
Abstract
Asphaltene aggregation is a subject under vivid discussion: There are several parameters one needs to determine before its behavior can be mastered and better target solutions can be tailored. The nature of asphaltene aggregation (colloidal or supramolecular) and the role of solvents and their mixtures are among the least understood parameters in asphaltene science. This paper addresses molecular dynamic simulations to correlate the aggregation properties of asphaltenes, their molecular structure and the concentration of these solvents. We show that the formation of the nanoaggregate depends, primarily, on the size of the conjugated core and on the eventual presence of polar groups capable of forming H-bonds. Heteroatoms on the conjugated core do not change their shape or type of aggregation but may induce stronger \(\uppi - \uppi\) interactions. The macroaggregation formation depends upon the length of the lateral chains of asphaltenes and also on the presence of polar groups at its end. Moreover, n-heptane and water may interact selectively with asphaltenes in function of their molecular architecture. Given this fact and the aggregation behavior observed, we advocate toward the assumption that a colloidal behavior of asphaltenes might be a particular case of a more general model, based on a supramolecular description.
Similar content being viewed by others
1 Introduction
Among the challenges the oil industry must face, understanding the physical–chemical behavior of heavyweight fractions is crucial. These fractions have a high tendency to self-assemble and can precipitate out of solution when various crudes of different natures are mixed together, or even when pressure/temperature conditions are changed, blocking well-bores, pipelines and even entire refining processing lines (Akbarzadeh et al. 2007). They can also be responsible for coke deposits, catalysis deactivation, low yield in fuels or chemicals, significant content of sulfur or metals in the output and energy consumption during the upgrading process (Merdrignac and Espinat 2007). Both the chemical composition and the molecular cartography (i.e, how the chemical elements form the various structures) play a key role in the understanding of this problematic phenomenon (Headen et al. 2017). In this way, a clear and well-determined structure–function link between molecular structure and aggregation behavior of asphaltenes is highly sought after so that these nasty effects can be avoided, and new refining techniques, asphaltene inhibitors and catalysts can be developed, and so on (Murgich et al. 1996; Takanohashi et al. 2003; Ortega-Rodriguez et al. 2004).
Traditionally, asphaltenes refer to molecules constituted of a conjugated carbon core, having heteroatoms under the form of heterorings, with lateral chains grafted directly to these cores (Dutta Majumdar et al. 2013; Schuler et al. 2015, 2017). If there is only one core per asphaltene molecule, one calls it an “island” (or, also, a “continent”), but if there are two or more conjugated cores linked together by aliphatic chains, they are called “archipelago” molecules. More importantly, asphaltenes are experimentally defined as the n-heptane-insoluble material in the SARA fractionation framework (Fan and Buckley 2002; Qiao et al. 2017). In this top-down approach, one may be missing important molecules that have somehow a molecular architecture similar to what is expected from asphaltenes but, due to a single characteristic (chain length, for instance), may become n-heptane soluble and not be considered as an asphaltene. In the present work, we do not do any pre-assumption considering the n-heptane-insoluble character of the molecules we study as being asphaltenes. The consequences of such “semantic shift” concerning the definition of asphaltenes were already highlighted, in another context, by Qiao et al. (2017).
Their self-interaction and self-assembly processes are then believed to follow two “distinct, antagonist” mechanisms:
-
(1)
The model of hierarchical asphaltene self-assembly in which nanoaggregates display a colloidal behavior in solution is called the Yen–Mullins model (Mullins et al. 2007, 2012). Asphaltene molecules form stacks governed by \(\uppi - \uppi\) interactions (\(\uppi\)-stacking), irrespective of their island/archipelago character, even though much evidence has been given toward the predominance of island molecules (Headen et al. 2009; Dutta Majumdar et al. 2013; Ungerer et al. 2014; Ghosh et al. 2016). These stacks are constituted of < 10 asphaltene molecules (Headen et al. 2009; Pomerantz et al. 2015), each having 4–9 pericondensed aromatic rings on the conjugated core (Dutta Majumdar et al. 2013; Ghosh et al. 2016), even if larger ones have been proposed (Schuler et al. 2017). These conjugated cores are mainly responsible for asphaltene self-interactions via the formation of van der Waals-driven \(\uppi - \uppi\) interactions. The alkyl lateral chains grafted to this core induce repulsive-only, steric hindrance, interactions with other chains of neighbor asphaltene molecules (Mullins et al. 2007) and have a length comprised between three and eight carbon atoms. In this way, the so-formed asphaltene nanoaggregates do not grow in size if one adds more asphaltene molecules to the solution, since the number of high-energy surfaces available to stabilize additional asphaltene molecules (the conjugated cores) are limited. Instead, only the number of aggregates is increased (Mullins et al. 2007). Nanoaggregates, on their turn, can also interact among them and form macroaggregates, also called clusters. Interactions between nanoaggregates are considerably less intense than the ones measured within the nanoaggregate themselves (Takanohashi et al. 2003; Pomerantz et al. 2015).
-
(2)
In opposition, Gray et al. (2011) argue that the Yen–Mullins model is not capable of describing/accepting experimental observations such as (a) the molecular complexity (the presence of porphyrins, carboxylic acids, nitrogen bases, etc.); (b) the heterogeneous distribution of nanoaggregates sizes; (c)the presence of trapped molecules in asphaltene aggregates, mainly nanoaggregates; (d)the porosity of asphaltene aggregates; (e)the asphaltene film formation at oil–water interfaces; (f) the elastic properties of asphaltenes aggregates; (g) and the surfactant behavior of nanoaggregates. A supramolecular model would then be a better description, providing a more complete context to understand asphaltene’s behavior. In this model, the driving forces are, among others, the molecular recognition and host–guest interactions in three-dimensional porous networks. In this context, \(\uppi - \uppi\) interactions are a contributing factor rather than a dominant one. Moreover, even if the several physical–chemical interactions that can take place in this porous, three-dimensional network are weak, their cumulative effects yield strongly associated structures. Taking into account polar, Coulomb-driven interactions that are not well accommodated in the Yen–Mullins model, may help in describing some physical–chemical properties such as the formation of H-bond networks, for instance, as well as introducing acid/base, metal–organic and “recognition” interactions in the asphaltene’s description. One of the clear consequences of such model would be the “suppression” of the nanoaggregates’ size limitation, since nanoaggregates, if they still can be called like that, could continue increasing size by forming H-bonds or acid–base interactions, for instance, with other asphaltene molecules. Particularly, we have recently demonstrated the possibility of existence of such H-bond networks (Santos Silva et al. 2017a).
