
Abstract
Primary familial brain calcification (PFBC) is an inherited neurodegenerative disorder mainly characterized by progressive calcium deposition bilaterally in the brain, accompanied by various symptoms, such as dystonia, ataxia, parkinsonism, dementia, depression, headaches, and epilepsy. Currently, the etiology of PFBC is largely unknown, and no specific prevention or treatment is available. During the past 10 years, six causative genes (SLC20A2, PDGFRB, PDGFB, XPR1, MYORG, and JAM2) have been identified in PFBC. In this review, considering mechanistic studies of these genes at the cellular level and in animals, we summarize the pathogenesis and potential preventive and therapeutic strategies for PFBC patients. Our systematic analysis suggests a classification for PFBC genetic etiology based on several characteristics, provides a summary of the known composition of brain calcification, and identifies some potential therapeutic targets for PFBC.
Similar content being viewed by others
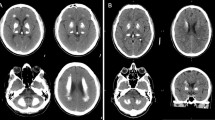
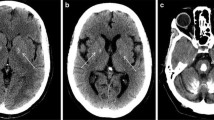

Introduction
Brain calcification is a common neuropathological phenomenon in the clinic. Its prevalence increases with age (from ~1% in young individuals to 20%–30% in the elderly >60 years) [1,2,3,4]. Other than genetic defects, a variety of factors, including endocrine disorders (hypo/hyperparathyroidism and hypothyroidism), intracranial atherosclerosis, infections, brain neoplasms, neurotoxicity, physical injury, and inflammation have been reported to promote or even cause brain calcification [5, 6].
Primary familial brain calcification (PFBC), formerly known as Fahr’s disease or idiopathic basal ganglia calcification, is an inherited and intractable disorder mainly characterized by progressive bilateral calcification distributed in the basal ganglia region and/or other areas of the brain [7]. PFBC can serve as an ideal model in which to study the pathogenesis and potential prevention and treatment of brain calcification. PFBC can result in a variety of clinical symptoms, ranging from occasional migraines to serious symptoms including motor disorders (parkinsonism, tremor, and dystonia), cognitive disorders (memory impairment, executive dysfunction symptoms, and mental retardation), and neurological disorders (depression, affective disorder, and insanity), but nearly one-third of the carriers of causative gene mutation are asymptomatic [3, 8, 9]. Due to the high degree of clinical heterogeneity, the clinical symptoms are not suitable criteria for a diagnosis of PFBC. In contrast, the clinical diagnosis mainly relies on brain calcification identified by computed tomography (CT). The most typical feature in the neuroimages of PFBC patients is symmetrical bilateral calcification in the basal ganglia, thalamus, frontal cortex, or cerebellum. However, the levels of calcium (Ca), phosphorus (P), alkaline phosphatase, parathyroid hormone, and other serum biochemical indicators are normal in PFBC patients [3, 10, 11]. The overall prevalence of PFBC is estimated to be 0.21%–0.66% [12, 13].
It has been >170 years since the initial report of a case of bilateral basal ganglia calcification by Delacour in 1850 [14]. The genetic etiology of PFBC was largely unknown before the identification of SLC20A2 as the first associated gene in 2012 by Prof. Jing-Yu Liu’s lab [11]. A few pathological studies found that the main component of brain calcification is hydroxyapatite [15, 16]. Calcification particles mainly occur in adventitial vessel cells and sometimes in glial cells, as observed by transmission electron microscopy. Some spherical and hemispherical calcium deposits have been located in the vascular adventitia and connected to the filamentous processes of surrounding cells, as observed by scanning electron microscopy [17]. Furthermore, in some PFBC cases, large spherical calcification particles, mainly calcium phosphate, have been reported to be attached to the capillary wall and located in the media of some large arteries, and reactive astrocytes and microglia have been found to accumulate around the calcification sites [18, 19]. A few neurofibrillary tangles and dystrophic neurites in the medial temporal lobe, rarely spreading senile plaques, and a few amyloid deposits in vascular walls have been detected by silver staining and immunohistochemical staining [20]. Overall, these studies demonstrate that brain calcifications are mostly associated with blood vessels and sometimes involve neurons or glial cells, but the calcification process is still largely unknown.
Since 2012, mutations in 6 genes have been associated with PFBC, including the 4 autosomal dominant genes SLC20A2, PDGFRB, PDGFB, and XPR1, and the 2 autosomal recessive genes MYORG and JAM2 [11, 21,22,23,24,25]. Recent cohort studies have expanded the causative gene mutation spectrum by identifying 248 different variants, including 125 in SLC20A2 (57 missense, 15 nonsense, 11 splicings, 30 small deletions, 4 small insertions, 1 intronic, and 7 gross deletions), 15 in PDGFRB (14 missense and 1 start loss), 26 in PDGFB (9 missense, 4 nonsense, 5 splicings, 1 small deletion, 1 small insertion, 2 start loss, 2 stop loss, 1 gross deletion, and 1 complete deletion), 14 in XPR1 (13 missense and 1 small insertion), 59 in MYORG (34 missense, 9 nonsense, 9 small deletions, 6 small insertions, and 1 small indel), and 8 in JAM2 (2 missense, 1 nonsense, 1 splicing, 2 small deletions, 1 start loss, and 1 gross deletion). The frequencies of these genes counted from 555 individuals with PFBC were 59.78%, 5.98%, 12.68%, 5.98%, 13.59%, and 1.99%, respectively [9, 26,27,28,29,30,31,32,33,34,35,36,37,38,39] (http://www.hgmd.cf.ac.uk/ac/index.php), but the spectrum may vary among different cohorts and countries. Some mechanistic studies of these causative genes at the cellular level and in animals have been reported, contributing to the understanding of PFBC pathogenesis.
In this review, we focus on studies of the biological functions of the causative genes SLC20A2, PDGFRB, PDGFB, XPR1, MYORG, and JAM2 to summarize PFBC pathogenesis and potential strategies for PFBC prevention and treatment.
The Genetic Etiology of PFBC
Based on the functions of the reported causative genes (SLC20A2, PDGFRB, PDGFB, XPR1, MYORG, and JAM2), the genetic etiology of PFBC can be classified into two categories: imbalance of inorganic phosphate (Pi) and dysfunction of the neurovascular unit (NVU) in the brain. The Pi levels in the cerebrospinal fluid (CSF) from PFBC patients are known to be an overall increase compared with controls [40], implying that impairment of cerebral Pi homeostasis is a major factor in brain calcification. Mutations of SLC20A2 and XPR1 resulting in cellular Pi imbalance can be direct contributors to the increased CSF Pi levels, and mutations of PDGFRB, PDGFB, MYORG, and JAM2 leading to NVU dysfunction may be indirect contributors to the increased CSF Pi levels.
Pi Imbalance
In the brain, Pi homeostasis is closely related to changes in Pi levels in CSF, brain interstitial fluid (ISF), cerebral blood, and intracellular fluid, the regulation of which is mainly dependent on Pi transporters. Since CSF occurs in the ventricles and subarachnoid space, it is easier to acquire and test than other cerebral fluids, and therefore is the currently best choice to reflect changes in Pi levels in the brain. In both blood and CSF, there is a dynamic shuttle of phosphate between its free inorganic form and various Pi-containing compounds, which results in different outcomes in the measurement of inorganic Pi concentrations by different kits from different suppliers. Using a clinical spectrophotometry method and a malachite green and ammonium molybdate-based colorimetric approach, the concentration of CSF Pi is reported to be maintained at ~0.6 mmol/L in humans and ~0.9 mmol/L in mice [41, 42].
SLC20A2
SLC20A2 encodes type III sodium-dependent phosphate (Na+/Pi) transporter 2 (PiT2), which was originally identified as a receptor for amphotropic murine retroviruses and soon shown to be able to transport extracellular Pi into the cell depending on excess sodium ions with a 2:1 Na+:Pi transport stoichiometry [43,44,45]. In vitro, SLC20A2 mutations impair the cellular Pi transport activity of PiT2, leading to the accumulation of extracellular Pi [11, 46], indicating that the formation of brain calcification may be associated with cerebral Pi dyshomeostasis. Recently, changes in PiT2 expression levels have been shown to affect not only Pi uptake but also some Pi efflux [47]. In animal models, Drosophila cannot be used for mechanistic studies of PFBC due to embryonic lethality resulting from a deficiency of dPiT (the protein homologous to human PiT2) [48]. Mice with PiT2 deficiency develop widespread brain calcification and exhibit abnormal multisystem phenotypes, including placental calcification, fetal growth restriction, developmental delay, lean body mass, lower bone quality and strength, skeletal malformation, a high likelihood of eye defects, impaired spatial learning, memory, and sensorimotor gating [49,50,51,52].
Furthermore, compared with wild-type mice, homozygous Slc20a2-knockout (Slc20a2−/−) mice display a dramatically elevated Pi level in CSF [42, 53], consistent with the increase in CSF Pi level in PFBC patients with SLC20A2 mutations [40]. Considering that CSF flowing along the paravascular spaces exchanges metabolites with parenchyma ISF [54, 55], it is reasonable to expect that the ISF Pi level also undergoes an increase similar to CSF Pi. In addition, PiT2 is widely and strongly expressed in neurons, astrocytes, oligodendrocytes, microglia, vascular smooth muscle cells (VSMCs), and vascular fibroblast-like cells (Fig. 1) [56] (https://www.proteinatlas.org/), and deficiencies or mutations of PiT2 in these cells probably have impaired Pi transport activity leading to the accumulation of extracellular ISF Pi. Moreover, PiT2-deficient VSMCs have been shown to present high Pi-induced calcification and elevated expression of Runx2 and osteopontin (OPN) [57]. Thus, the high extracellular Pi environment may induce intracranial VSMC calcification, possibly via transdifferentiation of VSMCs into osteoblast-like cells, an initial phase similar to the mineralization process during bone development [57,58,59].

