
Abstract
In this study, the prevalence of different enteric viruses in commercial mussels was evaluated at the retail level in three European countries (Finland, Greece and Spain). A total of 153 mussel samples from different origins were analysed for human norovirus (NoV) genogroups I and II, hepatitis A virus (HAV) and hepatitis E virus (HEV). Human adenovirus (HAdV) was also tested as an indicator of human faecal contamination. A full set of controls (such as sample process control, internal amplification controls, and positive and negative controls) were implemented during the process. The use of a sample process control allowed us to calculate the efficiencies of extraction, which ranged from 79 to 0.5 %, with an average value of 10 %. Samples were positive in 41 % of cases, with HAdV being the most prevalent virus detected (36 %), but no significant correlation was found between the presence of HAdV and human NoV, HAV and HEV. The prevalences of human norovirus genogroup II, HEV and human NoV genogroup I were 16, 3 and 0.7 %, respectively, and HAV was not detected. The estimated number of PCR detectable units varied between 24 and 1.4 × 103 g−1 of digestive tract. Interestingly, there appeared to be a significant association between the type of mussel species (M. galloprovincialis) and the positive result of samples, although a complete overlap between country and species examined required this finding to be confirmed including samples of both species from all possible countries of origin.
Similar content being viewed by others

Introduction
Due to their filtre-feeding nature, bivalve molluscs tend to accumulate human pathogens (Rippey 1994) in their stomach and their digestive glands (Schwab et al. 1998; Rodríguez-Lázaro et al., in press). In one study, Lees (2000) observed that shellfish grown in sewage polluted waters tend to bioaccumulate environmentally stable enteric viruses, such as norovirus (NoV), hepatitis A virus (HAV) and enterovirus (EV). Processing interventions such as depuration do not completely eliminate viral particles (Loisy et al. 2005; Schwab et al. 1998) and the habit of eating bivalve mollusks raw or slightly cooked increases the health risk related to shellfish consumption (Butt et al. 2004; Rippey 1994).
For the detection of enteric viruses in shellfish, molecular methods such as reverse transcription-polymerase chain reaction (RT-PCR) are widely used (Le Guyader et al. 2000; Bosch et al. 2011). However, the low quantity of virus in environmental samples such as shellfish renders them a difficult and variable matrix that is also known to cause amplification inhibition (Lowther et al. 2008) increasing the risk of false negative results. For this reason, effective preliminary sample treatment steps such as elution and concentration of viruses from the shellfish tissue and RNA extraction and purification are essential for final PCR accuracy and reproducibility (Le Guyader et al. 2000). To overcome those issues the utilization of several controls throughout the process is necessary (Rodríguez-Lázaro et al. 2007, in press; Bosch et al. 2011; D’Agostino et al. 2011). Sample process controls (SPCV) and internal amplification controls (IAC) must be used to verify the accuracy of the results obtained (D’Agostino et al. 2011; Diez-Valcarce et al. 2011a, b). An SPCV is used to verify whether the sample treatment has operated correctly and also allow us to estimate the efficiency of extraction for each individual sample analysed. The IAC is used to monitor the possible inhibition of the reaction due to inhibitory compounds in the sample, avoiding any false negative interpretation of the analysis.
The increasing amount of data on virus detection in shellfish (Le Guyader et al. 2000.; Formiga-Cruz et al. 2002; Myrmel et al. 2004; Croci et al. 2007) and shellfish-borne viral outbreaks (Svraka et al. 2007; Le Guyader et al. 2008; Vilariño et al. 2009; Pintó et al. 2009; Baker et al. 2011) points out the necessity of a constant surveillance system in European countries. The European FP7 project VITAL (Integrated Monitoring and Control of Foodborne Viruses in European Food Supply Chains), which ran from 2008 to 2011, aimed to gather data on virus contamination of food sources for quantitative viral risk assessment and development of virus-specific guidance for food supply chain operators. In this project, different European laboratories have investigated the shellfish supply chain for NoVGI, NoVGII, HAV and hepatitis E virus (HEV). Human adenovirus (HAdV) was also investigated to demonstrate the potential existence of a route of viral faecal contamination from human sources to the sampling point within the food supply chain. In this study a survey was performed to acquire information on viral prevalence in mussels across Europe at the retail level. Methods used have been previously validated through ring trials in order to have comparable quantitative data (D’Agostino et al. 2012).