A great number of experimental data advocate toward the colloidal, Yen–Mullins model, as indicated by Dutta Majumdar et al. (2017). These findings indicate that aggregation among individual asphaltene molecules is primarily driven by \(\uppi - \uppi\) interactions, in a hierarchical way. Oppositely, experimental (Varadaraj and Brons 2012; Schulze et al. 2015; Subramanian et al. 2017) as well as simulation-based evidence (Headen et al. 2009, 2017) exist corroborating the supramolecular, Gray’s model. These works showed that asphaltene self-interaction depends also on acid–base and polar interactions. More particularly, Headen et al. (2009, 2017), using molecular dynamics simulations, highlight some physical–chemical phenomena that cannot be described by the Yen–Mullins model, mainly entrained solvent within asphaltene nanoaggregates. In their simulations, they observed loose, rather than rigid, stacking of asphaltenes, meaning that the nanoaggregates can be formed and unformed during the simulations. Even if this observation could be linked to the chosen molecular structure of their asphaltene molecules (conjugated cores that contain non-aromatic rings), this interesting result indicates that the conformation of asphaltene aggregates is somehow dynamic. The same authors also observed that very commonly solvent molecules get entrained within the nanoaggregates, changing considerably their density and number of asphaltene molecules constituting the nanoaggregate depending on the solvent being used. These authors conclude that a continuous distribution of cluster sizes would better describe these observations than the Yen–Mullins model could.
Another point of interest for the asphaltene aggregation process is to determine whether resins aid the stabilization of asphaltenes or not. Resins are molecules displaying physical–chemical behavior and structurally in-between the aromatics and asphaltenes molecules, as defined by the SARA (saturates, aromatics, resins and asphaltenes) fractionation (Fan and Buckley 2002; Qiao et al. 2017). If this comes to be the case, in the context of the Yen–Mullins model, resins would be then found in-between asphaltene nanoaggregates, the former acting like a natural surfactant for the latter. On the other hand, in Gray’s model, resins would be expected to be found within nanoaggregates, interacting closely with the asphaltene molecules, probably even disturbing their aggregation.
Interestingly though, after years of investigation to answer this question, both models agree on the fact that resin molecules do not solvate or solubilize the asphaltene clusters. Mullins et al. (2007) showed that samples from different heights in the oil column exhibit little changes in the resins concentrations, whereas it is not the case for asphaltenes. Headen et al. (2017) showed that the formation of resin–asphaltene aggregates is only favorable in n-heptane solutions, induced by the entrainment/intrusion of resins within asphaltene aggregates. In toluene, resins and asphaltenes do not interact favorably probably because resins, having small conjugated cores and more polar structures, are more soluble than asphaltenes and have a little affinity with them. This “resin dichotomy” is evidence that both models have common points and that they can finally describe some physical–chemical behaviors in a similar way.
Regardless of the model being considered to study asphaltene aggregation, the fact that their ultimate physical–chemical behavior is dictated by their molecular structure is a solid consensus (Dutta Majumdar et al. 2017). The calculations of the major part of molecular properties of asphaltenes (average molecular weight, interaction energies, aggregation behavior, etc.) are dependent on their molecular structure and can differ significantly if different molecular structures are considered, as proved by the first works done by Rogel (1995, 2000). This is not only problematic for molecular modeling approaches, but also experimental techniques that use asphaltenes from various sources and maybe using even slightly, different fractionation procedures (Fan and Buckley 2002; Kharrat et al. 2007; Sabbah et al. 2011; Langevin and Argillier 2016; Qiao et al. 2017).
In this sense, the access to the molecular structure of asphaltene molecules is a great leap toward the full comprehension of their physical–chemical behavior. Recently, Schuler et al. (2015, 2017), using atomic force microscopy (AFM), unveiled the molecular structure of some asphaltene molecules from various sources. The structures proposed in this work also agree with experimental, NMR-aided, proposed structures (Dutta Majumdar et al. 2013). This pioneer work establishes a new basis for in-silico asphaltene research since now one can easily study the behavior of the molecules for different molecular architectures in a true molecular cartography strategy.
Taking advantage of this information and using molecular dynamics simulations, we have undertaken the construction of a rational link between the molecular structure of asphaltenes and their aggregation properties, regardless of any prior consideration of which aggregation model we should follow to describe and understand our results. Following this strategy, our group has already:
-
Studied the role of heteroatom substitution on the conjugated core and the effect of mixing different asphaltenes together, showing that such substitutions do not modify the shape of the nanoaggregate but change considerably the energy of interaction between asphaltene molecules (Santos Silva et al. 2016);
-
Presented solid arguments allowing one to identify the role of some heteroatoms when they are found in the lateral chains: Oxygen easily induces the formation of hydrogen bonds that change drastically how nanoaggregates interact; sulfur can also induce S–O interactions (Sodero et al. 2016), and so on;
-
Demonstrated that porphyrins, when they have no lateral chain grafted to their core, have interactions with asphaltenes that are similar to asphaltene–asphaltene interactions as well, besides the fact that vanadyl porphyrins can also form hydrogen bonds with the asphaltenes, even those which have no polar lateral chain (Santos Silva et al. 2017b); and
-
Shown that asphaltene nanoaggregation depends on the shape and size of the conjugated core and on the possibility of forming hydrogen bonds with their environment (Santos Silva et al. 2017a).
Some of these observations/conclusions have been demonstrated by other research groups as well (Dutta Majumdar et al. 2017; Headen et al. 2017; Samieadel et al. 2018; Biktagirov et al. 2017). In any case, the rational and consistent modification of the asphaltenes molecular structures and the conditions to which they are submitted to allow us to establish the structure–function link we intend to determine.
Even though our previous studies have been able to provide insightful trends, these molecular cartography results are only valid for toluene and asphaltene solutions. One has also to screen these properties against the presence of n-heptane and water alongside toluene as solvent, in order to gain in complexity and to describe asphaltene aggregation phenomena observed during exploration and refining in a more precise and closer-to-the-reality way. Even if studies on asphaltene aggregation are common in the literature, our strategy is based on rationally modifying experimentally observed asphaltene molecules so that their behavior toward aggregation can be studied if slightly different molecular parameters are changed. Concerning the study of asphaltene aggregation in different solvents, in our case, we aimed to investigate the role of low concentrations of the co-solvents (at the limit of water solubility in toluene, for instance). In this way, this paper presents molecular dynamics simulations in order to answer the following questions:
-
(1)
Which is the influence of the conjugated core size on asphaltene’s nano- and macroaggregation in the presence of water, n-heptane or water and n-heptane in low-concentration regimes?
-
(2)
Which is the influence of the lateral chain length on asphaltene’s nano- and macroaggregation in the presence of water, n-heptane or water and n-heptane?
-
(3)
Which is the role played by polar chain ends in such different solvents?
-
(4)
Which are the roles of water and n-heptane concentrations and how they are coupled together?
With such questioning, we also expect to corroborate one or other asphaltene aggregation model based on the molecular structures herein studied. This document is organized into three main blocks: simple solvent systems (toluene, water or n-heptane as solvents); binary solvent systems (toluene/water or toluene/n-heptane); and ternary solvent systems (toluene/n-heptane/water). For the systems with more than one solvent, the relative concentrations have been screened in an interval having some physical–chemical representativity as explained in the following section.
The aim of this paper is not to be another molecular dynamics simulation study showing how asphaltenes aggregate. Its aim is to use these results to elucidate if asphaltenes can indeed be understood as a solubility class or if they would be rather defined by a molecular structure class. This is clearly of utmost importance if one wants to generalize their behavior in crude oil samples, where they have not yet been precipitated by n-heptane addition. The results herein presented also corroborate the fact that the model describing asphaltene’s aggregation is highly dependent on this definition, which may not be a good approximation to the behavior in real oil.