Expression of genes in mouse NVU-related cells [56] (https://www.proteinatlas.org/). A–F Single cell RNA-seq of the mouse brain database showing the expression of (A) Slc20a2, (B) Xpr1, (C) Pdgfrb, (D) Pdgfb, (E) Myorg, and (F) Jam2 in NVU-related cells. PC, pericytes; vSMC, venous smooth muscle cells; aaSMC, arteriolar smooth muscle cells; aSMC, arterial smooth muscle cells; MG, microglia; FB1, type 1 vascular fibroblast-like cells; FB2, type 2 vascular fibroblast-like cells; OL, oligodendrocytes; EC1, type 1 endothelial cells; EC2, type 2 endothelial cells; EC3, type 3 endothelial cells; vEC, venous endothelial cells; capilEC, capillary endothelial cells; aEC, arterial endothelial cells; AC, astrocytes.
How do Pi levels increase in CSF in response to PiT2 deficiency or SLC20A2 mutations? CSF is predominantly secreted by the choroid plexus in the lateral, third, and fourth ventricles, and may also be associated with some poorly-defined sources such as ISF, ependyma, and capillaries [60,61,62]. In the brain, type III transporters (PiT1 and PiT2) are the major detectable Na+/Pi transporters [63, 64], which are highly tissue-specifically expressed in the choroid plexus. PiT1 is largely expressed in the vascular endothelium of the choroid plexus facing the blood, while PiT2 is mainly expressed in the apical membrane region facing the CSF [53, 63], implying that PiT1 contributes to the transport of Pi from blood to CSF and PiT2 contributes to the transport of Pi from CSF to blood. A single impairment of PiT2 may largely reduce Pi transportability from CSF to blood, which leads to Pi accumulation in the CSF. On the other hand, the likely increased ISF Pi level may also contribute to the increased CSF Pi level via substance exchange in the CSF-ISF system.
Mechanistically, at the plasma membrane, PiT2 and PiT1 are mostly in the form of homodimers, but some are in the form of Pi-regulated heterodimers, which may mediate extracellular Pi signaling [65]. Under high extracellular Pi conditions, PiT2 deficiency increases VSMC calcification by reducing the expression of osteoprotegerin (OPG), supplementation of which can attenuate calcification [57]. OPG is an anti-calcification factor that inhibits NF-κB/RANKL/RANK (nuclear factor kappa B/receptor activator of NF-κB ligand/receptor activator of NF-κB) signaling, which plays important roles in Pi-induced osteochondrogenic differentiation and vascular calcification [66, 67]. In contrast, PiT1 plays a role in promoting high Pi-induced osteochondrogenic differentiation and VSMC calcification via extracellular regulated protein kinases 1/2 (ERK1/2) phosphorylation independent of Pi uptake. Knockdown of PiT1 abrogates high Pi-induced ERK1/2 signaling, which is rescued by supplementation with transport-deficient PiT1 mutants or wild-type PiT1 (Fig. 2) [68, 69].

Potential mechanism of Pi transport and calcification regulated by Pi transporters at the cellular level. Intracellular Pi levels are in a dynamic balance regulated by the Pi importers PiT2/PiT1 and the Pi exporter XPR1. Under physiological conditions, PiT2 inhibits osteochondrogenic differentiation and calcification by activating OPG (osteoprotegerin) and inhibiting NF-κB/RANKL/RANK signaling. However, increased extracellular Pi can induce osteochondrogenic differentiation and calcification by activating PiT1/ERK1/2 signaling. XPR1-dependent Pi efflux is regulated by XPR1/InsP8 signaling, deficiency of which increases intracellular Pi and induces osteochondrogenic differentiation and calcification. Excessive intracellular Pi can be stored by the synthesis of polyP via IP6Ks and 5-InsP7. ERK1/2, extracellular regulated protein kinases 1/2; DIPP, diphosphoinositol polyphosphate phosphohydrolase; InsP8, 1,5-bis-diphosphoinositol 2,3,4,6-tetrakisphosphate; PPIP5K, diphosphoinositol pentakisphosphate kinase; IP6Ks, inositol hexakisphosphate kinases; 5-InsP7, 5-diphosphoinositol 1,2,3,4,6-pentakisphosphate; 1-InsP7, 1-diphosphoinositol 1,2,3,4,6-pentakisphosphate; InsP6, inositol hexakisphosphate; polyP, intracellular inorganic polyphosphate.
XPR1
XPR1 encodes xenotropic and polytropic murine virus receptor 1 (XPR1), which was originally identified as a retroviral receptor [70, 71], and later shown to be able to export intracellular Pi out of the cell; XPR1 is the only known Pi exporter in metazoans [72]. The amino terminus of XPR1 contains an SPX (named after the proteins SYG1, PHO81, and XPR1) domain that was recently identified as an intracellular sensor of Pi level and homeostasis in both plants and animals [73, 74]. In vitro studies found that mutations in the SPX domain of XPR1 or short interfering RNA-mediated knockdown of XPR1 lead to decreased Pi export [23], suggesting that missense mutations in XPR1 in PFBC patients may also be associated with impaired brain Pi homeostasis. Recently, knockout of XPR1 in human cell lines was reported to cause not only a significant reduction in Pi efflux but also to markedly down-regulate Pi uptake, suggesting a potential synergistic mechanism to coordinate Pi uptake and export. Moreover, when incubated in a high Pi medium, cultured human Saos-2 cells with XPR1 deficiency show increased expression of osteocalcin, osteonectin, and alkaline phosphatase, and finally accumulation of calcification deposits [75].
In animal models, xpr1b (the orthologue of human XPR1 in zebrafish) mutations in zebrafish lead to a lack of microglia in the brain and an osteopetrotic phenotype, but no data on brain calcification have been reported [76]. Heterozygous Xpr1-knockout mice older than 1 year fail to show a brain calcification phenotype (unpublished data), and the homozygotes die perinatally [77]. Complete knockout of Xpr1 seems to not be a good choice for generating a PFBC model, but knock-in or conditional knockout of Xpr1 in cerebral cells such as neurons and astrocytes may be worth attempting. In other tissue studies associated with Pi transport, XPR1 deficiency in mice results in severe placental calcification, reduced placental Pi exchange, lower Pi levels in amniotic fluid and serum, and decreased fetal skeletal mineral content [77]. Conditional inactivation of Xpr1 in the renal tubule of mice impairs renal Pi reabsorption, which is accompanied by glycosuria, aminoaciduria, calciuria, albuminuria, hypophosphatemic rickets, reduction in NaPi-IIa/NaPi-IIc expression, and vertebral osteomalacia [78]. These results indicate that XPR1 deficiency impairs tissue Pi homeostasis and may be an inducible factor of ectopic calcification.
In the brain, the only known Pi exporter, XPR1, is widely expressed in various cerebral cells and is particularly highly expressed in neurons, astrocytes, and microglia (Fig. 1) [56] (https://www.proteinatlas.org/). It must be expected that mutations in XPR1 impair Pi balance in cerebral cells, but the detailed effects of Pi levels in CSF and ISF are largely unknown. In addition to the lack of direct measurements of CSF and ISF Pi levels, studies of the tissue-specific expression of XPR1 in the structures of substance exchange systems, such as the choroid plexus of the blood-CSF barrier (BCB) and blood-brain barrier (BBB), are lacking.
How does XPR1 regulate Pi homeostasis and calcification at the cellular level? Recent studies have shown that XPR1-dependent Pi efflux is specifically regulated by a member of the inositol pyrophosphate (PP-InsP) signaling family, 1,5-bis-diphosphoinositol 2,3,4,6-tetrakisphosphate (InsP8), which binds to the SPX domain of XPR1 [75]. The multiple upstream pathways of InsP8, including diphosphoinositol pentakisphosphate kinases (PPIP5Ks), 5-diphosphoinositol 1,2,3,4,6-pentakisphosphate (5-InsP7), inositol hexakisphosphate kinases (IP6Ks), and inositol hexakisphosphate (InsP6), indirectly affect XPR1-dependent Pi efflux [47, 75, 79]. Deficiency of XPR1/InsP8 signaling results in an elevated intracellular Pi environment, which contributes to the induction of an osteoblastic phenotype and calcification in Saos-2 cells [75]. In addition, inhibition of XPR1 leads to the accumulation of intracellular inorganic polyphosphate (polyP) [80], a major Pi storage molecule, the synthesis of which is also associated with IP6Ks and 5-InsP7 (Fig. 2) [81,82,83,84].
NVU Dysfunction
The concept of the NVU was first described in 2001 to emphasize the unique relationship between brain cells and cerebral blood vessels (https://www.ninds.nih.gov/About-NINDS/Strategic-Plans-Evaluations/Strategic-Plans/Stroke-Progress-Review-Group). The NVU comprises neurons, glial cells (including astrocytes, microglia, and oligodendroglia), and vascular cells (including endothelial cells, pericytes, and VSMCs) [85, 86]. Specifically, the tubular structure of the cerebral capillaries is composed of endothelial cells. Adjacent endothelial cells are connected by tight junctions and adherens junctions. Tight junctions are mainly composed of occludin, claudins, and junctional adhesion molecules (JAMs). The outer edges of endothelial tubes are surrounded by pericytes and astrocytic end-feet (Fig. 3) [85, 87, 88]. The BBB is centrally located within the NVU and consists of a continuous endothelial cell membrane structure and coverage structures of endothelial cells by the basement membrane and pericytes [89,90,91].

Structural diagram of an NVU (neurovascular unit). Endothelial cells form the tubular structure of the cerebral capillaries via the connection of tight junctions and adherens junctions. The basement membrane is embedded between endothelial cells and pericytes. Astrocytic end-foot processes ensheath the vascular wall. Microglia and neurons surround the vascular wall.
An NVU, as the minimal functional unit in the brain, plays a vital role in regulating cerebral blood flow and maintaining BBB integrity, dysfunction of which impairs the normal substance exchange between blood and cerebral parenchyma. In humans, the Pi level in CSF (~0.6 mmol/L) is significantly lower than that in serum (~1.1 mmol/L) [41]. If the BBB is impaired, the increased BBB permeability probably leads to a high concentration of blood Pi leaking into the brain.
