
Abstract
Coronaviruses (CoVs) are important human and animal pathogens that cause respiratory and gastrointestinal diseases. Porcine epidemic diarrhoea (PED), characterized by severe diarrhoea and vomiting in pigs, is a highly lethal disease caused by porcine epidemic diarrhoea virus (PEDV) and causes substantial losses in the swine industry worldwide. However, currently available commercial drugs have not shown great therapeutic effects. In this study, a fluorescence resonance energy transfer (FRET)-based assay was applied to screen a library containing 1,590 compounds and identified two compounds, 3-(aminocarbonyl)-1-phenylpyridinium and 2,3-dichloronaphthoquinone, that target the 3C-like protease (3CLpro) of PEDV. These compounds are of low molecular weight (MW) and greatly inhibited the activity of this enzyme (IC50 values were obtained in this study). Furthermore, these compounds exhibited antiviral capacity against another member of the CoV family, feline infectious peritonitis virus (FIPV). Here, the inhibitory effects of these compounds against CoVs on Vero cells and feline kidney cells were identified (with EC50 values) and cell viability assays were performed. The results of putative molecular docking models indicate that these compounds, labeled compound 1 and compound 2, contact the conserved active sites (Cys144, Glu165, Gln191) of 3CLpro via hydrogen bonds. These findings provide insight into the antiviral activities of compounds 1 and 2 that may facilitate future research on anti-CoV drugs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Coronaviruses (CoVs) are positive-sense, single-stranded enveloped RNA viruses that can cause respiratory, enteric, hepatic, or neurological infections in a variety of animal species, including bats, birds, cats, dogs, pigs, mice, horses, whales, and humans (St John et al. 2015). The Coronaviridae family is divided into four genera (Alpha-, Beta-, Gamma-, and Deltacoronavirus). Porcine epidemic diarrhoea virus (PEDV) and feline CoV (FCoV) belong to the genus Alphacoronavirus (α-CoV) (Cui et al. 2019).
PEDV is the causative agent of porcine epidemic diarrhoea (PED), which is characterized by acute gastroenteritis in neonatal piglets, the clinical signs of which include vomiting, diarrhoea, and dehydration. PED was first reported in Belgium and the United Kingdom in 1978 (Wood 1977; Pensaert and de Bouck 1978) and was described then as “epidemic viral diarrhoea (EVD)”; it subsequently emerged across Europe and Asia (Choudhury et al. 2016; Pensaert and Martelli 2016). The last outbreak of PED in China occurred in 2010 and was caused by a variant strain that led to tremendous economic losses in the swine industry (Li et al. 2012). The efficiencies of vaccines based on classic strains are greatly challenged due to the hypervariability of PEDV, which has made field pandemics complex (Langel et al. 2016; Wang et al. 2016; Gerdts and Zakhartchouk 2017). Feline infectious peritonitis virus (FIPV) is a virulent mutant of FCoV (Pedersen 2009). In cats, FIPV causes a fatal infectious disease characterized by phlebitis and granulomatous to pyogranulomatous lesions in various tissues and organs (Kipar and Meli 2014). The ability of CoVs to transmit across species barriers has been evident since the outbreaks of severe acute respiratory syndrome (SARS) in 2003 and Middle-East respiratory syndrome (MERS) in 2012 (Schoeman and Fielding 2019). In addition, several research groups have revealed that the genome of SARS-CoV-2 is highly similar to that of bat CoV (Guo et al. 2020). The emergence and re-emergence of CoVs strongly signal that coronaviruses are able to escape from current vaccination protocols and biosecurity and control systems (Jung and Saif 2015; Wang et al. 2019). With the ongoing COVID-19 situation, the development and optimization of new broad-spectrum antiviral drugs cannot be delayed. Attempts have been made to discover effective treatments for CoV infections, but there are few effective commercial veterinary drugs available (Pedersen 2014; Kim et al. 2016).
The nonstructural protein nsp5 is a 3C-like protease (3CLpro) of CoV that cleaves the majority of synthesized polyproteins at 11 conserved sites (Ziebuhr 2005; Stobart et al. 2012; Yang et al. 2014). Because of its highly conserved sequence and the necessity for viral replication, the active site of 3CLpro represents an attractive target for anti-CoV drug development to treat infectious diseases caused by CoV infections, including SARS, MERS and COVID-19 (Grum-Tokars et al. 2008; Kankanamalage et al. 2018; Jo et al. 2020). One successful approach in treating animal infectious diseases by targeting 3CLpro is a peptidyl compound (GC376) that effectively inhibits FCoV (Kim et al. 2013; Perera et al. 2019); this compound has recently been reported to be broadly effective against ferret and mink CoVs as well (Perera et al. 2018). However, the increasing need to develop novel strategies to prevent and treat fatal diseases caused by CoVs has promoted additional vaccine and drug research.
In this study, we established a FRET-based in vitro system to screen a library containing 1590 compounds and identified two hit compounds, compound 1 and compound 2, that target the 3CLpro of PEDV and inhibit its enzyme activity. Our results revealed broad-spectrum inhibitory effects of compounds 1 and 2 against PEDV and FIPV replication in cell culture, and docking in the 3CL protease to predict the possible binding model provides new insight into structure-based design and synthesis of new antiviral drugs to treat swine CoVs and feline CoVs.
