
Abstract
Low-level detection and quantification of genetically engineered (GE) traits with polymerase chain reaction (PCR) is challenging. For unapproved GE events, any level of detection is not acceptable in some countries because of zero tolerance. Droplet digital PCR (ddPCR) has been successfully used for absolute quantification of GE events. In this study, reliability of low level quantification of GE events with ddPCR was assessed using a total of 50, 100, 200, 400, and 600 ng DNA spiked at 0.01% and 0.1% concentration levels. Genetically engineered canola (GT73 and MON88302 events) and soybean (A2704-12 and DP305423 events) events were used for the study. For samples spiked at 0.1% level, reliable quantification was achieved for the four GE events using 50 or 100 ng DNA. Few target droplets were generated for 0.01% spiked GE samples using 50 and 100 ng DNA. Increasing the amount of DNA for ddPCR generated more number of target droplets. For GE canola events, the use of 400 and 600 ng DNA for ddPCR resulted in saturation. The use of multiple wells of 200 ng DNA (instead of 400 and 600 ng per well) helped to overcome the saturation problem. Overall, the use of high amount of DNA for ddPCR was helpful for the detection and quantification of 0.01% GE samples.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Accuracy of genetically engineered (GE) trait identification and quantification in non-processed and processed food samples is affected by factors such as sampling/sub-sampling, DNA extraction, DNA quality/purity, DNA amount, and PCR conditions (Alarcon et al. 2019; Demeke and Dobnik 2018). The minimum required performance limit (MRPL), which is the lowest level of GE material for satisfactorily reproducible validation of quantitative methods, has been set at 0.1% (European Commission Regulation No. 619/2011). However, for some countries, any level of detection of unapproved GE events is not acceptable. Unapproved GE events on the European market are reported through the Rapid Alert System for Food and Feed (European Commission, RASFF Portal). For unauthorized FP967 GE flax, sampling and testing protocol was set to achieve a level of detection of 0.01% (Canadian Grain Commission 2018).
One of the challenges for accurate quantification of GE events below 0.1% is obtaining repeatable or reliable results. Transgenic concentrations of 0.1% (m/m) certified reference materials resulted in bias and uncertainty (Cottenet et al. 2019). Concentrations such as 1% and 10% resulted in less bias. Use of high amount of DNA for real-time quantitative PCR enabled reliable detection of GE events at low concentration levels (Mano et al. 2018).
Effect of amount of DNA on quantitative detection of GE events using droplet digital PCR (ddPCR) results has not been investigated. The objective of the study was to determine the effect of amount of DNA on quantification of GE traits using ddPCR. The study will help to determine if high amount of DNA can be used for accurate quantification of low GE concentration using ddPCR. Real-time qualitative PCR assay was also used for comparison.
Materials and Methods
Source of Samples
The sources of canola samples were Legend (non-GE variety), AOCS 1011-A (MON88302 reference material), and Conquest breeder seed (GT73 event). Legend and Conquest seeds were received from Oilseeds Program of the Canadian Grain Commission. For soybean, Colby (non-GE variety), PS 2295 LL (A2704-12 variety, Breeder seed), and ERM-BF426d (DP305423 event, certified reference material) were used. Certified seeds of Colby were obtained from WG Thompson & Sons (Ontario, Canada), while certified seeds of PS 2295 LL were obtained from Bayer Crop Science. GE and non-GE seed samples were ground with a RETSCH model ZM 100 Centrifugal Grinding Mill (Fischer Scientific, Ottawa, Canada) using a 1-mm sieve.
DNA Extraction and Quantification
DNeasy mericon Food kit (QIAGEN Life Sciences, LLC, Louisville, KY) was used for DNA extraction. The quality of DNA was checked by agarose gel electrophoresis (1.2%). 1× TAE (Tris-Acetate-EDTA) buffer containing 0.44 mM GelRed dye (Biotium, Cedarlane Laboratories, Hornby, Ontario) was used for gel electrophoresis. The amount of DNA was determined with a Pico Green® assay (Molecular Probes, Eugene, OR, USA), and λ DNA was used to generate calibration curve. The Pico Green® dsDNA reagent was added to diluted DNA in a 96-well plate. SpectraMax M5 Microplate Reader (Molecular Devices, Toronto, Canada) was used to measure the fluorescence in duplicates. 0.1% and 0.01% GE samples were prepared by mixing non-GE and GE DNA.
Droplet Digital PCR
Digital PCR was carried out using the QX200 ddPCR system (Bio-Rad, Pleasanton, CA). The primer and probe DNA sequences for the target and reference genes as well as the concentrations used for ddPCR and RT-qPCR assays are listed in Table 1. Duplex ddPCR (mixing of target and reference primers and probes in the same reaction well) was used to generate target and reference droplets at the same time. 12.5 μL Bio-Rad ddPCR Supermix for Probes (no dUTP) and 50, 100, 200, 400, and 600 ng total template DNA were used for the ddPCR assays. The DG-32 cartridge for automated droplet generator (Cat. No. 186-4108) was used to produce droplets of 20 μL volume. The generated droplets were transferred to semi-skirted 96-well Eppendorf plates (Cat. No. 12001925) and sealed with a pierceable foil heat seal (Cat. No. 1814040). PCR amplification of the generated droplets was carried out using a MJ Thermal Cycler (PTC 200). The thermal cycling conditions used were an initial denaturation step of 95 °C for 10 min, followed by 50 cycles of 95 °C for 15 s, 60 °C for 1 min, and a final step of 98 °C for 10 min. A ramp rate of 0.6 °C/s was used between the cycling steps, and a ramp rate of 0.3 °C/s was used at the last step to maintain the reaction at 15 °C. The droplets were counted using the droplet reader on the QX200 system. QuantaSoft version 1.7.4.0917 and automatic threshold were used for ddPCR data analysis.
Real-Time Qualitative PCR
Real-time qualitative PCR (RT-PCR) was carried out using the ABI 7500 PCR instrument (Thermo Fisher Scientific). The RT-PCR contained DNA template (50, 100, 200, 400, and 600 ng), primers, and probes (concentrations provided in Table 1), and 1× (12.5 μL) TaqMan Universal Master Mix II with UNG (Applied Biosystems). The thermal profile used for the real-time PCR had an initial hold for 2 min at 50 °C, 10 min at 95 °C, and then 40 cycles of 15 s at 95 °C and 1 min annealing at 60 °C.
Results and Discussion
Effect of Amount of DNA on ddPCR Quantification of GE Canola and Soybean Events
Reliable quantification of GE events in cereals, grains, and food samples is important. Fifty to 100 ng DNA is generally used for the detection, identification, and quantification of GE events by PCR. Accurate quantification of GE events is challenging for concentrations below 0.1% (European Commission Regulation No. 619/2011; Kay and Van den Eede 2001). For this study, 0.01% and 0.1% spiked GE canola and soybean DNA were prepared for ddPCR analysis using total DNA concentrations of 50, 100, 200, 400, and 600 ng. For 100 ng DNA samples of 0.1% GT73 and MON88302 canola, an average of 45 and 69 target droplets were observed, respectively (Table 2). The number of target droplets generated steadily increased as the amount of DNA sample was increased. The haploid genome weight of Brassica napus (canola) is 1.13 pg (Johnston et al. 2005). Thus, 100 ng DNA spiked at 0.1% will have approximately 90 copies of canola target DNA; and the chance of PCR detection of GE materials at 0.1% level is generally high. The measured concentrations were mostly within the expected range (Table 2). Overall, reliable ddPCR detection was achieved for the two GE canola events at 0.1% level using 50 or 100 ng input DNA, and there is no need to use higher amount of DNA.
For 0.01% GE concentration, an average of two to eight target droplets were observed for 50, 100, and 200 ng DNA for the two GE canola events (Table 2). Bio-Rad recommends at least three positive droplets in order to call a sample positive. The three positive droplets can be either in a single well or across merged wells (Droplet Digital™ PCR Applications Guide, Bio-Rad). Even though the measured concentrations were close to the 0.01% expected value (Table 2), repeatability can be an issue with the generation of few number of target droplets.
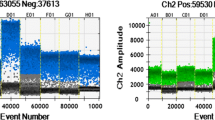
For 0.1% GE soybean DNA, the number of target droplets increased as more amount of DNA was used for ddPCR (Table 3). Dual-positive target and reference droplets were more common than single droplets, due to high occupancy of the reference gene (Fig. 1). The measured target concentrations of GE samples were similar to the expected values. The number of positive target droplets proportionally increased up to 600 ng DNA. For 0.01% GE DNA, less than four positive target droplets were obtained on average for 50, 100, and 200 ng DNA for both GE soybean events. In comparison, relatively higher number of target droplets were generated for 0.01% spiked 400 and 600 ng soybean DNA samples (Table 3).

Distribution of droplets for 0.1% DP305423 soybean. A, target droplets; B, dual droplets; C, negative droplets; and D, reference droplets. I, II, III, IV, and V indicate 50, 100, 200, 400, and 600 ng DNA
Saturation Problem with the Use of 400 and 600 ng Canola DNA
The use of 400 and 600 ng canola DNA for ddPCR resulted in saturation of the system for both 0.1% and 0.01% concentrations. Excess number of reference droplets was generated, whereas no negative droplets were present. When there are no negative droplets, Poisson correction cannot be applied, and it is not possible to calculate concentration (Droplet Digital™ PCR Applications Guide, Bio-Rad). To overcome the saturation problem, multiple wells (2 wells of 200 ng for 400 ng total DNA and 3 wells of 200 ng for 600 ng total DNA) were used, and the droplets were pooled. For pooled results of 400 ng and 600 ng DNA, 21 to 30 and 30 to 47 target droplets were obtained for 0.01% spiked GT73 and MON88302 GE canola DNA, respectively (Table 2). The use of multiple wells of 200 ng canola DNA and pooling the droplets enabled reliable ddPCR detection at 0.01% level. In contrast to canola, droplet saturation with soybean reference gene was not observed when 400 and 600 ng DNA was used for ddPCR. The use of multiple wells of 200 ng soybean DNA and pooling the results (for total of 400 and 600 ng DNA) slightly increased the number of target droplets compared with single well results with the exception of 400 ng DP305423 soybean DNA (Table 3).
Qualitative PCR Results for 0.01% Spiked Samples
For qualitative RT-PCR, amplification was observed for most samples with the exception of low amount of DNA for A2704-12 and DP305423 soybean. Decreasing average threshold (Ct) values were observed as the DNA concentration was increased for 0.01% spiked samples for three of the four GE events (Table 4). Generally, reliability of detection increases if the Ct values are lower than 40. For DP305423 soybean event, the decrease in Ct value was not consistent. Mano et al. (2018) reported that a tenfold increase in sensitivity was achieved as a result of using 500 ng DNA instead of 50 ng DNA for real-time PCR. According to Fernandes et al. (2016), the amount of template DNA used in the PCR may be increased when the number of available gene copies is not sufficient for PCR amplification. However, there can be PCR inhibition because of using high amount of DNA.
Conclusions
Absolute quantification of GE canola (GT73 and MON88302 events) and soybean (A2704-12 and DP305423 events) events was achieved at 0.1% level using 50 and 100 ng DNA. For 0.01% spiked samples, the addition of 400 and 600 ng DNA in ddPCR generated meaningful data points for absolute quantification of GE canola and soybean events. The use of 400 and 600 ng canola DNA for ddPCR resulted in saturation of the system because of generation of excessive number of reference droplets and absence of negative droplets. The saturation problem was overcome by the use of multiple wells of 200 ng DNA. For qualitative PCR of 0.01% spiked samples, lower threshold values were obtained for 400 and 600 ng DNA compared to 50 or 100 ng DNA.
References
Alarcon CM, Shan G, Layton DT, Bell TA, Whipkey S, Shillito RD (2019) Application of DNA- and protein-based detection methods in agricultural biotechnology. J Agric Food Chem 67:1019–1028
Canadian Grain Commission (2018) Sampling and testing protocol for bulk shipments of flaxseed exported to the European Union https://www.grainscanada.gc.ca/en/industry/policy/protocols/european/pdf/stpf-peevl.pdf Accessed 16 June 2020
Cottenet G, Blancpain C, Chuah PF (2019) Performance assessment of digital PCR for the quantification of GM-maize and GM-soya events. Anal Bioanal Chem 411:2461–2469
Demeke T, Dobnik D (2018) Critical assessment of digital PCR for the detection and quantification of genetically modified organisms. Anal Bioanal Chem 410:4039–4050
Droplet Digital™ PCR Applications guide. BioRad. http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6407.pdf. Accessed 16 June 2020
European Commission (Regulation No. 619/2011) Laying down the methods of sampling and analysis for the official control of feed as regards presence of genetically modified material for which an authorisation procedure is pending or the authorisation of which has expired. Official Journal of the European Union. Vol. 54 L166/9
European Commission - The Rapid Alert System for Food and Feed (RASFF) Portal. https://webgate.ec.europa.eu/rasff-window/portal. Accessed 16 June 2020
Fernandes TJR, Costa J, Plácido A, Villa C, Grazina L et al (2016) Genetically modified organism analysis as affected by DNA degradation. In: Watson RR, Preedy VR (eds) Genetically modified organisms in food. Production, safety, regulation and public health. Academic Press, Amsterdam
Johnston JS, Pepper AE, Hall AE, Chen ZJ, Hodnett G, Drabek J, Lopez R, Price HJ (2005) Evolution of genome size in Brassicaceae. Ann Bot 95:229–223
Kay S, Van den Eede G (2001) The limits of GMO detection. Nat Biotechnol 19:405
Mano J, Hatano S, Nagatomi Y, Futo S, Takabatake R, Kitta K (2018) Highly sensitive GMO detection using real-time PCR with a large amount of DNA template: single-laboratory validation. J AOAC Int 101:507–514
Acknowledgments
Dr. Veronique Barthet, Program Manager of the Oilseeds Program of the Grain Research Laboratory of the Canadian Grain Commission is acknowledged for supply of seeds.
Author information
Authors and Affiliations
Contributions
Conceptualization and methodology were performed by Tigst Demeke and Sun-Jong Lee. Material preparation, data collection, and analysis were performed by Monika Eng and Michelle Holigroski. The first draft of the manuscript was written by Tigst Demeke, and all authors have commented and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Tigst Demeke declares that he has no conflict of interest. Monika Eng declares that she has no conflict of interest. Michelle Holigroski declares that she has no conflict of interest. Sung-Jong Lee declares that he has no conflict of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Not applicable.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Demeke, T., Eng, M., Holigroski, M. et al. Effect of Amount of DNA on Digital PCR Assessment of Genetically Engineered Canola and Soybean Events. Food Anal. Methods 14, 372–379 (2021). https://doi.org/10.1007/s12161-020-01889-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12161-020-01889-y