PDGFRB and PDGFB
PDGFRB encodes platelet-derived growth factor receptor beta (PDGFR-β), and its ligand protein, platelet-derived growth factor B (PDGF-B), is encoded by PDGFB [89, 92,93,94]. PDGFR-β is a transmembrane tyrosine kinase receptor for platelet-derived growth factor family members (PDGF-A, B, C, and D). In the mammalian brain, PDGFR-β is expressed mainly by pericytes, VSMCs, and vascular fibroblast-like cells [92, 95, 96]. On the contrary, PDGFB is predominantly expressed by vascular endothelial cells (vECs) and neurons (Fig. 1) [56]. As a paracrine factor, secreted PDGF-B homodimer enables vECs to recruit chemotactic PDGFR-β-expressing pericytes and VSMCs, thereby promoting the envelopment or wrapping of the cerebral vessel network by pericytes and VSMCs to consolidate the cellular basis of BBB integrity [97, 98]. Deficiency of PDGF-B/PDGFR-β signaling leads to microvascular pericyte deficiency, endothelial hyperplasia, increased vessel diameter, abnormal endothelial cell shape and ultrastructure, the abnormal cellular distribution of junctional proteins (occludin and vascular endothelial-cadherin), and morphological signs of increased vascular permeability [92, 93]. Adult mice with partial inactivation of Pdgfb or Pdgfrb also present with defects in pericyte generation, which impairs BBB integrity and increases BBB permeability due to the activation of endothelial transcytosis and astrocytic end-foot polarization [89, 91].
In vitro, missense mutations in PDGFR-β interfere with the activation of PDGFR-β and its downstream effectors via reduced autophosphorylation or protein levels [99]. Similarly, several functional studies subsequently demonstrated that PDGFR-β autophosphorylation, in response to PDGF-BB (the dimer of PDGF-B) stimulation, is abolished/reduced due to PDGFB or PDGFRB mutations [99,100,101], indicating that a functional loss of PDGF-B/PDGFR-β may be the cause of PFBC. Interestingly, several studies have reported that PDGF-BB induces calcification in VSMC lines accompanied by increased expression of PiT1 in the endoplasmic reticulum and an increased Pi transport rate [102, 103]. Based on these findings, Nicolas et al. put forward the opposite hypothesis that an activating mutation in PDGF-B/PDGFR-β may lead to brain calcification due to alterations in the PDGF-PiT1 pathway directly inducing vascular calcification [22]. However, no further evidence has supported this hypothesis. Recently, PDGF-BB was shown to increase intracellular Pi uptake by regulating the membrane migration of PiT-1 by activating protein kinase B (AKT) signaling in human neuroblastoma SH-SY5Y cells [104], indicating that increased PDGF-B/PDGFR-β signaling may contribute to intracellular Pi uptake in the central nervous system (CNS), but it is unclear whether decreased PDGF-B/PDGFR-β signaling in the CNS has an influence similar to SLC20A2 mutations on normal intracellular Pi uptake and leads to extracellular Pi accumulation.
As both homozygous Pdgfb- and Pdgfrb-knockout mice die perinatally [105, 106], these mouse lines are not good models in which to simulate brain calcification. However, the mutant line Pdgfbret/ret, with a disrupted PDGF-B retention motif that impairs its maturation and binding to the membrane [107], develops progressive calcification in the brain [21]. Another mouse line, Pdgfb−/−; R26P+/0 mice, with a 50% level of rescue by transgenic re-expression of human PDGF-B in the endothelium [89], develop smaller and fewer lesions but with a similar histological appearance and anatomical location [21], suggesting a strong correlation of endothelial PDGFB deficiency with brain calcification. Pathologic studies have shown that with respect to the levels in control mice, Pdgfbret/ret mice display a more profound reduction in pericyte coverage and a greater increase in BBB permeability than Pdgfb+/−; Pdgfrb+/− or Pdgfrbredeye/redeye mice [101]. These results imply that brain calcification resulting from a reduction in PDGF-B/PDGFR-β signaling may be associated with the degree of pericyte loss and/or BBB impairment, which contributes to the high concentration of blood Pi leaking into the brain. In a neuropathological study of one PFBC case, impaired BBB integrity with substance leakage from blood to the brain was identified in calcified brain areas [18], which supports the above speculation. However, the possibility that the integrity of the BBB in this case was destroyed by increased calcification cannot be excluded. Another study showed that PFBC patients carrying PDGFB mutants display a slight increase in the CSF Pi level, further supporting the aforementioned phenomena and speculation [40].
MYORG
MYORG encodes a putative myogenesis-regulating glycosidase (MYORG), which contains an N-terminal transmembrane domain and a C-terminal predictive glycosidase domain. It is widely expressed in many tissues [108], but its molecular function remains largely unknown. The homozygous nonsense mutations in PFBC patients suggest a causal association of the functional loss of MYORG with brain calcification [24]. In animal models, homozygous Myorg-knockout (Myorg−/−) mice are viable and display calcification in the thalamus at 9 months of age, which is much later than that reported in Slc20a2−/− and Pdgfbret/ret mice. In contrast to the enriched expression in perivascular cell types by other causative genes, MYORG has been found to be predominantly expressed in astrocytes (Fig. 1) [56], and is mainly localized to the endoplasmic reticulum [24]. Astrocytes function as essential metabolic intermediates between neuron-centralized intra-parenchyma circumstances and NVU-connected blood circulation, the dysfunction of which is frequently found to cause abnormal BBB function and neuroinflammation in neurodegenerative diseases [109]. Thus, it is interesting to explore whether the function of astrocytes is impaired in both Myorg-deficient mice and human patients, which in turn might result in the disturbance of NVU permeability, or otherwise decoy causative changes of surrounding pericytes and smooth muscle cells, paving the way for calcification development.
JAM2
JAM2 encodes junctional adhesion molecule 2 (JAM2), a member of the JAM family, and is a key component of the tight junctions between adjacent endothelial cells in the NVU [25]. In the brain, JAM2 is expressed mainly in endothelial cells and astrocytes and to a lesser degree in vascular fibroblast-like cells (Fig. 1) [56]. Functional studies of JAM2 variants have demonstrated that JAM2-associated PFBC results from the effects of JAM2 loss-of-function [25, 110]. Homozygous Jam2-knockout (Jam2−/−) mice present with no calcification but prominent vacuolation with reactive astrogliosis and neuronal density reduction in the brain at the age of 6 or 18 months. Interestingly, in the spinal cord of Jam2−/− mice, in addition to vacuolation, mineralized deposits are widely evident in the grey matter [110]. In addition to mutations in JAM2, biallelic mutations in the tight junction proteins occludin and JAM3 (a member of the JAM family and a counter-receptor of JAM2 [111]) have also been reported to result in brain calcification in congenital syndromes in humans [112, 113]. JAM3 mutations or deficiencies in humans and mice also result in hemorrhages in the brain and hydrocephalus [114, 115], suggesting that JAM3 is associated with the regulation of BBB integrity. Taken together, we speculate that JAM2 mutants probably impair the tight junctions between adjacent endothelial cells in the BBB and/or BCB, leading to increased BBB and/or BCB permeability with high Pi leakage from the blood to the brain, which contributes to an increased Pi level in the CSF.
In addition to regulating the formation of tight junctions, JAMs participate in the immune cell response by regulating the transendothelial migration of leukocytes [116, 117], but this has not yet been linked to calcification. Much more research on the relationship between the functional duality of JAMs and brain calcification is needed.
In summary, two characteristics of PFBC initiation have been identified: Pi imbalance (SLC20A2 and XPR1) and NVU dysfunction (PDGFRB, PDGFB, MYORG, and JAM2). Cerebral Pi dyshomeostasis seems to be a common cause of PFBC. Mutations in SLC20A2 and XPR1 directly impair the Pi transport capacity of cerebral cells. Mutations in PDGFRB, PDGFB, MYORG, and JAM2 impair the integrity of the BBB and/or BCB, leading to blood Pi leakage into the brain (Figs. 4, 5).

Putative mechanism of brain calcification induced by increased Pi levels. In the brain, increased Pi levels may induce the ossification of NVU cells contributing to the production of organic substances for calcification. On the other hand, increased Pi levels may contribute to combination with Ca2+ to form hydroxyapatite. After a series of combinations of organic substances and inorganic substances including hydroxyapatite, Cu2+, Zn2+, Fe2+, and Mg2+, calcified crystals form and grow larger. HAP, hydroxyapatite; APP, amyloid precursor protein.

Structural diagram of the potential pathogenesis and potential prevention/treatment of PFBC. BBB, blood-brain barrier; BCB, blood-CSF barrier; OPN, osteopontin; AHSG, fetuin A; MGP, matrix gla protein; SPARCL1, an ancestral mineralization protein sparc-like 1; APP, amyloid precursor protein; APLP2, amyloid precursor-like protein 2; RUNX2, runt-related transcription factor-2; RANK, receptor activator of nuclear factor kappa B.
The Basis of Calcification in PFBC
The above studies on the genetic etiology of PFBC revealed that it originally results from a Pi imbalance and NVU dysfunction in the brain, but the further mechanisms by which these defects lead to brain calcification remain unclear. Here, we further analyze the possible explanations by summarizing the composition of calcification in PFBC, which may also provide some potential biological markers and therapeutic targets for PFBC.
Early pathological examination revealed that the main component of brain calcification in PFBC patients is hydroxyapatite (Ca10[PO4]6[OH]2) [15, 16]. In addition, trace metals such as Al, Ar, Co, Cu, Mo, Fe, Pb, Mn, Mg, Ag, and Zn have also been detected in the calcium deposits in PFBC patients [16, 118, 119]. In 1987, Kobayashi et al. found that the calcium deposits were composed of a mixture of calcium salts, iron, glycoproteins, and mucopolysaccharides in one PFBC patient [17]. In 2005, Miklossy et al. identified the elements of calcium deposits via combined scanning electron microscopy and X-ray spectrometry, revealing a high level of Ca (19.97%) and P (10.08%), indicative of hydroxyapatite; C (18.11%) and O (50.49%), indicative of organic substances; and a small amount of Na (0.51%), K (0.24%), S (0.20%), and Mg (0.39%) [18]. In 2018, Jensen et al. analyzed calcium deposits in Slc20a2−/− mice and found increased contents of Ca, P, Fe, Zn, Al, Mg, S, O, and N compared with the levels in non-calcified areas [120]. These studies indicate that calcium deposits in PFBC patients include a mixture of inorganic and organic substances (Fig. 4).