Materials and Methods
Expression and Purification of 3CLpro of PEDV
The full-length 3CLpro was encoded by cDNAs of the PEDV YN144 strain (Chen et al. 2015) (NCBI accession No. KT021232) with a sequence encoding 6 His residues at the C-terminus and was inserted between the NdeI and BamHI restriction sites. The synthesized gene was PCR amplified and subcloned into pET-42b vector. Each construct was validated through DNA sequencing, and each 3CLpro was expressed and purified following standard procedures previously described by our laboratory (Ye et al. 2016). In brief, the integrated vector was transformed into Escherichia coli BL21, and the cells were cultured at 37 °C in LB medium with 50 mg/L kanamycin. Once the OD600 reached 0.8, we added 1 mmol/L IPTG to the culture. The culture was then incubated at 37 °C for 4–5 h before harvesting. The harvested protein was concentrated and filtered using a Superdex 200 gel filtration column (GE Healthcare). The storage volume of 3CLpro was 10 μL per tube.
Cells and Viruses
African green monkey cells (Vero cells; CCL-81) and crandell reese feline kidney feline kidney cells (CRFK; CCL-94) were purchased from the American Type Culture Collection (ATCC, Manassas, VA). Cells were grown in Dulbecco’s minimum essential medium (DMEM, HyClone) supplemented with 10% foetal bovine serum (FBS, Gibco), 100 U/mL penicillin and 100 µg/mL streptomycin and maintained in a 5% CO2 incubator at 37 °C. The PEDV YN144 strain (GenBank Accession No. KT021232) and FIPV WSU-79/1146 strain (GenBank Accession No. DQ010921.1) were used in all assays.
Compounds
For high-throughput antiviral inhibitor screening, we obtained a low-molecular-weight (low-MW) fragment-based library containing over 1,500 compounds (Specs, Delf, Netherlands). Each of the compounds in the library is defined as a small molecule that obeys the Congreve ‘rule of three’, for which the MW is < 300, the number of hydrogen bond donors and acceptors is ≤ 3, and cLogP is ≤ 3 (Congreve et al. 2003).
FRET assay and IC50 Measurement
The screening of protease inhibitors was based on a FRET assay, following previously described procedures (Shi et al. 2016). The polypeptide substrate was Dabcyl-TSAVLQ↓SGFRKM-Edans, which was synthesized by GenScript Biotech Corporation, Jiangsu, PRC. The cleavage activity with a series of PEDV 3CLpro concentrations was determined with FRET assays. We confirmed the final protease concentration to be 100 nmol/L. Compounds and substrate were used at final concentrations of 1.25 mmol/L and 40 µmol/L, respectively. The mixtures of 3CLpro, substrate and compounds were added to black 96-well plates and incubated for 30 min at 37 °C. Fluorescence readings were recorded at 0 min and 30 min, with 340 nm excitation and 485 nm emission. The relative fluorescence values were calculated by subtracting background fluorescence (fluorescence of a control well without protease). The percentage of inhibition was calculated as follows: percentage of inhibition (%) = 100 × [1 − RFU of the experimental group (30–0 min)/RFU of the control group (30–0 min)]. The calculation of the 50% inhibitory concentration (IC50) was performed in GraphPad Prism software 7.0 (GraphPad Software Inc., San Diego, CA, USA) following the methods described in our previous report.
Antiviral Effects of Compounds in Cell Culture
Vero cells and CRFK cells were grown in 6-well plates to 80%–90% confluency. The medium of Vero cells was changed to DMEM (5% trypsin) without serum immediately prior to infection. Serial dilutions of compound (25, 12.5, 6.25, and 3.125 µmol/L) and mock treatment (medium only) were added to the wells, and the cells were immediately inoculated with PEDV/FIPV at a multiplicity of infection (MOI) of 0.1. Vero cells and CRFK cells developed visible cytopathic effects (CPEs), which were characterized by multiple regional cell fusion and syncytium formation (Hofmann and Wyler 1988; Chen et al. 2015). The infected cells were then incubated at 37 °C for 16–20 h until CPEs appeared. The supernatant of each well was harvested and frozen at −80 °C for virus titre determination. Briefly, Vero cells were seeded in 96-well plates and infected with tenfold dilutions of the thawed supernatants collected earlier. Each sample was analyzed in 8 replicates. The plates were incubated for 72–96 h until no further CPE was observed. The viral titres were measured using the Spearman-Karber 50% tissue culture infectious dose (TCID50) method. The 50% effective concentration (EC50) values were calculated using GraphPad Prism software 7.
Indirect Fluorescence Assay
To observe the antiviral effects of the compounds on infected cells, we performed indirect immunofluorescence assays (IFAs). Vero cells and CRFK cells were seeded in 96-well plates. Infection with PEDV/FIPV was performed following the same method used in the TCID50 assays. The positive controls were infected cells without compound treatment (exposure to medium only). The cells were washed with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde solution for 15 min, and the membrane was permeabilized using 0.2% Triton X-100. Cells were then blocked with 5% bovine serum albumin (BSA) at 37 °C for 30 min. Subsequently, an anti-nucleocapsid (N) protein rabbit monoclonal antibody prepared and preserved by our laboratory (Xie et al. 2019; Ye et al. 2020) was added (1:1000), and the cells were incubated for 1 h at 37 °C. After the monoclonal antibody was removed, goat anti-rabbit IgG with a fluorescein isothiocyanate (FITC) tag (ThermoFisher, Waltham, MA, USA) was added (1:500), and the samples were incubated for 45 min. The nuclei were stained with 4',6-diamidino-2-phenylindole (DAPI; 1:5000; Sigma) for 5 min at room temperature before being washed with PBS. The samples were observed and imaged under a fluorescence microscope.
