10.1186/s13578-019-0298-7
Saha B, Borgohain MP, Dey C, Thummer RP (2018a) iPS cell generation: current and future challenges. Annals Stem Cell Res Ther 1:4
Saha B, Krishna Kumar H, Borgohain MP, Thummer RP (2018b) Prospective applications of induced pluripotent stem cells in military medicine. Med J Armed Forces India 74:313–320. https://doi.org/10.1016/j.mjafi.2018.03.005
Sánchez-Danés A, Richaud-Patin Y, Carballo-Carbajal I et al (2012) Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol Med 4:380–395. https://doi.org/10.1002/emmm.201200215
Santos R, Vadodaria KC, Jaeger BN et al (2017) Differentiation of inflammation-responsive astrocytes from glial progenitors generated from human induced pluripotent stem cells. Stem Cell Rep 8:1757–1769. https://doi.org/10.1016/j.stemcr.2017.05.011
Sareen D, O’Rourke JG, Meera P et al (2013) Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci Transl Med 5:208ra149–208ra149. https://doi.org/10.1126/scitranslmed.3007529
Schmidt O, Teis D (2012) The ESCRT machinery. Curr Biol 22:R116–R120. https://doi.org/10.1016/j.cub.2012.01.028
Schmitt CE, Morales BM, Schmitz EMH et al (2017) Fluorescent tagged episomals for stoichiometric induced pluripotent stem cell reprogramming. Stem Cell Res Ther 8:132. https://doi.org/10.1186/s13287-017-0581-7
Schulte J, Littleton JT (2011) The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr Trends Neurol 5:65–78. https://doi.org/10.1038/nrn1806
Seirafi M, Kozlov G, Gehring K (2015) Parkin structure and function. FEBS J 282:2076–2088. https://doi.org/10.1111/febs.13249
Selkoe DJ, Peter J Lansbury J (1999) Alzheimer’s disease is the most common neurodegenerative disorder. Basic Neurochemistry: Molecular, Cellular and Medical Aspects 6th edition
Seminary ER, Sison SL, Ebert AD (2018) Modeling protein aggregation and the heat shock response in ALS iPSC-derived motor neurons. Front Neurosci 12:86. https://doi.org/10.3389/fnins.2018.00086
Shi C, Mao C, Zhang S et al (2016) CHCHD2 gene mutations in familial and sporadic Parkinson’s disease. Neurobiol Aging 38:217.e9-217.e13. https://doi.org/10.1016/j.neurobiolaging.2015.10.040
Shin JW, Kim K-H, Chao MJ et al (2016) Permanent inactivation of Huntington’s disease mutation by personalized allele-specific CRISPR/Cas9. Hum Mol Genet ddw286. https://doi.org/10.1093/hmg/ddw286
Shulman JM, De Jager PL, Feany MB (2011) Parkinson’s disease: genetics and pathogenesis. Annu Rev Pathol Mech Dis 6:193–222. https://doi.org/10.1146/annurev-pathol-011110-130242
Sidransky E, Lopez G (2012) The link between the GBA gene and parkinsonism. Lancet Neurol 11:986–998. https://doi.org/10.1016/S1474-4422(12)70190-4
Siller R, Greenhough S, Park I-H, Sullivan GJ (2013) Modelling human disease with pluripotent stem cells. CGT 13:99–110. https://doi.org/10.2174/1566523211313020004
Singh PK, Muqit MMK (2020) Parkinson’s: a disease of aberrant vesicle trafficking. Annu Rev Cell Dev Biol 36:237–264. https://doi.org/10.1146/annurev-cellbio-100818-125512
Singh VK, Kalsan M, Kumar N et al (2015) Induced pluripotent stem cells: applications in regenerative medicine, disease modeling, and drug discovery. Front Cell Dev Biol 3. https://doi.org/10.3389/fcell.2015.00002
Smith KR, Damiano J, Franceschetti S et al (2012) Strikingly different clinicopathological phenotypes determined by progranulin-mutation dosage. Am J Hum Genet 90:1102–1107. https://doi.org/10.1016/j.ajhg.2012.04.021
Soldner F, Hockemeyer D, Beard C et al (2009) Parkinson’s disease patient-derived induced pluripotent stem cells free of viral reprogramming factors. Cell 136:964–977. https://doi.org/10.1016/j.cell.2009.02.013
Soldner F, Stelzer Y, Shivalila CS et al (2016) Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature 533:95–99. https://doi.org/10.1038/nature17939
Sonninen T-M, Hämäläinen RH, Koskuvi M et al (2020) Metabolic alterations in Parkinson’s disease astrocytes. Sci Rep 10:14474. https://doi.org/10.1038/s41598-020-71329-8
Spataro R, Kousi M, Farhan SMK et al (2019) Mutations in ATP13A2 (PARK9) are associated with an amyotrophic lateral sclerosis-like phenotype, implicating this locus in further phenotypic expansion. Hum Genomics 13:19. https://doi.org/10.1186/s40246-019-0203-9
Sriram S, Kang N-Y, Subramanian S et al (2021) Novel live cell fluorescent probe for human-induced pluripotent stem cells highlights early reprogramming population. Stem Cell Res Ther 12:113. https://doi.org/10.1186/s13287-021-02171-6
Srivastava T, Raj R, Dubey A et al (2020) Fast kinetics of environmentally induced α-synuclein aggregation mediated by structural alteration in NAC region and result in structure dependent cytotoxicity. Sci Rep 10:18412. https://doi.org/10.1038/s41598-020-75361-6
Stefanis L (2012) α-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 2:a009399–a009399. https://doi.org/10.1101/cshperspect.a009399
Stoklund Dittlau K, Krasnow EN, Fumagalli L et al (2021) Human motor units in microfluidic devices are impaired by FUS mutations and improved by HDAC6 inhibition. Stem Cell Rep S2213671121001600. https://doi.org/10.1016/j.stemcr.2021.03.029
Strong MJ (ed) (2012) Amyotrophic lateral sclerosis and the frontotemporal dementias. Oxford University Press
Sugai K, Sumida M, Shofuda T et al (2021) First-in-human clinical trial of transplantation of iPSC-derived NS/PCs in subacute complete spinal cord injury: study protocol. Regen Ther 18:321–333. https://doi.org/10.1016/j.reth.2021.08.005
Sundaravadivelu PK, Raina K, Thool M et al (2021) Tissue-restricted stem cells as starting cell source for efficient generation of pluripotent stem cells: an overview. Advances in Experimental Medicine and Biology. Springer International Publishing, Cham, pp 1–30
Tada M, Coon EA, Osmand AP et al (2012) Coexistence of Huntington’s disease and amyotrophic lateral sclerosis: a clinicopathologic study. Acta Neuropathol 124:749–760. https://doi.org/10.1007/s00401-012-1005-5
Takahashi J (2020) iPS cell-based therapy for Parkinson’s disease: a Kyoto trial. Regen Ther 13:18–22. https://doi.org/10.1016/j.reth.2020.06.002
Takahashi K, Tanabe K, Ohnuki M et al (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872. https://doi.org/10.1016/j.cell.2007.11.019
Takahashi K, Yamanaka S (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676. https://doi.org/10.1016/j.cell.2006.07.024
Tang C, Han J, Dalvi S et al (2021) A human model of batten disease shows role of CLN3 in phagocytosis at the photoreceptor–RPE interface. Commun Biol 4:161. https://doi.org/10.1038/s42003-021-01682-5
Tang S, Xie M, Cao N, Ding S (2016) Patient-specific induced pluripotent stem cells for disease modeling and phenotypic drug discovery: miniperspective. J Med Chem 59:2–15. https://doi.org/10.1021/acs.jmedchem.5b00789
Tian R, Gachechiladze MA, Ludwig CH et al (2019) CRISPR interference-based platform for multimodal genetic screens in human iPSC-derived neurons. Neuron 104:239-255.e12. https://doi.org/10.1016/j.neuron.2019.07.014
Tran J, Anastacio H, Bardy C (2020) Genetic predispositions of Parkinson’s disease revealed in patient-derived brain cells. npj Parkinsons Dis 6:8. https://doi.org/10.1038/s41531-020-0110-8
Trapecar M, Wogram E, Svoboda D et al (2021) Human physiomimetic model integrating microphysiological systems of the gut, liver, and brain for studies of neurodegenerative diseases. Sci Adv 7:eabd1707. https://doi.org/10.1126/sciadv.abd1707
Überbacher C (2019) Application of CRISPR/Cas9 editing and digital droplet PCR in human iPSCs to generate novel knock-in reporter lines to visualize dopaminergic neurons. Stem Cell Res 10. https://doi.org/10.1016/j.scr.2019.101656
Ugbode C, West RJH (2021) Lessons learned from CHMP2B, implications for frontotemporal dementia and amyotrophic lateral sclerosis. Neurobiol Dis 147:105144. https://doi.org/10.1016/j.nbd.2020.105144
Valadez-Barba V, Cota-Coronado A, Hernández-Pérez OR et al (2020) iPSC for modeling neurodegenerative disorders. Regen Ther 15:332–339. https://doi.org/10.1016/j.reth.2020.11.006
Valdez C, Wong YC, Schwake M et al (2017) Progranulin-mediated deficiency of cathepsin D results in FTD and NCL-like phenotypes in neurons derived from FTD patients. Hum Mol Genet 26:4861–4872. https://doi.org/10.1093/hmg/ddx364
van Es MA, Hardiman O, Chio A et al (2017) Amyotrophic lateral sclerosis. Lancet 390:2084–2098. https://doi.org/10.1016/S0140-6736(17)31287-4
Vázquez-Vélez GE, Zoghbi HY (2021) Parkinson’s disease genetics and pathophysiology. Annu Rev Neurosci 44:87–108. https://doi.org/10.1146/annurev-neuro-100720-034518
Verma A, Verma M, Singh A (2020) Animal tissue culture principles and applications. In: Animal Biotechnology. Elsevier, pp 269–293
Vethe H, Bjørlykke Y, Ghila LM et al (2017) Probing the missing mature β-cell proteomic landscape in differentiating patient iPSC-derived cells. Sci Rep 7:4780. https://doi.org/10.1038/s41598-017-04979-w
Wang C, Najm R, Xu Q et al (2018) Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med 24:647–657. https://doi.org/10.1038/s41591-018-0004-z
Wang C, Zhang M, Garcia G et al (2021) ApoE-isoform-dependent SARS-CoV-2 neurotropism and cellular response. Cell Stem Cell 28:331-342.e5. https://doi.org/10.1016/j.stem.2020.12.018
Wang L, Yi F, Fu L et al (2017) CRISPR/Cas9-mediated targeted gene correction in amyotrophic lateral sclerosis patient iPSCs. Protein Cell 8:365–378. https://doi.org/10.1007/s13238-017-0397-3
Wang T, Zhang J, Liao J et al (2020) Donor genetic backgrounds contribute to the functional heterogeneity of stem cells and clinical outcomes. Stem Cells Transl Med 9:1495–1499. https://doi.org/10.1002/sctm.20-0155
White RB, Thomas MG (2012) Moving beyond tyrosine hydroxylase to define dopaminergic neurons for use in cell replacement therapies for Parkinson’s disease. CNSNDDT 11:340–349. https://doi.org/10.2174/187152712800792758
Wördehoff MM, Bannach O, Shaykhalishahi H et al (2015) Single fibril growth kinetics of α-synuclein. J Mol Biol 427:1428–1435. https://doi.org/10.1016/j.jmb.2015.01.020
Wu L, Rosa-Neto P, Hsiung G-YR et al (2012) Early-onset familial Alzheimer’s disease (EOFAD). Can J Neurol Sci 39:436–445. https://doi.org/10.1017/S0317167100013949
Xiao Y, Zhang J, Shu X et al (2020) Loss of mitochondrial protein CHCHD10 in skeletal muscle causes neuromuscular junction impairment. Hum Mol Genet 29:1784–1796. https://doi.org/10.1093/hmg/ddz154
Xu X, Tay Y, Sim B et al (2017) Reversal of phenotypic abnormalities by CRISPR/Cas9-mediated gene correction in Huntington disease patient-derived induced pluripotent stem cells. Stem Cell Rep 8:619–633. https://doi.org/10.1016/j.stemcr.2017.01.022
Yagi T, Ito D, Okada Y et al (2011) Modeling familial Alzheimer’s disease with induced pluripotent stem cells. Hum Mol Genet 20:4530–4539. https://doi.org/10.1093/hmg/ddr394