Anomalous Deposition of Inorganic Substances
The inorganic substances in brain calcification are mainly hydroxyapatite, as well as low-level incorporation of metal elements, including Cu2+, Zn2+, Fe2+, and Mg2+. Intriguingly, constituent analysis of CSF from three PFBC patients detected increased concentrations of Cu2+, Zn2+, Fe2+, and Mg2+, with Ca2+, Na+, and Cl− levels remaining unchanged [40, 121]. Although increased Pi levels have been shown to be a major driving factor of PFBC pathogenesis, Ca2+ seems to be a passive component in hydroxyapatite formation. Increased levels of Cu2+, Zn2+, Fe2+, and Mg2+ in CSF are also commonly found in some other neurodegenerative diseases such as amyotrophic lateral sclerosis, Alzheimer’s disease (AD), and Parkinson’s disease [122,123,124]. Whether and how an abnormality of metal minerals is engaged in the onset and/or progression of these diseases is an interesting question. Cu2+ and Zn2+ are potentially associated with amyloid beta protein (Aβ) 42 plaque formation, oxidative stress, and neurodegeneration [124, 125]. Similarly, Fe2+ has been suggested to be involved in the release of oxidative factors and inflammation leading to neurodegeneration [126,127,128]. In humans, plasma Cu2+, Zn2+, and Fe2+ concentrations are much higher than those in CSF [124]. Disrupted BBB integrity is also associated with increased levels of Cu2+, Zn2+, and Fe2+ in the brains of AD patients [129]. Thus, combined with studies showing BBB impairment in a PFBC patient and Pdgfrb/Pdgfb-deficient mice [18, 89, 91], increased levels of Cu2+, Zn2+, and Fe2+ in PFBC patients might be associated with increased BBB permeability leading to blood leakage. Interestingly, amyloid precursor protein (APP) and amyloid precursor-like protein 2 (APLP2), which harbor binding sites for Cu2+ and Zn2+ [130], accumulate in calcified nodules [131]. This feature may contribute to Cu2+ and Zn2+ deposition in calcifications.
Anomalous Changes in Organic Substances
Many studies have shown that ectopic calcification is not a simple passive process but a complex and highly-regulated active process involving the activation of cell signaling pathways and the production of calcification inhibitors and hormone regulation [132, 133], some of which may be detected in the organic components of calcium deposits. Few early reports described organic substances in brain calcifications, and systematic studies are particularly rare. Recently, Nahar et al., using liquid chromatography with tandem mass spectrometry, identified the protein components in brain calcifications in Pdgfbret/ret mice, and found 10 proteins exclusively in calcified nodules, including three bone-formation inhibitors, fetuin A (alpha 2-Heremans–Schmid glycoprotein, AHSG), matrix gla protein (MGP), and OPN [134,135,136]; an ancestral mineralization protein sparc-like 1 (SPARCL1) [137]; three neuronal function-associated proteins APP, APLP2, and the neurosecretory protein nerve growth factor inducible (VGF) [138, 139]; two hormonal activity-associated proteins secretogranin-1 (CHGB) and chromogranin A (CHGA) [140]; and the lysosomal proteinase cathepsin Z (CTSZ) [141]. Immunohistochemical staining has confirmed the presence of OPN, APP, APLP2, SPARCL1, VGF, and CHGA in calcium deposits in Pdgfbret/ret mice, and similarly, APP, APLP2, and SPARCL1 have also been found in calcium deposits in Slc20a2−/− mice [131].
Among the organic content in calcium deposits, four proteins (AHSG, MGP, OPN, and SPARCL1) are well-known to modulate bone formation and homeostasis [136, 142,143,144], implying that brain calcification undergoes similar pathological processes. In addition, Zarb et al. analyzed the cellular circumstances of calcified nodules in Pdgfbret/ret mice and showed the presence of bone matrix proteins (collagen I, osteocalcin, and OPN) in calcification deposits. Moreover, the cells around calcium deposits were found to express markers of osteoblasts (RUNX2), osteoclasts (cathepsin K and RANK), and osteocytes (sclerostin). Consistent with this, osteogenic niches surrounding brain calcium deposits have also been reported in 3 PFBC patients carrying mutations in PDGFRB (p.Arg695Cys), SLC20A2 (p.Met1_Val652del), or SLC20A2 (p.Ser113*) [145]. In addition, accumulated reactive astrocytes and microglia have been observed around calcium deposits in one PFBC case and Pdgfbret/ret mice [18, 131, 145]. Reactive astrocytes abnormally express the bone matrix protein OPN and the osteocyte marker podoplanin accompanied by a neurotoxic response and oxidative damage [145, 146], indicating that reactive astrocytes play an important role in the formation of an osteogenic environment but remain difficult to identify as promoters or inhibitors of brain calcification. Reactive microglia abnormally express the osteoclast markers cathepsin K and RANK and have been demonstrated to be beneficial for pathological brain calcification; their deficiency intensifies brain calcification via triggering receptors expressed on myeloid cells 2 (TREM2) [146]. Considering that VSMCs can be induced by high Pi to undergo differentiation to osteoblast-like cells and form calcification [58, 59], brain calcification associated with osteogenic environments and vessels suggests that ossification of some cerebral cells induced by increased Pi levels in the brain might be a common process in PFBC.
The proteins associated with neuronal function APP, APLP2, and VGF are associated with AD, especially APP, which is key to the formation of Aβ plaques in AD [147,148,149]. However, these proteins deposited in calcified areas lack a β-pleated sheet conformation and structural regularity [146], in contrast to Aβ plaques in patients with AD. Recently, APP and APLP2 messenger RNAs have been shown to be highly expressed in bone [150]. APP and Aβ (a small proteolytic fragment of APP) have also been identified in the bone, largely in osteocytes and the bone matrix. Furthermore, Aβ enhances the differentiation and activation of osteoclasts [151, 152]. Thus, the accumulation of APP and APLP2 in brain calcification seems to be caused by a mechanism similar to that of AHSG, MGP, and OPN accumulation, contributing to the formation of osteogenic environments. Another protein component of calcium deposits, CTSZ, a lysosomal proteinase cathepsin, is produced in both osteoclast and osteoblast lineages [153,154,155], suggesting that CTSZ in calcification areas also contributes to the formation of osteogenic environments. Nahar et al. hypothesized that VGF and the hormonal activity-associated proteins CHGB and CHGA passively adhere to calcifications because the expression of these proteins is mostly restricted to neurons [131]. However, more experiments are needed to identify the key proteins because ectopic calcification is highly regulated by hormones.
In summary, analysis of the composition of calcification in PFBC reveals that brain calcification formation is a complex process involving various inorganic and organic substances. The discovery of osteogenic environments in the calcified brain suggests that cell ossification induced by increased Pi levels in the brain seems to be a common process of calcification in PFBC (Fig. 4).
Prevention and Treatment of PFBC
Although PFBC has been studied for >170 years, no specific prevention or treatment has been discovered. The identification of causative genes (SLC20A2, PDGFRB, PDGFB, XPR1, MYORG, and JAM2) and studies of its pathogenesis may provide some potential therapeutic targets for the prevention and treatment of PFBC.
Treatment Cases
Currently, the clinical treatment of PFBC patients is mainly symptomatic-oriented. Due to the large clinical heterogeneity of PFBC symptoms, different medications are used, including antipsychotics, anticonvulsants, antidepressants, mood stabilizers, antiparkinsonism-directed and anti-incontinence-directed medications, analgesics, and benzodiazepines [156]. However, only a few patients have shown improved control of their symptoms (e.g., Tololeski et al. reported an adolescent patient for whom quetiapine treatment successfully attenuated acute psychosis, and Uno et al. reported a middle-aged patient who was relieved of recurrent psychosis with risperidone) and no positive effect on brain calcification has been shown; moreover, the risk of symptom relapse is high [26, 157].
Effective therapies to control the progress of brain calcification have rarely been reported. Treatment with nimodipine, a calcium channel blocker in the CNS, has been unsuccessful in attenuating PFBC [10]. Bisphosphonates, with a structure analogous to that of pyrophosphoric acid, are mainly used in the clinical treatment of osteoporosis, and they can bind to hydroxyapatite and preferentially localize to the site of active bone remodeling, reducing the process of bone resorption by inhibiting osteoclast activity [158]. In a small number of PFBC patients, oral bisphosphonate administration has been reported to improve symptoms, but brain calcification was not reduced, as indicated by CT imaging. Loeb described a middle-aged patient treated with disodium etidronate, a bisphosphonate, which produced a twofold improvement in his speech and gait rate but without affecting spasticity, dystonia, ataxia, or brain calcification [159]. In addition, Loeb et al. reported that two patients treated with disodium etidronate presented with a significant reduction in seizure frequency and headaches but no reduction in calcium deposit size, as determined by CT imaging [160]. Recently, Oliveira et al. reported 7 patients treated with alendronate, another bisphosphonate, and reported improvements and stability without obvious side-effects in some of these patients, particularly younger patients, but no specific change in brain calcification was identified by CT imaging [161]. Although the effectiveness of bisphosphonates for PFBC needs to be confirmed with more evidence, they may be potentially effective drug options for the nonspecific treatment of PFBC conditions.
Speculation on Potential Prevention and Treatment
Hypovitaminosis D was reported in a PFBC patient carrying an SLC20A2 mutation [162], and vitamin D receptor (VDR) knockout in mice resulted in symmetrical thalamic calcification [163], suggesting that deficient vitamin D or its receptor is associated with brain calcification. Furthermore, SLC20A2 expression is positively regulated by vitamin D, which reduces calcification in vitro [164]. Thus, vitamin D may be a potential treatment for PFBC patients, but the detrimental effects of high levels of vitamin D should be noted.
In general, the frequency of nonsense variants in SLC20A2, PDGFRB, PDGFB, XPR1, MYORG, and JAM2 reaches 13.2% [9]. Considering that PFBC patients carry these variants, Peters et al. suggested that ataluren, an agent with the potential for treating a broad range of genetic diseases caused by nonsense variants [165], might be a potential option [166].