Cytotoxicity Assay
The viabilities of Vero cells and CRFK cells were measured by cytotoxicity assay kit (Beyotime Biotechnology, Shanghai, PRC) following the manufacturer’s protocol. Briefly, cells were seeded in 96-well plates at 1 × 105 cells and incubated for up to 16 h. Upon reaching confluence, the cells were treated with the compounds at a series of concentrations for 72 h. Next, 100 µL/well CellTiter-Lumi™ reagent was added, and the samples were incubated for 10 min at room temperature (approximately 25 °C). The 50% cell death toxic concentrations (CC50) were calculated using GraphPad Prism software 7.0.
Western Blot Analysis
Vero cells and CRFK cells were cultured in 6-well plates. When the density of cells reached 5 × 105–1 × 106 cells per well, PEDV/FIPV was added at an MOI of 0.1. Cells were then treated with compounds at various concentrations (25, 12.5, 6.25, and 3.125 µmol/L). The positive control well contained virus and 0.4% DMSO. The plates were incubated for 16 h. When CPEs were observed under the microscope, both the infected cells and control cells were lysed in lysis buffer (20 mmol/L Tris [pH 7.5], 150 mmol/L NaCl, 1% Triton X-100) containing proteinase inhibitors (Beyotime Biotechnology, Shanghai, PRC). Then, the supernatants were harvested, and each sample was analyzed by 12% SDS-PAGE. After transfer to PVDF membranes, the protein samples were blocked using 5% dried skim milk. The membranes were incubated overnight with an anti-GAPDH monoclonal antibody (1:5000; Proteintech, Hubei, PRC). A goat anti-mouse secondary antibody (1:5000; Boster Biological Technology Co., Ltd., PRC) was used to detect GAPDH. Then, the membranes were incubated for 1 h with an anti-N protein rabbit monoclonal antibody (1:5000). A goat anti-rabbit monoclonal antibody was used as the secondary antibody to detect the N protein. The monoclonal antibodies were prepared and preserved by our laboratory (Xie et al. 2019; Ye et al. 2020). Antigen–antibody reactions were detected using an enhanced chemiluminescence system (Amersham Imager 600, GE Healthcare).
Results
Identification of Two Inhibitors against PEDV 3CLpro
Two low-MW compounds, 3-(aminocarbonyl)-1-phenylpyridinium (compound 1, Specs ID number: AC-907/25005189) and 2,3-dichloronaphthoquinone (compound 2, Specs ID number: AQ-390/13304011) were identified after the preliminary screening of 1,590 compounds from the library. The final concentrations of protease and compounds were 100 nmol/L and 1.25 mmol/L, respectively. During the FRET assays, these two compounds exhibited the ability to inhibit over 90% of the protease activity of PEDV 3CLpro. The percentage of inhibition for the 1,590 compounds is shown in Fig. 1A. The chemical structures of compounds 1 and 2 are illustrated in Fig. 1B.

Screening of and information on compounds 1 and 2: A High-throughput screening for inhibitors targeting PEDV 3CLpro from a library containing 1590 low-MW compounds was conducted. Each dot represents the percentage of inhibition of a compound. Compounds and substrate were used at final concentrations of 1.25 mmol/L and 40 µmol/L, respectively. B The structures and chemical names of compounds 1 and 2.
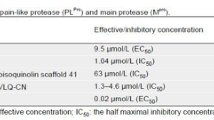
We then evaluated the inhibitory effects of the compounds at various concentrations against PEDV 3CLpro, with a DMSO control. The IC50 values of compounds 1 and 2 against 3CLpro were determined at a protease concentration of 100 nmol/L (Fig. 2A). The results indicated that both compounds exhibited concentration-dependent inhibitory activities. Compound 1 showed inhibitory activity against 3CLpro at concentrations of 0.39–12.5 µmol/L, and compound 2 exhibited inhibitory activity at 3.125–200 µmol/L. In addition, the IC50 values of compounds 1 and 2 were 0.1877 µmol/L and 6.765 µmol/L, respectively, indicating that compound 1 exhibited stronger inhibitory activity than compound 2.

Dose response for compounds 1 and 2 in suppressing PEDV 3CLpro activity and PEDV replication in Vero cells. Each concentration in the dose–response experiment was assayed in triplicate: A FRET assays were performed to measure the IC50 values of compounds 1 and 2, reflecting their ability to inhibit PEDV 3CLpro activity. B Cell viability analysis of compound 1 (6.25–100 µmol/L) and compound 2 (3.125–50 µmol/L) in Vero cells. C Inhibitory effects of compound 1 (3.125–25 µmol/L) and compound 2 (3.125–25 µmol/L) in PEDV-infected cells at an MOI of 0.1. Cell culture supernatant was harvested for TCID50 assays after incubation with serial dilutions of the test compounds for 16 h. D Dose–response curves determing the IC50 and EC50 values of compounds 1 and 2 by nonlinear regression. Bars represent the SD from triplicate trials (* represents a significant difference between test concentration and control; *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001).
Antiviral Ability of Compounds 1 and 2 against PEDV
To further study the antiviral activities of compounds 1 and 2 against PEDV, each compound was added to PEDV-infected Vero cells at serial concentrations of 3.125 µmol/L, 6.25 µmol/L, 12.5 µmol/L, and 25 µmol/L (Fig. 2C). We then obtained the TCID50 values of each concentration (with analyses performed in triplicate) and identified the dose-dependent inhibition of each compound against PEDV in Vero cells. Since 3CLpro plays an important role in PEDV replication and is the protein targeted by compounds 1 and 2, we concluded that these two compounds effectively decreased the viral titre at 3.125–25 µmol/L by blocking the enzyme activity of 3CLpro during PEDV replication. Significantly, the EC50 value of compound 1 (28.63 µmol/L) was lower than its CC50 value (73.8 µmol/L) obtained in Vero cells (Fig. 2B), which indicated its ability to suppress CoV replication with little cytotoxicity. Compound 2 showed moderate inhibitory effects against PEDV, with an EC50 value of 38.45 µmol/L; its CC50 value (21.79 µmol/L) was lower than its EC50 value in Vero cells, indicating potential cytotoxicity of compound 2 to Vero cells. However, the EC50 value of compound 1 was 28.63 µmol/L, and cell viability was higher than 80% in the presence of this compound at 25 µmol/L. Hence, 25 µmol/L might be an effective working concentration of compound 1 for inhibiting viral replication in vivo.
We performed an IFA and Western blotting to verify the results mentioned above. The images captured under fluorescence microscope (Fig. 3A) showed that compounds 1 and 2 each inhibited PEDV replication in a dose-dependent manner. There was no typical CPE of PEDV infection observed at a concentration of 25 µmol/L after 16 h of infection, and the amounts of infected cells at 12.5, 6.25 and 3.125 µmol/L significantly decreased compared to the amount with mock treatment (i.e., without compound 1 or 2 added). Additionally, the cytotoxic effect of compound 2 was evident, as the amount of DAPI-stained nuclei with compound 1 present was less than that with compound 2. The supernatant from infected cells in the presence or absence of the test compounds was collected to perform Western blotting (Fig. 3B). PEDV N protein level was decreased by compounds 1 and 2 in a dose-dependent manner, and no N protein was detectable with either compounds at 25 µmol/L or with compound 1 at 12.5 µmol/L. The concentration of N protein was moderately reduced in the presence of compound 2 at 12.5 µmol/L.

Dose response for compounds 1 and 2 in PEDV-infected Vero cells: A Immunofluorescence assay of compounds 1 and 2 against PEDV in Vero cells. PEDV-infected cells were used as a control group. Serial dilutions of the test compounds from 3.125 µmol/L to 25 µM were assessed independently. Nuclei were stained with DAPI (blue fluorescence), and PEDV infection was detected using a monoclonal antibody followed by FITC-tagged goat anti-rabbit IgG (green fluorescence). B Western blot analysis of the effects of compounds 1 and 2 on the production of CoV N protein in PEDV-infected cells. GAPDH was used as an internal control.
Compounds 1 and 2 are Potential Antivirals against Other α-CoVs
Given the presence of 3CLpro in all CoVs, we sought to determine the antiviral effects of compounds 1 and 2 against other members of the Alphacoronavirus genus. We performed similar antiviral assays with feline kidney cells (CRFK cells) against FIPV (Fig. 4). The EC50, CC50 and selectivity index (SI) values are presented in Table 1. In feline kidney cells (CRFK; CCL-94), compound 2 showed a greater capacity to inhibit FIPV replication than compound 1; the EC50 value of compound 2 was 46.03 µmol/L, whereas that of compound 1 was 59.66 µmol/L (Fig. 4A). The percentage inhibition achieved by compound 2 against FIPV at 25 µmol/L was 35%, whereas cell viability remained at 80% (Fig. 4A and 4B). Western blotting and immunofluorescence assays verified the ability of compounds 1 and 2 to suppress α-CoV replication (Figs. 3A and 3B, 4C and 4D).

A Cell viability analyses of compound 1 (6.25–100 µmol/L) and compound 2 (3.125–50 µmol/L) in CRFK cells. B Inhibitory effects of compound 1 (3.125–25 µmol/L) and compound 2 (3.125–25 µmol/L) in FIPV-infected cells at an MOI of 0.1. Cell culture supernatant was harvested for TCID50 assays after incubation with serial dilutions of test compounds for 20 h (* represents a significant difference between test concentration and control; *, P < 0.05; **, P < 0.01; ***, P < 0.001; and ****, P < 0.0001). (C) Immunofluorescence assay of compounds 1 and 2 against FIPV in CRFK cells. FIPV-infected cells were tested as a control group. Serial dilutions of the test compounds from 3.125 µM to 25 µmol/L were assessed independently. Nuclei were stained with DAPI (blue fluorescence), and FIPV infection was detected using a monoclonal antibody followed by FITC-tagged goat anti-rabbit IgG (green fluorescence). Bars represent the SD from triplicate trials. (D) Western blot analysis of the effects of compounds 1 and 2 on the production of CoV N protein in FIPV-infected cells. GAPDH was used as an internal control.
Potential Binding Sites
To elucidate the inhibitory mechanism of the inhibitors against the protease, PEDV 3CLpro crystals were soaked with various concentrations of compound 1 and compound 2 to obtain the protein-inhibitor complex. Unfortunately, we were unable to obtain complex structures. Thus, the potential binding sites were analyzed by molecular simulations based on the crystal structure of PEDV 3CLpro obtained by our laboratory (PDB code: 4ZUH) and the structure of the protein in complex with the effective 3CLpro inhibitor GC376 (PDB code: 6L70) (Kim et al. 2013; Ye et al. 2020a). Compounds 1 and 2 were docked into the PEDV 3CLpro structure, covering the S1 specificity binding pocket composed of Phe139, Ile140, Asn141, Gly142, Ala143, Cys144, His162, Gln163 and Glu165 (Ye et al. 2016). To simulate the most likely conformation between the ligand and the macromolecule (3CLpro), we selected the lowest energy conformations for further analysis. The simulated molecular docking results showed that compounds 1 and 2 formed extensive hydrogen bonds with the residues of the protease binding pocket (Fig. 5A and 5B).

In silico molecular docking of compounds 1 and 2 with PEDV 3CLpro. A Docked conformation of compound 1 with PEDV 3CLpro. The protease is shown via a charge-potential surface. The possible hydrogen bond interactions are presented as yellow dashed lines, and the bond distances are shown. The S1 and S4 pockets are labelled. B Docked conformation of compound 2 with PEDV 3CLpro. The compounds and interactions between compounds and binding sites are represented as in A. C Sequence alignment of various coronavirus 3CLpros. The amino acid sequence information was retrieved from the NCBI Database, and the sequence alignments are based on one virus strain each (PEDV-YN144: KT021232.1; HCoV-229E: KU291448.1; HCoV-NL63: NC_005831.2; FIPV-WSU-79/1146: AAY32595.1; TGEV-PUR-MAD: NC_038861.1; SARS-CoV-Tor2: NP_828863.1; SARS-CoV-2-Wuhan-Hu-1: YP_009742612.1; MERS-CoV isolate HCoV-EMC/2012: NC_019843.3; IBV-Beaudette: NP_740626.1; PDCoV/USA/Nebraska145/2015: ANI85845.1; SADS-CoV: QJF53984.1). The key residues for 3CLpro activity sites and the compounds' potential binding sites on PEDV are marked with blue arrows at the bottom. The sequences were aligned using ClustalW2, and images were constructed with ESPript3.0.
In the lowest energy conformations, compound 1 formed hydrogen bonds with Glu165, Gln187, Thr189, and Gln191, and compound 2 bound the active site residues Cys144 and Gly142. Because Glu165 consists of the S1 specificity binding pocket and Cys144 is a conserved active site of 3CLpro (Ye et al. 2016), the two compounds may inhibit PEDV replication via blockage of the recognition and binding of 3CLpro and its substrate.
Sequence alignment of the 3CLpros of multiple CoVs, including PEDV, human coronaviruses (HCoV), FIPV, transmissible gastroenteritis coronavirus (TGEV), SARS-CoV, SARS-CoV-2, MERS-CoV, infectious bronchitis virus (IBV), porcine deltacoronavirus (PDCoV) and swine acute diarrhea syndrome coronavirus (SADS-CoV), indicates that the active sites (His 41, Cys144) of CoV 3CLpros and the potential binding sites (Cys144, Glu165, Gln191) of the compounds in PEDV 3CLpro are conserved (Fig. 5C). This conservation explains the antiviral activity of compounds 1 and 2 against swine and feline CoVs.
Discussion
The viral proteases of CoVs that play essential roles in CoV replication are commonly recognized as ideal active sites in antiviral drug research. Several approaches have identified potent therapeutics for SARS-CoV and MERS-CoV in humans (Yang et al. 2006; Pfefferle et al. 2011; Kumar et al. 2017) and for FCoV (Kim et al. 2013; Theerawatanasirikul et al. 2020). However, studies on antiviral drugs against PEDV that target replication steps are very limited.
In this study, we used a FRET-based system to screen a low-MW compound library and identified two compounds, 3-(aminocarbonyl)-1-phenylpyridinium (compound 1) and 2,3-dichloronaphthoquinone (compound 2), that effectively inhibit 3CLpro activity in vitro, with IC50 values of 0.1877 µmol/L and 6.765 µmol/L, respectively. However, both EC50 values and Western blot results reflecting inhibitory activity against PEDV on cells were higher than protease inhibition concentrations. Subsequently, the two inhibitors were applied to FIPV-infected cells to determine the inhibitory activities of the compounds against other α-CoVs. We found that although compound 2 showed less inhibitory efficacy than compound 1 against PEDV replication (Figs. 2C, 3A and 3B), it exhibited greater inhibitory capacity than compound 1 against FIPV (Fig. 4B–4D). We speculate that these differences in antiviral effects are due to the different potential binding positions of the two inhibitors against 3CLpro (Fig. 5A and 5B) and the different pharmacokinetics of compounds in different cells (The CC50 values are presented in Table 1.). Toxicity predictions provided by the compounds library (Specs, Delf, Netherlands) suggest that compound 1 may have a lower toxic effect than compound 2 (rat oral LD50: compound 1, 3210 mg/kg; compound 2, 288 mg/kg). This suggestion is in line with our results regarding the cytotoxicity of the compounds in Vero cells and CRFK cells (Table 1). Animal experiments are required to evaluate the therapeutic effects and toxicity of both compounds to determine the possibility of modifying these protease inhibitors for novel drug development.
In 3CLpro, the substrate-binding site is located in a cleft between domain I and II, and the substrate-binding subsite S1 in protomer A confers absolute specificity for the substrate P1 residue on the enzyme (Pillaiyar et al. 2016). To elucidate the binding mechanism of each compound and 3CLpro, we docked compounds 1 and 2 into the substrate-binding pocket and simulated the most likely binding conformations. A previous study demonstrated that Cys144 and His41 residues act as the active site of PEDV 3CLpro, creating a catalytic dyad in which the cysteine functions as a common nucleophile in the proteolytic process (Ye et al. 2016). Indeed, our docking analysis results suggested that compound 2 potentially binds in the S1 subsite of PEDV 3CLpro and that the oxygen of the carbonyl group forms hydrogen bonds with residues Cys144 and Gly142 (Fig. 5B). Hence, compound 2 theoretically blocks the substrate combination and catalysis process, thereby inhibiting the enzyme's activity. In contrast, the molecular docking model of the compound 1 bound to 3CLpro showed that the binding orientation of compound 1 differs from that of compound 2 (Fig. 5A). The amido group of compound 1 on the side chain of the benzene ring occupies the S4 subsite of the substrate-binding pocket and forms hydrogen bonds with residues Gln187, Thr189, Gln191 and Glu165 (Fig. 5B). In light of these observations, we speculate that these two compounds serve as competitive inhibitors that replace the substrate at different substrate-binding sites of 3CLpro. Compared with compound 2, compound 1 potentially forms more covalent bonds with 3CLpro substrate-binding sites such that its dissociation from the protease is more difficult than that of compound 2. This mechanism may explain the different inhibitory effects of the two compounds against 3CLpro.
The inhibitors targeting the 3CLpro of CoVs that have been reported to date are mainly peptide inhibitors that mimic natural peptide substrates (Ghosh et al. 2005; Shie et al. 2005; Zhu et al. 2011). In particular, a series of tripeptide a,b-unsaturated esters and ketomethylene isosteres have been found to exhibit potent inhibitory activity, with IC50 values of 11–39 µmol/L against SARS-CoV 3CLpro and EC50 values of 0.11–18.86 µmol/L against SARS-CoV (Shie et al. 2005). However, compounds 1 and 2 identified in this study are previously unreported lead compounds with greater potency than other inhibitors against 3CLpro, with IC50 values of 0.1877 µmol/L and 6.765 µmol/L, respectively. The EC50 values of these two inhibitors, although higher than those of peptide-derived 3CLpro inhibitors, are nonetheless in the low micromolar range. Thus, we believe that these two novel lead compounds suggest possibilities for the design and optimization of peptidomimetic inhibitors targeting CoV 3CLpro. Among nonpeptidic 3CLpro inhibitors, amentoflavone is reported to be effective against SARS-CoV 3CLpro (IC50 = 8.3 µmol/L; MW = 538.46) (Ryu et al. 2010). In this regard, the inhibitors identified in this study show potential as a starting point in the development of new 3CLpro inhibitors with IC50 values and MWs that are lower than those of other nonpeptidic 3CLpro inhibitors (compound 1 MW: 199.23; compound 2 MW: 277.05). However, it should be mentioned that the use of drugs targeting specific viral proteases may induce viral mutations, therefore reduced the susceptibility of drugs (Mokaya et al. 2018; Omoto et al. 2018). In order to prevent drug resistance caused by virus mutations, consideration should be given to the combined use of drugs and post-treatment monitoring in the clinical studies. In addition, findings in this study, together with our previous report of structural details of PEDV 3CLpro in complex with the potent broad-spectrum inhibitor GC376 (Ye et al. 2020), experiments of optimization on GC376 and other relevant inhibitors are on the way of our future anti-CoV therapeutic study.
References
Chen F, Zhu Y, Wu M, Ku X, Ye S, Li Z, Guo X, He Q (2015) Comparative genomic analysis of classical and variant virulent parental/attenuated strains of porcine epidemic diarrhea virus. Viruses 7:5525–5538
Choudhury B, Dastjerdi A, Doyle N, Frossard JP, Steinbach F (2016) From the field to the lab-an European view on the global spread of PEDV. Virus Res 226:40–49
Congreve M, Carr R, Murray C, Jhoti H (2003) A ‘Rule of Three’ for fragment-based lead discovery? Drug Discov Today 8:876–877
Cui J, Li F, Shi ZL (2019) Origin and evolution of pathogenic coronaviruses. Nat Rev Microbiol 17:181–192
Gerdts V, Zakhartchouk A (2017) Vaccines for porcine epidemic diarrhea virus and other swine coronaviruses. Vet Microbiol 206:45–51
Ghosh AK, Xi K, Ratia K, Santarsiero BD, Fu W, Harcourt BH, Rota PA, Baker SC, Johnson ME, Mesecar AD (2005) Design and synthesis of peptidomimetic severe acute respiratory syndrome chymotrypsin-like protease inhibitors. J Med Chem 48:6767–6771
Grum-Tokars V, Ratia K, Begaye A, Baker SC, Mesecar AD (2008) Evaluating the 3C-like protease activity of SARS-Coronavirus: recommendations for standardized assays for drug discovery. Virus Res 133:63–73
Guo YR, Cao QD, Hong ZS, Tan YY, Chen SD, Jin HJ, Tan KS, Wang DY, Yan Y (2020) The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak - an update on the status. Mil Med Res 7:11
Hofmann M, Wyler R (1988) Propagation of the virus of porcine epidemic diarrhea in cell culture. J Clin Microbiol 26:2235–2239
Jo S, Kim S, Shin DH, Kim MS (2020) Inhibition of SARS-CoV 3CL protease by flavonoids. J Enzyme Inhib Med Chem 35:145–151
Jung K, Saif LJ (2015) Porcine epidemic diarrhea virus infection: Etiology, epidemiology, pathogenesis and immunoprophylaxis. Vet J 204:134–143
Kankanamalage ACG, Kim Y, Damalanka VC, Rathnayake AD, Fehr AR, Mehzabeen N, Battaile KP, Lovell S, Lushington GH, Perlman S, Chang KO, Groutas WC (2018) Structure-guided design of potent and permeable inhibitors of MERS coronavirus 3CL protease that utilize a piperidine moiety as a novel design element. Eur J Med Chem 150:334–346
Kim Y, Mandadapu SR, Groutas WC, Chang KO (2013) Potent inhibition of feline coronaviruses with peptidyl compounds targeting coronavirus 3C-like protease. Antiviral Res 97:161–168
Kim Y, Liu H, Kankanamalage ACG, Weerasekara S, Hua DH, Groutas WC, Chang KO, Pedersen NC (2016) Reversal of the progression of fatal coronavirus infection in cats by a broad-spectrum coronavirus protease inhibitor. PLoS Pathog 12:e1005531
Kipar A, Meli ML (2014) Feline infectious peritonitis: still an enigma? Vet Pathol 51:505–526
Kumar V, Shin JS, Shie JJ, Ku KB, Kim C, Go YY, Huang KF, Kim M, Liang PH (2017) Identification and evaluation of potent Middle East respiratory syndrome coronavirus (MERS-CoV) 3CL(Pro) inhibitors. Antiviral Res 141:101–106
Langel SN, Paim FC, Lager KM, Vlasova AN, Saif LJ (2016) Lactogenic immunity and vaccines for porcine epidemic diarrhea virus (PEDV): historical and current concepts. Virus Res 226:93–107
Li W, Li H, Liu Y, Pan Y, Deng F, Song Y, Tang X, He Q (2012) New variants of porcine epidemic diarrhea virus, China, 2011. Emerg Infect Dis 18:1350–1353
Mokaya J, McNaughton AL, Hadley MJ, Beloukas A, Geretti AM, Goedhals D, Matthews PC (2018) A systematic review of hepatitis B virus (HBV) drug and vaccine escape mutations in Africa: a call for urgent action. PLoS Negl Trop Dis 12:e0006629
Omoto S, Speranzini V, Hashimoto T, Noshi T, Yamaguchi H, Kawai M, Kawaguchi K, Uehara T, Shishido T, Naito A, Cusack S (2018) Characterization of influenza virus variants induced by treatment with the endonuclease inhibitor baloxavir marboxil. Sci Rep 8:9633
Pedersen NC (2009) A review of feline infectious peritonitis virus infection: 1963–2008. J Feline Med Surg 11:225–258
Pedersen NC (2014) An update on feline infectious peritonitis: diagnostics and therapeutics. Vet J 201:133–141
Pensaert MB, de Bouck P (1978) A new coronavirus-like particle associated with diarrhea in swine. Arch Virol 58:243–247
Pensaert MB, Martelli P (2016) Porcine epidemic diarrhea: a retrospect from Europe and matters of debate. Virus Res 226:1–6
Perera KD, Kankanamalage ACG, Rathnayake AD, Honeyfield A, Groutas W, Chang KO, Kim Y (2018) Protease inhibitors broadly effective against feline, ferret and mink coronaviruses. Antiviral Res 160:79–86
Perera KD, Rathnayake AD, Liu H, Pedersen NC, Groutas WC, Chang KO, Kim Y (2019) Characterization of amino acid substitutions in feline coronavirus 3C-like protease from a cat with feline infectious peritonitis treated with a protease inhibitor. Vet Microbiol 237:108398
Pfefferle S, Schöpf J, Kögl M, Friedel CC, Müller MA, Carbajo-Lozoya J, Stellberger T, von Dall’Armi E, Herzog P, Kallies S, Niemeyer D, Ditt V, Kuri T, Züst R, Pumpor K, Hilgenfeld R, Schwarz F, Zimmer R, Steffen I, Weber F, Thiel V, Herrler G, Thiel HJ, Schwegmann-Wessels C, Pöhlmann S, Haas J, Drosten C, von Brunn A (2011) The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog 7:e1002331