Materials and Methods
Sampling Strategy
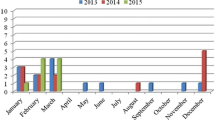
This study was conducted in three European countries (Spain, Greece and Finland) during the period of summer–winter 2010 (from May to December). Mussel species collected were Mytilus galloprovincialis in Spain and Greece (102 samples) and Mytilus edulis in Finland (51 samples). The origin of samples was also different: in Finland they were imported from Denmark; in Spain, all samples were locally collected in the Galicia region; while in Greece, samples were imported from Chile (40 samples), New Zealand (5 samples), Spain (4 samples) and also collected locally (2 samples).
In each country, a total of 51 mussel samples were taken at local retail stores during ten independent sampling times separated by at least 1 week. On each sampling occasion, five mussels (six on one sampling occasion) were randomly selected for subsequent analysis. One hundred and two samples were purchased fresh and 51 were purchased frozen, all samples being cultured mussels. The sampling plan was developed on a rationale assuming that if a batch of mussels was contaminated, it was likely that the growing waters were contaminated and that a large proportion of the batch would carry at least 1 virus particle. With the detection system used in this study we were able to detect contamination in retail stores with 95 % confidence when 50 % or more of the mussels were contaminated. This strategy increases the probability of detecting virus when low virus concentrations were expected since extraction and inhibition controls were used, and analyses were performed in duplicate and in serial dilutions of nucleic acids. Aiming for most accurate estimates of prevalence given the total fixed number of samples, the priority was to detect the virus when 50 % or more of the samples in a batch were contaminated.
Sample Process Control Virus
The SPCV was murine norovirus 1 (MNV-1) (Diez-Valcarce et al. 2011b), which had been propagated in RAW264.7 cells to a concentration of 108 plaque forming units (pfu) ml−1. MNV-1 stocks were kindly provided by the group of Dr. Franco Ruggeri at the Istituto Superiore de Sanità, Rome, Italy by agreement with Washington University, St. Louis, MO, USA.
Positive Controls
Positive controls were nucleic acids extracted from the target viruses or chimerical standards provided in the project (Martínez-Martínez et al. 2011). Nucleic acid sequences of these chimerical standards were identical to the sequence of the target viruses.
Virus Concentration and Extraction from Shellfish
Mussels were selected and any mud from the shell was washed off using tap water. The sample was then processed using the method of Henshilwood et al. (2003). Briefly, one shellfish was placed on a rubber shucking block and the shells opened with a clean shucking knife. The digestive gland was dissected out aseptically using scissors and forceps (or equivalent tools), transferred to a clean Petri dish, and chopped finely with a razor blade to homogenate the sample. The chopped glands were then weighed, and transferred into a centrifuge tube. The SPCV (10 μl; ca. 106 pfu) was added. One ml of 3 U ml−1 proteinase K solution (prepared in molecular grade water) was added and mixed well. The sample was incubated at 37 °C in a shaking incubator or equivalent for 60 min, ensuring that the speed setting for the shaker induced continual gentle movement of the enzyme/gland mixture. A second incubation was carried out by placing the tube in a water bath at 65 °C for 15 min with shaking. The sample was then centrifuged at 3,000×g for 5 min, and 500 μl of supernatant was transferred to a clean microcentrifuge tube and was immediately used for nucleic acid extraction or stored at −20 °C. Nucleic acids (500 μl) were extracted using a NucliSENS® miniMAG® kit (bioMérieux) according to the manufacturer’s instructions. The final elution was performed twice with 150 μl elution buffer, resulting in a 300 μl nucleic acid extract. The nucleic acid extract was assayed immediately or stored at −70 °C.