PDGFR-β is a tyrosine kinase receptor, and its activation further activates many downstream signaling pathways. Considering this, Lemos et al. speculated that PDGFRB might be an interesting therapeutic target for PFBC patients with PDGFB or PDGFRB mutations and that drugs modulating the PDGF-B/PDGFR-β signaling pathways might be potential treatments [156]. However, animal experiments and case studies to confirm this possibility are lacking, and the mechanism by which the downstream pathways affected by PDGF-B/PDGFR-β impairment cause calcification also needs further study.
Analyses of the genetic etiology of PFBC have prompted the hypothesis that cerebral Pi dyshomeostasis may be a common cause of PFBC. In addition, in SLC20A2-deficient brains of both humans and mice, significant elevation of CSF Pi levels has been detected and is increasingly accepted as a marker for PFBC [40, 42, 53]. Thus, we inferred that a high level of brain Pi might be a therapeutic target for PFBC prevention. Reducing Pi intake through reasonable dietary intervention or restoring the normal Pi transport by supplementation of PiT2 expression with viral vectors and gene editing technology in the brain may be promising research directions.
Cell ossification occurs in PFBC and seems to be a common process. Reactive microglia have been shown to be beneficial for inhibiting pathological brain calcification, possibly through matrix degradation mediated by the TREM2-dependent protein cathepsin K (Fig. 4). Hence, inhibiting cell ossification and increasing bone resorption-promoting protein release might be interesting lines of research for exploring PFBC treatment.
Here, we have summarized the current research progress of prevention and treatment for PFBC in two aspects, reported treatment cases and potential prevention and treatment.
Conclusions and Perspectives
In conclusion, the studies analyzed three aspects of PFBC research mainly based on functional research on the reported causative genes in the past decade. First, two characteristics of the genetic etiology of PFBC are classified: Pi imbalance and NVU dysfunction in the brain; in addition, cerebral Pi dyshomeostasis seems to be a common cause of PFBC that can be directly influenced by mutations in SLC20A2 and XPR1 and may be indirectly influenced by mutations in PDGFRB, PDGFB, MYORG, and JAM2. Second, based on studies of the composition of calcified nodules, cell ossification induced by increased Pi levels in the brain seems to be a common process of calcification in PFBC. Third, the current research status of PFBC treatment is summarized as follows: no specific prevention or treatment is available, and we highlight several potential prevention/treatment options and therapeutic targets: (1) bisphosphonates target calcification and may be effective against some PFBC symptoms; (2) vitamin D may be effective for PFBC patients with SLC20A2 mutations and hypovitaminosis D because it increases SLC20A2 expression; (3) ataluren is an agent with the potential for suppressing nonsense mutations and may be effective for PFBC patients with nonsense mutations; (4) as an interesting therapeutic against PDGFR-β, drugs modulating PDGF-B/PDGFR-β signaling may be potential treatments for PFBC patients with PDGFB or PDGFRB mutations; (5) as a therapeutic target to re-establish cerebral Pi homeostasis, reducing Pi intake or restoring the normal Pi transport in the brain may be interesting research directions; and (6) inhibiting the process of cerebral cell ossification may be an interesting therapeutic option to inhibit calcification in PFBC (Fig. 5).
To effectively control the progress of brain calcification and accompanying brain insults, more studies are needed. First, the genetic basis in ~40% of PFBC patients remains unclear [9, 26,27,28,29,30,31,32,33,34,35,36,37,38,39, 167]; the application of novel genetic sequencing technology is critical to identify new causative genes to expand the genetic spectrum of PFBC. Second, dissection of the cellular and molecular mechanisms underlying brain calcification requires inter-disciplinary collaborative efforts to establish a systematic understanding, especially of the signaling network of PFBC-associated genes and the osteogenic hypothesis. Third, in addition to mouse models of PFBC, novel animal models, especially large animal models (e.g., rat and monkey), are needed for the simulation of PFBC pathogenesis and translational research. Fourth, more studies on the mechanism of action of potential drugs and the identification of the targets of potential drugs will help apply the treatment of monogenic PFBC to the treatment of common brain calcification treatments.
References
Förstl H, Krumm B, Eden S, Kohlmeyer K. Neurological disorders in 166 patients with basal ganglia calcification: A statistical evaluation. J Neurol 1992, 239: 36–38.
Simoni M, Pantoni L, Pracucci G, Palmertz B, Guo X, Gustafson D. Prevalence of CT-detected cerebral abnormalities in an elderly Swedish population sample. Acta Neurol Scand 2008, 118: 260–267.
Nicolas G, Pottier C, Charbonnier C, Guyant-Maréchal L, Le Ber I, Pariente J, et al. Phenotypic spectrum of probable and genetically-confirmed idiopathic basal ganglia calcification. Brain 2013, 136: 3395–3407.
Yamada M, Asano T, Okamoto K, Hayashi Y, Kanematsu M, Hoshi H, et al. High frequency of calcification in basal ganglia on brain computed tomography images in Japanese older adults. Geriatr Gerontol Int 2013, 13: 706–710.
Baba Y, Broderick DF, Uitti RJ, Hutton ML, Wszolek ZK. Heredofamilial brain calcinosis syndrome. Mayo Clin Proc 2005, 80: 641–651.
Saade C, Najem E, Asmar K, Salman R, El Achkar B, Naffaa L. Intracranial calcifications on CT: An updated review. J Radiol Case Rep 2019, 13: 1–18.
Lemos RR, Ramos EM, Legati A, Nicolas G, Jenkinson EM, Livingston JH, et al. Update and mutational analysis ofSLC20A2: A major cause of primary familial brain calcification. Hum Mutat 2015, 36: 489–495.
Tadic V, Westenberger A, Domingo A, Alvarez-Fischer D, Klein C, Kasten M. Primary familial brain calcification with known gene mutations: A systematic review and challenges of phenotypic characterization. JAMA Neurol 2015, 72: 460–467.
Balck A, Schaake S, Kuhnke NS, Domingo A, Madoev H, Margolesky J, et al. Genotype-phenotype relations in primary familial brain calcification: Systematic MDSGene review. Mov Disord 2021, 36: 2468–2480.
Manyam BV. What is and what is not ‘Fahr’s disease.’ Park Relat Disord 2005, 11: 73–80.
Wang C, Li YL, Shi L, Ren J, Patti M, Wang T, et al. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat Genet 2012, 44: 254–256.
Nicolas G, Charbonnier C, Campion D, Veltman JA. Estimation of minimal disease prevalence from population genomic data: Application to primary familial brain calcification. Am J Med Genet B Neuropsychiatr Genet 2018, 177: 68–74.
Chen S, Cen Z, Fu F, Chen Y, Chen X, Yang D, et al. Underestimated disease prevalence and severe phenotypes in patients with biallelic variants: A cohort study of primary familial brain calcification from China. Park Relat Disord 2019, 64: 211–219.
Delacour A. Ossification des capillaires du cerveau. Ann MedPsychol 1850, 2: 458–461.
Michotte Y, Smeyers-Verbeke J, Ebinger G, Maurus R, Pelsmaekers J, Lowenthal A, et al. Brain calcification in a case of acute lymphoblastic leukaemia. J Neurol Sci 1975, 25: 145–152.
Smeyers-Verbeke J, Michotte Y, Pelsmaeckers J, Lowenthal A, Massart DL, Dekegel D, et al. The chemical composition of idiopathic nonarteriosclerotic cerebral calcifications. Neurology 1975, 25: 48–57.
Kobayashi S, Yamadori I, Miki H, Ohmori M. Idiopathic nonarteriosclerotic cerebral calcification (Fahr’s disease): An electron microscopic study. Acta Neuropathol 1987, 73: 62–66.
Miklossy J, MacKenzie IR, Dorovini-Zis K, Calne DB, Wszolek ZK, Klegeris A, et al. Severe vascular disturbance in a case of familial brain calcinosis. Acta Neuropathol 2005, 109: 643–653.
Wider C, Dickson DW, Schweitzer KJ, Broderick DF, Wszolek ZK. Familial idiopathic basal ganglia calcification: A challenging clinical-pathological correlation. J Neurol 2009, 256: 839–842.
Wszolek ZK, Baba Y, MacKenzie IR, Uitti RJ, Strongosky AJ, Broderick DF, et al. Autosomal dominant dystonia-plus with cerebral calcifications. Neurology 2006, 67: 620–625.
Keller A, Westenberger A, Sobrido MJ, García-Murias M, Domingo A, Sears RL, et al. Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat Genet 2013, 45: 1077–1082.
Nicolas G, Pottier C, Maltête D, Coutant S, Rovelet-Lecrux A, Legallic S, et al. Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 2013, 80: 181–187.
Legati A, Giovannini D, Nicolas G, López-Sánchez U, Quintáns B, Oliveira JRM, et al. Mutations in XPR1 cause primary familial brain calcification associated with altered phosphate export. Nat Genet 2015, 47: 579–581.
Yao XP, Cheng XW, Wang C, Zhao M, Guo XX, Su HZ, et al. Biallelic mutations in MYORG cause autosomal recessive primary familial brain calcification. Neuron 2018, 98: 1116-1123.e5.
Cen Z, Chen Y, Chen S, Wang H, Yang D, Zhang H, et al. Biallelic loss-of-function mutations in JAM2 cause primary familial brain calcification. Brain 2020, 143: 491–502.
Uno A, Tamune H, Kurita H, Hozumi I, Yamamoto N. SLC20A2-associated idiopathic basal ganglia calcification-related recurrent psychosis response to low-dose antipsychotics: A case report and literature review. Cureus 2020, 12: e12407.
Arteche-López A, Álvarez-Mora M, Sánchez Calvin M, Lezana Rosales J, Palma Milla C, Gómez Rodríguez MJ, et al. Biallelic variants in genes previously associated with dominant inheritance: CACNA1A, RET and SLC20A2. Eur J Hum Genet 2021, 29: 1520–1526.
Duan RN, Zhao DD, Liu YM, Yan CZ. A heterozygous deletion of PDGFB gene causes paroxysmal kinesigenic dyskinesia with primary familial brain calcification. Park Relat Disord 2021, 92: 83–87.
Fei BN, Su HZ, Yao XP, Ding J, Wang X. Idiopathic basal ganglia calcification associated with new MYORG mutation site: A case report. World J Clin Cases 2021, 9: 7169–7174.