Pillaiyar T, Manickam M, Namasivayam V, Hayashi Y, Jung SH (2016) An overview of severe acute respiratory syndrome-coronavirus (SARS-CoV) 3CL protease inhibitors: peptidomimetics and small molecule chemotherapy. J Med Chem 59:6595–6628
Ryu YB, Jeong HJ, Kim JH, Kim YM, Park JY, Kim D, Nguyen TT, Park SJ, Chang JS, Park KH, Rho MC, Lee WS (2010) Biflavonoids from Torreya nucifera displaying SARS-CoV 3CL(pro) inhibition. Bioorg Med Chem 18:7940–7947
Schoeman D, Fielding BC (2019) Coronavirus envelope protein: current knowledge. Virol J 16:69
Shi Y, Li Y, Lei Y, Ye G, Shen Z, Sun L, Luo R, Wang D, Fu ZF, Xiao S, Peng G (2016) A dimerization-dependent mechanism drives the endoribonuclease function of porcine reproductive and respiratory syndrome virus nsp11. J Virol 90:4579–4592
Shie JJ, Fang JM, Kuo TH, Kuo CJ, Liang PH, Huang HJ, Wu YT, Jan JT, Cheng YS, Wong CH (2005) Inhibition of the severe acute respiratory syndrome 3CL protease by peptidomimetic alpha, beta-unsaturated esters. Bioorg Med Chem 13:5240–5252
St John SE, Tomar S, Stauffer SR, Mesecar AD (2015) Targeting zoonotic viruses: Structure-based inhibition of the 3C-like protease from bat coronavirus HKU4–The likely reservoir host to the human coronavirus that causes Middle East Respiratory Syndrome (MERS). Bioorg Med Chem 23:6036–6048
Stobart CC, Lee AS, Lu X, Denison MR (2012) Temperature-sensitive mutants and revertants in the coronavirus nonstructural protein 5 protease (3CLpro) define residues involved in long-distance communication and regulation of protease activity. J Virol 86:4801–4810
Theerawatanasirikul S, Kuo CJ, Phetcharat N, Lekcharoensuk P (2020) In silico and in vitro analysis of small molecules and natural compounds targeting the 3CL protease of feline infectious peritonitis virus. Antiviral Res 174:104697
Wang D, Fang L, Xiao S (2016) Porcine epidemic diarrhea in China. Virus Res 226:7–13
Wang Q, Vlasova AN, Kenney SP, Saif LJ (2019) Emerging and re-emerging coronaviruses in pigs. Curr Opin Virol 34:39–49
Wood EN (1977) An apparently new syndrome of porcine epidemic diarrhoea. Vet Rec 100:243–244
Xie W, Ao C, Yang Y, Liu Y, Liang R, Zeng Z, Ye G, Xiao S, Fu ZF, Dong W, Peng G (2019) Two critical N-terminal epitopes of the nucleocapsid protein contribute to the cross-reactivity between porcine epidemic diarrhea virus and porcine transmissible gastroenteritis virus. J Gen Virol 100:206–216
Yang S, Chen SJ, Hsu MF, Wu JD, Tseng CT, Liu YF, Chen HC, Kuo CW, Wu CS, Chang LW, Chen WC, Liao SY, Chang TY, Hung HH, Shr HL, Liu CY, Huang YA, Chang LY, Hsu JC, Peters CJ, Wang AH, Hsu MC (2006) Synthesis, crystal structure, structure-activity relationships, and antiviral activity of a potent SARS coronavirus 3CL protease inhibitor. J Med Chem 49:4971–4980
Yang DQ, Ge FF, Ju HB, Wang J, Liu J, Ning K, Liu PH, Zhou JP, Sun QY (2014) Whole-genome analysis of porcine epidemic diarrhea virus (PEDV) from eastern China. Arch Virol 159:2777–2785
Ye G, Deng F, Shen Z, Luo R, Zhao L, Xiao S, Fu ZF, Peng G (2016) Structural basis for the dimerization and substrate recognition specificity of porcine epidemic diarrhea virus 3C-like protease. Virology 494:225–235
Ye G, Wang X, Tong X, Shi Y, Fu ZF, Peng G (2020) Structural basis for inhibiting porcine epidemic diarrhea virus replication with the 3C-like protease inhibitor GC376. Viruses 12:240
Zhu L, George S, Schmidt MF, Al-Gharabli SI, Rademann J, Hilgenfeld R (2011) Peptide aldehyde inhibitors challenge the substrate specificity of the SARS-coronavirus main protease. Antiviral Res 92:204–212
Ziebuhr J (2005) The coronavirus replicase. Curr Top Microbiol Immunol 287:57–94
Acknowledgements
We thank Professor Yunfeng Song for his valuable suggestions on the manuscript. This work was supported by the National Key R&D Plan of China (grant no. 2018YFD0500102) and the Natural Science Foundation of Hubei Province of China (grant no. 2016CFA069).
Author information
Authors and Affiliations
Contributions
GP and MZ and YH designed the experiments. MZ, YH, ML and GY performed the experiments. GP, MZ, and ML prepared the manuscript. MZ, ML and GP proofed the manuscript. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
The authors declare that they have no conflict of interest.
Ethical approval
The authors have no ethical issues relevant to this work to declare.
Rights and permissions
About this article

Cite this article
Zhou, M., Han, Y., Li, M. et al. Two Inhibitors Against the 3C-Like Proteases of Swine Coronavirus and Feline Coronavirus. Virol. Sin. 36, 1421–1430 (2021). https://doi.org/10.1007/s12250-021-00415-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12250-021-00415-6