Detection of Viruses
The presence of enteric pathogenic viruses—HEV, HAV, NoVGI and NoVGII—were evaluated using reverse transcription real-time PCR (RT-qPCR). Detection of SPCV was also conducted by RT-qPCR. In addition, the presence of HAdV was also evaluated using real-time PCR (qPCR) in Spain and Greece. In all the cases, a neat and a 10-fold dilution of the virus nucleic acid extract were tested; all samples were tested in duplicate (two neat and two diluted). An internal amplification control (IAC) (Diez-Valcarce et al. 2011a) and its probe labelled with VIC (50 nM) were included in every assay.
All RT-qPCR assays were performed using the RNA Ultrasense reaction mix (Invitrogen), the qPCR assays were performed using TaqMan Universal PCR Master Mix (Applied Biosystems) and a carry-over contamination prevention system, uracil N-glycosylase. In each assay, 10 μl sample of nucleic acid extract was added, to make a final reaction volume of 20 μl, except in case of HAdV in which the final reaction volume was 25 μl. All oligonucleotides were purchased from MWG Biotech AG (Ebersberg, Germany) except the minor-groove binder (MGB) TaqMan probes HAV150(-) and MGB-ORF1/2 that were acquired from Applied Biosystems (Warrington, UK) and NV1LCpr that was acquired from Sigma-Aldrich (St. Louis, MO, USA). Virus assays were performed using the oligonucleotides and the conditions described in Table 1.
Extraction and Theoretic Efficiencies
The SPCV was employed as a control of the virus concentration and nucleic acid extraction. Prior to virus recovery from the mussel homogenates, the samples were spiked with a known quantity (ca. 106 pfu) of MNV-1. Viral RNA extracted from mussels was tested for target viruses undiluted and 10-fold diluted to evaluate the effect of potential qRT-PCR inhibitors. If MNV-1 signal was negative for a sample, it was retested from the beginning due to the PCR inhibition or the sample inhibition of the process. The extraction efficiency value was calculated by comparing the C q value (quantification cycle, previously known as the threshold cycle) for the 10-fold dilution of MNV-1 (not extracted) with that obtained for the SPCV in the tested samples. The result was classified as poor (extraction efficiency <1 %), acceptable (1–10 %), or good (>10 %) (da Silva et al. 2007).
The theoretic efficiency was calculated by comparing the C q value of a mussel sample containing the control (SPCV) with the C q value of the SPCV alone, just spiked in the reagents used for concentration and extraction of the sample but without any matrix (chopped mussel); the formula used was: \( 2^{{C_{\text{q}} \;{\text{SPCV}} - C_{\text{q}} \;{\text{sample}}}} \times 100 \). This efficiency was also classified in the same three categories (poor, acceptable and good).
Reporting and Interpretation of Data
For a proper interpretation of the results, four different signals were assayed: The target virus, the SPCV control, the target IAC and the SPCV IAC (D’Agostino et al. 2011). When at least one of the two replicate targets (for HAV, HEV, NoVGI, NoVGII and HAdV) was detected, these mussel samples were considered to be positive. Twelve of the 153 (7.8 %) samples were inhibited when neat samples were assayed, but diluted samples amplified for the target. When an assay showed a C q value ≤45, independently of the corresponding IAC C q value, the result was interpreted as positive. When an assay showed no C q value for the target with the corresponding IAC C q value ≤45 and at least one of the four replicates of MNV-1 (two neat and two diluted) assayed positive, the result was interpreted as negative. When an assay showed both the target and its corresponding IAC C q values absent, the reaction was considered to have failed.
Virus Quantification
The number of viruses per gram of mussel tissue was estimated using the most probable number-like approach (Teunis et al. 2005; De Roda Husman et al. 2009). Presence/absence profiles for target viruses were generated per mussel by examining neat and serial 10-fold dilutions of nucleic extracts of samples until the end-point dilution, in duplicate. It was assumed that viruses, if present, were distributed homogeneously in samples. The unit of quantification was a PCR detectable unit (PDU), which represents an unknown number of target genomes (under ideal amplification conditions and a perfect assay, a single PDU would represent a single virus genome).
Statistical Analysis
Statistical analysis was performed by Pearson’s Chi-square test to test the significance between various categorical variables: mussel species and presence rate and fresh or frozen mussel and presence rate. p < 0.05 was considered significant and p < 0.001, highly significant. Odds ratios were also calculated. Statistical analysis was performed by using SPSS software version 17 (SPSS Inc., Chicago, IL, USA).
Results
Efficiencies of Extraction
The mean virus extraction efficiency of the process was 10 % with a standard deviation of 22. Values ranged from 79 to 0.5 %, and the mean virus theoretic efficiency was 6 % with a standard deviation of 15, with values ranging from 51 to 0.3 %. Overall: 92 % of the samples showed acceptable or good extraction efficiency (45 and 47 %, respectively) and only 8 % showed poor extraction efficiency (<1 %). Similarly, most of the samples (88 %) showed acceptable or good theoretic efficiency (55 and 33 %, respectively) and only 8 % showed poor theoretic efficiency (<1 %).
Detection of Viruses
Enteric viruses were detected in 41 % of the tested samples (62/153): only one type of enteric virus was detected in 38 % of samples (58/153), and two types of enteric viruses were detected in 3 % (4/153) of the samples (Table 2). HAdV was the most prevalent virus, detected in 36 % of the samples (37/102), followed by NoVGII (16 %; 25/153), HEV (3 %; 3/102) and NoVGI (0.7 %; 1/153). However, HAV was not found in any of the samples analysed. Interestingly, none of the samples tested positive for HAdV was positive for any of the other human pathogenic viruses, indicating no significant correlation between the presence on HAdV and any of the pathogenic viruses studied. No significant differences were observed in the sensitivity of the assays among the three laboratories, since previous ring trials tests were done in the laboratories involved in the study to overcome these possible issues before the actual study was performed. The most likely estimates for PDU concentrations ranged between 24 and 1.4 × 103 g−1 of mussel tissue for NoV GII, between 127 and 348 for HEV and was estimated to be ~260 for NoV GI (Table 3).
A high percentage of the 102 M. galloprovincialis were positive for enteric viruses compared to the 51 M. edulis (Table 2). There was a highly significant association between the type of mussel species and the analytical outcome of the sample (p < 0.001): a sample was ~25 times more likely to be positive if the shellfish species was M. galloprovincialis than if it was M. edulis. Among the 102 fresh mussels 25 % (25/102), 6 % (3/51), 3 % (3/102) and 0.98 % (1/102) were positive for NoVGII, HAdV, HEV and NoVGI, respectively, whereas only HAdV was detected in 34 samples (67 %) of the frozen mussels samples (Table 4). Therefore, no significant association was found between the storage conditions of the mussels and whether or not samples were positive (p > 0.05).
Discussion
The results obtained showed that 41 % (62/153) of samples were contaminated with at least one of the enteric viruses studied. This percentage rose up to 59 % (60/102) if we consider only Mytillus galloprovincialis species. In studies conducted in countries close to those of this study, the prevalence of enteric viruses varies from around 15 % for NoV (Terio et al. 2010), 34.4 % for NoV (Suffredini et al. 2011) in Italy, 4.5 % for NoVGI in Turkey (Yilmaz et al. 2010), 6.8 % for NoV and 18.6 % for HAdV (Myrmel et al. 2004) in Norway and 37 % for NoV and 33 % for HAV in Portugal (Mesquita et al. 2011). Factors such as decreased shellfish activity at lower temperature and differential retention of viruses by distinct mollusc species cannot be overlooked (Lees 2000). M. galloprovincialis was harvested in areas from Spain, Greece, Chile and New Zealand, whereas M. edulis was harvested in Denmark, so factors such as mussel species may have influence the final prevalence observed. Despite the highly significant association between the mussel species and the analytical outcome, the origin of the mussels can also play a part in this association, more samples of both species and from all different origins would be required to more deeply understand this association. A similar result was found regarding the storage conditions of samples: we found pathogenic viruses only in fresh purchased mussels whereas all frozen samples were negative for the pathogenic viruses analyzed (Table 4). But as all those negative samples were from the same species (M. edulis), more samples of different species also stored frozen are needed to get any conclusion about the possible effect of freezing in elimination of pathogenic viruses in shellfish.
The effectiveness of monitoring programmes based on bacteriological indicators such as Escherichia coli to determine the sanitary quality of molluscs and their harvesting areas (Council Directive 91/492/EEC, EC Reg No 854/2004) has been previously questioned (Mesquita et al. 2011; Silva et al. 2010). Consequently, we evaluated the use of HAdV as indicator of faecal contamination and to link its presence to that of other enteric pathogenic viruses such as human NoV as previously suggested (Silva et al. 2011; Wyn-Jones et al. 2011). Our results show that HAdV was the virus most frequently detected (36 %; 37/102). This could indicate that the shellfish, independently of the species and the country of origin, were in contact with waters polluted with human faeces during their production. However, there was not a direct relationship between the presence of HAdV and the detection of the pathogenic viruses assayed (NoV, HAV and HEV), this finding being in accordance with previous results (Myrmel et al. 2004).
An interesting result from our study is the total absence of HAV in the tested samples. Shellfish is considered a main route of contamination for enteric viruses (Rodíguez-Lázaro et al., in press), but HAV is not as commonly detected as NoV (Vilariño et al. 2009). Rotaviruses and astroviruses were also analysed in the molluscs collected in Spain in this study, but none of the samples were positive (data not shown), similar to other studies (Vilariño et al. 2009). One explanation may be that the bioaccumulation of NoVs is not only based on passive filtration but also an active process of fixation on shellfish tissues (Maalouf et al. 2011; Le Guyader et al. 2006).
Simultaneous presence of different viruses or virus strains could lead to more severe symptoms, the occurrence of two episodes of the same or different diseases, and also potentially facilitate emergence of new recombinant strains (Lees 2000). In this study, the simultaneous presence of two or more enteric viruses was found in four samples (3 %), but only one (0.7 %), was contaminated with both human NoV genogroups (NoVGI and NoVGII). Interestingly, the possibility has also been suggested that coexistence of NoV genogroups in an outbreak could be a good indicator for a shellfish-related origin of the outbreak (Hamano et al. 2005). However, due to the lack of information on potential outbreaks originated from the batches of samples analysed in the current study, this hypothesis cannot be corroborated here. No actions were taken when positive samples were found since this was out of the scope of this study, and no current legislation applies for enteric viruses in shellfish.
In conclusion, this study provides relevant information on the presence of potentially pathogenic enteric viruses in shellfish, especially NoVGII. Regarding the potential value of HAdV as indicator virus in routine screening, there was no significant correlation between the viral indicator HAdV and the target viruses.
References
Baert, L., Wobus, C. E., Van Coillie, E., Thackray, L. B., Debevere, J., & Uyttendaele, M. (2008). Detection of murine norovirus 1 by using plaque assay, transfection assay, and real-time reverse transcription-PCR before and after heat exposure. Applied and Environmental Microbiology, 74(2), 543–546.
Baker, K., Morris, J., McCarthy, N., Saldana, L., Lowther, J., Collinson, A., et al. (2011). An outbreak of norovirus infection linked to oyster consumption at a UK restaurant, February 2010. Journal of Public Health (Oxford), 33(2), 205–211.
Bosch, A., Sánchez, G., Abbaszadegan, M., Carducci, A., Guix, S., Le Guyader, F. S., et al. (2011). Analytical methods for virus detection in water and food. Food Analytical Methods, 4(1), 4–12.