Gao L, Chen J, Dong H, Li X. A novel mutation in MYORG leads to primary familial brain calcification and cerebral infarction. Int J Neurosci 2021: 1–5.
Lenglez S, Sablon A, Fénelon G, Boland A, Deleuze JF, Boutoleau-Bretonnière C, et al. Distinct functional classes of PDGFRB pathogenic variants in primary familial brain calcification. Hum Mol Genet 2022, 31: 399–409.
Shen Y, Shu S, Ren Y, Xia W, Chen J, Dong L, et al. Case report: Two novel frameshift mutations in SLC20A2 and one novel splice donor mutation in PDGFB associated with primary familial brain calcification. Front Genet 2021, 12: 643452.
Sun H, Cao Z, Gao R, Li Y, Chen R, Du S, et al. Severe brain calcification and migraine headache caused by SLC20A2 and PDGFRB heterozygous mutations in a five-year-old Chinese girl. Mol Genet Genomic Med 2021, 9: e1670.
Tang LO, Hou BH, Zhang XN, Xi ZY, Li CX, Xu L. Biallelic XPR1 mutation associated with primary familial brain calcification presenting as paroxysmal kinesigenic dyskinesia with infantile convulsions. Brain Dev 2021, 43: 331–336.
Tekin Orgun L, Besen Ş, Sangün Ö, Bisgin A, Alkan Ö, Erol İ. First pediatric case with primary familial brain calcification due to a novel variant on the MYORG gene and review of the literature. Brain Dev 2021, 43: 789–797.
Zeng YH, Lin BW, Su HZ, Guo XX, Li YL, Lai LL, et al. Mutation analysis of MYORG in a Chinese cohort with primary familial brain calcification. Front Genet 2021, 12: 732389.
Sakai K, Ishida C, Hayashi K, Tsuji N, Kannon T, Hosomichi K, et al. Familial idiopathic basal ganglia calcification with a heterozygous missense variant (c.902C>T/p.P307L) in SLC20A2 showing widespread cerebrovascular lesions. Neuropathology 2022, 42: 126–133.
Chen SY, Lin WC, Chang YY, Lin TK, Lan MY. Brain hypoperfusion and nigrostriatal dopaminergic dysfunction in primary familial brain calcification caused by novel MYORG variants: Case report. BMC Neurol 2020, 20: 329.
Woo KA, Yoo D, Lee JY, Kim MJ, Seong MW, Park SS, et al. SLC20A2 mutation manifesting as very late-onset orofacial dyskinesia. Neurol Sci 2021, 42: 2561–2564.
Hozumi I, Kurita H, Ozawa K, Furuta N, Inden M, Sekine SI, et al. Inorganic phosphorus (Pi) in CSF is a biomarker for SLC20A2-associated idiopathic basal ganglia calcification (IBGC1). J Neurol Sci 2018, 388: 150–154.
Idland AV, Wyller TB, Støen R, Dahl GT, Frihagen F, Brækhus A, et al. Cerebrospinal fluid phosphate in delirium after hip fracture. Dement Geriatr Cogn Dis Extra 2017, 7: 309–317.
Jensen N, Autzen JK, Pedersen L. Slc20a2 is critical for maintaining a physiologic inorganic phosphate level in cerebrospinal fluid. Neurogenetics 2016, 17: 125–130.
Kavanaugh MP, Miller DG, Zhang W, Law W, Kozak SL, Kabat D, et al. Cell-surface receptors for gibbon ape leukemia virus and amphotropic murine retrovirus are inducible sodium-dependent phosphate symporters. Proc Natl Acad Sci U S A 1994, 91: 7071–7075.
Miller DG, Edwards RH, Miller AD. Cloning of the cellular receptor for amphotropic murine retroviruses reveals homology to that for gibbon ape leukemia virus. Proc Natl Acad Sci U S A 1994, 91: 78–82.
Ravera S, Virkki LV, Murer H, Forster IC. Deciphering PiT transport kinetics and substrate specificity using electrophysiology and flux measurements. Am J Physiol Cell Physiol 2007, 293: C606–C620.
Sekine SI, Nishii K, Masaka T, Kurita H, Inden M, Hozumi I. SLC20A2 variants cause dysfunctional phosphate transport activity in endothelial cells induced from Idiopathic Basal Ganglia Calcification patients-derived iPSCs. Biochem Biophys Res Commun 2019, 510: 303–308.
López-Sánchez U, Tury S, Nicolas G, Wilson MS, Jurici S, Ayrignac X, et al. Interplay between primary familial brain calcification-associated SLC20A2 and XPR1 phosphate transporters requires inositol polyphosphates for control of cellular phosphate homeostasis. J Biol Chem 2020, 295: 9366–9378.
Ma XX, Li X, Yi P, Wang C, Weng J, Zhang L, et al. PiT2 regulates neuronal outgrowth through interaction with microtubule-associated protein 1B. Sci Rep 2017, 7: 17850.
Jensen N, Schrøder HD, Hejbøl EK, Füchtbauer EM, de Oliveira JRM, Pedersen L. Loss of function of Slc20a2 associated with familial idiopathic basal ganglia calcification in humans causes brain calcifications in mice. J Mol Neurosci 2013, 51: 994–999.
Wallingford MC, Gammill HS, Giachelli CM. Slc20a2 deficiency results in fetal growth restriction and placental calcification associated with thickened basement membranes and novel CD13 and lamininα1 expressing cells. Reprod Biol 2016, 16: 13–26.
Beck-Cormier S, Lelliott CJ, Logan JG, Lafont DT, Merametdjian L, Leitch VD, et al. Slc20a2, encoding the phosphate transporter PiT2, is an important genetic determinant of bone quality and strength. J Bone Miner Res 2019, 34: 1101–1114.
Ren Y, Shen Y, Si N, Fan S, Zhang Y, Xu W, et al. Slc20a2-deficient mice exhibit multisystem abnormalities and impaired spatial learning memory and sensorimotor gating but normal motor coordination abilities. Front Genet 2021, 12: 639935.
Wallingford MC, Chia JJ, Leaf EM, Borgeia S, Chavkin NW, Sawangmake C, et al. SLC20A2 deficiency in mice leads to elevated phosphate levels in cerbrospinal fluid and glymphatic pathway-associated arteriolar calcification, and recapitulates human idiopathic basal ganglia calcification. Brain Pathol 2017, 27: 64–76.
Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, et al. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid β. Sci Transl Med 2012, 4: 147ra111.
Iliff JJ, Wang M, Zeppenfeld DM, Venkataraman A, Plog BA, Liao YH, et al. Cerebral arterial pulsation drives paravascular CSF-interstitial fluid exchange in the murine brain. J Neurosci 2013, 33: 18190–18199.
Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, del Gaudio F, et al. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554: 475–480.
Yamada S, Leaf EM, Chia JJ, Cox TC, Speer MY, Giachelli CM. PiT-2, a type III sodium-dependent phosphate transporter, protects against vascular calcification in mice with chronic kidney disease fed a high-phosphate diet. Kidney Int 2018, 94: 716–727.
Steitz SA, Speer MY, Curinga G, Yang HY, Haynes P, Aebersold R, et al. Smooth muscle cell phenotypic transition associated with calcification: upregulation of Cbfa1 and downregulation of smooth muscle lineage markers. Circ Res 2001, 89: 1147–1154.
Chen NX, O’Neill KD, Duan D, Moe SM. Phosphorus and uremic serum up-regulate osteopontin expression in vascular smooth muscle cells. Kidney Int 2002, 62: 1724–1731.
Abbott NJ. Evidence for bulk flow of brain interstitial fluid: Significance for physiology and pathology. Neurochem Int 2004, 45: 545–552.
Moody DM. The blood-brain barrier and blood-cerebral spinal fluid barrier. Semin Cardiothorac Vasc Anesth 2006, 10: 128–131.
Sakka L, Coll G, Chazal J. Anatomy and physiology of cerebrospinal fluid. Eur Ann Otorhinolaryngol Head Neck Dis 2011, 128: 309–316.
Guerreiro PM, Bataille AM, Parker SL, Renfro JL. Active removal of inorganic phosphate from cerebrospinal fluid by the choroid plexus. Am J Physiol Renal Physiol 2014, 306: F1275–F1284.
Inden M, Iriyama M, Zennami M, Sekine SI, Hara A, Yamada M, et al. The type III transporters (PiT-1 and PiT-2) are the major sodium-dependent phosphate transporters in the mice and human brains. Brain Res 2016, 1637: 128–136.
Bon N, Couasnay G, Bourgine A, Sourice S, Beck-Cormier S, Guicheux J, et al. Phosphate (Pi)-regulated heterodimerization of the high-affinity sodium-dependent Pi transporters PiT1/Slc20a1 and PiT2/Slc20a2 underlies extracellular Pi sensing independently of Pi uptake. J Biol Chem 2018, 293: 2102–2114.
van Campenhout A, Golledge J. Osteoprotegerin, vascular calcification and atherosclerosis. Atherosclerosis 2009, 204: 321–329.
Lee SJ, Lee IK, Jeon JH. Vascular calcification-new insights into its mechanism. Int J Mol Sci 2020, 21: 2685.
Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res 2006, 98: 905–912.
Chavkin NW, Chia JJ, Crouthamel MH, Giachelli CM. Phosphate uptake-independent signaling functions of the type III sodium-dependent phosphate transporter, PiT-1, in vascular smooth muscle cells. Exp Cell Res 2015, 333: 39–48.
Battini JL, Rasko JE, Miller AD. A human cell-surface receptor for xenotropic and polytropic murine leukemia viruses: Possible role in G protein-coupled signal transduction. Proc Natl Acad Sci U S A 1999, 96: 1385–1390.
Tailor CS, Nouri A, Lee CG, Kozak C, Kabat D. Cloning and characterization of a cell surface receptor for xenotropic and polytropic murine leukemia viruses. Proc Natl Acad Sci U S A 1999, 96: 927–932.
Giovannini D, Touhami J, Charnet P, Sitbon M, Battini JL. Inorganic phosphate export by the retrovirus receptor XPR1 in metazoans. Cell Rep 2013, 3: 1866–1873.
Secco D, Wang C, Arpat BA, Wang Z, Poirier Y, Tyerman SD, et al. The emerging importance of the SPX domain-containing proteins in phosphate homeostasis. New Phytol 2012, 193: 842–851.
Secco D, Wang C, Shou H, Whelan J. Phosphate homeostasis in the yeast Saccharomyces cerevisiae, the key role of the SPX domain-containing proteins. FEBS Lett 2012, 586: 289–295.
Li X, Gu C, Hostachy S, Sahu S, Wittwer C, Jessen HJ, et al. Control of XPR1-dependent cellular phosphate efflux by InsP8 is an exemplar for functionally-exclusive inositol pyrophosphate signaling. Proc Natl Acad Sci U S A 2020, 117: 3568–3574.
Meireles AM, Shiau CE, Guenther CA, Sidik H, Kingsley DM, Talbot WS. The phosphate exporter xpr1b is required for differentiation of tissue-resident macrophages. Cell Rep 2014, 8: 1659–1667.
Xu X, Li X, Sun H, Cao Z, Gao R, Niu T, et al. Murine placental-fetal phosphate dyshomeostasis caused by an Xpr1 deficiency accelerates placental calcification and restricts fetal growth in late gestation. J Bone Miner Res 2020, 35: 116–129.
Ansermet C, Moor MB, Centeno G, Auberson M, Hu DZ, Baron R, et al. Renal fanconi syndrome and hypophosphatemic rickets in the absence of xenotropic and polytropic retroviral receptor in the nephron. J Am Soc Nephrol 2017, 28: 1073–1078.
Wilson MS, Jessen HJ, Saiardi A. The inositol hexakisphosphate kinases IP6K1 and-2 regulate human cellular phosphate homeostasis, including XPR1-mediated phosphate export. J Biol Chem 2019, 294: 11597–11608.
Mailer RK, Allende M, Heestermans M, Schweizer M, Deppermann C, Frye M, et al. Xenotropic and polytropic retrovirus receptor 1 regulates procoagulant platelet polyphosphate. Blood 2021, 137: 1392–1405.
Ghosh S, Shukla D, Suman K, Lakshmi BJ, Manorama R, Kumar S, et al. Inositol hexakisphosphate kinase 1 maintains hemostasis in mice by regulating platelet polyphosphate levels. Blood 2013, 122: 1478–1486.
Wild R, Gerasimaite R, Jung JY, Truffault V, Pavlovic I, Schmidt A, et al. Control of eukaryotic phosphate homeostasis by inositol polyphosphate sensor domains. Science 2016, 352: 986–990.
Gerasimaite R, Pavlovic I, Capolicchio S, Hofer A, Schmidt A, Jessen HJ, et al. Inositol pyrophosphate specificity of the SPX-dependent polyphosphate polymerase VTC. ACS Chem Biol 2017, 12: 648–653.
Abbasian N, Harper MT. High extracellular phosphate increases platelet polyphosphate content. Platelets 2021, 32: 992–994.
Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci 2011, 12: 723–738.
Niu B, Zhang T, Hu H, Cao B. Transcriptome sequencing reveals astrocytes as a therapeutic target in heat-stroke. Neurosci Bull 2017, 33: 627–640.
Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57: 178–201.
Yu X, Ji C, Shao A. Neurovascular unit dysfunction and neurodegenerative disorders. Front Neurosci 2020, 14: 334.
Armulik A, Genové G, Mäe M, Nisancioglu MH, Wallgard E, Niaudet C, et al. Pericytes regulate the blood–brain barrier. Nature 2010, 468: 557–561.
Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, et al. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68: 409–427.
Daneman R, Zhou L, Kebede AA, Barres BA. Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature 2010, 468: 562–566.
Lindahl P, Johansson BR, Levéen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997, 277: 242–245.
Hellström M, Gerhardt H, Kalén M, Li X, Eriksson U, Wolburg H, et al. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol 2001, 153: 543–553.
Tallquist MD, French WJ, Soriano P. Additive effects of PDGF receptor beta signaling pathways in vascular smooth muscle cell development. PLoS Biol 2003, 1: E52.
Hutchins JB, Jefferson VE. Developmental distribution of platelet-derived growth factor in the mouse central nervous system. Brain Res Dev Brain Res 1992, 67: 121–135.
Ishii Y, Oya T, Zheng L, Gao Z, Kawaguchi M, Sabit H, et al. Mouse brains deficient in neuronal PDGF receptor-beta develop normally but are vulnerable to injury. J Neurochem 2006, 98: 588–600.
Tallquist MD, Klinghoffer RA, Heuchel R, Mueting-Nelsen PF, Corrin PD, Heldin CH, et al. Retention of PDGFR-beta function in mice in the absence of phosphatidylinositol 3’-kinase and phospholipase Cgamma signaling pathways. Genes Dev 2000, 14: 3179–3190.
Fredriksson L, Li H, Eriksson U. The PDGF family: Four gene products form five dimeric isoforms. Cytokine Growth Factor Rev 2004, 15: 197–204.
Sanchez-Contreras M, Baker MC, Finch NA, Nicholson A, Wojtas A, Wszolek ZK, et al. Genetic screening and functional characterization of PDGFRB mutations associated with basal ganglia calcification of unknown etiology. Hum Mutat 2014, 35: 964–971.
Arts FA, Velghe AI, Stevens M, Renauld JC, Essaghir A, Demoulin JB. Idiopathic basal ganglia calcification-associated PDGFRB mutations impair the receptor signalling. J Cell Mol Med 2015, 19: 239–248.
Vanlandewijck M, Lebouvier T, Andaloussi Mäe M, Nahar K, Hornemann S, Kenkel D, et al. Functional characterization of germline mutations in PDGFB and PDGFRB in primary familial brain calcification. PLoS One 2015, 10: e0143407.
Kakita A, Suzuki A, Nishiwaki K, Ono Y, Kotake M, Ariyoshi Y, et al. Stimulation of Na-dependent phosphate transport by platelet-derived growth factor in rat aortic smooth muscle cells. Atherosclerosis 2004, 174: 17–24.
Villa-Bellosta R, Levi M, Sorribas V. Vascular smooth muscle cell calcification and SLC20 inorganic phosphate transporters: Effects of PDGF, TNF-alpha, and Pi. Pflugers Arch 2009, 458: 1151–1161.
Takase N, Inden M, Murayama Y, Mishima A, Kurita H, Hozumi I. PDGF-BB is involved in phosphate regulation via the phosphate transporters in human neuroblastoma SH-SY5Y cells. Biochem Biophys Res Commun 2022, 593: 93–100.
Levéen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes Dev 1994, 8: 1875–1887.
Soriano P. Abnormal kidney development and hematological disorders in PDGF beta-receptor mutant mice. Genes Dev 1994, 8: 1888–1896.
Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, et al. Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev 2003, 17: 1835–1840.
Datta K, Guan T, Gerace L. NET37, a nuclear envelope transmembrane protein with glycosidase homology, is involved in myoblast differentiation. J Biol Chem 2009, 284: 29666–29676.
McConnell HL, Li Z, Woltjer RL, Mishra A. Astrocyte dysfunction and neurovascular impairment in neurological disorders: Correlation or causation? Neurochem Int 2019, 128: 70–84.
Schottlaender LV, Abeti R, Jaunmuktane Z, MacMillan C, Chelban V, O’Callaghan B, et al. Bi-allelic JAM2 variants lead to early-onset recessive primary familial brain calcification. Am J Hum Genet 2020, 106: 412–421.
Arrate MP, Rodriguez JM, Tran TM, Brock TA, Cunningham SA. Cloning of human junctional adhesion molecule 3 (JAM3) and its identification as the JAM2 counter-receptor. J Biol Chem 2001, 276: 45826–45832.
Mochida GH, Ganesh VS, Felie JM, Gleason D, Hill RS, Clapham KR, et al. A homozygous mutation in the tight-junction protein JAM3 causes hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. Am J Hum Genet 2010, 87: 882–889.
O’Driscoll MC, Daly SB, Urquhart JE, Black GCM, Pilz DT, Brockmann K, et al. Recessive mutations in the gene encoding the tight junction protein occludin cause band-like calcification with simplified gyration and polymicrogyria. Am J Hum Genet 2010, 87: 354–364.
Wyss L, Schäfer J, Liebner S, Mittelbronn M, Deutsch U, Enzmann G, et al. Junctional adhesion molecule (JAM)-C deficient C57BL/6 mice develop a severe hydrocephalus. PLoS One 2012, 7: e45619.
Akawi NA, Canpolat FE, White SM, Quilis-Esquerra J, Morales Sanchez M, Gamundi MJ, et al. Delineation of the clinical, molecular and cellular aspects of novel JAM3 mutations underlying the autosomal recessive hemorrhagic destruction of the brain, subependymal calcification, and congenital cataracts. Hum Mutat 2013, 34: 498–505.
Lamagna C, Meda P, Mandicourt G, Brown J, Gilbert RJC, Jones EY, et al. Dual interaction of JAM-C with JAM-B and alpha(M)beta2 integrin: Function in junctional complexes and leukocyte adhesion. Mol Biol Cell 2005, 16: 4992–5003.
Garrido-Urbani S, Bradfield PF, Imhof BA. Tight junction dynamics: The role of junctional adhesion molecules (JAMs). Cell Tissue Res 2014, 355: 701–715.
Neumann MA. Iron and calcium dysmetabolism in the brain with special predilection for globus pallidus and cerebellum. J Neuropathol Exp Neurol 1963, 22: 148–163.
Duckett S, Galle P, Escourolle R, Poirier J, Hauw JJ. Presence of zinc, aluminum, magnesium in striopalledodentate (SPD) calcifications (Fahr’s disease): Electron probe study. Acta Neuropathol 1977, 38: 7–10.
Jensen N, Schrøder HD, Hejbøl EK, Thomsen JS, Brüel A, Larsen FT, et al. Mice knocked out for the primary brain calcification-associated gene Slc20a2 show unimpaired prenatal survival but retarded growth and nodules in the brain that grow and calcify over time. Am J Pathol 2018, 188: 1865–1881.
Hozumi I, Kohmura A, Kimura A, Hasegawa T, Honda A, Hayashi Y, et al. High levels of copper, zinc, iron and magnesium, but not calcium, in the cerebrospinal fluid of patients with Fahr’s disease. Case Rep Neurol 2010, 2: 46–51.
Cuajungco MP, Lees GJ. Zinc metabolism in the brain: Relevance to human neurodegenerative disorders. Neurobiol Dis 1997, 4: 137–169.
Harris ED. Basic and clinical aspects of copper. Crit Rev Clin Lab Sci 2003, 40: 547–586.
Hozumi I, Hasegawa T, Honda A, Ozawa K, Hayashi Y, Hashimoto K, et al. Patterns of levels of biological metals in CSF differ among neurodegenerative diseases. J Neurol Sci 2011, 303: 95–99.
Craddock TJ, Tuszynski JA, Chopra D, Casey N, Goldstein LE, Hameroff SR, et al. The zinc dyshomeostasis hypothesis of Alzheimer’s disease. PLoS One 2012, 7: e33552.
Penke L, Valdés Hernandéz MC, Maniega SM, Gow AJ, Murray C, Starr JM, et al. Brain iron deposits are associated with general cognitive ability and cognitive aging. Neurobiol Aging 2012, 33: 510-517.e2.
Daugherty AM, Haacke EM, Raz N. Striatal iron content predicts its shrinkage and changes in verbal working memory after two years in healthy adults. J Neurosci 2015, 35: 6731–6743.
Acosta-Cabronero J, Betts MJ, Cardenas-Blanco A, Yang S, Nestor PJ. In vivo MRI mapping of brain iron deposition across the adult lifespan. J Neurosci 2016, 36: 364–374.
Shams M, Martola J, Charidimou A, Granberg T, Ferreira D, Westman E, et al. Cerebrospinal fluid metals and the association with cerebral small vessel disease. J Alzheimer’s Dis 2020, 78: 1229–1236.
August A, Schmidt N, Klingler J, Baumkötter F, Lechner M, Klement J, et al. Copper and zinc ions govern the trans-directed dimerization of APP family members in multiple ways. J Neurochem 2019, 151: 626–641.
Nahar K, Lebouvier T, Mäe MA, Konzer A, Bergquist J, Zarb Y, et al. Astrocyte-microglial association and matrix composition are common events in the natural history of primary familial brain calcification. Brain Pathol 2020, 30: 446–464.
Giachelli C. Vascular calcification mechanisms. J Am Soc Nephrol 2004, 15: 2959–2964.
Leopold JA. Vascular calcification: Mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc Med 2015, 25: 267–274.
Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, et al. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature 1997, 386: 78–81.
Boskey AL, Spevak L, Paschalis E, Doty SB, McKee MD. Osteopontin deficiency increases mineral content and mineral crystallinity in mouse bone. Calcif Tissue Int 2002, 71: 145–154.
Jahnen-Dechent W, Heiss A, Schäfer C, Ketteler M. Fetuin-A regulation of calcified matrix metabolism. Circ Res 2011, 108: 1494–1509.
Kawasaki K, Weiss KM. Evolutionary genetics of vertebrate tissue mineralization: The origin and evolution of the secretory calcium-binding phosphoprotein family. J Exp Zool B Mol Dev Evol 2006, 306: 295–316.
Alder J, Thakker-Varia S, Bangasser DA, Kuroiwa M, Plummer MR, Shors TJ, et al. Brain-derived neurotrophic factor-induced gene expression reveals novel actions of VGF in hippocampal synaptic plasticity. J Neurosci 2003, 23: 10800–10808.
Weyer SW, Klevanski M, Delekate A, Voikar V, Aydin D, Hick M, et al. APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J 2011, 30: 2266–2280.
Feldman SA, Eiden LE. The chromogranins: Their roles in secretion from neuroendocrine cells and as markers for neuroendocrine neoplasia. Endocr Pathol 2003, 14: 3–23.
Turk V, Stoka V, Vasiljeva O, Renko M, Sun T, Turk B, et al. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim Biophys Acta 2012, 1824: 68–88.
Cowles EA, DeRome ME, Pastizzo G, Brailey LL, Gronowicz GA. Mineralization and the expression of matrix proteins during in vivo bone development. Calcif Tissue Int 1998, 62: 74–82.
Feng X. Chemical and biochemical basis of cell-bone matrix interaction in health and disease. Curr Chem Biol 2009, 3: 189–196.
Houben E, Neradova A, Schurgers LJ, Vervloet M. The influence of phosphate, calcium and magnesium on matrix Gla-protein and vascular calcification: A systematic review. G Ital Nefrol 2016, 33: gin/33.65.
Zarb Y, Weber-Stadlbauer U, Kirschenbaum D, Kindler DR, Richetto J, Keller D, et al. Ossified blood vessels in primary familial brain calcification elicit a neurotoxic astrocyte response. Brain 2019, 142: 885–902.
Zarb Y, Sridhar S, Nassiri S, Utz SG, Schaffenrath J, Maheshwari U, et al. Microglia control small vessel calcification via TREM2. Sci Adv 2021, 7: eabc4898.
Lee HN, Jeong MS, Jang SB. Molecular characteristics of amyloid precursor protein (APP) and its effects in cancer. Int J Mol Sci 2021, 22: 4999.
Quinn JP, Kandigian SE, Trombetta BA, Arnold SE, Carlyle BC. VGF as a biomarker and therapeutic target in neurodegenerative and psychiatric diseases. Brain Commun 2021, 3: fca261.
Zhang X, Zhao F, Wang C, Zhang J, Bai Y, Zhou F, et al. AVP(4–8) improves cognitive behaviors and hippocampal synaptic plasticity in the APP/PS1 mouse model of Alzheimer’s disease. Neurosci Bull 2020, 36: 254–262.
Stapledon CJM, Stamenkov R, Cappai R, Clark JM, Bourke A, Solomon LB, et al. Relationships between the bone expression of Alzheimer’s disease-related genes, bone remodelling genes and cortical bone structure in neck of femur fracture. Calcif Tissue Int 2021, 108: 610–621.
Cui S, Xiong F, Hong Y, Jung JU, Li XS, Liu JZ, et al. APPswe/Aβ regulation of osteoclast activation and RAGE expression in an age-dependent manner. J Bone Miner Res 2011, 26: 1084–1098.
Li S, Liu B, Zhang L, Rong L. Amyloid beta peptide is elevated in osteoporotic bone tissues and enhances osteoclast function. Bone 2014, 61: 164–175.
Udagawa N, Takahashi N, Akatsu T, Tanaka H, Sasaki T, Nishihara T, et al. Origin of osteoclasts: Mature monocytes and macrophages are capable of differentiating into osteoclasts under a suitable microenvironment prepared by bone marrow-derived stromal cells. Proc Natl Acad Sci U S A 1990, 87: 7260–7264.
Kos J, Sekirnik A, Premzl A, Zavašnik Bergant V, Langerholc T, Repnik U, et al. Carboxypeptidases cathepsins X and B display distinct protein profile in human cells and tissues. Exp Cell Res 2005, 306: 103–113.
Staudt ND, Aicher WK, Kalbacher H, Stevanovic S, Carmona AK, Bogyo M, et al. Cathepsin X is secreted by human osteoblasts, digests CXCL-12 and impairs adhesion of hematopoietic stem and progenitor cells to osteoblasts. Haematologica 2010, 95: 1452–1460.
Lemos RR, Ferreira JBMM, Keasey MP, Oliveira JRM (2013) An update on primary familial brain calcification. International Review of Neurobiology, Elsevier, Amsterdam, pp 349–371.
Plemeniti Tololeski B, Debeljak M, Perkovič Benedik M, Rigler T, Kyriakopoulos M, Kotnik P, et al. The use of quetiapine in treatment of acute psychotic symptoms in an adolescent patient with primary brain calcification: A case report. BMC Psychiatry 2019, 19: 67.
Räkel A, Boucher A, Ste-Marie LG. Role of zoledronic acid in the prevention and treatment of osteoporosis. Clin Interv Aging 2011, 6: 89–99.
Loeb JA. Functional improvement in a patient with cerebral calcinosis using a bisphosphonate. Mov Disord 1998, 13: 345–349.
Loeb JA, Sohrab SA, Huq M, Fuerst DR. Brain calcifications induce neurological dysfunction that can be reversed by a bone drug. J Neurol Sci 2006, 243: 77–81.
Oliveira JR, Oliveira MF. Primary brain calcification in patients undergoing treatment with the biphosphanate alendronate. Sci Rep 2016, 6: 22961.
Ferreira JB, Pimentel L, Keasey MP, Lemos RR, Santos LM, Oliveira MF, et al. First report of a de novo mutation at SLC20A2 in a patient with brain calcification. J Mol Neurosci 2014, 54: 748–751.
Kalueff A, Loseva E, Haapasalo H, Rantala I, Keranen J, Lou YR, et al. Thalamic calcification in vitamin D receptor knockout mice. Neuroreport 2006, 17: 717–721.
Keasey MP, Lemos RR, Hagg T, Oliveira JRM. Vitamin-D receptor agonist calcitriol reduces calcification in vitro through selective upregulation of SLC20A2 but not SLC20A1 or XPR1. Sci Rep 2016, 6: 25802.
Peltz SW, Morsy M, Welch EM, Jacobson A. Ataluren as an agent for therapeutic nonsense suppression. Annu Rev Med 2013, 64: 407–425.
Peters MEM, de Brouwer EJM, Bartstra JW, Mali W, Koek HL, Rozemuller AJM, et al. Mechanisms of calcification in Fahr disease and exposure of potential therapeutic targets. Neurol Clin Pract 2020, 10: 449–457.
Westenberger A, Balck A, Klein C. Primary familial brain calcifications: Genetic and clinical update. Curr Opin Neurol 2019, 32: 571–578.
Acknowledgements
This review was supported by the National Natural Science Foundation of China (31871262), the Shanghai Municipal Science and Technology Major Project (2018SHZDZX05), and the Innovation Incentive Foundation (Center for Excellence in Brain Science and Intelligence Technology, Chinese Academy of Sciences, Shanghai, China).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Xu, X., Sun, H., Luo, J. et al. The Pathology of Primary Familial Brain Calcification: Implications for Treatment. Neurosci. Bull. 39, 659–674 (2023). https://doi.org/10.1007/s12264-022-00980-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-022-00980-0