Ye T, Duan Y, Tsang HWS et al (2021) Efficient manipulation of gene dosage in human iPSCs using CRISPR/Cas9 nickases. Commun Biol 4:195. https://doi.org/10.1038/s42003-021-01722-0
Yin R-H, Yu J-T, Tan L (2015) The role of SORL1 in Alzheimer’s disease. Mol Neurobiol 51:909–918. https://doi.org/10.1007/s12035-014-8742-5
Yoshihara M, Hayashizaki Y, Murakawa Y (2017) Genomic instability of iPSCs: challenges towards their clinical applications. Stem Cell Rev Rep 13:7–16. https://doi.org/10.1007/s12015-016-9680-6
Young W (2012) Patient-specific induced pluripotent stem cells as a platform for disease modeling, drug discovery and precision personalized medicine. J Stem Cell Res Ther 1. https://doi.org/10.4172/2157-7633.S10-010
Yu J, Vodyanik MA, Smuga-Otto K et al (2007) Induced pluripotent stem cell lines derived from human somatic cells. Science 318:1917–1920. https://doi.org/10.1126/science.1151526
Yu J-T, Tan L (2012) The role of clusterin in Alzheimer’s disease: pathways, pathogenesis, and therapy. Mol Neurobiol 45:314–326. https://doi.org/10.1007/s12035-012-8237-1
Yucel N, Blau HM (2019) Skeletal muscle stem cells. In: Principles of Regenerative Medicine. Elsevier, pp 273–293
Yun Y, Hong S-A, Kim K-K et al (2020) CRISPR-mediated gene correction links the ATP7A M1311V mutations with amyotrophic lateral sclerosis pathogenesis in one individual. Commun Biol 3:33. https://doi.org/10.1038/s42003-020-0755-1
Yusa K (2013) Seamless genome editing in human pluripotent stem cells using custom endonuclease–based gene targeting and the piggyBac transposon. Nat Protoc 8:2061–2078. https://doi.org/10.1038/nprot.2013.126
Zeng L, Zhang D, McLoughlin HS et al (2018) Loss of the Spinocerebellar Ataxia type 3 disease protein ATXN3 alters transcription of multiple signal transduction pathways. PLoS ONE 13:e0204438. https://doi.org/10.1371/journal.pone.0204438
Zhang N, An MC, Montoro D, Ellerby LM (2010) Characterization of human Huntington’s disease cell model from induced pluripotent stem cells. PLoS Curr 2. https://doi.org/10.1371/currents.RRN1193
Zhang X-H, Tee LY, Wang X-G et al (2015) Off-target effects in CRISPR/Cas9-mediated genome engineering. Mol Ther - Nucleic Acids 4:e264. https://doi.org/10.1038/mtna.2015.37
Zhang Y, Schmid B, Nikolaisen NK et al (2017) Patient iPSC-derived neurons for disease modeling of frontotemporal dementia with mutation in CHMP2B. Stem Cell Rep 8:648–658. https://doi.org/10.1016/j.stemcr.2017.01.012
Zhou M, Hu Z, Qiu L et al (2018) Seamless genetic conversion of SMN2 to SMN1 via CRISPR/Cpf1 and single-stranded oligodeoxynucleotides in spinal muscular atrophy patient-specific induced pluripotent stem cells. Hum Gene Ther 29:1252–1263. https://doi.org/10.1089/hum.2017.255
Acknowledgements
This work was supported by the North Eastern Region – Biotechnology Programme Management Cell (NERBPMC), Department of Biotechnology, Government of India (BT/PR16655/NER/95/132/2015), and MHRD fellowship (TS). The figure was created using BioRender.com.
Author information
Authors and Affiliations
Contributions
TS and RPT were responsible for conception and design, collection and/or assembly of data, data analysis and interpretation, manuscript writing, and final approval of the manuscript. All the authors gave consent for publication.
Corresponding author
Ethics declarations
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sen, T., Thummer, R.P. CRISPR and iPSCs: Recent Developments and Future Perspectives in Neurodegenerative Disease Modelling, Research, and Therapeutics. Neurotox Res 40, 1597–1623 (2022). https://doi.org/10.1007/s12640-022-00564-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-022-00564-w