2 Methodology
To determine the influence of the conjugated core size on the aggregation of asphaltenes, we have modeled different asphaltene molecules belonging to the same class and family. Additionally, polar and apolar ends of the chain were considered by including or groups at the end of each chain. This portion of the analysis highlights any possible link between the aggregation and the lateral chain length and/or ability to form H-bonds. It is also important to stress that these molecules were proposed in a bottom-up scheme, and it means that no pre-assumption was made on their solubility in n-heptane, but we took into account the characteristics of their molecules structures which could match the molecular (and not solubility) definition of asphaltenes.
The computational details behind the molecular dynamics (MD) simulations are fully presented in electronic supplementary information (ESI). In brief, the simulations consist of using the GROMOS96 force field with the 53a6 parameter set (Van Gunsteren et al. 2002) within the Gromacs 5.0 (Van Der Spoel et al. 2005) package. This united-atom (UA) force field allows one to increase the size of the systems being treated without compromising accuracy. Moreover, bond vibrations have been constrained since they occur at timescales that have orders of magnitude faster than the physical–chemical process of interest.
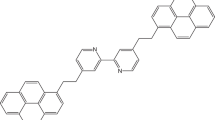
The studied molecular systems are based on the so-called CA22 (Fig. 1a) molecule identified by Schuler et al. (2015) to which we have grafted two lateral chains on the opposite sides. Then, the length of this chain was set to be n-C6H13 or n-C16H33, and the number of fused rings on the conjugated core was set to 7 or 11, as shown in Fig. 1a, b. The so-called PA3 molecule type described by the above authors was also studied with two different lateral chains. Toluene was used as a solvent in order to mimic the infinite dissolution effect and to avoid the formation of the aggregates due to differences in polarity. The so-formed solutions have an asphaltene concentration on the order of ~5 wt%. These molecules are defined as presented in Table 1 alongside the labeling system used. Other physical–chemical properties of these molecules are presented in Table S1, found in ESI material.

Molecular structure of the studied molecules with increasing conjugated core size and lateral chain length. a Represents molecules of the type AAX (derived from CA22), having 25 \(\uppi\)-conjugated carbon atoms; b ADX molecules, 35 \(\uppi\)-conjugated carbon atoms. Finally, c the PA3-type molecule, having 32 \(\uppi\)-conjugated carbon atoms (A13 and A14). \(R1\) chains are defined in Table 1
Finally, the following points need to be stressed out: In comparing interaction energies, as it is performed in this work, one cannot use them to correlate with experimentally determined ones, since it lacks pressure–volume (pV) and temperature–entropy (TS) terms. However, given the close-to-zero compressibility of liquids, the former term can be considered as constant and vanishes when comparing two energies. The latter, one the other side, is commonly obtained by analyzing the radial distribution function (RDF) plots by means of potential mean force analysis, for instance. Even if this is not performed in this work, the conclusions obtained from analyzing the RDFs and these internal energies are coherent with each other, meaning that TS terms also vanish when comparing the energies of two closely related systems. Also, it is worth saying that the RDFs herein used are not center-of-mass ones, but rather calculated with reference to the surface (-surf mol option in gromacs rdf module) of the asphaltene molecules, whenever they happen to be the reference molecule. Also, in this text, nanoaggregation and macroaggregation do not have the same meaning as they traditionally do in the framework of the Yen–Mullins model: The former is understood as the compact agglomerate of asphaltene molecules interacting by \(\uppi\)-stacking, and the latter is understood as the interaction between two or more of these agglomerates, mostly probably due to the formation of H-bonds between them.
For each asphaltene molecule, the construction of the simulation boxes was performed as follows:
-
45 asphaltene molecules were placed randomly within the box;
-
They were solvated using a pre-equilibrated toluene box using the gromacs solvate tool;
-
The average of the number of toluene molecules over all the asphaltene systems was calculated;
-
This averaged number of toluene molecules (5482) was introduced in a copy of the configuration obtained in step 1;
-
The so-formed systems were simulated as is or additional water and/or n-heptane molecules were added, keeping constant the number of toluene molecules.
The introduction of water and n-heptane molecules followed the quantities is presented in Table S3.
3 Results and discussion
3.1 Simple solvent systems: toluene, n-heptane or water
To construct a baseline for further comparisons, simple solvent systems were firstly studied. They are constituted of 45 asphaltene molecules of the same type with toluene, n-heptane or water as the unique solvent in the simulation boxes. Systems containing only toluene were already extensively studied, and a thorough analysis of the structure–property link in this solvent can be found in our latest work (Santos Silva et al. 2017a). Figure 2 presents the calculated \(CN/N\left( \% \right)\) (coordination number over number of molecules) ratios for every molecule in each solvent system as well as the calculated Kernel density estimation (KDE) associated with them. The ratio \(CN/N\left( \% \right)\) estimates the proportion of the molecules that have a \(\uppi\)-stacking aggregation pattern among the first neighbors compared to all the other molecules present in the system, i.e., this ratio is an index that compares the proportion of the number of asphaltene molecules within the nanoaggregates to the number of nanoaggregates in the simulation box (an index of macroaggregation). Mathematically, it is calculated as the ratio between the integral of the radial distribution function (RDF) up to the first minimum (\(r_{m}\)) after the first \(\uppi\)-stacking peak (\(r_{m} =\) 0.6 nm) and the full RDF integral. More details can be found in our previous work (Santos Silva et al. 2017a). KDE plots are obtained as a sum of the Gaussian curves enveloping the histogram of \(CN/N\left( \% \right)\) values. The RDFs are used for such computations, and further details can be found in ESI material (Fig. S2).

\(CN/N\left( \% \right)\) for every molecule a and the calculated Kernel b for each simple solvent system
Straightforwardly, the trends observed for the \(CN/N\left( \% \right)\) ratios in water do not follow those calculated for either toluene or n-heptane. Although expected, it is interesting to note that molecules having longer lateral chains have lower \(CN/N\left( \% \right)\) ratios in toluene and n-heptane, whereas they present the highest ratios when water is the sole solvent. The mechanism behind such phenomenon can be attributed to the equilibrium between \(\uppi\)–\(\sigma\) and \(\sigma\)–\(\sigma\) interactions, as reported by Jian et al. (2013). In the same way, the \(CN/N\left( \% \right)\) distributions from the KDE plots are bimodal with modes associated with the length of the lateral chain (six or 16 linear carbon atoms long). One can easily deduct that the main parameter behind the macroaggregation process in these solvents is the length of the lateral chains: With longer chains, nanoaggregates can stay in suspension longer without any coalescence.
Figures 3, 4 and 5 present the final snapshots for each solvent system for eight among the ten studied molecules. In toluene, molecules having larger conjugated cores tend to form larger nanoaggregates than their equivalents with smaller conjugated cores and same lateral chains. On the other hand, molecules having the same conjugated core but different lateral chains behave differently with respect to the macroaggregation: Molecules having shorter lateral chains, as AAC, tend to form nanoaggregates more packed together than molecules having longer lateral chains, such as AAF. The same is true for the other pairs: ADC/ADF, AAH/AAI and ADH/ADI. These findings agree well with the results found by Takanohashi et al. (2003) several years ago.

Snapshots of 45 asphaltene molecules after 60 ns of simulation in toluene. Toluene molecules are not shown for clarity

Snapshots of 45 asphaltene molecules after 60 ns of simulation in n-heptane. n-Heptane molecules are not shown for clarity

Snapshots of 45 asphaltene molecules after 60 ns of simulation in water. Water molecules are not shown for clarity
These observations are also true for n-heptane, even though in this situation the conjugated core size seems to have a less important role (Fig. 4). Molecules having long lateral chains clearly form defined nanoaggregates, more separated than the ones formed by molecules having shorter lateral chains. This is also the case when the lateral chain has a polar group.
Generally speaking, nanoaggregates are less sparse in n-heptane than in toluene solutions, i.e, toluene seems to be a better solvent for asphaltenes, even though the solubility in n-heptane is increased with increasing asphaltene’s lateral chain lengths, in agreement with Wang et al. (2017b). This was already shown by the KDE plots (Fig. 2) that depicted the center of mass of the probability distributions shifted toward lower solubilities for n-heptane compared to toluene. Inversely, in water, molecules having longer lateral chains are the most sensible and they coalesce faster. All the studied molecules form one or two very distinct macroaggregates with all the lateral chains folded over themselves, which is typical of non-solubility in this solvent (Kuznicki et al. 2008). Visually (Fig. 5), the most compact structures correspond to the molecules with longer lateral chains, also agreeing with the results obtained by Headen et al. (2009, 2017). Surprisingly though, the polar groups of shorter chains seem to give the molecules a very low but visible affinity with water, as one can see on Fig. 5 (AAH). This seems not to be the case with a longer chain (AAI).
Once the parameter ruling macroaggregation is determined, the asphaltene–asphaltene interaction energies are a good indicator of the nanoaggregation process and its relation with the molecular architecture. This is so since in molecular dynamics simulations, Coulomb and vdW interactions are calculated within a sphere around each atom whose radius is called the cutoff radius, which equals 12 Å for our simulations. The choice of this value is done in a way that the simulation’s results are not impacted by it. Imposing such cutoff restrains the quantity of pair interactions to be calculated during the simulation, reducing the computational cost. In this way, the fact that energies are calculated within a given distance sphere assures that the analysis we make out of it concerns more the nanoaggregates rather than the macroaggregates.
Figure 6 presents asphaltene–asphaltene interaction energies averaged over the whole dynamics and the total KDE for each individual solvent. The energies can be normalized in two ways:

Energy of interaction per asphaltene molecule in each solvent system as a probability distribution function plot (PDF—colored curves) alongside KDE curves (dashed) related to these energies for each solvent case
-
(1)
Dividing them by the number of asphaltenes within the simulation box; or
-
(2)
Dividing this result further by the double bond equivalent (DBE) of each molecule type.
Whereas the first strategy highlights the energy of interaction in function of the molecular structure of the asphaltene, the second goes even further and correlates this with a more intrinsic parameter: the number of \(\uppi\)-electrons in the structure. Both strategies were pursued: The first one is herein presented, and the second can be found in ESI material (Sect. 3.1).
In toluene, the asphaltene–asphaltene interaction energies spread from −20 up to −80 kcal/mol per molecule, whereas in n-heptane, it goes from − 25 up to − 95 kcal/mol and from − 30 up to − 100 kcal/mol in water, in line with other results found in the literature (Pacheco-Sánchez et al. 2004; Headen et al. 2009; Sedghi et al. 2013; Headen et al. 2017). The shifts for the average and median values of these energies indicate that solvents have also an impact on the formation of the nanoaggregate. In poorer solvents, as macroaggregation occurs, the aggregate becomes denser and denser, increasing the interaction energy between asphaltenes.
Concerning the molecules individually, the asphaltenes having larger conjugated cores form more cohesive nanoaggregates (Takanohashi et al. 2003; Pacheco-Sánchez et al. 2004; Sedghi et al. 2013; Liu et al. 2017; Headen et al. 2017; Dutta Majumdar et al. 2017). In toluene, there seem to be three different groups:
-
(1)
ADI and ADH: molecules having large conjugated cores and polar chain ends, regardless of its length (Sedghi et al. 2013; Liu et al. 2017; Wang et al. 2017b);
-
(2)
AAI, ADF, ADC, A14, AAH and A13: molecules having large conjugated cores with apolar chains regardless of their length; molecules having small conjugated cores with polar chains regardless of their lengths; molecules having heteroatoms on the conjugated core;
-
(3)
AAC and AAF: molecules having small conjugated cores with apolar lateral chains regardless of their length.
This demonstrates that in this solvent, the effect of the size of the conjugated core is coupled with the one of the polar chain end. This seems also to be the case for n-heptane (Headen et al. 2017). However, in water, this order slightly changes and this solvent seems to concern more intensive molecules having long lateral chains than shorter ones. In this way, the size of the conjugated core and the presence of polar groups in chain ends rule nanoaggregation in such conditions.
When the DBE renormalization condition is employed (see ESI—Sect. 3.1), i.e, once the size of the conjugated core is discarded as a parameter, the lateral chain length also has a role on the formation of the nanoaggregates even though this role is secondary. In toluene, molecules having long lateral chains with polar groups at the end have a tendency to interact more strongly than molecules having shorter chains with the same groups or molecules having no polar group at all (Sedghi et al. 2013; Liu et al. 2017; Wang et al. 2017b). In n-heptane, the same trend is found even though the distribution of the energies is now tighter, indicating that n-heptane should discriminate more molecules in function of their chain length (Headen et al. 2009). In water, molecules with long lateral chains have the highest asphaltene–asphaltene interaction energies very probably because these chains prefer to display \(\sigma\)–\(\sigma\) interactions among themselves rather than with water (Jian et al. 2013).
This indicates that at a second level, the chain length is coupled with the presence of polar groups in the formation of nanoaggregates. This conclusion is corroborated by the analysis of the asphaltene–solvent interaction, as it is presented in Fig. S22. Asphaltenes interact more strongly with toluene, then n-heptane and finally with water. Neither the conjugated core size nor the polarity of the chain has a role on this type of interactions. However, the short and polar chains (such is the case of AAH and ADH molecules) present Coulomb interaction with water through the formation of H-bonds (see ESI, Figs. S54–S56). The dispersion of these energies (Fig. S23) shows that, for toluene and n-heptane, these interactions are divided into two groups (the two “modes” of the curve): one formed by the molecules having long lateral chains and the other formed by the molecules having short ones. This means that asphaltene–solvent interactions are basically a function of the lateral chain length. In water, the polarity of the chain is coupled with its length in the determination of the strength of such interactions.
3.2 Binary solvent systems: toluene/water or toluene/n-heptane
3.2.1 Toluene/water
As already indicated in the Methodology section, stepwise amounts of water were added to the simulation boxes with a fixed quantity of toluene molecules. The lowest water amount was chosen to match the solubility of water in toluene at these T, P conditions (0.5 g/L at T = 298 K and P = 1 bar) (Kirchnerová and Cave 1976). Then, this concentration was increased 2, 10 and 20 times. The snapshots of the final configurations of these simulations are given in ESI material, Figs. S3–S5 and section 4.4.1.
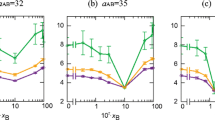
Similarly to the simple solvent systems, we analyzed the \(CN/N\left( \% \right)\) ratio, the associated KDE plots and the asphaltene–asphaltene energy of interaction. These results are reported in Fig. 7. The asphaltene–solvent energy of interaction was also taken into consideration and is presented in ESI (Sect. 3.2.1).

\(CN/N\left( \% \right)\) for every molecule (a), the calculated Kernel density estimation for each concentration of the co-solvent (b) and the asphaltene–asphaltene interaction energy by asphaltene molecule (c). Error bars are calculated from the standard deviation
To recall, both the \(CN/N\left( \% \right)\) ratios and the KDE plots give insightful information on the macroaggregation behavior, whereas the asphaltene–asphaltene energy of interaction concerns more particularly the nanoaggregation process, as already discussed. The \(CN/N\left( \% \right)\) ratio, regardless of the water concentration, follows approximately the same trend already observed for toluene-only solutions (see Fig. 2), indicating that the presence of water in the simulation box, at these concentrations, may have a little or no effect on the formation of macroaggregates. The KDE plots exhibit the same bimodal distribution observed for the case of pure toluene, only observed for the lowest concentration (i.e., the one in which water is soluble in toluene). When the water concentration is increased, this bimodal behavior is continuously lost in detriment of a shift toward lower \(CN/N\left( \% \right)\) ratios. The “mode” being shifted is associated with the molecules having short lateral chains with ratio values close to the ones found for molecules having long lateral chains. This means that the more water we add, the more the nanoaggregates formed by molecules having short lateral chains move away from each other, which is very surprising. A possible explanation for this observation is: As we add water over its solubility limit, some small water droplet(s) are formed and intercalate in between the nanoaggregates, forcing them to keep away from each other (Khvostichenko et al. 2004), as one can note from Figs. S4.4.1–4.4.4.
The energy of interaction between asphaltene and water molecules (Fig. S30) shows that only the molecules having polar groups at the lateral chain end have a chance to interact with water, in agreement with the results of Kuznicki et al. (2008) and also corroborated by the analysis of the H-bonds formed throughout the simulation (Figs. S54–56). It is interesting to note that, as classical molecular dynamics simulations do not allow bond-breaking, these interactions exist even when the –OH bond is not dissociated, as it is the case in our simulations. For the simulations done by Kuznicki et al. (2008), this was not the case: They considered a deprotonated –COOH group. We believe that, in such toluene-rich simulation, it is advisable to treat polar groups as not dissociated, in order to not induce obvious observations. Although measurable, this interaction is very small and equals ~1.4 kcal/mol per asphaltene molecule in average over the different water concentrations. The other asphaltene molecules, even the ones having heteroatoms on the conjugated core, have no interaction at all with water.
Concerning the formation of the nanoaggregate itself, the presence of water, at these concentrations, does not change its behavior since we retrieve the same trends and energy values as already observed for the toluene-only case (Fig. 6). All these results show that the water concentration, within the limits of this study, has no effect on the formation of the nanoaggregates and only a little effect on the interaction between different nanoaggregates (during macroaggregation). This is probably the case since toluene and water being immiscible, water droplets are formed within the toluene solution and do not interact with asphaltenes that are dispersed in the latter. In the next section, we will focus on toluene/n-heptane solutions which are miscible and can then present different behaviors.
3.2.2 Toluene/n-heptane
Similarly, we have studied systems with the stepwise increase in n-heptane concentration in toluene solutions. Being aliphatic, n-heptane has a higher affinity with asphaltene’s lateral chains. Moreover, it is miscible with toluene and can thus be in closer contact with asphaltenes rather than water can. The lowest n-heptane concentration was chosen so that the number of n-heptane molecules within the simulation box equals the number of asphaltene molecules. Then, 2, 10 and 20 times this concentration value were also studied. The final snapshots of these simulations are provided in ESI, Section 4.5.1. Figure 8 presents the \(CN/N\left( \% \right)\) ratio, the KDE associated plots and the asphaltene–asphaltene interaction energy for each molecule and for each concentration.

\(CN/N\left( \% \right)\) for every molecule (a), the calculated Kernel density estimation for each concentration of the co-solvent (b) and the asphaltene–asphaltene interaction energy by asphaltene molecule (c). Error bars are calculated from the standard deviation
In this case, even if the evolution of the \(CN/N\left( \% \right)\) ratio follows the same trends already observed for toluene or n-heptane alone, it seems now that the n-heptane concentration has a more important impact on the macroaggregation behavior than previously. One can observe from the KDE plots that, with the increase in n-heptane concentration, the \(CN/N\left( \% \right)\) ratio decreases for all the studied molecules. This is even more evident for molecules AAC and AAF (small conjugated core, apolar lateral chains regardless of their length). KDE plots shift progressively to the left. The explanation is the same as for water in toluene, the difference being that now n-heptane is miscible with toluene. But even so the same behavior of reducing macroaggregation is also observed.
Taking into account the asphaltene–asphaltene interaction energies, one can observe that differences are present mainly for AAC and AAF molecules, for which the energy of interaction lowers with the increase in the n-heptane concentration. This fact, also observed in the \(CN/N\left( \% \right)\) curves, indicates that these molecules are slightly soluble in n-heptane and with the increased concentration of this solvent, they interact more with the solvent and less with each other. For the other molecules, within the concentration limits studied, adding n-heptane to the toluene solution has no effect on the nanoaggregation formation and stabilization, in line with other works (Rogel 1995; Sedghi et al. 2013; Wang et al. 2017a; Headen et al. 2017) In this way, helped by its miscibility with toluene, n-heptane can interact selectively with the asphaltene molecules in function of their lateral chain length. This fact indicates that during fractionation of asphaltene molecules following the “precipitation in n-heptane” procedure, it is possible that the asphaltenes are unwillingly selected in function of their lateral chain lengths (Fan and Buckley 2002; Kharrat et al. 2007; Sabbah et al. 2011; Langevin and Argillier 2016; Qiao et al. 2017). Experimental studies that are then performed on these samples might be biased by the fact that the asphaltenes have a narrower chain length distribution than real asphaltenes in crude oil conditions would have.
Figure S39 shows the asphaltene–n-heptane interaction energies as function of the number of \(\sigma\)-carbon atoms of the lateral chains of asphaltene molecules. This figure shows that this interaction is selective toward two groups of molecules:
-
(1)
A14, ADI, AAF, AAI and ADF
-
(2)
A13, ADH, AAC, AAH and ADC
Yet, the common characteristic to each group is the length of their lateral chains, meaning that n-heptane interacts more strongly with molecules having longer lateral chains than shorter ones, whose consequences were above-mentioned.
3.3 Ternary solvent systems: toluene/n-heptane/water
The effect of both water and n-heptane present simultaneously in the simulation boxes was studied by screening together both concentrations. In this way, 160 (4 × 4 × 10) different, full-size molecular dynamics simulations were performed additionally to those realized for the study of simple and binary solvents. Given the number of simulations and the results already obtained for the simple and binary solvent systems, we rely, mainly, on the analysis of the \(CN/N\left( \% \right)\) ratio for these systems, presented in Fig. 9, under the form of heat maps for each asphaltene molecule. All of them share the same \(z\) scale. Each pixel of this figure was obtained after a full simulation of a system with very particular solvent and asphaltene compositions.

\(CN/N\left( \% \right)\) for every molecule for each concentration of the co-solvent
These \(CN/N\left( \% \right)\) ratio heat maps show that, in a general way, no direct trend in the macroaggregation can be deduced by the increase in both water and n-heptane concentrations in toluene. For molecules having the same conjugated core, longer lateral chains coincides with the decrease in the \(CN/N\left( \% \right)\) ratio, corroborating the fact that they reduce the probability of cluster formation (Takanohashi et al. 2003; Pacheco-Sánchez et al. 2004; Sedghi et al. 2013; Liu et al. 2017). However, polar lateral chains, compared to same-length apolar analogs, do induce such clusterization effect (Wang et al. 2017a, b). Instead, with different conjugated cores for the same lateral chain, no significant/steep trends are observed, comforting the observation that the lateral chains are indeed the parameter responsible for controlling macroaggregation in such solvation conditions.
The asphaltene–n-heptane interactions do not vary with the increase in n-heptane concentration. The same behavior is observed for asphaltene–water interactions. This indicates that no permanent trapping of solvent molecules within asphaltene nanoaggregates is observed (which is sensibly different from trapping in macroaggregates), while the initial states of the simulations are the completely dissociated states. If such was the case, these would be dynamic interactions that are somehow independent of the solvent concentration. Furthermore, as already observed, the intensities of asphaltene–n-heptane and asphaltene–water interactions are higher for molecules having longer lateral chains and molecules having polar groups at chain ends, respectively. This indicates that even in the presence of water, n-heptane continues to be selective toward the length of the asphaltene’s lateral chains.
Concerning the nanoaggregates, asphaltene–asphaltene interaction energies, presented in ESI material, Fig. S53, do not vary considerably as a function of water/n-heptane concentration, as is the case of these solvents alone. When renormalized, one can verify that the large conjugated core and the presence of polar groups on lateral chain ends continue to be the essential factor ruling the formation of the nanoaggregate, inversely to what is observed for the formation of the macroaggregate, governed by the length of the lateral chain (Sedghi et al. 2013).
3.4 Proposition of a structure–function link
To summarize all these results obtained for different solvent conditions, nanoaggregates formation depends on:
-
(1)
The size of the conjugated core;
-
(2)
The eventual presence of polar groups capable of forming H-bonds.
It does not depend on the polarity of the conjugated core (the presence of heteroatoms) (Sedghi and Goual 2009; Sedghi et al. 2013), even if the stability (energy of interaction) of the nanoaggregate indeed does (Santos Silva et al. 2016). However, given the range of energies found for the \(\uppi - \uppi\) interactions, asphaltene decomposition (coke formation) would take place before any dissociation of this interaction could be achieved (Takanohashi et al. 2003). In this way, controlling macroaggregation is the sole opportunity one has to reduce asphaltene flocculation.
Given this, the macroaggregation depends upon:
-
(1)
The length of the lateral chains of asphaltenes;
-
(2)
The presence of polar groups at its end;
In the same way, it is the lateral chains the responsible parameter for the interactions with the solvent.
The presence of polar groups in asphaltene chain ends is somewhat subject to discussions (Rogel 2000), even if some authors have already highlighted their presence in real-world samples (Wang et al. Wang et al. 2017a, b). The formation of H-bonds is not taken into account in the Yen–Mullins model of asphaltene aggregation, even though aliphatic and aromatic esters have already been found in heavy fractions of crude oil (Strausz and Lown 2003). In this model, the aliphatic chains are considered uniquely as responsible for repulsion between nanoaggregates, but it is clearly not the case for chains capable of doing H-bonds and other polar interactions.
Nevertheless, when simulating “well-behaved” asphaltenes (medium number of aromatic cores, low number of aliphatic carbons on the lateral chain and absence of polar groups at its end), the aggregation process of asphaltenes is undoubtedly well described by the Yen–Mullins model. One can clearly corroborate this model by simply looking at a molecular dynamics snapshot of such systems. As we proposed following the indications found by several authors (Fan and Buckley 2002; Kharrat et al. 2007; Sabbah et al. 2011; Langevin and Argillier 2016; Qiao et al. 2017), the presence of these “well-behaved” asphaltenes might be the result of the purification process used to isolate the asphaltene molecules out of the crude oil. Being selective toward specific molecular structures, the solvent fractionation can induce samples that inevitably display a colloidal behavior even though this is not the case in the crude oil sample.
In such a way, given the results obtained in our work, we believe that the Yen–Mullins model describes well asphaltenes molecules having small conjugated cores (4–8 fused rings) and apolar lateral chains with a lower number of carbons (~6). The inclusion of molecules having larger conjugated cores and polar lateral chains is out of this model and could then be better described by Gray’s model. In our understanding, this does not mean that these two models of asphaltene aggregation are in opposition to each other but only that the Yen–Mullins model seems to be a particular case of Gray’s model for a more precise class of molecules.
Changing this paradigm induces assuming that nanoaggregates (defined then as an aggregate of a few asphaltene molecules) could eventually be bigger than what is imaged up to now, and this would be a function of the asphaltene composition and the procedure used to isolate the asphaltene samples. Giving its natural origin, the geological time and the presence of potential catalysts (metallic porphyrins), there is no reason why asphaltenes would not have polar groups that induce polar interactions and H-bond formation. Assuming this is true, it comes as evidence that the colloidal understanding of asphaltenes is a concept reserved to a sub-fraction of the asphaltene’s zoology present in the crude oil. This also implies that the asphaltene’s molecular structure and associated physical–chemical behavior should meet, under real conditions, a continuum distribution. The discrete case, typical of the Yen–Mullins picture, would be, according to our understanding, a particular case of a more general one. The large number of experimental results in agreement with this model would be caused by the molecular selection done during asphaltene fractionation.
Finally, our results give evidence toward both the Yen–Mullins and Gray’s models of asphaltene aggregation. “Choosing” which model is better suited to explain the data depends on the molecular structure of the asphaltenes under study. Coupled to the fact that solvents as n-heptane and water are selective toward these molecular structures, this indicates that, very probably, the experimental observation of a colloidal behavior comes from the molecular selection unwillingly performed during asphaltene fractionation. Studies on samples not submitted to fractionation would then be necessary as well as molecular dynamics simulations done on a wide range of asphaltene molecular structures in order to access deeper information on the trouble-making asphaltene aggregation process and how to suppress it.
4 Conclusions
We gave indications toward the establishment of a structure–property link between asphaltene molecular structure and their aggregation properties in different solvent mixtures. In simple solvent solutions (toluene, n-heptane or water), the length of the lateral chains is the parameter behind the macroaggregation process in these solvents. With longer chains, nanoaggregates can stay in suspension longer without any coalescence. Moreover, the polarity of the lateral chain taken into account as possible formation of H-bonds with other similar molecules also helps the formation of the macroaggregates, in opposition to the previsions of the Yen–Mullins model. Inversely, the formation and stabilization of the nanoaggregates are dependent on the size of the conjugated core and the polarity of the lateral chain, both factors being coupled.
In binary (toluene/n-heptane and toluene/water) and ternary (toluene/n-heptane/water) solvent systems, the same observations were also retrieved. No permanent solvent entrainment effect was observed with increasing concentration of the solvent. Asphaltenes interactions with the co-solvent occur regardless of the concentration. The asphaltene/n-heptane interactions are selective versus the length of the lateral chain. Longer lateral chains interact more with n-heptane molecules. Upon n-heptane addition, the first nanoaggregates to form clusters and probably precipitate, most probably, contain the more asphaltenes with short lateral chains. This effect could be responsible for selecting asphaltene molecules during the fractionation to obtain experimental samples. On the other hand, interactions with water are mediated by the presence of polar groups at chain ends able to form H-bonds. This is also independent on the concentration of water, indicating that polar-chain-ended asphaltenes do not have a role in “solubilizing” water molecules in toluene solutions, even though some small interactions could be observed. Moreover, asphaltene molecules with heteroatoms on the conjugated core do not seem to have particular interactions with water. This indicates that the water–asphaltene interactions are mediated uniquely by the lateral chains of the latter.
Last, but not least, the properties observed in our studies seem to be outside the scope of the predictions made by the Yen–Mullins model concerning the formation of nanoaggregates when different molecular architectures are considered, for instance. This model could be a particular case of a more general one: the Gray’s model.
References
Akbarzadeh K, Hammami A, Kharrat A, Zhang D, Allenson S, Creek J, Kabir S, et al. Asphaltenes—problematic but rich in potential. Oilfield Rev. 2007;19(2):22–43.
Biktagirov T, Gafurov M, Mamin G, Gracheva I, Galukhin A, Orlinskii S. In situ identification of various structural features of vanadyl porphyrins in crude oil by high-yield (3.4 T) electron–nuclear double resonance spectroscopy combined with density functional theory calculations. Energy Fuels. 2017;31(2):1243–9. https://doi.org/10.1021/acs.energyfuels.6b02494.
Dutta Majumdar R, Gerken M, Mikula R, Hazendonk P. Validation of the Yen-Mullins model of Athabasca oil-sands asphaltenes using solution-state 1H NMR relaxation and 2D HSQC spectroscopy. Energy Fuels. 2013;27(11):6528–37. https://doi.org/10.1021/ef401412w.
Dutta Majumdar R, Montina T, Mullins OC, Gerken M, Hazendonk P. Insights into asphaltene aggregate structure using ultrafast MAS solid-state 1H NMR spectroscopy. Fuel. (2017);193;359–368.
Fan T, Buckley JS. Rapid and accurate SARA analysis of medium gravity crude oils. Energy Fuels. 2002;16(6):1571–5. https://doi.org/10.1021/ef0201228.
Ghosh AK, Chaudhuri P, Kumar B, Panja SS. Review on aggregation of asphaltene vis-a-vis spectroscopic studies. Fuel. 2016;185:541–54. https://doi.org/10.1016/j.fuel.2016.08.031.
Gray MR, Tykwinski RR, Stryker JM, Tan X. Supramolecular assembly model for aggregation of petroleum asphaltenes. Energy Fuels. 2011;25(7):3125–34. https://doi.org/10.1021/ef200654p.
Headen TF, Boek ES, Skipper NT. Evidence for asphaltene nanoaggregation in toluene and heptane from molecular dynamics simulations. Energy Fuels. 2009;23(3):1220–9. https://doi.org/10.1021/ef800872g.
Headen TF, Boek ES, Jackson G, Totton TS, Müller EA. Simulation of asphaltene aggregation through molecular dynamics: insights and limitations. Energy Fuels. 2017;31(2):1108–25. https://doi.org/10.1021/acs.energyfuels.6b02161.
Jian C, Tang T, Bhattacharjee S. Probing the effect of side-chain length on the aggregation of a model asphaltene using molecular dynamics simulations. Energy Fuels. 2013;27(4):2057–67. https://doi.org/10.1021/ef400097h.
Kharrat AM, Zacharia J, Cherian VJ, Anyatonwu A. Issues with comparing SARA methodologies. Energy Fuels. 2007;21(6):3618–21. https://doi.org/10.1021/ef700393a.
Khvostichenko DS, Andersen SI, Viktorov AI. Solubility and binding of water in toluene solutions of asphaltenes. Russ J Appl Chem. 2004;77(6):1013–8. https://doi.org/10.1023/B:RJAC.0000044135.29973.03.
Kirchnerová J, Cave GCB. The solubility of water in low-dielectric solvents. Can J Chem. 1976;54(24):3909–16. https://doi.org/10.1139/v76-562.
Kuznicki T, Masliyah JH, Bhattacharjee S. Molecular dynamics study of model molecules resembling asphaltene-like structures in aqueous organic solvent systems. Energy Fuels. 2008;22(4):2379–89.
Langevin D, Argillier JF. Interfacial behavior of asphaltenes. Adv Colloid Interface Sci. 2016;233:83–93. https://doi.org/10.1016/j.cis.2015.10.005.
Liu L, Zhang R, Wang X, Simon S, Sjöblom J, Xu Z, Jiang B. Interactions of polyaromatic compounds. Part 1: nanoaggregation probed by electrospray ionization mass spectrometry and molecular dynamics simulation. Energy Fuels. 2017;31(4):3465–74. https://doi.org/10.1021/acs.energyfuels.6b03029.
Merdrignac I, Espinat D. Physicochemical characterization of petroleum fractions: the state of the art. Oil Gas Sci Technol Rev. 2007;62(1):7–32. https://doi.org/10.2516/ogst:2007002.
Mullins OC, Betancourt SS, Cribbs ME, Dubost FX, Creek JL, Andrews AB, Venkataramanan L. The colloidal structure of crude oil and the structure of oil reservoirs. Energy Fuels. 2007;21(5):2785–94. https://doi.org/10.1021/ef0700883.
Mullins OC, Sabbah H, Eyssautier J, Pomerantz AE, Barré L, Andrews AB, Ruiz-Morales Y, et al. Advances in asphaltene science and the Yen-Mullins model. Energy Fuels. 2012;26(7):3986–4003. https://doi.org/10.1021/ef300185p.
Murgich J, Rodríguez J, Aray Y. Molecular recognition and molecular mechanics of micelles of some model asphaltenes and resins. Energy Fuels. 1996;10(1):68–76. https://doi.org/10.1021/ef950112p.
Ortega-Rodriguez A, Duda Y, Guevara-Rodriguez F, Lira-Galeana C. Stability and aggregation of asphaltenes in asphaltene- resin- solvent mixtures. Energy Fuels. 2004;18(3):674–81. https://doi.org/10.1021/ef0301699.
