
Abstract
Vesicular glutamate transporters (VGLUTs) control quantal size of glutamatergic transmission and have been the center of numerous studies over the past two decades. VGLUTs contain two independent transport modes that facilitate glutamate packaging into synaptic vesicles and phosphate (Pi) ion transport into the synaptic terminal. While a transmembrane proton electrical gradient established by a vacuolar-type ATPase powers vesicular glutamate transport, recent studies indicate that binding sites and flux properties for chloride, potassium, and protons within VGLUTs themselves regulate VGLUT activity as well. These intrinsic ionic binding and flux properties of VGLUTs can therefore be modulated by neurophysiological conditions to affect levels of glutamate available for release from synapses. Despite their extraordinary importance, specific and high-affinity pharmacological compounds that interact with these sites and regulate VGLUT function, distinguish between the various modes of transport, and the different isoforms themselves, are lacking. In this review, we provide an overview of the physiologic sites for VGLUT regulation that could modulate glutamate release in an over-active synapse or in a disease state.
Similar content being viewed by others

Introduction
Glutamate (Glu) is the major excitatory neurotransmitter in the mammalian central nervous system and is involved in all brain functions and in various neurological pathologies [1,2,3,4,5]. Excessive and sustained release of glutamate from synapses triggers glutamate-induced, NMDA receptor-dependent excitotoxicity. The prolonged presence of glutamate in the extrasynaptic space in the hippocampus has been proposed to underlie the onset of a variety of cognitive neurological disorders including those observed after cardiac arrest-induced global or focal brain ischemia, traumatic brain injury, epilepsy, and Alzheimer’s disease (AD) [6,7,8,9,10,11,12,13,14], among others. Mechanisms to reduce excessive synaptic glutamate release under these conditions could potentially prevent/reduce excitotoxic damage to vulnerable hippocampal neurons. In addition, glutamate is suspected to be at the core of major psychiatric disorders, such as schizophrenia, depression, addiction, and compulsive disorders [15,16,17,18,19,20,21,22,23,24,25]. Treatment options to modulate glutamatergic transmission are limited. Currently, in human studies, most post-synaptic glutamatergic interventions (with the exception of esketamine in major depressive disorders [26,27,28] and memantine in AD [29,30,31]) have been disappointing because of poor efficacy or unacceptable side effects [32, 33]. Compounds selectively modulating the presynaptic release of glutamate could constitute a novel pharmacological approach for the prevention of glutamate excitotoxicity and modulation of behavior under various disorders. Possible sites to regulate presynaptic glutamate release are modulation of synaptic glutamate synthesis, VGLUT expression level, or sites that mediate the transport of glutamate into synaptic vesicles. Three subtypes of vesicular glutamate transporters (VGLUT1–3) have been identified that package glutamate into vesicles [34,35,36,37,38,39,40,41,42] reviewed in [43,44,45,46]. The three VGLUTs share a high degree of structural homology and, so far, their functional activity cannot be distinguished by their bioenergetic or pharmacological profiles. Nevertheless, identifying the structural and functional sites for VGLUT regulation and understanding the differential molecular and cellular modes of VGLUT regulation themselves is critical to recognize novel potential targets to modulate presynaptic glutamatergic transmission in normal and aberrant states.
VGLUTs: Markers for Glutamatergic Transmission
VGLUT1 and VGLUT2 are expressed in distinct and complementary subsets of neurons in the CNS that display differences in release probability [36, 39]. VGLUT1 is the most abundant subtype in the CNS [47]. Unlike, vesicular transporters for monoamines (VMAT1 and VMAT2) and acetycholine (VAChT) that are found in both cell bodies and nerve terminals, VGLUT1 and VGLUT2 proteins are restricted to nerve endings where they continuously recycle between the plasma membrane, endosomes, and newly formed synaptic vesicles [48,49,50]. Thus, VGLUT1 and VGLUT2 likely recycle in synapses for an extended period of time and are therefore unique synaptic markers for select glutamatergic terminals. VGLUT1 is found in asymmetric synapses in the cerebral cortex, the hippocampus, the cerebellum, and the amygdala (for review, see [44]). VGLUT2 is primarily, though not exclusively, used by subcortical excitatory neurons [36, 38, 39, 51,52,53]. VGLUT1 and VGLUT2 are also co-expressed in some thalamic neurons, layer IV cortical interneurons and pinealocytes [38, 54,55,56,57]. Co-expression of VGLUT1 and VGLUT2 in synapses could afford these cells with two distinct modes of release, if they are sorted to different vesicles. VGLUT3, the atypical subtype, is sparingly expressed compared to VGLUT1 and VGLUT2 [44, 58] and is often present in neurons that use other “classic” neurotransmitters, such as serotonin, acetylcholine or GABA [40,41,42, 59]. Indeed, in some neuronal populations, such as striatal cholinergic interneurons, VGLUT3 is abundantly present in the somato-dendritic compartment, although its function there is not yet elucidated [44]. However, VMAT2 sorting to the somatodendritic compartment in neurons confers the activity-dependent release of monoamines as well as multiple retrograde signals involved in synaptic function, growth, and plasticity [60, 61]. VGLUT1–3 are also expressed in sensory nerves from the ventral horn of the spinal cord, suggesting their involvement in nociception [62,63,64,65,66,67,68]. The distribution of VGLUT1–3 is conserved between humans and rodents [69].
Mouse lines with deleted VGLUTs demonstrate the importance of VGLUTs for glutamatergic transmission in normal brain function and facilitate recognizing roles VGLUTs could contribute to brain disorders [47, 70,71,72,73,74,75,76,77,78]. In mice, VGLUT1 or VGLUT2 deletion (VGLUT1-KO mice and VGLUT2-KO mice, respectively) is lethal. VGLUT1-KO mice die 2 to 3 weeks after birth, which is a time that normally follows a strong up-regulation of VGLUT1 [55, 79, 80] and increased synaptic vesicle clustering in VGLUT1 synapses [81, 82]. The post-natal up-regulation of VGLUT1 also replaces the VGLUT2 isoform that is predominant in early cerebellar, hippocampal, and cortical synapses [83]. VGLUT2-KO mice succumb to respiratory failure immediately after birth [71, 72] as VGLUT2 is abundantly expressed in descending and in local brainstem glutamatergic systems that control respiration [39, 52, 84, 85]. Reduced expression of VGLUT2 during neuronal development results in reduced pyramidal neuron plasticity, dendritic refinement, and spatial learning [76]. Unlike VGLUT1-KO mice and VGLUT2-KO mice, VGLUT3-KO mice survive [73, 86]. However, VGLUT3 null mice are deaf, hyperactive, and demonstrate increased anxiety [73,74,75, 87]. VGLUT3 may also provide protection against neonatal hypoxic stress [88] and be critically involved in reward regulation [78]. Selective modulation of specific VGLUT-encoded systems will be required to repair any alteration in glutamatergic transmission in specific VGLUT pathways that may contribute to excitotoxic or disease pathology.
VGLUT expression levels have been considered as potential pathological or diagnostic markers for impaired or over-active glutamatergic transmission. In humans, altered expression of VGLUT1 is associated with anxiety and mood disorder [15, 17,18,19], and in neurological conditions, such as Parkinson’s disease, AD, and epilepsy [89,90,91,92]. Modulation of VGLUT2 expression levels have been observed in schizophrenia and neuropathic pain [21, 64, 72]. Reduced expression of VGLUT2 is associated with decreased motoneuron degeneration in a mouse model of amyotrophic lateral sclerosis (ALS) [93], yet these mice are more susceptible to clonic seizures [94]. A marked increase in VGLUT1 expression and glutamate release (+ 40%) has been reported in a tau animal model of AD during the early stages of the pathology [95]. Neuronal hyperactivity and increased functional connectivity have been confirmed in preclinical AD, mild cognitive impairment (MCI), and early AD stages at various levels [96,97,98]. Later stages of AD in humans and animal models of AD [99] may include outright loss of excitatory synaptic terminals [100,101,102,103,104,105,106]. In humans, initial studies pointed to a marked decrease of VGLUT1 expression in the cortex of AD patients [89, 90, 107]. However, recent work suggests that synapse loss is probably not a hallmark specific to AD [108] and only minimal alterations of VGLUT1 are observed in the prefrontal cortex of demented individuals [109]. Instead, Alzheimer’s disease may be a result of presynaptic glutamatergic dysfunction induced by tau and oligomeric β-amyloid [33, 110,111,112].
Molecular Sites for VGLUT Regulation
VGLUTs are specific molecular and functional markers of glutamatergic transmission as their presence in synaptic vesicles in neurons is sufficient to convey exocytotic glutamate release [35]. Excitatory synaptic vesicles in mammalian synapses are thought to contain between 4 and 14 molecules of VGLUT each [113, 114]. While alterations in levels of VGLUTs leads to multiple altered or pathological behaviors in humans and in mouse models, it is not entirely clear how alterations in synaptic VGLUT levels impact glutamate transmission. Primary hippocampal autaptic cultures from VGLUT1- and VGLUT2-KO mice reveal a decrease in quantal size that can be rescued by transgene over-expression of VGLUT1 or VGLUT2, respectively [70, 72]. However, miniature EPSC amplitude, reflecting the amount of glutamate released per vesicle (as well as the postsynaptic response) does not differ in acute hippocampal slices from VGLUT1-KO mice relative to wild-type littermates [115]. Likewise, severe reduction of VGLUT3 (up to 80%) does not alter glutamatergic signaling [116]. Liu and colleagues verified biophysically that increasing the number of VGLUT1 molecules at hippocampal excitatory synapses in dissociated neuronal cultures results in an increase in the amount of glutamate released per vesicle into the synaptic cleft [117]. Control of the neurotransmitter content by transporter copy number has been interpreted as a result of an equilibrium between glutamate uptake and leakage. The modulation of synaptic strength by VGLUT1 expression is endogenously regulated, both across development to coincide with a maturational increase in vesicle cycling and quantal amplitude and by excitatory and inhibitory receptor activation in mature neurons to provide an activity-dependent scaling of quantal size via a presynaptic mechanism [117,118,119]. Indeed, presynaptic scaling of VGLUT1 and VGLUT2 levels in synapses is observed at the molecular and synaptic level [55, 120]. Presynaptic scaling also occurs with the vesicular GABA transporter (VIAAT/VGAT) [55, 121]. Work in Drosophila suggests that a single copy of VGLUT on a vesicle is sufficient to load a vesicle [122]. While increasing VGLUT levels in Drosophila also results in increased quantal size (and synaptic vesicle volume) a compensatory decrease is observed in the number of synaptic vesicles released that maintains normal levels of synaptic excitation [123]. Molecular mechanisms of VGLUT regulation for homeostasis may differ in Drosophila, which only express a single VGLUT type, and higher organisms that express 3 VGLUTs in the brain.
Original findings revealed that a reduction of VGLUT1 expression results in the loss of synaptic vesicles in nerve terminals [115]. More recent studies indicate that synaptic vesicle clustering in VGLUT1 terminals is mediated through a tripartite interaction of VGLUT1, endophilinA1, and intersectin1 resulting in a combined reduction of axonal synaptic vesicle super-pool size and miniature excitatory events frequency [124, 125]. Indeed, low glutamate release probability is a characteristic feature of VGLUT1-encoded synaptic terminals [126,127,128,129]. Similarly, using high-resolution stimulated emission depletion (STED) microscopy, decreased VGLUT3 protein levels seems to be accompanied by a reduction in the number of VGLUT3-positive vesicles in varicosities [116]. VGLUT expression in mammalian synapses may therefore not only contribute to quantal size, but also to the availability of vesicles for release, which could explain the different release properties of VGLUT1- and VGLUT2-encoded synapses.
Molecular regulation of VGLUT synthesis and degradation represent powerful targets to control glutamate availability at the glutamate site on VGLUTs for transport into vesicles and subsequent exocytotic release at synapses. Regulation of VGLUT expression is used endogenously to provide resistance against glutamate-induced neurodegeneration. For instance, ischemic tolerance is a well-known phenomenon in which brief ischemic insults (ischemic preconditioning) confer robust neuroprotection to hippocampal CA1 neurons against a subsequent severe ischemic challenge [130,131,132,133]. Similarly, one or more brief seizures can serve to activate endogenous protective programs which render brain regions temporarily less susceptible to damage following an otherwise harmful episode of status epilepticus (i.e., a prolonged seizure) [78, 134, 135]. Furthermore, ischemic/hypoxic preconditioning can protect the brain from seizure-induced damage while epileptic preconditioning can protect vulnerable neurons to ischemia-induced injury [136, 137] suggesting some common mechanisms for neuroprotection. Although the molecular mechanisms underlying ischemic/epileptic tolerance are not yet fully delineated, the considerable delay (~ 24 h) from the preconditioning stimulus until onset of tolerance is consistent with a role for transcriptional changes in such neuroprotection. In neuronal cortical and hippocampal culture models, preconditioning induces tolerance to exocytotic injury by suppressing vesicular glutamate release and increasing vesicular release of GABA [138,139,140,141]. Accordingly, preconditioning stimuli result in the presynaptic down-regulation of VGLUT1 expression in excitatory neurons and up-regulation of VIAAT and the GABA synthesizing enzymes GAD65 and GAD67 in inhibitory neurons [55, 118, 120, 121, 142, 143]. Interestingly, preconditioning stimuli also up-regulate VGLUT2 in select VGLUT1-encoded synapses in cortical neurons that synapse onto GABAergic neurons [120], suggesting that selective trafficking of VGLUT2 in these neurons to synapses that target GABAergic inhibitory neurons could promote glutamate-induced feed-forward inhibitory transmission as neuroprotective strategy for neural circuit stability.
Molecular sites for selective VGLUT regulation are not yet well defined. A critical challenge moving forward is to be able to selectively modulate discrete VGLUT-driven pathways in the brain. Understanding the genetic controls and physiologic factors that regulate VGLUT expression is therefore critical [120, 144,145,146,147,148]. In addition, the development of viral vectors that allow efficient glutamatergic-selective gene expression or knockdown would permit the selective modification of VGLUT levels in defined neuronal cell populations [149,150,151]. Indeed, restoration of hearing in the VGLUT3 knockout mouse has been accomplished using virally mediated gene therapy, which is an important step towards gene therapy of human deafness [152].
VGLUT Structural Sites
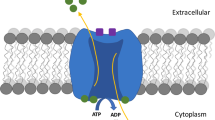
The superfamily of solute carrier transmembrane transporters (SLC) is encoded by more than 300 genes organized into 52 families. The substrates used by these carriers are very diverse, including charged or neutral organic molecules and various ions. Currently, in the SLC superfamily, nine genes divided into three families (SLC17, SLC18, SLC32) have been identified as coding for vesicular transporters and are divided according to their natural substrates (Fig. 1) [43]. These three SLC family members predominantly use the transmembrane proton electrochemical gradient (ΔμH+) generated by a vacuolar-type H+ pump (V-ATPase) to translocate substrates. The SLC17 family includes (i) VGLUT1–3 (substrate: glutamate, Km ~ 1 mM) [153], (ii) Sialin (substrate: sialic acid, Km ~ 0.2 mM) [154], and (iii) a vesicular nucleotide transporter VNUT (substrate: ATP, Km ~ 1 mM) [155]. The SLC17 family also includes the Na+-dependent inorganic phosphate (Pi) transporters NPT-1, NPT-3, NPT-4, and NPT-5 (substrate: Pi, Km ~ 3–6 mM) [156]. The SLC18 family includes the vesicular polyamine transporter VPAT, (SLC18B1, substrate: spermine and spermidine, Km ~ 100 μM) [157], vesicular amine transporters for adrenaline, dopamine, norepinephrine, histamine, and serotonin (VMAT1 and VMAT2, SLC18A1 and SLC18A2, Km ~ 1 μM) [158,159,160,161] and acetylcholine (VAChT, Km ~ 1 mM) [162, 163]. SLC32 includes a single member, the vesicular inhibitory amino acid transporter (VIAAT or VGAT) [164, 165] that can transport GABA or glycine (Km ~ 5–10 mM) [166, 167].

H+-dependent vesicular neurotransmitter transport. Specific H+-dependent transporters are responsible for neurotransmitter vesicular uptake and belong to different families depending on the global charge of their respective substrates: SLC17 for glutamate and ATP, SLC18 for monoamines and acetylcholine, and SLC32 for GABA and glycine. The driving force for the vesicular accumulation of all neurotransmitters is an H+ electrochemical gradient (∆μH+) generated by the vacuolar V-ATPase
VGLUTs are composed of 560 to 582 amino acids with 12-membrane spanning segments and with the N- and C-termini in the cytoplasm. No crystal structures for these proteins are currently available, but technical progress in transporter crystallography of distantly related bacterial transporters provides some clues to the resolution of VGLUT structure. VGLUTs show primary sequence homology with the major facilitator superfamily (MFS), the second major family of transmembrane transporters involved in the translocation of small solutes using the driving force of an electrochemical gradient [168]. The crystallographic 3D structures of the lactose bacterial permease, also known as glycerol-3-phosphate transporter (GlpT), as well as D-galactonate transporter (DgoT) led to the conclusion that these transporters consist of 12 α-helices organized into two groups of 6 (two halves) [169, 170]. The two groups of 6 α-helices are connected by a cytoplasmic flexible loop, forming a hydrophilic cavity at their center deep in the transporter for translocation of hydrophilic substrates. The amino acid residues responsible for the specificity of the transporter are located on the walls of this polar pocket [170,171,172]. Because of the distant, yet distinct, homology between GlpT, DgoT, and VGLUTs, a putative 3D homology model of VGLUTs can be postulated (Fig. 2).

Putative 3D structure of GlpT and VGLUTs. The glycerol-3-phosphate transporter (GlpT) shares distant homology with members of the SLC17 family. Its 3D structure possesses two groups of 6 transmembrane helices (H1–12) linked by a large cytosolic and a luminal loop [169] (a, b). VGLUT 3D homology model based on glpT structure (pdb code 1PW4) is shown in (c, d). Key residues for glutamate transport identified so far are H128 (blue), R184 (blue), and E191 (red) located on TM2, TM4, and TM4, respectively. The software that use used to make the models was Discovery Studio Modeling Environment, release 4.5; Dassault Systems BIOVIA: San Diego, 2015
Site-directed mutagenesis of VGLUTs has identified several transmembrane charged residues important for the recognition and translocation of substrates (i.e., Arg184, His128, and Glu191) [173]. Arg184 is located on transmembrane domain 4 (H4). This residue is conserved in all the members of the SLC17 family, suggesting a common and essential role, such as recognition of anionic substrates (Glu, Asp, ATP), and Cl− ions. Its mutation into a neutral residue (e.g., alanine), or a positively or negatively charged amino acid (e.g., glutamate, or even lysine), leads to complete inhibition of transport [173]. Interestingly, Na+-dependent Pi transport of this mutant was normal [173]. More recent studies show that the arginine residue Arg184 in TMD4 selectively controls the allosteric binding of Cl− in the vesicle lumen, which might be important to VGLUT activation [174] (see below). His128 and Glu191 reside on transmembrane domain 2 and 4 (TMD2 and TMD4), respectively and are conserved in the 3 subtypes of VGLUTs—mutation of these residues also inhibit glutamate transport [173]. However, His128 is not conserved in the anionic transporter sialin [175], suggesting an important role for His128 in the selectivity of substrate transport. In this regard, sialin transports both aspartate and glutamate, while VGLUTs only recognize glutamate. In the VGLUT 3D putative model, these three charged residues are located deep in the hydrophilic binding pocket created by α helices [173, 176]. In alternate access model of transport, the interaction of Cl− ions with a basic residue may induce a conformational change that facilitates the translocation of glutamate (Fig. 3).

Model of transport and ion binding of VGLUTs. One model that has been proposed to describe the uptake of glutamate by VGLUTs is based on the alternate access model [177]. a VGLUTs have several binding sites for transported ions: glutamate, Cl−, K+ and H+ (and Na+ and Pi). b In high chloride concentrations and in the cytosol upon open conformation, VGLUT is loaded with one glutamate, one Cl−, and partially by one K+. Glutamate and K+ are released into the lumen when VGLUT shifts to a lumen open conformation (step 1). At this stage, the anionic binding site is filled with another Cl− while the cationic site loads one proton (step 2). Finally, VGLUT reopens in the cytosol where it releases one Cl− and one proton (step 3). c In the presence of low luminal concentrations of chloride, VGLUT follows the same steps as in b but during step 2, the partially filled cationic site is fully occupied by a proton, while only a few chloride ions are released into the cytosol in step 3
The molecular and functional methods available today will enable the identification of additional structural sites important for VGLUT-specific transport function. A clever idea to understand basic mechanisms of ΔμH+-driven substrate translocation across intracellular brain vesicles is to understand these mechanisms in simpler organisms, such as their bacterial homologs. For example, Shuldiner and colleagues examined SARs between various transmembrane embedded charged amino acids in the bacterial multiresistant transporters for the study of their mammalian counterparts, such as VMATs [178,179,180,181]. Recently, Edwards and colleagues used a similar approach with DgoT showing an interesting cooperation of H+ transfer between transmembrane embedded glutamate (Glu133) and arginine (Arg47) residues that suggest a mechanism that couples H+ flux to substrate recognition [170]. That is, protonation of this bacterial transmembrane glutamate residue effectively releases a transmembrane arginine residue to bind and translocate substrate. If there is no substrate bound, the glutamate residue must give up its H+ so that it can form a charge pair with the arginine residue and reorient empty to complete the transport cycle [170].
Genetic tools may also be of great value to identify important structural residues involved in vesicular transporter function. Rand and colleagues identified numerous point mutants in Caenorhabditis elegans that map to highly conserved regions of the VAChT gene and exhibit behavioral phenotypes consistent with a reduction in vesicular transport activity and neurosecretion [182]. These mutants display selective defects in initial acetycholine transport velocity with Km values ranging from 2- to 8-fold lower than that of wild-type. This indicates that specific structural changes in VAChT translate into specific alterations in the intrinsic parameters of transport and in the storage and synaptic release of acetycholine in vivo [182]. Similar work in Drosophila [183] or other organisms [184] where genetic manipulation can be performed with relative ease could identify additional important structural sites in VGLUT important for transport function and synaptic release of glutamate.
VGLUT Functional Sites
Glutamate accumulates in synaptic vesicles by virtue of one of the three VGLUT subtypes and considerable efforts have been made to understand how VGLUTs operate compared to the other vesicular neurotransmitter transporters in the brain. In the early 1980s, two independent groups showed, using purified synaptic vesicles from rat or bovine brain, that vesicular glutamate transport is dependent on a transmembrane H+ gradient generated by the vacuolar type (V-ATPase) proton pump [185, 186]. Shortly, thereafter it was discovered that Cl− ions greatly stimulates glutamate uptake into synaptic vesicles in vitro [187]. Several teams rapidly confirmed these initial findings [188,189,190]. VGLUTs have relatively low affinity for glutamate (Km ~ 1–2 mM) but are highly selective for glutamate compared to other structurally similar amino acids, such as aspartate or glutamine. Estimates of glutamate levels in synaptic vesicles suggest between 60 and 120 mM concentrations [191, 192].
Inorganic Phosphate Site for VGLUT Regulation
VGLUT1–3 belong to the family of Na+-dependent inorganic phosphate transporters (NPTs) forming the SLC17 subfamily and were initially shown to transport inorganic phosphate (Pi) [193, 194]. Interestingly, upon originally cloning of this brain-specific inorganic Pi transporter (formally called BNPI), it was revealed that it has strong sequence similarity to EAT-4, a Caenorhabditis elegans protein implicated in glutamatergic transmission and localized almost exclusively to mammalian brain terminals forming asymmetric excitatory-type synapses [195]. Although BNPI (now called VGLUT1) [34, 35] depends on a Na+ gradient for Pi transport across the plasma membrane, surprisingly BNPI associated preferentially with the membranes of small synaptic vesicles [195]. Since phosphate-activated glutaminase (PAG) in nerve terminals produces glutamate from glutamine for release as a neurotransmitter [196], it was proposed that BNPI (VGLUT1) may augment excitatory transmission following vesicle exocytosis by increasing its expression at the plasma membrane and thereby increase cytoplasmic Pi concentrations within the nerve terminal to activate PAG and hence replenish glutamate synthesis lost by neurotransmission [195]. Such intrasynaptic sequestration of transport proteins involved in the Ca2+-dependent expression on the plasma membrane, and in replenishment of transmitter synthesis and/or vesicle sequestration for release, has previously been reported in cholinergic and GABAergic neurons [197,198,199]. When expressed in artificial liposomes VGLUTS transport Pi in a Na+-coupled manner with glutamate competing for binding, but at lower affinity [173, 200]. Pi transport did not require Cl− and was not inhibited by Evans blue, a competitive inhibitor of VGLUTs [173]. Pi transport into vesicles via a transmembrane ΔμH+ has also been reported [200], but also with lower affinity than glutamate. This Pi may also be transported out of the vesicle in a Na+-coupled manner, which may be involved in Pi homeostasis within glutamatergic neurons (Fig. 4) [200]. That VGLUTs possess intrinsic transport machineries that are independent of each other: Na+-dependent Pi flux and H+-dependent vesicular glutamate uptake, would suggest dual mechanisms to support glutamatergic neurotransmission, especially under high activity. However, other transporters have been proposed as the major Pi transporters in the brain [201]. The identification of activity-regulated transport of Pi across the synaptic membrane following exocytosis and glutamate release could reveal a specialized role for VGLUTs in regulating Pi transport and synaptic glutamate synthesis.

VGLUTs transport glutamate as well as multiple ions. In addition to their capacity to transport glutamate into synaptic vesicles, VGLUTs also display a Cl− transport mode and a K+/H+ antiport mode, the later could support maintenance of ionic and charge balance during glutamate transport. The Na+/Pi co-transport mode of VGLUTs may be important in vesicles and when expressed on the plasma membrane following exocytosis to regulate Pi homeostasis in glutamatergic terminals
Proton Site for VGLUT Regulation
Classic studies have concluded that cytosolic ATP activates the vacuolar-type ATPase in the membrane of intracellular storage organelles to transport H+ ions into the vesicle lumen creating a transmembrane electrochemical gradient (ΔμH+) (Fig. 4) [202, 203]. Compounds that dissipate the transmembrane ΔμH+, such as FCCP (H+ ionophore), were originally used to show H+-dependent glutamate transport by synaptic vesicles [185, 186] to differentiate this activity from Na+-coupled glutamate transport across the plasma membrane [204]. The electrical component (Δψ) of the total ΔμH+ generated by the V-ATPase is the primary driving force for the transport of glutamic acid and subsequent acidification of excitatory vesicles by glutamate itself [188]. Indeed, selective inhibition of the ΔpH component of the transmembrane ΔμH+ gradient (thus, favoring Δψ) by A23187 (a divalent cation ionophore) [205], nigericin (an electroneutral cation that exchanges H+ against K+) [206], or ammonium ion (NH4+) [205, 207] increases vesicular glutamate uptake at low (4 mM) cytoplasmic chloride concentrations. Inhibition of the V-ATPase with bafilomycin A1 reduces the amplitude of glutamatergic miniature excitatory postsynaptic currents (mEPSCs) in vitro, indicating that decreasing the amount of glutamate in synaptic vesicles reduces quantal size [208]. However, direct inhibition of the V-ATPase or the dissipation of the transmembrane ΔμH+ would inhibit transport of all neurotransmitters into synaptic vesicles. While blocking the transmembrane ΔpH component may be selective to VGLUT stimulation in synaptic vesicles, this too would abolish transport by vesicular amine transporters that rely on protons for H+ antiport (e.g., VMAT1 and 2, VAChT).
Whether VGLUTs operate as glutamate/H+ exchangers [192, 209], glutamate/Cl− exchangers [205, 209], or as electrogenic uniporters [210] remains controversial [174, 177, 211]. However, the luminal H+ contribution to Δψ activates VGLUTs to drive the filling of synaptic vesicles with glutamate. For an electrogenic transport mechanism, the presence of a chloride-H+ exchanger (e.g., CIC3) [212] and a cation-H+ exchanger (e.g., NHE) [213] in the synaptic vesicle membrane could also contribute to charge and H+ balance. For example, K+/H+ exchange could play a role in sustaining glutamate uptake as it would maintain Δψ and decrease ΔpH [213]. Recent evidence using reconstituted systems indicate that VGLUTs have cation (K+) and H+ binding sites on the cytoplasmic and vesicle lumen sides, respectively [177, 211]. The localization of the H+ site on the luminal side of VGLUT required for VGLUT activation is not known. Whether this proton binding site allows for H+ antiport that is coupled to electrogenic glutamate transport into vesicles as suggested [192] is not entirely clear [177, 211]. Nevertheless, VGLUTs themselves can exchange K+ for H+ (Fig. 4) and while this may only loosely couple to glutamate transport [177], this property could be influenced by physiologic conditions in the brain to modulate vesicular glutamate storage.
Following exocytosis of synaptic vesicles VGLUTs are positioned in the plasma membrane such that glutamate could leak out of the cytoplasm into the synaptic cleft. It is not clear how much or how long VGLUTs could reside on plasma membranes following exocytosis in normal or disease conditions before they are endocytosed [48], although kiss-and-run exocytosis is obviously short-lived [214]. But, under conditions of massive depolarization (high extracellular K+) or energy failure such as that observed following transient cerebral ischemia and traumatic brain injury, the extent and duration of VGLUT expression on the plasma membrane may increase. Extracellular H+ (ΔpH) can increase the release of glutamate from cytoplasm through the plasma membrane via VGLUTs [211, 215] and this could occur with ischemic/seizure insults in vivo [172, 216,217,218,219,220]. Whether this H+ binding site on the extracellular facing VGLUTs is the same site as the internal H+ binding site in the vesicle lumen exposed following activation of the V-ATPase (and controlling glutamate transport into vesicles) is not presently known. However, such a H+ binding site could restrict glutamate flux to synaptic vesicles under normal conditions [211]. Drugs that selectively interfere with H+ binding to this site may therefore have importance to inhibit the leakage of glutamate from synaptic terminals under conditions of acidic extracellular pH such as following ischemia or traumatic brain injury [172, 219, 220] where extracellular glutamate levels dramatically increase [216,217,218]. Drug binding to this H+ site on VGLUTs before engulfing by endocytosis and reformation of vesicles may also serve to limit H+ activation of VGLUTs in the vesicle lumen and thus affect vesicular glutamate sequestration and release.
Chloride Site for VGLUT Regulation
Early work had established a biphasic dependence of glutamate transport on extravesicular Cl− ion concentrations with low levels (4 mM) greatly stimulating and higher (> 20 mM) levels inhibiting transport [187, 188]. In the absence of a permeable anion, the V-ATPase generates only a transmembrane electrical gradient (i.e., Δψ). However, in the presence of Cl− (> 20 mM), a measurable ∆pH is generated by the luminal accumulation of H+ and Cl− (i.e., HCl), which limits vesicular glutamate transport [188, 206]. Jahn and colleagues also described the presence of a Cl− binding site on the cytoplasmic side of the transporter, distinct from the substrate binding site, and that low Cl− concentrations (e.g., 4 mM) stimulate transport activity directly through this site [221]. Wolosker’s work confirmed that Cl− affects glutamate transport by two different mechanisms: one is related to a change of the proportions between the transmembrane Δψ and ∆pH components of the total ΔμH+, and the other involves a direct stimulatory interaction of Cl− with the glutamate transporter [205].
The molecular mechanism by which chloride ions could differentially affect transvesicular Δψ and · pH gradients was thought to occur by Cl− influx (at higher > 20 mM cytoplasmic levels) through exogenous Cl− channels (e.g., ClC-3) that are present on synaptic vesicles [212]. However, upon molecular identification of VGLUT1, Edwards and colleagues demonstrated that the transporter itself displays a Cl− conductance (Fig. 4) and that glutamate and Cl− ion could compete for the same transport site [34]. Thus, the lumen of glutamatergic vesicles will acidify by both glutamate and Cl− flux [188, 205, 206]. Indeed, glutamate or Cl− transported into the vesicle may further stimulate the V-ATPase to maintain a positive Δψ [222]. Glutamate-induced acidification of vesicles also enables the ∆pH-dependent uptake of monoamines, acetylcholine (ACh) or GABA by H+ antiport in neurons that co-express co-express VGLUT3 and VMAT2, VAChT or VGAT/VIAAT (see [223] for review).
To clarify the role of Cl− in VGLUT regulation, simplified in vitro transport flux studies were developed with proteoliposomes containing purified recombinant VGLUT and the H+-ATPase of Bacillus stearothermophilus (TF0F1 = F-ATPase) [173]. Using this system, contributions of any endogenous vesicular components that would affect transmembrane Δψ and ΔpH gradients and various ion fluxes (e.g., K+, Cl−, Pi) due to other proteins are eliminated. These studies clearly show that VGLUT-mediated glutamate influx into vesicles exhibits a biphasic dependence on Cl− ion [177, 209]. Thus, VGLUTs (and not the chloride channel CIC-3 [212]) may represent the major Cl− permeation pathway in glutamatergic synaptic vesicles. Importantly, high luminal Cl− concentrations markedly enhance loading of glutamate by Δψ-driven uptake [209]. This is consistent with the idea that upon glutamate release into the synaptic cleft, vesicles are regenerated by endocytosis where they may contain high levels of Na+ and Cl− ions (~ 130 mM) [190]. Using live-cell imaging with pH- and Cl−-sensitive fluorescent probes in cultured hippocampal neurons or wild-type and VGLUT1-deficient mice, Martineau and colleagues confirmed that export of Cl− ion from the vesicle lumen into the cytoplasm can drive vesicular glutamate transport [192]. It is not clear what happens to luminal Na+ following endocytosis, but it is possible that an increase in Δψ following Cl− efflux could also be a result of residual Na+ ions left in the vesicle lumen, or maybe they just leak out of the vesicle into the cytoplasm (Fig. 4).
A novel approach to study VGLUT without the activity of an endogenous H+ pump, and its link to Δψ and various ion gradients associated with it, is by the electrophysiological analysis of VGLUT-associated currents. Recently, Edwards and colleagues used patch clamp recording from enlarged endosomes of VGLUT-transfected cells and confirmed once again that Cl− interacts with VGLUTs as both a permeant ion and allosteric activator [174]. Remarkably, in addition to the allosteric activation of VGLUTs by low cytosolic Cl− as described above, endosome recording revealed allosteric regulation of glutamate transport by luminal Cl− as well [174]. Interestingly, neutralization of the highly conserved arginine residue in TM4 is sufficient to confer the activation normally provided by Cl−. This basic residue is predicted to face the vesicle lumen and that in the absence of luminal Cl−, it prevents both vesicular glutamate transport and the associated Cl− conductance [174]. Thus, in the presence of luminal Cl−, this intravesicular Cl− binding site activates glutamate transport into vesicles and Cl− efflux from vesicles into the cytoplasm (Fig. 4). The Cl− trapped in recycling vesicles by endocytosis (or transported into the vesicle from the cytoplasm) by VGLUTs, could therefore confer additional allosteric activation required for glutamate transport into vesicles. As glutamate entry into the vesicle dissipates Δψ the resulting drop in luminal pH would activate Cl− efflux that would support the maintenance of Δψ, and so maintain the driving force for glutamate uptake. These results support the idea that Cl− permeates through a low affinity conduction pathway of a channel, and glutamate through the alternating access mechanisms of a transporter [174, 224], similar to the Na+-coupled excitatory amino acid transporters (EAATs) [225, 226].
Understanding the physiological role of the Cl− allosteric binding sites and the Cl− conductance’s among all VGLUT isoforms and the structural and functional sites involved will require more specific tools to inhibit Cl− binding/permeation by the VGLUTs. Inhibitors, such as DIDS (EC50 = 0.7 μM) and SITS (EC50 = 0.2 μM) inhibit glutamate uptake by competing with their Cl− binding site [227]. DIDs also potently inhibits Cl− dependent vesicular ATP uptake with an IC50 of 1.5 μM [155]. On the other hand, DIDS and SITS are also potent Cl−-HCO3-exchange blockers [228] and also block monoamine transport into synaptic vesicles [229] (Scheme 1).

Inhibitors of the electrochemical gradient
Ketone bodies (acetoacetate and 3-hydroxybutyrate) and α-keto acids have been proposed to compete with Cl− for VGLUT activation at a cytoplasmic-exposed site and therefore inhibit glutamate uptake into vesicles and release from synapses [227, 230, 231]. Metabolic derivatives such as acetoacetate, pyruvate, phenylpyruvate, kynurenate, α-keto-β-methyl valerate, and α-keto-isovalerate ketones bind to this Cl− site to modulate the activity of VGLUTs by negative cooperation. One explanation for the beneficial effect of ketogenic diets for the treatment of young patients with epilepsy then could be that these compounds modulate glutamatergic neurotransmission by reducing vesicular glutamate accumulation and quantal size by an allosteric mechanism [228289]. Whether VGLUTs can affect metabolism (or at least metabolic pools of glutamate in synapses) in addition to their synaptic function is not known. Further exploration of endogenous and dietary compounds that could modulate Cl− binding in VGLUTs and whether these compounds interfer with Cl− flux properties, as well, could lead to the discovery of novel pharmacological tools to treat neurological disorders caused by excessive synaptic release of glutamate.
Cl− fluxes across the plasma membranes of glutamatergic synapses can be mediated by axo-axonal GABAergic synapses [232], neuron-specific cation-Cl− co-transporters [233], or other modulators [234]. Thus, modulation of cytoplasmic Cl− concentrations could be important effectors of the cytoplasmic Cl− binding and permeation sites of VGLUTs, in some way. However, varying intracellular levels of Cl− in the ‘giant’ synapses from the calyx of Held in the auditory brainstem has little bearing on quantal size of glutamate released [235]. However, more recent studies have shown that presynaptic Cl− levels play a biphasic regulatory role in the process of glutamate refilling into those vesicles via VGLUTs [236], similar to that observed with in vitro transport experiments. Excitability of hippocampal neurons could be greatly affected by intracellular Cl− levels, especially under potential excitotoxic conditions [237, 238]. Since extracellular Cl− levels (or various drugs) that are present in the extrasynaptic space are endocytosed into newly formed vesicles they could also potentially modulate intravesicular Cl−, glutamate, and H+ binding sites and fluxes across vesicles and thus, intracellular storage of vesicular glutamate available for synaptic release.
Glutamate Site for VGLUT Regulation
Under normal conditions, cytoplasmic glutamate levels for vesicular loading are likely mostly derived from glucose metabolism, pyruvate carboxylation and tricarboxylic acid (TCA) intermediates such as α-ketoglutarate [239,240,241,242]. Neuronal glutamate reuptake after release may also occur to some extent in forebrain regions [243,244,245]. Whole-cell recording of the presynaptic terminals in the calyx of Held reveal that the content of a single vesicle is insufficient to saturate AMPA receptors such that the release of several vesicles is required [246,247,248,249]. Indeed, direct injection of glutamate into this synapse increases quantal size and thereby post-synaptic receptor saturation by single vesicle release of glutamate [246, 249]. Single vesicle release also may not saturate post-synaptic glutamate receptors at hippocampal synapses either [250] and since vesicle release probability is very low in hippocampal synapses [126, 129] single vesicle release and nonsaturation of post-synaptic glutamate receptors likely predominates there too [251, 252].
Regulation of vesicle filling levels can be mediated by the cytoplasmic glutamate concentration that is available to the glutamate site on VGLUTs for uptake into synaptic vesicles (Fig. 4). The cytoplasmic concentration of glutamate in glutamatergic synaptic terminals is not precisely known. Semiquantitative electron microscopic analysis of the distribution of glutamate-like immunoreactivity in excitatory hippocampal pathways have estimated the glutamate concentration in synapses to range from 1 to 10 mM [253,254,255,256,257]. Transporter flux experiments using brain synaptic vesicles in vitro indicate that maximal vesicle filling in vitro saturates at ~ 4 mM external glutamate concentrations [117], which is similar to cholinergic synaptic vesicles that also exhibit low affinity for acetylcholine [162, 258]. Thus, if vesicular glutamate storage capacity is not maximal under normal conditions [246, 247, 249, 250] then endogenous cytoplasmic glutamate concentrations may lie somewhere below 4 mM; likely around the Km of VGLUTs for glutamate (1–2 mM) to enable activity-stimulated regulation. Sub-saturating levels of glutamate at the glutamate binding site on VGLUTs in the cytoplasm under normal conditions suggests that regulation of vesicle filling by modulating biosynthesis or degradation of glutamate in the terminal would affect vesicular glutamate filling. That the vesicle fill level is dependent on the cytoplasmic neurotransmitter levels available for filling in synaptic terminals may be a common feature of vesicular transmitter storage, in general, as the extravesicular level of all other transmitters such as acetylcholine [162, 259], GABA [142], and the monoamines [260] in synaptic vesicles can also be scaled pharmacologically by interfering with the metabolism or by providing precursors for transmitter synthesis.
Rapid replenishment of neurotransmitter glutamate synthesis under conditions of high neural activity in various neurological conditions (e.g., epileptic seizures, traumatic brain injury, cerebral ischemia, and others) would be required to maintain synaptic cytoplasmic glutamate levels at levels optimal for continued release [261]. Original suggested that glutamine transport into synapses (derived from surrounding astroglial cells [262]) is a preferred precursor for transmitter glutamate released following K+-depolarization (56 mM KCl) of slices of the molecular layer of the dentate gyrus [263,264,265]. Thus, the availability of glutamine to nerve terminals could affect synaptic glutamate synthesis and vesicular glutamate storage for release [266,267,268,269], especially under conditions of excessive synaptic glutamate release. However, Kam and Nicoll reported that excitatory transmission can persist for hours without glutamine in young neuronal cultures and slices [270]. Yet, synaptic transmission is impaired under intense stimulation by preventing the conversion of glutamine to glutamate in phosphate-activated glutaminase (PAG) knockout mice [271]. Indeed, the enzymatic activity of PAG has been detected in neurons and in nerve endings, suggesting local glutamate biosynthesis in axon terminals [272,273,274]. Whether Na+-dependent glutamine import into neuronal synapses from astroglial synapses to replenish synaptic cytoplasmic glutamate stores under high synaptic activity is a potential novel target to prevent excessive glutamate release under conditions of excitotoxicity is currently not known. While glutamine synthetase (GS) is a critical component of the glutamate/glutamine cycle, GS-KO mice develop epilepsy [275], likely because GS must inactivate synaptically released glutamate that is taken up in astrocytes, and cleared from the synaptic cleft, by the EAATs [276, 277] as well. However, activity-induced modulation of synaptic glutamate efficacy [278] and glutamate epileptiform activity [279,280,281] are significantly reduced by acute inhibition of glutamine synthesis in astrocytes with methionine sulfoximine or by application of 2-(methylamino-isobutyrate) (MeAIB), a competitive and reversible inhibitor of the neuronal Na+-coupled glutamine transporters (SNAT; system A) subtypes 1 and 2 [282]. However, SNAT1 and SNAT2 are confined to cell soma and proximal dendritic regions of neurons [283] and are excluded from axon terminals supporting the notion that an unidentified synaptic glutamine/MeAIB transporter could support activity-stimulated/excitotoxic glutamate release from synapses [284].
VGLUT Pharmacologic Sites
Despite their importance, the pharmacology of VGLUTs remains remarkably underdeveloped. The evaluation of VGLUT inhibitors has generally been performed using in vitro transport assays measuring the flux of radiolabeled glutamate across the membranes of whole brain synaptic vesicles and more recently using liposomes with engineered VGLUTs and transmembrane ΔμH+ gradients. Only a few VGLUT inhibitors have been shown to affect glutamatergic transmission in electrophysiologic recordings of rodent hippocampus and cerebral cortex [285, 286].
Competitive Inhibitors
Substrate Analogs
Among competitive modulators of glutamate uptake into synaptic vesicles, cyclic and noncyclic glutamate analogs, such as (2S, 4R)-4-methyl-glutamate (IC50 = 0.7 mM), L-trans-ACPD (IC50 = 0.23 mM), and DL-trans-ACHD (IC50 = 0.8 mM), are the most potent [287,288,289]. These compounds also modulate the activity of mGluR [290] (Scheme 2).

Cyclic and noncyclic glutamate analogues such as (2S, 4R)-4-methyl-glutamate (IC 50 = 0.7 mM), DL-trans-ACHD (IC50 = 0.8 mM) or L-trans-ACPD (IC 50 = 0.23 mM) (Thompson et al., 2005)
Quinolines and Quinoxaline Analogs
The capacity of quinoline and quinoxaline to inhibit glutamate uptake was reported 20 years ago [291]. Compounds such as kynurenate, xanthurenate, 7-chloro-kynurenate, and 2-quinoxaline carboxylate are competitive modulators of glutamate transport. However, these compounds also act at the postsynaptic level on ionotropic receptors [187, 287, 291, 292] (Scheme 3).

Structures of best motifs quinolines and quinoxalines able to inhibit VGLUTs
Kynurenate is a neuroactive endogenous compound (antiexcitotoxic and anticonvulsant) derived from tryptophan metabolism. Carrigan and coworkers developed a series of ligands from this motif [293] and SAR studies highlight conserved and crucial chemical functions to ensure potent inhibitory activity. This includes the importance of negative charges of the QDC motif, as well as the size of the hydrophobic aryl unit. Substitution of the phenyl moiety by biphenyl induces an affinity gain of one log [293,294,295]. The best modulators are 6-biphenyl-QDC and 6-(4-phenylstyryl)-QDC (affinities of 41 μM and 64 μM, respectively) (Scheme 4).

Structures of QDCs analogs as VGLUTs inhibitors (Carrigan et al., 2002)
Azoic Dyes
Azoic dyes are formed by a polar region connected to a hydrophobic body via an azo bond. These molecules have at least one negative charge provided by a sulfonate group. The following has been surmised of these molecules: (1) the negative charge is important for ligand activity and plays the same role as the COO-proximal glutamate moiety; (2) the NH2 group, present on the polar part of some inhibitors, plays the same role as the amino group of glutamate. This NH2 does not seem to be essential for glutamate affinity [296]. Currently, azo dyes that inhibit VGLUTs with the best affinity are as follows: Bright Yellow (BY, IC50 = 15 nM); Chrysophenine (CP, IC50 = 27 nM), which corresponds to the methylated analog of Bright Yellow; trypan blue (TB, IC50 = 50 nM); Evans blue (EB, IC50 = 90 nM); and Violet Direct 51 (VD51, IC50 = 148 nM) [224, 286, 296, 297] (Scheme 5).

Azo dyes (Favre-Besse et al., 2014; Omote et al., 2016; Tamura et al., 2013)
SAR studies have identified important pharmacophoric sites for the affinity of azo dyes (e.g., TB) [296, 297]. These studies highlighted the importance of the hydrophobic aryl motif. In particular, increased size (changing phenyl for a biphenyl, for example) or incorporation of halogen groups induced a significant increase in affinity to the VGLUT (Scheme 6).

Importance of the size of azo dyes for VGLUTs inhibition (Favre-Besse et al., 2014)
An SAR study was also conducted using BY and its derivatives [296]. These investigations confirmed the importance of the negative charge of the sulfonate group, the importance of the hydrophobic aryl body, the azo bond, and the H bond acceptor moiety (this time, in para position to the azo link). The OH group in BY or the OMe group in Chrysophenine (Scheme 5) establishes a key hydrogen bond with Tyr195 of VGLUTs, which prevents the binding of glutamate to charged residues (Arg184, His128, and, in particular, Glu191) in the VGLUT hydrophilic pocket [296]. The distance between the nitrogen atom of the azo bond and the negatively charged oxygen atom of the sulfonate group (distance N-O) is also an important factor regarding the affinity of azo dyes. This distance is similar in Brilliant Yellow (d (NO-) = 9.324 Å), trypan blue (d (NO-) = 9.799 Å) and Violet Direct 51 (d (NO-) = 8.274 Å, from (NO) = 10.354 Å) [296]. The negative charges of the naphthyl-disulphonic polar motif also play an important role in the inhibition of VGLUTs [297] (Scheme 7).

Pharmacophore of azo dyes and various potent inhibitors superposed to pharmacophore model of VGLUTs. The pharmacophore model allow the 3D representation of key features for potent VGLUT inhibitory activity. This model was generated based on the activity of various ligands (trypan blue (EC50 = 50 nM), Evans blue (EC50 = 90 nM), Brillant Yellow (EC50 = 15 nM, Chicago Blue Sky (EC50 = 330 nM), Direct violet 51 (EC50 = 148 nM), Chrysophenin (EC50 = 27 nM), four 2,4-dicarboxy-quinolines (EC50 = 41 μM; 64 μM; 167 μM; 288 μM), four monoazoic dyes (EC50 = 1.6 μM; 3.3 μM; 25 μM; 40 μM)). Structural key requirements to obtain a potent VGLUT inhibitor are represented by color spheres (green = H bond donor, slight blue = aromatic moiety, dark blue = negative charge); the putative ligand/VGLUT steric bump are represented by exclusion gray spheres
Evans blue was one of the first compounds found to inhibit vesicular transport of glutamate [298]. Evans blue also potently inhibits the Cl− dependent vesicular transport of ATP with an IC50 of 40 nM [155]. Recent studies indicate that Evans blue reduces both Cl− and glutamate currents in enlarged endosomes with a similar IC50 (~ 0.2 μM) [174]. Evans blue therefore appears to bind to the glutamate site on VGLUTs but it is not yet known if it (or other similar compounds) require a H+ gradient across the membrane, similar to reserpine for VMATs [299]. It should be noted that azo dyes do not remain VGLUT-specific at high concentrations [298] as high doses of Evans blue (> 20 μM) inhibit GABAergic vesicular transport [298, 300] and lysosomal ATP transport [301], but not vesicular transport of dopamine [298]. Evans blue is used as a means to assess the permeability of the blood-brain barrier (BBB) [302] and therefore cannot access VGLUTs in vivo. Comparable nontransported inhibitors of VGLUTs that can cross the BBB are not available.
Noncompetitive Inhibitors
Noncompetitive modulators of VGLUTs include polyhalogenated fluorescein analogs, among which Rose Bengal has the highest affinity (IC50 = 25 nM) [303,304,305] (Scheme ). SAR studies of Rose Bengal and its derivatives facilitated identification of the bioactive form of Rose Bengal derivatives (acid form of the molecule, see Scheme 8) [303]. The lactone form of Rose Bengal exhibits reduced affinity. The halogen substituents on xanthene and phenyl are particularly important in generating high affinity for VGLUTs (25 nM). However, the binding site for these types of ligands has not yet been identified. Some studies have suggested allosteric modulation of Rose Bengal on VGLUT [304], while others think indirect modulation occurs by blocking the V-ATPase [303]; although Rose Bengal strongly and specifically affects glutamatergic but not GABAergic boutons [192]. A recent study showed that Rose Bengal is a specific inhibitor of glutamate transport but not the VGLUT-associated Cl− conductance [192], so it could be an allosteric modulator of VGLUTs, like tetrabenazine for VMATs or vesamicol for VAChT. On the other hand, Evans blue inhibits both Cl− and glutamate currents with submicromolar potency [174]. A 50% inhibition of maximal uptake levels by submaximal concentrations of the competitive inhibitor trypan blue (like Evans blue), is eventually overwhelmed by glutamate competition (i.e., 4 mM) where vesicular levels reach control values [117]. On the other hand, a 50% knockdown of functional transporters by the noncompetitive inhibitor Rose Bengal produces a steady-state accumulation of glutamate that is approximately 50% less than control values at all external glutamate concentrations [117], yet maximal accumulation still occurs at the same external (4 mM) value. These data support evidence that the level of functional VGLUTs in vesicles is a critical determinant in vesicle filling.

Fluorescein analog inhibitor of VGLUTs
Conclusions
Multiple isoforms of VGLUT in mammals provide diversity in vesicular glutamate storage and release in discrete synapses as the three subtypes are differentially expressed in glutamatergic neurons in the brain and are likely subject to differential regulation at the level of biosynthesis and degradation. Indeed, the level of expression of VGLUTs in synapses could be a critical determinant that regulates both quantal size and the availability of vesicles for glutamate release. Changes in VGLUT expression levels in synapses are seen in a variety of neurological disorders and disease states. Adaptive changes in VGLUT expression levels may even be used as a neuroprotective strategy to support neural circuit stability to avoid excessive glutamate release from synapses and excitotoxicity. While we are not yet able to pharmacologically modify VGLUT biosynthetic pathways, molecular approaches to modify VGLUT expression in synapses using viral-mediated strategies are clearly a major avenue being explored to selectively target distinct glutamatergic systems that may affect aberrant synapses in neurologic disorders and disease.
The three subtypes of VGLUTs are so closely structurally related that none of the currently available inhibitors may be able to discriminate between them functionally. An inability to selectively interfere with vesicular glutamate storage and release in distinct glutamatergic neurons would render these compounds ineffective in any clinical setting. However, the inhibitory effects of these compounds have largely been performed with brain synaptic vesicle preparations that contain a mixed population of synaptic vesicles or in cortical/hippocampal neurons where VGLUT1 predominates. The evaluation of current inhibitors, and new pharmacological compounds, in recombinant VGLUT systems are therefore essential to determine if any are specific for VGLUT isoforms and could help distinguish between the sites in VGLUTs that are important for glutamate transport to better understand the mechanisms involved.
Biochemical transport flux experiments in vitro using radiolabeled tracers has long been instrumental in our understanding of vesicular neurotransmitter transport in brain synaptic vesicles for all of the classic neurotransmitters. Recent studies have shown important contributions are being made following the incorporation of recombinantly expressed transporters into artificial vesicles (liposomes) equipped with an H+-ATPase and individual VGLUTs without any interfering synaptic vesicle components. Indeed, these recent flux studies have provided important information to the existence of critical binding/flux sites for H+, monovalent cations, and Cl− on both sides of VGLUTs. Such assays could therefore also be used to screen established and novel VGLUT inhibitors to determine whether any of them are isoform-specific, or not. Surprisingly, the ionic binding/flux properties of VGLUTs that have recently been described may help generate transmembrane vesicular H+ gradients (i.e., Δψ) under physiologic conditions to facilitate glutamate transport into vesicles and maintain vesicular glutamate storage levels. In this regard, a unique feature of vesicle exocytosis is that endocytosed vesicles contain the content of the extracellular milieu (esp., Cl−) that can increase Δψ within vesicles (positive inside) during efflux from the vesicle once back inside the terminal. The unique ionic features of VGLUTs make them susceptible to regulation by physiologic factors such as changes in extracellular Cl−, K+, or H+ that occur under seizure, ischemic, and other acute and chronic excitotoxic conditions. Exposure of internal regulatory sites on VGLUTs to the extrasynaptic space following exocytosis allows for a potential means to introduce drugs to the vesicle interior, to modulate glutamate filling for synaptic release.
Glutamate-induced excitotoxicity is an important concern in a variety of detrimental brain injuries and disease. A powerful means to control vesicular glutamate filling and synaptic glutamate release is by regulating the availability of glutamate to the glutamate site on VGLUTs. However, the glutamate sites in the three VGLUT isoforms are likely highly conserved. Since, neurons expressing the different VGLUT isoforms display inherent differences in activity (esp., VGLUT1 and VGLUT2), it is likely that they also express differential requirements to supply and replenish cytoplasmic transmitter glutamate levels in synapses to sustain glutamatergic neurotransmission in an activity-dependent manner. It is not clear if differences in metabolic precursors (e.g., glucose, α-ketoglutarate, lactate, alanine, glutamine, etc.) and other potential pathways used for glutamate synthesis and neurotransmission exist in the different VGLUT-operated synapses. However, activity-dependent regulation of synaptic glutamate synthesis may be especially critical in VGLUT1-driven axon terminals, which are positioned in vulnerable brain areas such as the hippocampal CA subfields. VGLUT1-operated synapses in the hippocampus have low inherent release properties and are therefore subject to activity-stimulated regulation. Activity-stimulated glutamine and Pi transport across the plasma membrane in axon terminals of hippocampal neurons may therefore be novel sites to modulate phosphate-activated glutaminase activity and glutamate synthesis in synapses, activity-stimulated presynaptic glutamate release and neuronal glutamate-induced excitotoxicity.
Novel electrophysiologic analysis of VGLUT function has provided important information into understanding the molecular mechanisms of vesicular glutamate transporter regulation itself, similarly to the plasma membrane glutamate clearance transporters (i.e., EAATs), via independent ion conductance properties that are intrinsic to the transporters. Measurements of transport flux in recombinant systems in vitro and electrophysiologic analysis will be critical for in-depth analysis of new drugs and their specific interactions with the anionic and cationic binding sites and properties within VGLUTs. High through-put screens of potential drugs that modulate Cl− and H+ (both Δψ and ΔpH) changes are also likely to come from the use of novel fluorescent tags and probes in in vitro and in vivo studies.
References
Fonnum F (1984) Glutamate: a neurotransmitter in the mammalian brain. J Neurochem 42:1-11
Meldrum BS (2000) Glutamate as a neurotransmitter in the brain: review of physiology and pathology. J Nutr 130:1007S-1015S
Zhou Y, Danbolt NC (2014) Glutamate as a neurotransmitter in the healthy brain. J Neural Transm (Vienna) 121(8):799-817. https://doi.org/10.1007/s00702-014-1180-8
Lewerenz J, Maher P (2015) Chronic glutamate toxicity in neurodegenerative diseases - What is the evidence? Front Neurosci. https://doi.org/10.3389/fnins.2015.00469
Miladinovic T, Nashed MG, Singh G (2015) Overview of glutamatergic dysregulation in central pathologies. Biomolecules 5:3112-3141
Holmes GL (2002) Seizure-induced neuronal injury: animal data. Neurology 59:S3-S6
Dudek FE, Sutula TP (2007) Epileptogenesis in the dentate gyrus: a critical perspective. Prog Brain Res 163:755-773
Bittigau P, Ikonomidou C (1997) Glutamate in neurologic diseases. J Child Neurol 12:471-485
Dodd PR (2002) Excited to death: different ways to lose your neurons. Biogerontology 3:51-56
Fujimoto S, Katsuki H, Kume T, Kaneko S, Akaike A (2004) Mechanisms of oxygen glucose deprivation-induced glutamate release from cerebrocortical slice cultures. Neurosci Res 50:179-187
Benveniste H, BDrejer J, Schousboe A, Diemer NH (1984) Elevation of the extracellular concentrations of glutamate and aspartate in rat hippocampus during transient cerebral ischemia monitored by intracerebral microdialysis. J Neurochem 43:1369-1374
Tampellini D (2015) Synaptic activity and Alzheimer’s disease: a critical update. Front Neurosci 9:Article 9
Rudy CC, Hunsberger HC, Weitzner DS, Reed MN (2015) The role of the tripartite glutamatergic synapse in the pathophysiology of Alzheimer’s disease. Aging Dis 6:131-148
Kostandy BB (2012) The role of glutamate in neuronal ischemic injury: the role of spark in fire. Neurol Sci 33:223-237
Tordera RM, Totterdell S, Wojcik SM, Brose N, Elizalde N, Lasheras B, Del Rio J (2007) Enhanced anxiety, depressive-like behaviour and impaired recognition memory in mice with reduced expression of the vesicular glutamate transporter 1 (VGLUT1). Eur J Neurosci 25(1):281-290. https://doi.org/10.1111/j.1460-9568.2006.05259.x
Mark KA, Quinton MS, Russek S, Yamamoto BK (2007) Dynamic changes in vesicular glutamate transporter 1 function and expression related to methamphetamine-induced glutamate release. J Neurosci 27:6823-6831
Oni-Orisan A, Kristiansen LV, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE (2008) Altered vesicular glutamate transporter expression in the anteri or cingulate cortex in schizophrenia. Biol Psychiatry 63:766-775. https://doi.org/10.1016/j.biopsych.2007.10.020
Uezato A, Meador-Woodruff JH, McCullumsmith RE (2009) Vesicular glutamate transporter mRNA expression in the medial temporal lobe in major depressive disorder, bipolar disorder, and schizophrenia. Bipolar Disord 11:711-725. https://doi.org/10.1111/j.1399-5618.200900752.x
Eastwood SL, andHarrison,P.J. (2010) Markers of glutamate synaptic transmission and plasticity are increased in the anterior cingulate cortex in bipolar disorder. Biol Psychiatry 67:1010-1016. https://doi.org/10.1016/j.biopsych.2009.12.004
Zink M, Vollmayr B, Gebicke-Haerter PJ, Henn FA (2010) Reduced expression of glutamate transporters vGluT1, EAAT2 and EAAT4 in learned helpless rats, an animal model of depression. Neuropharmacology 58:465-473
Shen Y-C, Liao D-L, Lu C-L, Chen J-Y, Liou Y-J, Chen T-T, Chen C-H (2010) Resequencing of the vesicular glutamate transporter 2 gene (VGLUT2) reveals some rare genetic variants that may increase the genetic burden in schizophrenia. Schizophr Res 121(1):179-186
Shan D, Lucas EK, Drummond JB, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE (2013) Abnormal expression of glutamate transporters in temporal lobe areas in elderly patients with schizophrenia. Schizophr Res 144:1-8
Vrettou M, Nilsson KW, Tuvblad C, Rehn M, Aslund C, Andershed A-K (2019) VGLUT2 rs2290045 genotype moderates environmental sensitivity to alcohol-related problems in three samples of youths. Eur Child Adolesc Psychiatry. https://doi.org/10.1007/s00787-019-01293-w
Li C-T, Yang K-C, Lin W-C (2019) Glutamatergic dysfunction and glutamatergic compounds for major psychiatric disorders: evidence from clinical neuroimaging studies. Front Psych https://doi.org/10.3389/psyt.2018.00767
Birgner C, Nordenankar K, Lundblad M, Mendez JA, Smith C, le Greves M, Galter D, Olson L et al (2010) VGLUT2 in dopamine neurons is required for psychostimulant-induced behavioral activation. Proc Natl Acad Sci 107:389-394
Kirby T (2015) Ketamine for depression: the highs and lows. Lancet 2
Schwartz J, Murrough JW, Losifescu DV (2016) Ketamine for treatment-resistant depression: recent developments and clinical application. Evid Based Mental Health 19
Kraus C, Wasserman D, Henter ID, Acevedo-Diaz E, Kadriu B, Zarate CA Jr (2019) The influence of ketamine on drug discovery in depression. Drug Discov Today. https://doi.org/10.1016/j.drudis.2019.07.007
Atri A, Molinuevo JL, Lemming O, Wirth Y, Pulte I, Wilkinson D (2013) Memantine in patients with Alzheimer’s disease receiving donepezil: new analyses of efficacy and safety for combination therapy. Alzheimers Res Ther 5(6). https://doi.org/10.1186/alzrt1160
Wilkinson D, Wirth Y, Goebel C (2014) Memantine in patients with moderate to severe Alzheimer’s disease: meta-analysis using realistic definitions of response. Dement Geriatr Cogn Disord 37:71-85
Greig SL (2015) Memantine ER/Donepezil: a review in Alzheimer’s disease. CNS Drugs 29:963-970
Lipton SA (2004) Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx 1:101-110
Nimmrich V, Ebert U (2009) Is Alzheimer’s disease a result of presynaptic failure? Synaptic dysfunctions induced by oligomeric beta-amyloid. Rev Neurosci 20:1-12
Bellocchio EE, Reimer RJ, Fremeau RT, Edwards RH (2000) Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289:957-960
Takamori S, Rhee JS, Rosenmund C, Jahn R (2000) Identification of a vesicular glutamate transporter that defines a glutamatergic phenotype in neurons. Nature 407(6801):189-194. https://doi.org/10.1038/35025070
Fremeau RT, Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D et al (2001) The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 31:247-260
Takamori S, Rhee JS, Rosenmund C, Jahn R (2001) Identification of differentiation-associated brain-specific phosphate transporter as a second vesicular glutamate transporter (VGLUT2). J Neurosci 21:RC182
Herzog E, Bellenchi GC, Gras C, Bernard V, Ravassard P, Bedet C, Gasnier B, Giros B et al (2001) The existence of a second vesicular glutamate transporter specifies subpopulations of glutamatergic neurons. J Neurosci 21(22):RC181
Varoqui H, Schäfer MK, Zhu H, Weihe E, Erickson JD (2002) Identification of the differentiation-associated Na /Pi transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci 22:142-155
Gras C, Herzog E, Bellenchi GC, Bernard V, Ravassard P, Pohl M, Gasnier B, Giros B et al (2002) A third vesicular glutamate transporter expressed by cholinergic and serotoninergic neurons. J Neurosci 22(13):5442-5451
Fremeau RT Jr, Burman J, Qureshi T, Tran CH, Proctor J, Johnson J, Zhang H, Sulzer D et al (2002) The identification of vesicular glutamate transporter 3 suggests novel modes of signaling by glutamate. Proc Natl Acad Sci U S A 99(22):14488-14493. https://doi.org/10.1073/pnas.222546799
Schäfer MK, Varoqui H, Defamie N, Weihe E, Erickson JD (2002) Molecular cloning and functional identification of mouse vesicular glutamate transporter 3 and its expression in subsets of novel excitatory neurons. J Biol Chem 277:50734-50748
Omote H, Miyaji T, Juge N, Moriyama Y (2011) Vesicular neurotransmitter transporter: bioenergetics and regulation of glutamate transport. Biochemistry 50(25):5558-5565
El Mestikawy S, Wallén-Mackenzie Å, Fortin GM, Descarries L, Trudeau L-E (2011) From glutamate co-release to vesicular synergy: vesicular glutamate transporters. Nat Rev Neurosci 12:204-216. https://doi.org/10.1038/nrn2969
Anne C, Gasnier B (2014) Vesicular neurotransmitter transporters: mechanistic aspects. Curr Top Membr 73:149-174. https://doi.org/10.1016/B978-0-12-800223-0.00003-7
Reimer RJ (2013) SLC17: A functionally diverse family of organic anion transporters. Mol Asp Med 34:350-359. https://doi.org/10.1016/j.mam.2012.05.004
Fremeau RT, Voglmaier S, Seal RP, Edwards RH (2004) VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci 27:98-103. https://doi.org/10.1016/j.tins.2003.11.005
Voglmaier SM, Kam K, Yang H, Fortin DL, Nicoll RA, Edwards RH (2006) Distinct endocytic pathways control the rate and extent of synaptic vesicle protein recycling. Neuron 51:71-84
Vinatier J, Herzog E, Plamont MA, Wojcik SM, Schmidt A, Brose N, Daviet L, El Mestikawy S et al (2006) Interaction between the vesicular glutamate transporter type 1 and endophilin A1, a protein essential for endocytosis. J Neurochem 97:1111-1125
De Gois S, Jenclos E, Morris M, Grewal S, Varoqui H, Erickson JD (2006) Identification of endophillins 1 and 3 as selective binding partners for VGLUT1 and their co-localization in neocortical glutamatergic synapses: implications for vesicular glutamate transporter trafficking and excitatory vesicle formation. Cell Mol Neurobiol 26:679-693
Fujiyama F, Furuta T, Kaneko T (2001) Immunocytochemical localization of candidates for vesicular glutamate transporters in the rat cerebral cortex. J Comp Neurol 435(3):379-387
Kaneko T, Fujiyama F, Hioki H (2002) Immunohistochemical localization of candidates for vesicular glutamate transporters in the rat brain. J Comp Neurol 444(1):39-62
Kaneko T, Fujiyama F (2002) Complementary distribution of vesicular glutamate transporters in the central nervous system. Neurosci Res 42(4):243-250
Morimoto R, Hayashi M, Yatsushiro S, Otsuka M, Yamamoto A, Moriyama Y (2003) Co-expression of vesicular glutamate transporters (VGLUT1 and VGLUT2) and their association with synaptic-like microvesicles in rat pinealocytes. J Neurochem 84:382-391
De Gois S, Schafer MK-H, Defamie N, Chen C, Ricci A, Weihe E, Varoqui H, Erickson JD (2005) Homeostatic scaling of vesicular glutamate and GABA transporter expression in rat neocortical circuits. J Neurosci 25:7121-7133
Nakamura K, Hioki H, Fujiyama F, Kaneko T (2005) Postnatal changes of vesicular glutamate transporter VGLUT1 and VGLUT2 immunoreactivities and their colocalization in the mouse forebrain. J Comp Neurol 492:263-288
Nakamura K, Watakabe A, Hioki H, Fujiyama F, Tanaka Y, Yamamori T, Kaneko T (2007) Transiently increased colocalization of vesicular glutamate transporters 1 and 2 at single axon terminals during postnatal development of mouse neocortex: a quantitative analysis with correlation coefficient. Eur J Neurosci 26:3054-3067
Seal RP, Edwards RH (2006) The diverse roles of vesicular glutamate transporter 3. Handb Exp Pharmacol 175:137-150
Takamori S, Malherbe P, Broger C, Jahn R (2002) Molecular cloning and functional characterization of human vesicular glutamate transporter 3. EMBO Rep 3(8):798-803. https://doi.org/10.1093/embo-reports/kvf159
Li H, Waites CL, Staal RG, Dobryy Y, Park J, Sulzer DL, Edwards RH (2005) Sorting of vesicular monoamine transporter 2 to the regulated secretory pathway confers the somatodendritic exocytosis of monoamines. Neuron 48:619-633
Rice ME, Patel JC (2015) Somatodenritic dopamine release: recent mechanistic insights. Phil Trans R Soc A. https://doi.org/10.1098/rstb.2014.0185
Herzog E, Landry M, Buhler E, Bouali-Benazzouz R, Legay C, Henderson CE, Nagy F, Dreyfus P et al (2004) Expression of vesicular glutamate transporters, VGLUT1 and VGLUT2, in cholinergic spinal motoneurons. Eur J Neurosci 20(7):1752-1760. https://doi.org/10.1111/j.1460-9568.2004.03628.x
Li JL, Fujiyama F, Kaneko T, Mizuno N (2003) Expression of vesicular glutamate transporters, VGluT1 and VGluT2, in axon terminals of nociceptive primary afferent fibers in the superficial layers of the medullary and spinal dorsal horns of the rat. J Comp Neurol 457(3):236-249. https://doi.org/10.1002/cne.10556
Malet M, Brumovsky P (2015) VGLUTs and glutamate synthesis—focus on DRG neurons and pain. Biomolecules 5:3416-3437. https://doi.org/10.3390/biom5043416
Oliveira AL, Hydling F, Olsson E, Shi T, Edwards RH, Fujiyama F, Kaneko T, Hokfelt T et al (2003) Cellular localization of three vesicular glutamate transporter mRNAs and proteins in rat spinal cord and dorsal root ganglia. Synapse 50(2):117-129. https://doi.org/10.1002/syn.10249
Peirs C, Williams SP, Zhao X, Walsh CE, Gedeon JY, Cagle NE, Goldring AC, Hioki H et al (2015) Dorsal horn circuits for persistent mechanical pain. Neuron 87(4):797-812. https://doi.org/10.1016/j.neuron.2015.07.029
Seal RP, Wang X, Guan Y, Raja SN, Woodbury CJ, Basbaum AI, Edwards RH (2009) Injury-induced mechanical hypersensitivity requires C-low threshold mechanoreceptors. Nature 462(7273):651-655. https://doi.org/10.1038/nature08505
Seal RP (2016) Do the distinct synaptic properties of VGLUTs shape pain? Neurochem Int 98:82-88. https://doi.org/10.1016/j.neuint.2016.04.016
Vigneault É, Poirel O, Riad M, Prud'homme J, Dumas S, Turecki G, Fasano C, Mechawar N et al (2015) Distribution of vesicular glutamate transporters in the human brain. Front Neuroanat 9. https://doi.org/10.3389/fnana.2015.00023
Wojcik SM, Rhee JS, Herzog E, Sigler A, Jahn R, Takamori S, Brose N, Rosenmund C (2004) An essential role for vesicular glutamate transporter 1 (VGLUT1) in postnatal development and control of quantal size. Proc Natl Acad Sci U S A 101(18):7158-7163. https://doi.org/10.1073/pnas.0401764101
Wallen-Mackenzie A, Gezelius H, Thoby-Brisson M, Nygard A, Enjin A, Fujiyama F, Fortin D, Kullander K (2006) Vesicular glutamate transporter 2 is required for central respiratory rhythm generation but not for locomotor central pattern generation. J Neurosci Res 26:12294-12307. https://doi.org/10.1523/JNEUROSCI.3855
Moechars D, Weston MC, Leo S, Callaerts-Vegh Z, Goris I, Daneels G, Buist A, Cik M et al (2006) Vesicular glutamate transporter VGLUT2 expression levels control quantal size and neuropathic pain. J Neurosci 26(46):12055-12066. https://doi.org/10.1523/JNEUROSCI.2556-06.2006
Gras C, Amilhon B, Lepicard EM, Poirel O, Vinatier J, Herbin M, Dumas S, Tzavara ET, Wade MR, Nomikos GG, Hanoun N, Saurini F, Kemel ML, Gasnier B, Giros B, El Mestikawy S (2008) The vesicular glutamate transporter VGLUT3 synergizes striatal acetylcholine tone. Nat Neurosci 11(3):292-300. https://doi.org/10.1038/nn2052
Ruel J, Emery S, Nouvian R, Bersot T, Amilhon B, VanRybroek JM (2008) Impairment of SLC17A8 encoding vesicular glutamate transporter-3, VGLUT3, underlies non syndromic deafness DFNA25 and inner hair cell dysfunction in null mice. Am J Hum Genet 83:278-292. https://doi.org/10.1016/j.ajhg.2008.07.008
Amilhon B, Lepicard E, Renoir T, Mongeau R, Popa D, Poirel O, Miot S, Gras C et al (2010) VGLUT3 (vesicular glutamate transporter type 3) contribution to the regulation of serotonergic transmission and anxiety. J Neurosci Res 30:2198-2210. https://doi.org/10.1523/JNEUROSCI.5196-09.2010
He H, Mahnke AH, Doyle S, Fan N, Wang CC, Hall BJ, Tang YP, Inglis FM et al (2012) Neurodevelopmental role for VGLUT2 in pyramidal neuron plasticity, dendritic refinement, and in spatial learning. J Neurosci 32:15886-15901
Divito CB, Steece Collier K, Case DT, Williams SP, Stancati JA, Zhi L, Rubio ME, Sortwell CE et al (2015) Loss of VGLUT3 produces circadian-dependent hyperdopaminergia and ameliorates motor dysfunction and l-Dopa-mediated dyskinesias in a model of Parkinson’s disease. J Neurosci 35(45):14983-14999. https://doi.org/10.1523/JNEUROSCI.2124-15.2015
Sakae DY, Marti F, Lecca S, Vorspan F, Martin-Garcia E, Morel LJ, Henrion A, Gutierrez-Cuesta J et al (2015) The absence of VGLUT3 predisposes to cocaine abuse by increasing dopamine and glutamate signaling in the nucleus accumbens. Mol Psychiatry 20(11):1448-1459. https://doi.org/10.1038/mp.2015.104
Minelli A, Edwards RH, Manzoni T, Conti F (2003) Postnatal development of the glutamate vesicular transporter VGLUT1 in rat cerebral cortex. Brain Res Dev Brain Res 140(2):309-314
Boulland JL, Qureshi T, Seal RP, Rafiki A, Gundersen V, Bergersen LH, Fremeau RT, Edwards RH et al (2004) Expression of the vesicular glutamate transporters during development indicates the widespread corelease of multiple neurotransmitters. J Comp Neurol 480:264-280
Berry CT, Sceniak MP, Zhou L, Sabo SL (2012) Developmental up-regulation of vesicular glutamate transporter-1 promotes neocortical presynaptic terminal development. Plos one. https://doi.org/10.1371/journal.pone.0050911
Herzog E, Nadrigny F, Silm K, Biesemann C, Helling I, Bersot T, Steffens H, Schawatzmann R et al (2011) In vivo imaging of intersynaptic vesicle exchange using VGLUT1 Venus knock-in mice. J Neurosci 31:15544-15559
Miyazaki T, Fukaya M, Shimizu H, Watanabe M (2003) Subtype switching of vesicular glutamate transporters at parallel fibre-Purkinje cell synapses in developing mouse cerebellum. Eur J Neurosci 17(12):2563-2572. https://doi.org/10.1046/j.1460-9568.2003.02698.x
Guyenet PG, Stornetta RL, Abbott SB, Depuy SD, Kanbar R (2012) The retrotrapezoid nucleus and breathing. Adv Exp Med Biol 758:115-122
Koizumi H, Mosher B, Tariq MF, Zhang R, Koshiya N, Smith JC (2016) Voltage-dependent rhythmogenic property of respiratory pre-botzinger complex glutamatergic, Dbx1-derived, and somatostatin-expressing neuron populations revealed by graded optogenic inhibition. eNeuro 3(3):ENEURO.0081-ENEU16.2016
Nelson AB, Bussert TG, Kreitzer AC, Seal RP (2014) Striatal cholinergic neurotransmission requires VGLUT3. J Neurosci 34(26):8772-8777. https://doi.org/10.1523/JNEUROSCI.0901-14.2014
Seal RP, Akil O, Yi E, Weber CM, Grant L, Yoo J, Clause A, Kandler K et al (2008) Sensorineural deafness and seizures in mice lacking vesicular glutamate transporter 3. Neuron 57(2):263-275. https://doi.org/10.1016/j.neuron.2007.11.032
Miot S, Voituron N, Sterlin A, Vigneault E, Morel L, Matrot B, Ramanantsoa N, Amilhon B et al (2012) The vesicular glutamate transporter VGLUT3 contributes to protection against neonatal hypoxic stress. J Physiol 590(20):5183-5198. https://doi.org/10.1113/jphysiol.2012.230722
Kashani A, Lepicard E, Poirel O, Videau C, David JP, Fallet-Bianco C, Simon A, Delacourte A et al (2008) Loss of VGLUT1 and VGLUT2 in the prefrontal cortex is correlated with cognitive decline in Alzheimer disease. Neurobiol Aging 29(11):1619-1630. https://doi.org/10.1016/j.neurobiolaging.2007.04.010
Kirvell SL, Esiri M, Francis PT (2006) Down-regulation of vesicular glutamate transporters precedes cell loss and pathology in Alzheimer's disease. J Neurochem 98(3):939-950. https://doi.org/10.1111/j.1471-4159.2006.03935.x
Bell KFS, Bennett DA, Cuello AC (2007) Paradoxical upregulation of glutamatergic presynaptic boutons during mild cognitive impairment. J Neurosci 27:10810-10817
VanDerHel WS, Verlinde SA, Meijer DH, DeWit M, Rensen MG, Van Gassen KL, al e (2009) Hippocampal distribution of vesicular glutamate transporter1 in patients with temporal lobe epilepsy. . Epilepsia 50:1717-1728. https://doi.org/10.1111/j.1528-1167.2009.02054.x
Wootz H, Enjin A, Wallen-Mackenzie A, Lindholm D, Kullander K (2010) Reduced VGLUT2 expression increases motor neuron viability in Sod1(G93A) mice. Neurobiol Dis 37(1):58-66. https://doi.org/10.1016/j.nbd.2009.09.006
Schallier A, Massie A, Loyens E, Moechars D, Drinkenburg W, Michotte Y, Smolders I (2009) vGLUT2 heterozygous mice show more susceptibility to clonic seizures induced by pentylenetetrazol. Neurochem Int 55(1-3):41-44. https://doi.org/10.1016/j.neuint.2008.12.019
Hunsberger HC, Rudy CC, Batten SR, Gerhardt GA, Reed MN (2015) P301L tau expression affects glutamate release and clearance in the hippocampal trisynaptic pathway. J Neurochem 132(2):169-182. https://doi.org/10.1111/jnc.12967
Busche MA, Konnerth A (2007) Neuronal hyperactivity - a key defect in Alzheimer’s disease? BioEssays 37:624-632
Dickerson BC, Salat DH, Greve DN, Chua EF, Rand-Giovannetti E, Rentz DM, Bertram L, Millin K et al (2005) Increased hippocampal activation inmild cognitive impairment compared to normal aging and AD. Neurology 65:404-411
Putcha D, Brickhouse M, O’Keefe K, Sullivan C, Rentz D, Marshall G, Dickerson B, Sperling R (2011) Hippocampal hyperactivation associated with cortical thinning in Alzheimer’s disease signature regions in non-demented elderly adults. J Neurosci 31:17680-17688
Canas PM, Simoes AP, Rodrigues RJ, Cunha RA (2014) Predominant loss of glutamatergic terminal markers in a b-amyloid peptide model of Alzheimer’s disease. Neuropharmacology 76:51-56
Coleman PD, Yao PJ (2003) Synaptic slaughter in Alzheimer’s disease. Neurobiol Aging 24:1023-1027
DeKosky ST, Scheff SW (1990) Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27:457-464
DeKosky ST, Scheff SW, Styren SD (1996) Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration 5:417-421
Hamos JE, DeGennaro LJ, Drachman DA (1989) Synaptic loss in Alzheimer’s disease and other dementias. Neurology 39:355-361
Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ et al (2009) Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci 106:4012-4017
Terry RD (2000) Cell death or synaptic loss in Alzheimer’s disease. J Neuropathol Exp Neurol 59:1118-1119
Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572-580
Rodriguiz-Perdigon M, Tordera RM, Gil-Bea FJ, Gerenu G, Ramirez MJ, Solas M (2016) Down-regulation of glutamateric terminals (VGLUT1) driven by Ab in Alzheimer’s disease. Hippocampus 26:1303-1312
Scheff SW, Neltner JH, Nelson PT (2014) Is synaptic loss a unique hallmark of Alzheimer’s disease? Biochem Pharmacol 88:517-528
Poirel O, Mella S, Videau C, Ramet L, Davoli MA, Herzog E, Katsel P, Mechawar N et al (2018) Moderate decline in select synaptic markers in the prefrontal cortex (BA9) of patients with Alzheimer's disease at various cognitive stages. Sci Rep 8(1):938. https://doi.org/10.1038/s41598-018-19154-y
Benarroch EE (2018) Glutamatergic synaptic plasticity and dysfunction in Alzheimer’s disease: emerging mechanisms. Neurology 91:125-132
Forner S, Baglietto-Vargas D, Martini AC, Trujillo-Estrada L, LaFerla FM (2017) Synaptic impairment in Alzheimer’s disease: a dysregulated symphony. Trends Neurosci 40:347-357
Zhou L, McInnes J, Wierda K, Holt M, Herrmann AG, Jackson RJ, Wang YC, Swerts J et al (2017) Tau association with synaptic vesicles causes presynaptic dysfunction. Nat Commun 8:15295
Takamori S, Holt M, Stenius K, Lemke EA, Gronborg M, Riedel D, Urlaub H, Schenck S et al (2006) Molecular anatomy of a trafficking organelle. Cell 127(4):831-846
Mutch S, Kensel-Hammes P, Gadd J, Fujimoto B, Allen R, Schiro P, Lorenz R, Kuyper C et al (2011) Protein quantification at the single vesicle level reveals that a subset of synaptic vesicle proteins are trafficked with high precision. J Neurosci 31:1461-1470
Fremeau RT Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA et al (2004) Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science 304(5678):1815-1819. https://doi.org/10.1126/science.1097468
Ramet L, Zimmermann J, Bersot T, Poirel O, De Gois S, Silm K, Sakae DY, Mansouri-Guilani N et al (2017) Characterization of a human point mutation of VGLUT3 (p.A211V) in the rodent brain suggests a nonuniform distribution of the transporter in synaptic vesicles. J Neurosci 37(15):4181-4199. https://doi.org/10.1523/Jneurosci.0282-16.2017
Wilson NR, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G (2005) Presynaptic regulation of quantal size by the vesicular glutamate transporter VGLUT1. J Neurosci 25(26):6221-6234. https://doi.org/10.1523/JNEUROSCI.3003-04.2005
Wierenga CJ, Walsh MF, Turrigiano GG (2006) Temporal regulation of the expression locus of homeostatic plasticity. J Neurophysiol 96:2127-2133
Han EB, Stevens CF (2009) Development regulates a switch between post- and presynaptic strengthening in response to activity deprivation. Proc Natl Acad Sci 106:10817-10822
Doyle S, Pyndiah S, De Gois S, Erickson JD (2010) Excitation-transcription coupling via calcium/calmodulin-dependent protein kinase/ERK1/2 signaling mediates the coordinate induction of VGLUT2 and Narp triggered by a prolonged increase in glutamatergic activity. J Biol Chem 285:14366-14376
Hartman KN, Pal SK, Burrone J, Murthy VN (2006) Activity-dependent regulation of inhibitory synaptic transmission in hippocampal neurons. Nat Neurosci 9:642-649
Daniels RW, Collins CA, Chen K, Gelfand MV, Featherstone DE, DiAntonio A (2006) A single vesicular glutamate transporter is sufficient to fill a synaptic vesicle. Neuron:11-16
Daniels RW, Collins CA, Gelfand MV, Brooks ES, Krantz DE, DiAntonio A (2004) Increased expression of the Drosophila vesicular glutamate transporter leads to excess glutamate release and a compensatory decrease in quantal content. J Neurosci 24:10466-10474
Weston MC, Nehring RB, Wojcik SM, Rosenmund C (2011) Interplay between VGLUT isoforms and endophilin A1 regulates neurotransmitter release and short-term plasticity. Neuron 69:1147-1159
Zhang X-M, Francois U, Silm K, Angelo MF, Busch MVF, Maged M, Martin C, Cordelieres FP, Deshors M, Pons S, Maskos U, Bemelmans AP, Wojcik SM, El Mestikawy S, Humeau Y, Herzog E (2018) Mammalian vesicular glutamate transporter VGLUT1 reduces synaptic vesicle super-pool size and spontaneous release frequency. bioRxiv https://doi.org/10.I101/3688II
Hessler NA, Shirke AM, Malinow R (1993) The probability of transmitter release at a mammalian central synapse. Nature 366:569-572
Bolshakov VY, Siegelbaum SA (1995) Regulation of hippocampal transmitter release during development and long-term potentiation. Science 269:1730-1734
Dobrunz LE, Stevens CF (1997) Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron 18:995-1008
Hanse E, Gustafsson B (2001) Vesicle release probability and pre-primed pool at glutamatergic synapses in area CA1 of the rat neonatal hippocampus. J Physiol 531:481-493
Kirino T, Tsujita Y, Tamura T (1991) Induced tolerance to ischemia in gerbil hippocampal neurons. J Cereb Blood Flow Metab 11:299-307
Kitagawa K, Matsumoto M, Kuwabara K, Tagaya M, Ohtsuki T, Hata R, Ueda H, Handa N et al (1991) “Ischemic tolerance” phenomenon detected in various brain regions. Brain Res 561:203-211
Liu Y, Kato H, Nakata N, Kogure K (1992) Protection of rat hippocampus against ischemic neuronal damage by pretreatment with sublethal ischemia. Brain Res 586:121-124
Simon RP, Niiro M, Gwinn R (1993) Prior ischemic stress protects against experimental stroke. Neurosci Lett 163:135-137
Kelly ME, McIntyre DC (1994) Hippocampal kindling protects several structured from the neuronal damage from kainic acid-induced status epilepticus. Brain Res 634:245-256
Jimenez-Mateos EM, Henshall DC (2009) Seizure preconditioning and epileptic tolerance: models and mechanisms. Int J Physiol Pathophysiol Pharmacol 1:180-191
Emerson MR, Nelson SR, Samson FE, Pazdernik TL (1999) Hypoxia preconditioning attenuates brain edema associated with kainic acid-induced status epiliepticus in rats. Brain Res 825:189-193
Plamondon H, Blondeau N, Heurteaux C, Lazdunski M (1999) Mutually protective actions of kainic acid epileptic preconditioning and sublethal ischemia on hippocampal neuronal death: involvement of adenosine A1 receptors and KATP channels. J Cereb Blood Flow Metab 19:1296-1308
Grabb MC, Lobner D, Turetsky M, Choi DW (2002) Preconditioned resistance to oxygen-glucose deprivation-induced cortical neuronal death: alterations in vesicular GABA and glutamate release. Neuroscience 115:173-183
Dave KR, Lange-Asschenfeldt C, Raval AP, Prado R, Busto R, Saul I, Periz-Pinzon MA (2005) Ischemic preconditioning ameliorates excitotoxicity by shifting glutamate/gamma-aminobutyric acid release and biosynthesis. J Neurosci Res 82:665-673
Hogins J, Crawford DC, Jiang X, Mennerick S (2011) Presynaptic silencing is an endogenous neuroprotectant during excitotoxic insults. Neurobiol Dis 43:516-525
Tauskela JS, Aylsworth A, Hewitt M, Brunette E, Mealing GA (2012) Preconditioning induces tolerance by suppressing glutamate release in neuron culture ischemia models. J Neurochem 122:470-481
Lau CG, Murthy VN (2012) Activity-dependent regulation of inhibition via GAD67. J Neurosci 32:8521-8531
Hanno-Iijima Y, Tanaka M, Iijima T (2015) Activity-dependent bidirectional regulation of GAD expression in a homeostatic fashion is mediated by BDNF-dependent and independent pathways. PLoS One 10(8):e0134296. https://doi.org/10.1371/journal.pone.0134296.eCollection2015
Mattar P, Langevin LM, Markham K, Klenin N, Shivji S, Zinyk D, Schuurmas C (2008) Basic helix-loop-helix transcription factors cooperate to specify a cortical projection neuron identity. Mol Cell Biol 28:1456-1469
Serrano-Saiz E, Poole RJ, Felton T, Zhang F, De La Cruz ED, Hobert O (2013) Modular control of glutamatergic neuronal identity in C. elegans by distinct homeodomain proteins. Cell 155:659-673
Melo CV, Mele M, Curcio M, Comprido D, Silve CG, Duarte CB (2013) BDNF regulates the expression and distribution of vesicular glutamate transporters in cultured hippocampal neurons. Plos one. https://doi.org/10.1371/journal.pone.0053793
Zhang CR, Ho M-F, Shancez Vega MC, Burne THJ, Chong S (2015) Prenatal ethanol exposure alters adult hippocampal VGLUT2 expression with concomitant changes in promoter DNA methylation, H3K4 trimethylation and miR-467b-5p levels. Epigenetics Chromatin 8:40-52
Siano G, Varisco M, Caiazza MC, Quercioli V, Mainardi M, Iippolito C, Cattaneo A, Di Priimio C (2019) Tau modulates VGLUT1 expression. J Mol Biol 431:873-884
Li T, Bai L, Li J, Igarashi S, Ghishan FK (2008) Sp1 is required for glucose-induced transcriptional regulation of mouse vesicular glutamate transporter 2 gene. Gastroenterology 134:1994-2003
Zhang GR, Li X, Cao H, Zhao H, Geller AI (2011) The vesicular glutamate transporter-1 upstream promoter and first intron each support glutamatergic-specific expression in rat postrhinal cortex. Brain Res 1377:1-12
Egashira Y, Mori Y, Yanagawa Y, Takamori S (2018) Development of lentiviral vectors for efficient glutamatergic-selective gene expression in cultured hippocampal neurons. Sci Report 8:Article number: 15156
Akil O, Seal RP, Burke K, Wang CC, Alemi A, During M, Edwards RH, Lustig LR (2012) Restoration of hearing in the VGLUT3 knockout mouse using virally-mediated gene therapy. Neuron 75:283-293
Reimer RJ, Edwards RH (2004) Organic anion transport is the primary function of the SLC17/type I phosphate transporter family. Pflugers Arch 447:629-635
Morin P, Sagne C, Gasnier B (2004) Functional characterization of wild-type and mutant human sialin. EMBO J 23:4560-4570
Sawada K, Echigo N, Juge N, Miyaji T, Otsuka M, Omote H, Yamamoto A, Moriyama Y (2008) Identification of a vesicular nucleotide transporter. Proc Natl Acad Sci 105:5683-5686
Miyamoto K-I, Haito-Sugino S, Kuwahara S, Ohi A, Nomura K, Ito M, Kuwahata M, Kido S et al (2011) Sodium-dependent phosphate cotransporters: Lessons from gene knockout and mutation studies. J Pharm Sci 100:3719-3730. https://doi.org/10.1002/jps.22614
Hiasa M, Miyaji T, Haruna Y, Takeuchi T, Harada Y, Moriyama S, Yamamoto A, Omote H, Moriyama Y (2014) Identification of a mammalian vesicular polyamine transporter. Sci Reports Article number: 6836
Liu Y, Peter D, Roghani A, Schuldiner S, Prive GG, Eisenberg D, Brecha N, Edwards RH (1992) A cDNA that suppresses MPP+ toxicity encodes a vesicular amine transporter. Cell 70:539-551
Erickson JD, Eiden LE, Hoffman BJ (1992) Expression cloning of a reserpine-sensitive vesicular monoamine transporter. Proc Natl Acad Sci 89:10993-10997
Peter D, Jimenez J, Liu Y, Kim J, Edwards RH (1994) The chromaffin granule and synaptic vesicle amine transporters differ in substrate recognition and sensitivity to inhibitors. J BiolChem 269:7231-7237
Erickson JD, Schafer MK-H, Bonner TI, Eiden LE, Weihe E (1996) Distinct pharmacological properties and distribution in neurons and endocrine cells of two isoforms of the human vesicular monoamine transporter. Proc Natl Acad Sci 93:5166-5171
Varoqui H, Erickson JD (1996) Active transport of acetylcholine by the human vesicular acetylcholine transporter. J Biol Chem 271(44):27229-27232
Liu Y, Edwards RH (1997) Differential localization of vesicular acetylcholine and monoamine transporters in PC12 cells but not CHO cells. J Cell Biol 139:907-916
McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM (1997) Identification and characterization of the vesicular GABA transporter. Nature 389(6653):870-876. https://doi.org/10.1038/39908
Sagne C, El Mestikawy S, Isambert MF, Hamon M, Henry JP, Giros B, Gasnier B (1997) Cloning of a functional vesicular GABA and glycine transporter by screening of genome databases. FEBS Lett 417(2):177-183
Fykse EMFF (1988) Uptake of gamma-aminobutyric acid by a synaptic vesicle fraction isolated from rat brain. J Neurochem 50(1):237-1242
Apostolides PF, Trussell LO (2013) Rapid, activity-independent turnover of vesicular transmitter content at a mixed glycine/GABA synapse. J Neurosci 33:4768-4781
Quistgaard EM, Low C, Guettou F, Nordlund P (2016) Understanding transport by the major facilitator superfamily (MFS): structures pave the way. Nat Rev Mol Cell Biol 17(2):123-132. https://doi.org/10.1038/nrm.2015.25
Huang Y, Lemieux MJ, Song J, Auer M, Wang DN (2003) Structure and mechanism of the glycerol-3-phosphate transporter from Escherichia coli. Science 301:616-620
Leano JB, Batarni S, Eriksen J, Juge N, Pak JE, Kimura-Someya T, Robles-Colmenares Y, Moriyama Y et al (2019) Structures suggest a mechanism for energy coupling by a family of organic anion transporters. PLoS Biol 17(5):e3000260. https://doi.org/10.1371/journal.pbio.3000260
Abramson J, Smirnova I, Kasho V, Verner GH, Kaback R, Iwata S (2003) Structure and mechanism of the lactose permease of Escherichia coli. Science 301:610-615. https://doi.org/10.1126/science.1088196
Huang Y, McNamara JO (2004) Ischemic stroke: ‘acidotoxicity’ is a perpetrator. Cell Mol Neurobiol 118:665-666
Juge N, Yoshida Y, Yatsushiro S, Omote H, Moriyama Y (2006) Vesicular glutamate transporter contains two independent transport machineries. J Biol Chem 281(51):39499-39506. https://doi.org/10.1074/jbc.M607670200
Chang R, Eriksen J, Edwards RH (2018) The dual role of chloride in synaptic vesicle glutamate transport. eLife 7. https://doi.org/10.7554/eLife.34896
Hayashi M, Yamada H, Uehara S, Morimoto R, Muroyama A, Yatsushiro S, Takeda J, Yamamoto A et al (2003) Secretory granule-mediated co-secretion of L-glutamate and glucagon triggers glutamatergic signal transmission in islets of Langerhans. J Biol Chem 278(3):1966-1974. https://doi.org/10.1074/jbc.M206758200
Almqvist J, Huang Y, Laaksonen A, Wang D-N, Hovmöller S (2007) Docking and homology modeling explain inhibition of the human vesicular glutamate transporters. Protein Sci 16(9):1819-1829. https://doi.org/10.1110/ps.072944707
Preobraschenski J, Zander J-F, Suzuki T, Ahnert-Hilger G, Jahn R (2014) Vesicular gutamate tansporters use flexible anion and cation binding sites for efficient accumulation of neurotransmitter. Neuron 84(6):1287-1301. https://doi.org/10.1016/j.neuron.2014.11.008
Schuldiner S, Shirvan A, Linial M (1995) Vesicular neurotransmitter transporters: from bacteria to humans. Physiol Rev 75:369-392
Vardy E, Steiner-Mordoch S, Schuldiner S (2005) Characterization of bacterial drug antiporters homologous to mammalian neurotransmitter transporters. J Bacterol 187:7518-7525
Yaffe D, Vergara-Jaque A, Shuster Y, Listov D, Meena S, Singh SK, Forrest LR, Schuldiner S (2014) Functionally important carboxyls in a bacteial homologue of the vesicular monoamine transporter (VMAT). J Biol Chem 289:34229-34240
Yaffe D, Forrest LR, Schuldiner S (2018) The ins and outs of vesicular monoamine transporters. J Gen Physiol 150:671-682
Zhu H, Duerr JS, Varoqui H, McManus JR, Rand JB, Erickson JD (2001) Analysis of point mutants in the Caenorhabditis elegans vesicular acetylcholine transporter reveals domains involved in substrate translocation. J Biol Chem 276:41580-41587
Martin CA, Krantz DE (2014) Drosophila melanogaster as a genetic model system to study neurotransmitter transporters. Neurochem Int 73:71-88
Smear MC, Tao HW, Staub W, Orger MB, Gosse NJ, Liu Y, Takahshi K, Poo M-M et al (2007) Vesicular glutamate transport at a central synapse limits the acuity of visual perception in zebrafish. Neuron 53:65-77
Disbrow JK, Gershten MJ, Ruth JA (1982) Uptake of L-[3H] glutamic acid by crude and purified synaptic vesicles from rat brain. Biochem Biophys Res Commun 108(3):1221-1227
Naito S, Ueda T (1983) Adenosine triphosphate-dependent uptake of glutamate into protein I-associated synaptic vesicles. J Biol Chem 258:696-699
Naito S, Ueda T (1985) Characterization of Glutamate Uptake into Synaptic Vesicles. J Neurochem 44(1):99-109
Maycox PR, Deckwerth T, Hell JW, Jahn R (1988) Glutamate uptake by brain synaptic vesicles. Energy dependence of transport and functional reconstitution in proteoliposomes. J Biol Chem 263(30):15423-15428
Carlson MD, Kish PE, Ueda T (1989) Glutamate uptake into synaptic vesicles: competitive inhibition by bromocriptine. J Neurochem 53(6):1889-1894
Maycox PR, Hell JW, Jahn R (1990) Amino acid neurotransmission: spotlight on synaptic vesicles. Trends Neurosci 13:83-87
Burger PM, Mehl E, Cameron PL, Maycox PR, Baumert M, Lottspeich F, De Camilli P, Jahn R (1989) Synaptic vesicles immunoisolated from rat cerebral cortex contain high levels of glutamate. Neuron 3(6):715-720
Martineau M, Guzman RE, Fahlke C, Klingauf J (2017) VGLUT1 functions as a glutamate/proton exchanger with chloride channel activity in hippocampal glutamatergic synapses. Nature Comm 8:2279. https://doi.org/10.1038/s4167-2017-02367-02366
Ni BH, Rosteck PR, Nadi NS, Paul SM (1994) Cloning and expression of a cDNA-encoding a brain-specific Na+−dependent inorganic-phosphate cotransporter. Proc Natl Acad Sci 91(12):5607-5611
Aihara Y, Mashima H, Onda H, Hisano S, Kasuya H, Hori T, Yamada S, Tomura H et al (2000) Molecular cloning of a novel brain-type Na(+)-dependent inorganic phosphate cotransporter. J Neurochem 74(6):2622-2625
Bellocchio EE, Hu H, Pohorille A, Chan J, Pickel VM, Edwards RH (1998) The localization of the brain-specific inorganic phosphate transporter suggests a specific presynatic role in glutamatergic transmission. J Neurosci 18:8648-8659
Bradford HF, Ward HK (1976) On glutaminase activity in mammalian synaptosomes. Brain Res 110:115-125
Ferguson SM, Savchenko V, Apparsundaram S, Zqick M, Wright J, Heilman CJ, Yi H, Levey AI et al (2003) Vesicular localization and activity-dependent trafficking of presynaptic choline transporters. J Neurosci 23:9697-9709
Ribeiro FM, Pinthong M, Black SA, Gordon AC, Prado VF, Prado AM, Rylett RJ, Ferguson SM (2007) Regulated recycling and plasma membrane recruitment of the high-affinity choline transporter. Eur J Neurosci 26:3437-3448
Denken SL, Wang D, Quick MW (2003) Plasma membrane GABA transporters reside on distinct vesicles and undergo rapid regulated recycling. J Neurosci 23:1563-1568
Preobraschenski J, Cheret C, Ganzella M, Zander JF, Richter K, Schenck S, Jahn R, Ahnert-Hilger G (2018) Dual and direction-selective mechanisms of phosphate transport by the vesicular glutamate transporter. Cell Rep 23:535-545. https://doi.org/10.1016/j.celrep.2018.03.055
Inden M, Iriyama M, Zennami M, Sekine SI, Hara A, Yamada M, Hozumi I (2016) The type III transporters (PiT-1 and PiT-2) are the major sodium-dependent phosphate transporters in the mice and human brains. Brain Res 1637:128-136. https://doi.org/10.1016/j.brainres.2016.02.032
Johnson RG, Scarpa A (1979) Protonmotive force and catecholamine transport in isolated chromaffin granules. J Biol Chem 254:3750-3760
Cidon S, Sihra TS (1989) Characterization oif a H+-ATPase in rat brain synaptic vesicles: coupling to l-glutamate transport. J Biol Chem 264:8281-8288
Kanner BI, Sharon I (1978) Active transport of L-glutamate by membrane vesicles isolated from rat brain. Biochemistry 17:3949-3953
Wolosker H, de Souza DO, de Meis L (1996) Regulation of glutamate transport into synaptic vesicles by chloride and proton gradient. J Biol Chem 271:11726-11731
Tabb JS, Kish PE, Van Dyke R, Ueda T (1992) Glutamate transport into synaptic vesicles. Roles of membrane potential, pH gradient, and intravesicular pH. J Biol Chem 267(22):15412-15418
Moriyama Y, Maeda M, Futai M (1990) Energy coupling of L-glutamtae transport and vacuolar H(+)-ATPase in brain synaptic vesicles. J Biochem 108:689-693
Zhou Q, Petersen CC, Nicoll RA (2000) Effects of reduced vesicular filling on synaptic transmission in rat hippocampal neurons. J Physiol 525:195-206
Schenck S, Wojcik SM, Brose N, Takamori S (2009) A chloride conductance in VGLUT1 underlies maximal glutamate loading into synaptic vesicles. Nat Neurosci 12(2):156-162. https://doi.org/10.1038/nn.2248
Omote H, Moriyama Y (2013) Vesicular neurotransmitter transporters: An approach for studying transporters with purified proteins. Physiology 28(1):39-50. https://doi.org/10.1152/physiol.00033.2012
Eriksen J, Chang R, McGregor M, Silm K, Suzuki T, Edwards RH (2016) Protons regulate vesicular glutamate transporters through anallosteric mechanism. Neuron 90(4):768-780. https://doi.org/10.1016/j.neuron.2016.03.026
Stobrawa SM, Breiderhoff T, Takamori S, Engel D, Schweizer M, Zdebik AA, Bosl MR, Ruether K et al (2001) Disruption of ClC-3, a chloride channel expressed on synaptic vesicles, leads to a loss of the hippocampus. Neuron 29(1):185-196
Goh GY, Huang H, Ullman J, Borre L, Hnasko TS, Trussell LO, Edwards RH (2011) Presynaptic regulation of quantal size: K+/H+ exchange stimulates vesicular glutamate transport. Nat Neurosci 14:1285-1292
Alabi AA, Tsien RW (2013) Perspectives on kiss-and-run: role in exocytosis, endocytosis, and neurotransmission. Annu Rev Physiol 75:393-422
Mackenzie B, Illing AC, Morris ME, Varoqui H, Erickson JD (2008) Analysis of a vesicular glutamate transporter (VGLUT2) supports a cell leakage mode in addition to vesicular packaging. Neurochem Res 33:238-247
Bradford HF, Young AMJ, Crowder JM (1987) Continuous glutamate leakage from brain cells is balanced by compensatory high-affinity reuptake transporter. Neurocsci Lett 81:296-302
Katayama Y, Becker DP, Tamura T, Hovda DA (1994) Massive increases in extracellular potassium and the indiscriminant release of glutaamte following concussive brain injury. J Neurosurg 73:889-900
Pocock JM, Nicolls DG (1998) Exocytotic and nonexocytotic modes of glutamate release from cultured cerebellar granule cells during chemical ischemia. J Neurochem 70:806-813
Shiogai T, Nara I, Saruta K, Hara M, Saito I (1999) Continuous monitoring of cerebraospinal fluid acid-base balance and oxygen metabolism in patients with severe head injury: pathophysiology and treatments for cerebral acidosis and ischemia. Acta Neurochir Suppl 75:49-55
Clausen T, Khaldi A, Zauner A, Reinert M, Doppenberg E, Menzel M, al. e (2005) Cerebral acid-base homeostatisis after severe traumatic brain injury. J Neurosurg 3 (597-607)
Hartinger J, Jahn R (1993) An anion binding site that regulates the glutamate transporter of synaptic vesicles. J Biol Chem 268(31):23122-23127
Farsi Z, Jahn R, Woehler A (2017) Proton electrochemical gradient: Driving and regulating neurotransmitter uptake. Bioessays 39. https://doi.org/10.1002/bies.201600240
Trudeau LE, El Mestikawy S (2018) Glutamate Cotransmission in cholinergic, GABAergic and monoamine systems: contrasts and commonalities. Front Neural Circuits 12. https://doi.org/10.3389/fncir.2018.00113):113
Omote H, Miyaji T, Hiasa M, Juge N, Moriyama Y (2016) Structure, function, and drug interactions of neurotransmitter transporters in the postgenomic era. Annu Rev Pharmacol Toxicol 56(1):385-402. https://doi.org/10.1146/annurev-pharmtox-010814-124816
Wadiche JI, Amara SG, Kavanaugh MP (1995) Ion fluxes associated with excitatory amino acid transport. Neuron 15:721-728
Fairman WA, Vandenberg RJ, Arriza JL, Kavanaugh MP, Amara SG (1995) An excitatory amino-acid transporter with properties of a ligand-gated chloride channel. Nature 375:599-603 10.1038/
Juge N, Gray JA, Omote H, Miyaji T, Inoue T, Hara C, Uneyama H, Edwards RH et al (2010) Metabolic control of vesicular glutamate transport and release. Neuron 68(1):99-112. https://doi.org/10.1016/j.neuron.2010.09.002
Lai ZF, Liu J, Nishi K (1996) Effects of stilbene derivatives SITS and DIDS on development of intracellular acidosis during ischemia in isolated guinea pig ventricular papillary muscle in vitro. Jpn J Pharmacol 72(2):161-174
Erickson JD, Masserano JM, Barnes EM, Ruth JA, Weiner N (1990) Chloride ion increases [3H]dopamine accumulation by synaptic vesicles purified from rat striatum: inhibition by thiocyanate ion. Brain Res 516:155-160
Reiss M, Farage M, Wolosker H (2000) Chloride-dependent inhibition of vesicular glutamate uptake by alpha-keto acids accumulated in maple syrup urine disease. Biochim Biophys Acta 1475:114-118
Tavares RG, Santos CE, Tasca CI, Wajner M, Souza DO, Dutra-Filho CS (2000) Inhibitionof glutamate uptake into synaptic vesicles of rat brain by the metabolites accumulating in maple syrup urine disease. J Neurol Sci 181:44-49
Doyon N, Vinay L, Prescott SA, De Koninck Y (2016) Chloride regulation: a dynamic equilibrium crucial for synaptic inhibition. Neuron 89:1157-1172
Pellegrino C, Gubkina O, Schaefer M, Becq H, Ludwig A, Mukhtarov M, Chudotvorova I, