
Abstract
Introduction
From previous studies of pharmacodynamic data in mice, rats, beagle dogs and mini pigs, frequently in direct comparison to induction doses of propofol, ciprofol produced a rapid onset of anesthesia/sedation.
Methods
A phase 1 study suggested potential clinical advantages of ciprofol as a sedation/anesthetic agent, with no evidence of drug-related toxicity. However, the sedation effects and safety of ciprofol in intensive care unit (ICU) patients with mechanical ventilation should be further confirmed in a phase 3 study with a larger cohort of patients. During a phase 3, non-inferiority, multicenter, single-blind, randomized, propofol controlled trial, Chinese ICU patients undergoing mechanical ventilation and requiring endotracheal intubation will be sedated for 6–24 h after randomization. Considering a success rate for ICU sedation of 99% for ciprofol and the positive control drug propofol, a total sample size of 120 subjects with mechanical ventilation will be required to achieve 80% power to determine non-inferiority with a margin of 8%. Finally, taking into account 10% losses, 135 patients will be enrolled and randomly assigned to ciprofol (90 cases) and propofol (45 cases) groups in a 2:1 ratio. The primary outcome will be the success rate of sedation satisfied by the following conditions: the time within the range of Richmond Agitation and Sedation Score (+ 1 ~ − 2) must account for ≥ 70% of the study drug administration time and without other rescue treatments. Secondary outcomes will include the average time to reach the sedation goal, study drug usage, rescue medication given per unit weight, extubation time, recovery time to full consciousness and nursing scores. Safety endpoints will include adverse events (AEs), drug related AEs and serious AEs.
Planned Outcomes
The results of this study will provide crucial information on the use of ciprofol for sedation of patients in ICUs.
Trial Registration
ClinicalTrials.gov identifier, NCT04620031.
Similar content being viewed by others


Ciprofol is a new anesthetic 2,6-disubstituted phenolic derivative, which binds to the gamma-aminobutyric acid-A (GABAA) receptor as does propofol. |
Ciprofol has a stronger affinity for the GABAA receptor and produces greater GABAA-mediated cell currents in vitro. |
A phase 1 study suggested potential clinical advantages of ciprofol as a sedation/anesthetic agent, with no evidence of drug-related toxicity. |
Ciprofol might be effective as anesthetic agent for Chinese intensive care unit patients receiving mechanical ventilation requiring endotracheal intubation. |
Introduction
Analgesia and sedation are important procedures in intensive care units (ICUs), which can eliminate or reduce patient pain and physical discomfort and reduce inappropriate adverse stimulation and excessive excitation of the sympathetic nervous system during mechanical ventilation [1]. These procedures can also assist and improve patient sleep [2], induce amnesia and reduce or eliminate patients' memories of their pain during ICU treatment [3]. In addition, they reduce or eliminate anxiety and agitation thereby minimizing patients' unconscious behaviors (such as struggling) [4, 5] by intervening in their treatment. For example, agitation was observed in 36% of patients undergoing invasive mechanical ventilation and with high Richmond Agitation Sedation Scale (RASS), and the days on invasive mechanical ventilation were associated with agitation [6]. Despite pain and agitation, delirium is also prevalent in the ICU and associated with negative clinical outcomes, higher mortality rates, more complications, prolonged durations of mechanical ventilation and extended stays in the ICU [7]. The experience of stress, discomfort, pain and even delirium caused by invasive mechanical ventilation often occurs in ICU patients [8, 9]. To reduce pain, agitation and delirium, the search for a more appropriate anesthetic regimen has been the goal of clinical practice. It has been reported that benzodiazepine sedation drugs are associated with a longer ICU length of stay and prolonged dependence on mechanical ventilation compared to nonbenzodiazepine alternatives (i.e., propofol and dexmedetomidine) [10], but propofol also can produce a dose-dependent fall in blood pressure (BP), particularly in frail or elderly patients, and pain at the site of injection of propofol is a common adverse reaction that increases tension and anxiety in patients, which directly or indirectly affects the stability of the sedation regime [11, 12]. Ciprofol (HSK3486) is a new anesthetic 2,6-disubstituted phenolic derivative that binds to the gamma-aminobutyric acid-A (GABAA) receptor as does propofol [13]. However, compared to propofol, ciprofol has a stronger affinity for the GABAA receptor and produces greater GABAA-mediated cell currents in vitro. A phase 1 clinical trial [14] in Australia on healthy subjects reported that the tolerability and sedation effects of ciprofol were similar to those of propofol.
Recently, the findings of ciprofol effects in a phase 1 study in ICU patients (NCT04145596) showed that ciprofol as a 4- or 12-h infusion had good efficacy, rapid recovery, no significant accumulation and an excellent safety profile. A phase 2 ICU study (NCT04147416) revealed that 26 patients received satisfactory sedation over an average administration time of 11.25 h. The sedation success rate was 100% with low incidences of hypotension and bradycardia, rapid recovery and little influence on triglyceride concentrations during the period from drug withdrawal to extubation.
In the present study, based on the results of previous phase 1 and 2 clinical studies in ICU patients, we aim to confirm the efficacy and safety of ciprofol in patients undergoing mechanical ventilation and light sedation (RASS + 1 ~ − 2), with the aims of reducing the mechanical ventilation time and the duration of stays in ICU. We found in the previous study (NCT04147416) that the advantages of ciprofol applications are compared to propofol's lower lipid concentrations in infusions leading to lower blood lipid levels, and we hypothesize that this will lead to fewer adverse effects such as infusion syndrome in patients who require prolonged sedation, for example.
Methods
Study Design
This will be a multicenter, randomized, single-blind, propofol controlled non-inferiority study (ClinicalTrials.gov identifier: NCT04620031), which will involve 20 centers/hospitals. The study will be conducted in accordance with the clinical trial protocols (and any amendments), the Declaration of Helsinki (as currently revised), Chinese adult ICU analgesic and sedative treatment guidelines and the Clinical Practice Guidelines for the Management of Pain, Agitation and Delirium in Adult Patients in the ICU [15, 16].
Study Setting
Patients will be enrolled and treated in the ICUs of 27 centers/hospitals in China including: (1) The First Affiliated Hospital of Sun Yat-Sen University, (2) Zhongda Hospital Affiliated to Southeast University and (3) The West China Hospital of Sichuan University and others. For more detailed information, see Supplementary File 1.
Eligible patients according to the inclusion and exclusion criteria will be enrolled and randomly assigned to receive either ciprofol- or propofol-based intravenous sedation while undergoing mechanical ventilation in an ICU.
Inclusion Criteria
-
1.
Patients receiving endotracheal intubation and mechanical ventilation who will require sedation for 6 ~ 24 h after randomization;
-
2.
The desired sedation goal of a patient will be within the range of RASS (+ 1 ~ − 2);
-
3.
18 years old ≤ age < 80 years old, no gender restriction;
-
4.
18 kg/m2 ≤ BMI ≤ 30 kg/m2;
-
5.
Patients or their family members will have a full understanding of the purpose and significance of the trial, voluntarily participate and sign informed consent.
Exclusion Criteria
-
1.
Patients with known allergies to eggs, soy products or propofol as well as patients with contraindications to propofol, opioids and their relief drugs;
-
2.
Patients who will have received sedation for > 3 days in a general ward before being transferred to the ICU or were in an ICU before signing informed consent;
-
3.
Had medical histories or evidence that they were at increased risk during sedation/anesthesia in the screening period;
-
4.
Pregnant and lactating women;
-
5.
Participated in other clinical trials within 1 month prior to screening
-
6.
The investigators decide for various reasons that the patients are not eligible for inclusion in the clinical trial.
Randomization and Blinding
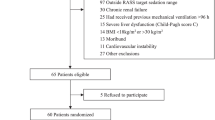
With an estimated success rate of ICU sedation of 99% for the study drug ciprofol and the positive control drug propofol, a total sample size of 120 patients receiving mechanical ventilation in the ICU will be required to achieve 80% power to determine non-inferiority, with a margin of 8%. Taking into account the possible withdrawal of 10% of patients, the sample size will be rounded up to 135 patients who will be randomly assigned at a 2:1 ratio to test and control groups, with about 90 patients in the ciprofol group and 45 patients in the propofol group. Before initiation of the study, an independent investigator who will have no contact with any participant will use SAS 9.4 software (SPSS Inc., Chicago, IL, USA) to generate a blocked random number table (block size = 5) to divide eligible participants in a 2:1 ratio into either the test drug group (ciprofol) or the control group (propofol). The study is designed as single blinded but only for the patients; due to ICU patients always having severe conditions, it can be difficult to blind the investigators. However, the study evaluator will be set as blind; both patients and investigator(s) will not be allowed to communicate with each other about study drug information. Moreover, the blind study evaluators will mainly be in charge of the decision about when to start drug administration, dose adjustment trends and the time to the end of drug administration. They will provide timely corresponding information to the non-blind study investigator to determine the dosage and drug administration according to the grouping. The study investigators (non-blind) will be required to calculate the initial administration dose and the top-up doses of patients in advance before drug administration. When giving the drug to patients in ICU, the study investigator will not disclose the administration dose, such as "initial administration of 5 mg," but only say "initial administration," "maintenance administration," "start of injection" or "end of administration."
Study Drug and Timelines
All patients will be given a loading dose of ciprofol (Haisco Pharmaceutical Group Co., Ltd, China) or propofol (AstraZeneca, UK). The total time of drug administration (including loading dose and maintenance dose) will be at least 6 h ± 30 min and the longest time should not be > 24 h ± 30 min.
The study will be divided into three periods: (1) a screening period (before drug administration on day − 1); (2) a drug administration period (on day 1 or days 1–2); (3) a follow-up period (0–24 h after the end of drug administration) (Table 1).
Sedation
Analgesia/Sedation Before Administration
Analgesic drug use will be given according to standardized procedures: all participants will receive continuous intravenous remifentanil analgesia prior to study drug administration during the screening period of the study. The loading dose will be 0.5 ~ 1 µg/kg (if necessary) and maintenance doses of 0.02 ~ 0.15 µg/kg/min. The Critical Care Pain Observation Tool (CPOT) score will be monitored during the maintenance period [17]. If the CPOT score is ≥ 3, the researchers can consider adjustment of the dosage; if the CPOT score is < 3 but patients complain of pain or the researchers judge the subjects' pain based on vital signs, the dosage can also be adjusted to allow the subject to reach the appropriate analgesia level. In addition, since sedation is recommended for mechanically ventilated (MV) patients [16], the investigator will need to confirm that each patient’s baseline sedation level reached RASS ≥ 2 before starting administration of the study drug. Before drug administration, the Confusion Assessment Method for the Intensive Care Unit (CAM-ICU) [18] will be employed for the evaluation of a patient’s delirium status.
Analgesia/Sedation During Study Drug Administration
Intravenous infusion of ciprofol or propofol at a loading dose of 0.1 mg/kg or 0.5 mg/kg, respectively, will be administered by the investigator within 4 min ± 30 s, based on the physical condition of the patient. Subsequently, 0.3 mg/kg/h or 1.5 mg/kg/h of propofol, ciprofol or propofol as the initial maintenance doses will be started immediately to achieve the goal of a sedation depth of RASS + 1 ~ − 2; the maintenance doses of ciprofol or propofol will be adjusted up and down as required. The range of the maintenance doses of ciprofol or propofol will be 0.06–0.8 mg/kg/h or 0.3–4 mg/kg/h, respectively. In addition, the investigator will be allowed to top-up ciprofol or propofol in 0.05 mg/kg or 0.25 mg/kg increments, respectively, during the study drug administration process. The top-up administration time will be 30 s to 1 min and the interval for each top-up administration ≥ 2 min.
Analgesia/Sedation After the End of Study Drug Administration
After administration of the study drug ceases, the investigator must wait until patients are fully conscious and carefully record the recovery time (RASS ≥ 0). If the time of the study drug administration is > 24 h + 30 min, but the investigator decides that patients still needed to be sedated and receive analgesia, the investigator can choose another sedation drug other than propofol or can use an appropriate dose of fentanyl or other analgesic to ease the pain, which will be recorded as a combined medication history.
If the investigator’s verdict is that patients still need to be sedated after extubation or need to be re-intubated with sedation, other sedation drugs except for propofol will be employed according to clinical routine practice, the use of which will again be recorded as a combined medication history.
If the investigator’s verdict is that a patient can complete sedation and prepare for extubation, but the patient cannot be extubated after discontinuing the study drug administration, sedation other than propofol will be selected according to routine clinical practice and recorded as a combined medication history.
Related tests including laboratory biochemical analyses, vital signs and a 12-lead electrocardiogram (ECG) will be completed before other sedation medication is given, and follow-up examinations will be performed 0–24 h after the administration of the study drug (Table 2).
Planned Outcomes
The primary endpoint: The success rate of sedation must be satisfied according to the following two conditions: (1) the time within the range of RASS + 1 to − 2 should account for ≥ 70% of the study drug administration time; (2) no rescue treatments.
The secondary endpoints will be the average rate of reaching the goal of sedation, study drug usage, rescue medication given per unit weight, extubation time, recovery time to full consciousness, nursing scores, agitation and delirium [19, 20] as well as safety endpoints including vital signs, adverse events (AEs), drug-related AEs and serious AEs.
The exploratory endpoints will be the pharmacokinetic analyses as well as ciprofol and propofol plasma concentrations.
Average Rate of Reaching the Goal of Sedation
Defined as the percentage of time in the RASS + 1 ~ − 2 range for the total study drug administration period.
Study Drug Usage
The loading and maintenance dose and the total dosage of ciprofol and propofol will be measured and the top-up times and number of additional study drugs required during the maintenance period analyzed.
Rescue Medication Given per Unit Weight
Defined as the average dose/h per unit body weight of other sedation medication that will be used within 24 h to maintain the target sedation (RASS + 1 ~ − 2) after the start of the study drug administration.
Extubation Time
This is defined as the time from intubation (suitable for patients with intubation after admission to ICU) and from admission to ICU (suitable for patients entering ICU with catheters) to extubation, and the time from withdrawal of the drug to extubation. Extubation time will not be applicable to patients who withdrew in advance and who we do not plan for extubation after withdrawal of medication.
Recovery Time to Full Consciousness
This will be defined as the time for a patient to return to full consciousness from sedation (RASS ≥ 0) after drug administration ceases, which should be accurate on a minute time scale. If RASS ≥ 0 when the drug is stopped (in + 30 s), the recovery time will be recorded as 0.
Safety Endpoint
Measurements to be carried out will include: vital signs (BP, systolic blood pressure [SBP], diastolic blood pressure [DBP], mean arterial pressure [MAP], heart rate [HR], respiratory rate [RR], oxygen saturation [SpO2], 12-lead ECG [including QT, QTcF, PR, QRS, RR]), physical examinations, laboratory measurements (routine blood tests, blood biochemistry, routine urine test), coagulation function, blood gas analysis, AEs, drug-related AE, serious AE as well as changes in triglyceride concentrations.
Sample Size
This trial is designed as a non-inferiority objective study for MV sedation within 24 h. Assuming the ciprofol study drug and propofol (the positive control drug) will have an ICU sedation success rate of 99%, the type I error (false positive) will be set at 0.025 (unilateral) and the power at 80%. The non-inferiority margin considered to be clinically acceptable will be set at 8%, with a required sample size of 120 patients, allowing for a 10% dropout rate. Thus, a total of 135 patients who will receive mechanical ventilation will be included from July 2020 to July 2022 in our ICU, based on random assignment at a ratio of 2:1 to the ciprofol (n = 90) or propofol control (n = 45) groups.
Data Management and Monitoring
All raw data will be recorded in case report forms by the appropriate investigator, and data input will be double checked for accuracy by two people. A data manager will performed database checking using SAS version 9.4 software. The decision to lock the database will be made by the principal investigator, the database manager and a statistician who will be responsible for statistical analyses. The conduct of the study will be monitored by a study coordinator in each center.
Data Analysis
General Analyses
Analyses will be conducted on an intention-to-treat dataset. Considering that an intention-to-treat analysis may not be appropriate for non-inferiority trials [21], a full analysis set or per protocol analysis, including all patients who have satisfactorily complied with the assigned treatment will be carried out for non-inferiority efficacy analysis. Safety set (SS) analysis will include all enrolled patients who received the study drug and have had safety evaluations after taking the drug. The pharmacokinetic analysis set (PKAS) will include all enrolled patients who have received the study drug and have blood concentration data that can be evaluated, which will be used for PK parameter analyses.
For continuous numeric variables, the number of cases, mean, median, standard deviation, minimum, maximum and coefficient of variation (CV, if applicable) will be presented after an independent sample t-test or Wilcoxon rank sum test. Categorical variables will be given as the number of cases (percentage) and will be analyzed using a X2 test or Fisher exact test. Baseline will be defined as the last non-missing observation data to be collected before first time use of the study drug.
Primary Efficacy Outcome Analysis
The aim of the trial is demonstrating that the study arm (ciprofol) with sedation is no less effective than the control arm (propofol). The clinical non-inferiority evaluation will be determined by the Newcombe-Wilson method, i.e., whether the low limit of the one-sided 95% CI of the difference in the sedation success rate in MV ICU will indicate that the experimental drug ciprofol is not inferior to the control drug propofol.
Secondary Efficacy Outcome Analysis
Means and standard deviation (or median and 25th and 75th percentile) of the average time to reach the goal of sedation, study drug usage, rescue medication given per unit weight, nursing scores as well as safety endpoints including vital signs, AEs, drug-related AEs and SAEs will be evaluated and compared using Student’s t-test or the Mann-Whitney test. Numbers and frequencies of alternative medicine administration for rescue and emergency intubation within 24 h and other outcomes will be calculated and compared using the X2 test or Fisher’s exact test. Extubation time and recovery time to full consciousness will be evaluated using a log-rank test to compare any differences between the two groups; details of other endpoint analyses are described in the statistical analysis plan (SAP).
Discussion
It is important to ameliorate injection pain felt by patients in the ICU. The injection pain of anesthetic and sedative drugs may cause discomfort and resistance of patients receiving mechanical ventilation. Ciprofol is known to produce a lesser degree of injection pain compared to propofol, as reported in a previous phase 1 study, since the aqueous phase concentration of ciprofol in a 1% lipid emulsion is significantly less than that of propofol [14]. Propofol has also been shown to have adverse effects on respiration (inhibition) [22], but only a few cases of AEs of respiratory inhibition occurred in ciprofol patients, indicating that ciprofol can reduce the occurrence of respiratory AEs in patients undergoing sedation/general anesthesia [14].
It has been reported that following a 2.5 mg/kg dose of propofol, SBP, DBP and MAP were all reduced by 25–40%, with hypotension being most marked in volume-depleted patients. It has been proposed that propofol inhibits the baroreceptor reflex thus reducing the tachycardiac response to hypotension. At the usual clinical dosages, propofol does not cause myocardial depression [23, 24]. Cardiac output may increase because of a reduction in afterload which suggests that propofol does not affect the conduction system in the heart, functions that will be considered and evaluated in the proposed study.
It was expected that ciprofol will have the same efficacy as propofol and elicit fewer complication in ICU patients. In particular, when ciprofol is used for light sedation in critically ill patients with mechanical ventilation, it may shorten the duration of mechanical ventilation and reduce the occurrence of delirium. It should be an ideal sedation/anesthetic agent for patients with mechanical ventilation in the ICU, because a too deep sedation is associated with unwanted AEs such as respiratory depression, hypotension, prolonged mechanical ventilation duration and even mortality [25,26,27].
The delirium of patients will also be monitored twice daily by using the CAM-ICU [28]. Delirium may not appear often in the ciprofol group, since delirium in critically ill patients is associated with higher mortality rate, more complications, longer duration of mechanical ventilation and a longer length of stay in the ICU and hospital [7]. It also needs to be demonstrated that ciprofol has similar effects to propofol, but compared to midazolam can decrease the risk of delirium [10, 29].
However, for our study, it will be difficult to conduct a double-blind design for reasons previously discussed (vide supra). It will be interesting when ciprofol is combined with other sedation medication, except solely propofol, in future studies of ICU patients.
In conclusion, in this study we will determine the sedation effects and safety of ciprofol for ICU patients with mechanical ventilation and confirm a non-inferiority to propofol with a larger cohort of patients. This study's results will provide evidence to confirm our hypothesis about the incidence of delirium and benefits of ciprofol regarding adverse event occurrences including infusion syndrome, hypotension, bradycardia and respiratory depression. Moreover, we believe that ciprofol will provide a novel choice for sedation of patients with mechanical ventilation in Chinese ICUs.
Ethics and Dissemination
The study protocol was approved by the Clinical Research Ethics Committees of The First Affiliated Hospital, Sun Yat-Sen University (2020-057-01) and all participating centers (see Supplementary Material for further information). Procedures will be performed following the principles described in the Declaration of Helsinki. All patients must provide a written informed consent form before being enrolled. The results of the study will be analyzed and reported separately.
References
Faust AC, Rajan P, Sheperd LA, Alvarez CA, McCorstin P, Doebele RL. Impact of an analgesia-based sedation protocol on mechanically ventilated patients in a medical intensive care unit. Anesth Analg. 2016;123(4):903–9.
Cooper AB, Thornley KS, Young GB, Slutsky AS, Stewart TE, Hanly PJ. Sleep in critically ill patients requiring mechanical ventilation. Chest. 2000;117(3):809–18.
Hall JE, Uhrich TD, Barney JA, Arain SR, Ebert TJ. Sedative, amnestic, and analgesic properties of small-dose dexmedetomidine infusions. Anesth Analg. 2000;90(3):699–705.
Rosenzweig AB, Sittambalam CD. A new approach to the prevention and treatment of delirium in elderly patients in the intensive care unit. J Community Hosp Intern Med Perspect. 2015;5(4):27950.
Vardon Bounes F, Pichon X, Ducos G, Ruiz J, Samier C, Silva S, et al. Remifentanil for procedural sedation and analgesia in central venous catheter insertion: a randomized. Control Trial Clin J Pain. 2019;35(8):691–5.
Zhang Z, Liu J, Xi J, Gong Y, Zeng L, Ma P. Derivation and validation of an ensemble model for the prediction of agitation in mechanically ventilated patients maintained under light sedation. Crit Care Med. 2021;49(3):e279–90.
Zhang Z, Pan L, Ni H. Impact of delirium on clinical outcome in critically ill patients: a meta-analysis. Gen Hosp Psychiatry. 2013;35(2):105–11.
Prime D, Arkless P, Fine J, Winter S, Wakefield DB, Scatena R. Patient experiences during awake mechanical ventilation. J Community Hosp Intern Med Perspect. 2016;6(1):30426.
Fink RM, Makic MBF, Poteet AW, Oman KS. The ventilated patient’s experience. Dimens Crit Care Nurs. 2015;34(5):301–8.
Fraser GL, Devlin JW, Worby CP, Alhazzani W, Barr J, Dasta JF, et al. Benzodiazepine versus nonbenzodiazepine-based sedation for mechanically ventilated, critically ill adults: a systematic review and meta-analysis of randomized trials. Crit Care Med. 2013;41(9 Suppl 1):S30–8.
Sun L, Ma W, Gao W, Xing Y, Chen L, Xia Z, et al. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis. 2019;10(8):542.
Marik PE. Propofol: therapeutic indications and side-effects. Curr Pharm Des. 2004;10(29):3639–49.
Qin L, Ren L, Wan S, Liu G, Luo X, Liu Z, et al. Design, synthesis, and evaluation of novel 2,6-disubstituted phenol derivatives as general anesthetics. J Med Chem. 2017;60(9):3606–17.
Teng Y, Ou M, Wang X, et al. Pharmacokinetic and pharmacodynamic properties of ciprofol emulsion in Chinese subjects: a single center, open-label, single-arm dose-escalation phase 1 study. Am J Transl Res. 2021 (In press).
Society Of Critical Care Medicine Chinese Medical Association. Guidelines for analgesia and sedation treatment in intensive care unit of Chinese adults. Chin Crit Care Med. 2018;30(6):497–514.
Devlin JW, Skrobik Y, Gélinas C, Needham DM, Slooter AJC, Pandharipande PP, et al. Clinical practice guidelines for the prevention and management of pain, agitation/sedation, delirium, immobility, and sleep disruption in adult patients in the ICU. Crit Care Med. 2018;46(9):e825–73.
Li Q, Wan X, Gu C, Yu Y, Huang W, Li S, et al. Pain assessment using the critical-care pain observation tool in Chinese critically ill ventilated adults. J Pain Symptom Manag. 2014;48(5):975–82.
Xing H, Zhou W, Fan Y, Wen T, Wang X, Chang G. Development and validation of a postoperative delirium prediction model for patients admitted to an intensive care unit in China: a prospective study. BMJ Open. 2019;9(11): e030733.
Khan BA, Guzman O, Campbell NL, Walroth T, Tricker JL, Hui SL, et al. Comparison and agreement between the Richmond Agitation-Sedation Scale and the Riker Sedation-Agitation Scale in evaluating patients’ eligibility for delirium assessment in the ICU. Chest. 2012;142(1):48–54.
Gusmao-Flores D, Salluh JI, Chalhub R, Quarantini LC. The confusion assessment method for the intensive care unit (CAM-ICU) and intensive care delirium screening checklist (ICDSC) for the diagnosis of delirium: a systematic review and meta-analysis of clinical studies. Crit Care (London, England). 2012;16(4):R115.
Schumi J, Wittes JT. Through the looking glass: understanding non-inferiority. Trials. 2011;12:106.
Ramgolam A, Hall GL, Zhang G, Hegarty M, von Ungern-Sternberg BS. Inhalational versus intravenous induction of anesthesia in children with a high risk of perioperative respiratory adverse events: a randomized controlled trial. Anesthesiology. 2018;128(6):1065–74.
Wodey E, Chonow L, Beneux X, Azzis O, Bansard JY, Ecoffey C. Haemodynamic effects of propofol vs thiopental in infants: an echocardiographic study. Br J Anaesth. 1999;82(4):516–20.
Graham MR, Thiessen DB, Mutch WA. Left ventricular systolic and diastolic function is unaltered during propofol infusion in newborn swine. Anesth Analg. 1998;86(4):717–23.
Noel C, Mallemat H. Sedation and analgesia for mechanically ventilated patients in the emergency department. Emerg Med Clin N Am. 2019;37(3):545–56.
Aragón RE, Proaño A, Mongilardi N, de Ferrari A, Herrera P, Roldan R, et al. Sedation practices and clinical outcomes in mechanically ventilated patients in a prospective multicenter cohort. Crit Care. 2019;23(1):130.
Pearson SD, Patel BK. Evolving targets for sedation during mechanical ventilation. Curr Opin Crit Care. 2020;26(1):47–52.
Pandharipande PP, Pun BT, Herr DL, Maze M, Girard TD, Miller RR, et al. Effect of sedation with dexmedetomidine vs lorazepam on acute brain dysfunction in mechanically ventilated patients. The MENDS randomized controlled trial. JAMA. 2007;298(22):2644–53.
Zhang Z, Chen K, Ni H, Zhang X, Fan H. Sedation of mechanically ventilated adults in intensive care unit: a network meta-analysis. Sci Rep. 2017;7:44979.
Acknowledgements
Funding
The study, the journal’s Rapid Service and Open Access Fees will be funded by Haisco Pharmaceutical Group Co., Ltd. The funding body had no role in the design of the study and will have no role in data collection, analysis, interpretation of data or writing of the manuscript.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship of this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authors’ Contributions
YJL, XL, WG drafted the manuscript. YJL, CXC, NL, LT, YN, JFW, XL, WG, LT and XDG all contributed to the development and design of the protocol, reviewed the manuscript and approved the final version.
Disclosures
Xiao Liu and Wei Gao are employees of Haisco Pharmaceutical Group Co., Ltd, China. Yongjun Liu, Chuanxi Chen, Ning Liu, Li Tong, Yao Nie, Jianfeng Wu, Lei Tang, Xiangdong Guan have no conflicts of interest.
Compliance with Ethics Guideline
The study protocol was approved by the Clinical Research Ethics Committees of The First Affiliated Hospital, Sun Yat-Sen University (2020-057-01) and all participating centers (see Supplementary Material for further information). Procedures will be performed following the principles described in the Declaration of Helsinki. All patients must provide a written informed consent form before being enrolled. The results of the study will be analyzed and reported separately.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Liu, Y., Chen, C., Liu, N. et al. Efficacy and Safety of Ciprofol Sedation in ICU Patients with Mechanical Ventilation: A Clinical Trial Study Protocol. Adv Ther 38, 5412–5423 (2021). https://doi.org/10.1007/s12325-021-01877-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-021-01877-6