Butt, A. A., Aldridge, K. E., & Sanders, C. V. (2004). Infections related to the ingestion of seafood Part I: Viral and bacterial infections. The Lancet Infectious Diseases, 4(4), 201–212.
Costafreda, M. I., Bosch, A., & Pintó, R. M. (2006). Development, evaluation, and standardization of a real-time TaqMan reverse transcription-PCR assay for quantification of hepatitis A virus in clinical and shellfish samples. Applied and Environmental Microbiology, 72(6), 3846–3855.
Croci, L., Losio, M. N., Suffredini, E., Pavoni, E., Di Pasquale, S., Fallacara, F., et al. (2007). Assessment of human enteric viruses in shellfish from the northern Adriatic sea. International Journal of Food Microbiology, 114(2), 252–257.
D’Agostino, M., Cook, N., Di Bartolo, I., Ruggeri, F. M., Martelli, F., Banks, M., et al. (2012). Multicenter collaborative trial evaluation of a method for detection of human adenoviruses in berry fruit. Food Analytical Methods, 5(1), 1–7.
D’Agostino, M., Cook, N., Rodríguez-Lázaro, D., & Rutjes, S. (2011). Nucleic acid amplification-based methods for detection of enteric viruses: Definition of controls and interpretation of results. Food and Environmental Virology, 3(2), 55–60.
da Silva, A. K., Le Saux, J. C., Parnaudeau, S., Pommepuy, M., Elimelech, M., & Le Guyader, F. S. (2007). Evaluation of removal of noroviruses during wastewater treatment, using real-time reverse transcription-PCR: different behaviors of genogroups I and II. Applied and Environmental Microbiology, 73(24), 7891–7897.
De Roda Husman, A. M., Lodder, W. J., Rutjes, S. A., Schijven, J. F., & Teunis, P. F. (2009). Long-term inactivation study of three enteroviruses in artificial surface and groundwaters, using PCR and cell culture. Applied and Environmental Microbiology, 75(4), 1050–1057.
Diez-Valcarce, M., Cook, N., Hernández, M., & Rodríguez-Lázaro, D. (2011a). Analytical application of a sample process control in detection of foodborne viruses. Food Analytical Methods, 4(4), 614–618.
Diez-Valcarce, M., Kovac, K., Cook, N., Rodríguez-Lázaro, D., & Hernández, M. (2011b). Construction and analytical application of internal amplification controls (IAC) for detection of foodborne viruses by (RT) real-time PCR. Food Analytical Methods, 4(3), 437–445.
EU. 1991. Council Directive 91/492/EEC of 15 July 1991 laying down the health conditions for the production and the placing on the market of live bivalve mollusks. Official Journal L268, 1–14.
EU. 2004. Commission regulation (EC) No. 854/2004 of 29 April 2004 laying down specific rules for the organisation of official controls on products of animal origin intended for human consumption. Official Journal L139, 83–127.
Formiga-Cruz, M., Tofiño-Quesada, G., Bofill-Mas, S., Lees, D. N., Henshilwood, K., Allard, A. K., et al. (2002). Distribution of human virus contamination in shellfish from different growing areas in Greece, Spain, Sweden, and the United Kingdom. Applied and Environmental Microbiology, 68(12), 5990–5998.
Hamano, M., Kuzuya, M., Fujii, R., Ogura, H., & Yamada, M. (2005). Epidemiology of acute gastroenteritis outbreaks caused by noroviruses in Okayama. Japan Journal of Medical Virology, 77(2), 282–289.
Henshilwood, K., Dore, W. J., Anderson, S., & Lees, D. N. (2003). Investigation of Norwalk like virus elimination during depuration using a real time quantitative PCR. In A. Villalba, J. L. Romalde, B. Reura, & R. Beiras (Eds.), Molluscan shellfish safety (pp. 451–465). Paris: UNESCO.
Hernroth, B. E., Conden-Hansson, A. C., Rehnstam-Holm, A. S., Girones, R., & Allard, A. K. (2002). Environmental factors influencing human viral pathogens and their potential indicator organisms in the blue mussel, Mytilus edulis: the first Scandinavian report. Applied and Environmental Microbiology, 68(9), 4523–4533.
Jothikumar, N., Cromeans, T. L., Robertson, B. H., Meng, X. J., & Hill, V. R. (2006). A broadly reactive one-step real-time RT-PCR assay for rapid and sensitive detection of hepatitis E virus. Journal of Virological Methods, 131(1), 65–71.
Le Guyader, F., Haugarreau, L., Miossec, L., Dubois, E., & Pommepuy, M. (2000). Three-year study to assess human enteric viruses in shellfish. Applied Environmental Microbiology, 66(8), 3241–3248.
Le Guyader, F. S., Le Saux, J. C., Ambert-Balay, K., Krol, J., Serais, O., Parnaudeau, S., et al. (2008). Aichi virus, norovirus, astrovirus, enterovirus, and rotavirus involved in clinical cases from a French oyster-related gastroenteritis outbreak. Journal of Clinical Microbiology, 46(12), 4011–4017.
Le Guyader, F., Loisy, F., Atmar, R. L., Hutson, A. M., Estes, M. K., Ruvoën-Clouet, N., et al. (2006). Norwalk virus-specific binding to oyster digestive tissues. Emerging Infectious Diseases, 12(6), 931–936.
Lees, D. (2000). Viruses and bivalve shellfish. International Journal of Food Microbiology, 59(1–2), 81–116.
Loisy, F., Atmar, R. L., Guillon, P., Le Cann, P., Pommepuy, M., & Le Guyader, F. S. (2005). Real-time RT-PCR for norovirus screening in shellfish. Journal of Virological Methods, 123(1), 1–7.
Lowther, J. A., Henshilwood, K., & Lees, D. N. (2008). Determination of norovirus contamination in oysters from two commercial harvesting areas over an extended period, using semiquantitative real-time reverse transcription PCR. Journal of Food Protection, 71(7), 1427–1433.
Maalouf, H., Schaeffer, J., Parnaudeau, S., Le Pendu, J., Atmar, R. L., Crawford, S. E., et al. (2011). Strain-dependent norovirus bioaccumulation in oysters. Applied and Environmental Microbiology, 77(10), 3189–3196.
Martínez-Martínez, M., Diez-Valcarce, M., Hernández, M., & Rodríguez-Lázaro, D. (2011). Design and application of nucleic acid standards for quantitative detection of enteric viruses by real-time PCR. Food and Environmental Virology, 3(2), 92–98.
Mesquita, J. R., Vaz, L., Cerqueira, S., Castilho, F., Santos, R., Monteiro, S., et al. (2011). Norovirus, hepatitis A virus and enterovirus presence in shellfish from high quality harvesting areas in Portugal. Food Microbiology, 28(5), 936–941.
Myrmel, M., Berg, E. M., Rimstad, E., & Grinde, B. (2004). Detection of enteric viruses in shellfish from the Norwegian coast. Applied and Environmental Microbiology, 70(5), 2678–2684.
Pintó, R. M., Costafreda, M. I., & Bosch, A. (2009). Risk assessment in shellfish-borne outbreaks of hepatitis A. Applied and Environmental Microbiology, 75(23), 7350–7355.
Rippey, S. R. (1994). Infectious diseases associated with molluscan shellfish consumption. Clinical Microbiology Reviews, 7(4), 419–425.
Rodríguez-Lázaro, D., Cook, N., Ruggeri, FM., Sellwood, J., Nasser, A., Nascimento, MS., D’Agostino, M., Santos, R., Saiz, JC., Rzeżutka, A., Bosch, A., Gironés, R., Carducci, A., Muscillo, M., Kovač, K., Diez-Valcarce, M., Vantarakis, A., von Bonsdorff, CH., de Roda Husman, A. M., Hernández, M., van der Poel, WH (2011). Virus hazards from food, water and other contaminated environments. FEMS Microbiology Reviews. doi:10.1111/j.1574-6976.2011.00306.x.
Rodríguez-Lázaro, D., Lombard, B., Smith, H., Rzezutka, A., D’Agostino, M., Helmuth, R., et al. (2007). Trends in analytical methodology in food safety and quality: monitoring microorganisms and genetically modified organisms. Trends in Food Science & Technology, 18(6), 306–319.
Schwab, K. J., Neill, F. H., Estes, M. K., Metcalf, T. G., & Atmar, R. L. (1998). Distribution of Norwalk virus within shellfish following bioaccumulation and subsequent depuration by detection using RT-PCR. Journal of Food Protection, 61(12), 1674–1680.
Silva, H. D., García-Zapata, M. T. A., & Anunciação, C. E. (2011). Why the use of adenoviruses as water quality virologic marker? Food and Environmental Virology, 3(3-4), 138–140.
Silva, A. M., Vieira, H., Martins, N., Granja, A. T., Vale, M. J., & Vale, F. F. (2010). Viral and bacterial contamination in recreational waters: a case study in the Lisbon bay area. Journal of Applied Microbiology, 108(3), 1023–1031.
Suffredini, E., Pepe, T., Ventrone, I., & Croci, L. (2011). Norovirus infection in shellfish using two Real-Time PCR methods. New Microbiologica, 34(1), 9-16
Svraka, S., Duizer, E., Vennema, H., de Bruin, E., van der Veer, B., Dorresteijn, B., et al. (2007). Etiological role of viruses in outbreaks of acute gastroenteritis in The Netherlands from 1994 through 2005. Journal of Clinical Microbiology, 45(5), 1389–1394.
Terio, V., Martella, V., Moschidou, P., Di Pinto, P., Tantillo, G., & Buonavoglia, C. (2010). Norovirus in retail shellfish. Food Microbiology, 27(1), 29–32.
Teunis, P. F. M., Lodder, W. J., Heisterkamp, S. H., & de Roda Husman, A. M. (2005). Mixed plaques: statistical evidence how plaque assays may underestimate virus concentrations. Water Research, 39(17), 4240–4250.
Vilariño, M. L., Le Guyader, F. S., Polo, D., Schaeffer, J., Kröl, J., & Romalde, J. L. (2009). Assessment of human enteric viruses in cultured and wild bivalve molluscs. International Microbiology, 12(3), 145–151.
Wyn-Jones, A. P., Carducci, A., Cook, N., D’Agostino, M., Diviza, M., Fleischer, J., et al. (2011). Surveillance of adenoviruses and noroviruses in European recreational waters. Water Research, 45(3), 1025–1038.
Yilmaz, H., Bostan, K., Turan, N., Muratoglu, K., Yilmaz, A., Ozkul, A., et al. (2010). Real-time PCR detection of norovirus in mussels collected from the Bosphorus in Istanbul, Turkey. Food and Environmental Virology, 2(2), 64–68.
Acknowledgments
This work was supported by the European Commission Framework Program 7 project “Integrated monitoring and control of foodborne viruses in European food supply chains (VITAL)” (Grant No. KBBE 213178) led by the coordination team of Nigel Cook (FERA, UK), Martin D’Agostino (FERA, UK) and Franco M. Ruggeri (ISS, Italy). MD-V received a Ph.D. studentship from the Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA), Spanish Ministry of Science and Innovation.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Marta Diez-Valcarce, Petros Kokkinos and Kirsi Söderberg contributed equally to the work.
Rights and permissions
About this article
Cite this article
Diez-Valcarce, M., Kokkinos, P., Söderberg, K. et al. Occurrence of Human Enteric Viruses in Commercial Mussels at Retail Level in Three European Countries. Food Environ Virol 4, 73–80 (2012). https://doi.org/10.1007/s12560-012-9078-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12560-012-9078-9