Pacheco-Sánchez JH, Alvarez-Ramirez F, Martínez-Magadán JM. Morphology of aggregated asphaltene structural models. Energy Fuels. 2004;18(6):1676–86. https://doi.org/10.1021/ef049911a.
Pomerantz AE, Wu Q, Mullins OC, Zare RN. Laser-based mass spectrometric assessment of asphaltene molecular weight, molecular architecture, and nanoaggregate number. Energy Fuels. 2015;29(5):2833–42. https://doi.org/10.1021/ef5020764.
Qiao P, Harbottle D, Tchoukov P, Masliyah J, Sjoblom J, Liu Q, Xu Z. Fractionation of asphaltenes in understanding their role in petroleum emulsion stability and fouling. Energy Fuels. 2017;31(4):3330–7. https://doi.org/10.1021/acs.energyfuels.6b02401.
Rogel E. Studies on asphaltene aggregation via computational chemistry. Colloids Surf A. 1995;104(1):85–93. https://doi.org/10.1016/0927-7757(95)03234-5.
Rogel E. Simulation of interactions in asphaltene aggregates. Energy Fuels. 2000;14(3):566–74. https://doi.org/10.1021/ef990166p.
Sabbah H, Morrow AL, Pomerantz AE, Zare RN. Evidence for island structures as the dominant architecture of asphaltenes. Energy Fuels. 2011;25(4):1597–604. https://doi.org/10.1021/ef101522w.
Samieadel A, Oldham D, Fini EH. Investigating molecular conformation and packing of oxidized asphaltene molecules in presence of paraffin wax. Fuel. 2018;220:503–12. https://doi.org/10.1016/j.fuel.2018.02.031.
Santos Silva H, Sodero ACR, Bouyssiere B, Carrier H, Korb J-P, Alfarra A, Vallverdu G, Bégué D, Baraille I. Molecular dynamics study of nanoaggregation in asphaltene mixtures: effects of the N, O, and S heteroatoms. Energy Fuels. 2016;30(7):5656–64. https://doi.org/10.1021/acs.energyfuels.6b01170.
Santos Silva H, Alfarra A, Vallverdu G, Bégué D, Bouyssiere B, Baraille I. Sensitivity of asphaltene aggregation toward the molecular architecture under desalting thermodynamic conditions. Energy Fuels. 2017a;32(3):2681–92. https://doi.org/10.1021/acs.energyfuels.7b02728.
Santos Silva H, Sodero ACR, Korb J-P, Alfarra A, Giusti P, Vallverdu G, Bégué D, Baraille I, Bouyssiere B. The role of metalloporphyrins on the physical-chemical properties of petroleum fluids. Fuel. 2017b;188:374–81. https://doi.org/10.1016/j.fuel.2016.10.065.
Schuler B, Meyer G, Peña D, Mullins OC, Gross L. Unraveling the molecular structures of asphaltenes by atomic force microscopy. J Am Chem Soc. 2015;137(31):9870–6. https://doi.org/10.1021/jacs.5b04056.
Schuler B, Fatayer S, Meyer G, Rogel E, Moir M, Zhang Y, Harper MR, et al. Heavy oil based mixtures of different origins and treatments studied by AFM. Energy Fuels. 2017;31(7):6856–61. https://doi.org/10.1021/acs.energyfuels.7b00805.
Schulze M, Lechner MP, Stryker JM, Tykwinski RR. Aggregation of asphaltene model compounds using a porphyrin tethered to a carboxylic acid. Org Biomol Chem. 2015;13(25):6984–91. https://doi.org/10.1039/C5OB00836K.
Sedghi M, Goual L. Role of resins on asphaltene stability. Energy Fuels. 2009;24(4):2275–80. https://doi.org/10.1021/ef9009235.
Sedghi M, Goual L, Welch W, Kubelka J. Effect of asphaltene structure on association and aggregation using molecular dynamics. J Phys Chem B. 2013;117(18):5765–76. https://doi.org/10.1021/jp401584u.
Sodero ACR, Santos Silva H, Level PG, Bouyssiere B, Korb J-P, Carrier H, Alfarra A, Bégué D, Baraille I. Investigation of the effect of sulfur heteroatom on asphaltene aggregation. Energy Fuels. 2016;30(6):4758–66. https://doi.org/10.1021/acs.energyfuels.6b00757.
Strausz OP, Lown EM. The chemistry of Alberta oil sands, bitumens and heavy oils. Calgary: Alberta Energy Research Institute (AERI); 2003.
Subramanian D, May N, Firoozabadi A. Functional molecules and the stability of water-in-crude oil emulsions. Energy Fuels. 2017;31(9):8967–77. https://doi.org/10.1021/acs.energyfuels.7b01039.
Takanohashi T, Sato S, Saito I, Tanaka R. Molecular dynamics simulation of the heat-induced relaxation of asphaltene aggregates. Energy Fuels. 2003;17(1):135–9. https://doi.org/10.1021/ef0201275.
Ungerer P, Rigby D, Leblanc B, Yiannourakou M. Sensitivity of the aggregation behaviour of asphaltenes to molecular weight and structure using molecular dynamics. Mol Simul. 2014;40(1–3):115–22. https://doi.org/10.1080/08927022.2013.850499.
Van Der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJC. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26(16):1701–18. https://doi.org/10.1002/jcc.20291.
Van Gunsteren WF, Daura X, Mark AE. GROMOS force field. In: Schleyer PVR, et al., editors. Encyclopedia of computational chemistry. Chichester: Wiley; 2002. https://doi.org/10.1002/0470845015.cga011.
Varadaraj R, Brons C. Molecular origins of crude oil interfacial activity. Part 4: oil–water interface elasticity and crude oil asphaltene films. Energy Fuels. 2012;26(12):7164–9. https://doi.org/10.1021/ef300830f.
Wang J, Gayatri MA, Ferguson AL. Mesoscale simulation and machine learning of asphaltene aggregation phase behavior and molecular assembly landscapes. J. Phys. Chem. B. 2017a;121(18):4923–44. https://doi.org/10.1021/acs.jpcb.7b02574.
Wang X, Zhang R, Liu L, Qiao P, Simon S, Sjoblom J, et al. Interactions of polyaromatic compounds. Part 2. Flocculation probed by dynamic light scattering and molecular dynamics simulation. Energy Fuels. 2017b;31(9):9201–12. https://doi.org/10.1021/acs.energyfuels.7b01511.
Acknowledgements
The authors would like to thank to the DN (Direction du Numérique) from Université de Pau et des Pays de l’Adour, MCIA (Mésocentre de Calcul Intensif Aquitain) and GENCI-CINES (Grant 2017-c2016087698) for providing the computation power needed for this project. Isifor-Carnot Institute and Total Refining & Chemicals are also acknowledged for their financial support to this research project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Edited by Xiu-Qin Zhu
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Santos Silva, H., Alfarra, A., Vallverdu, G. et al. Asphaltene aggregation studied by molecular dynamics simulations: role of the molecular architecture and solvents on the supramolecular or colloidal behavior. Pet. Sci. 16, 669–684 (2019). https://doi.org/10.1007/s12182-019-0321-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12182-019-0321-y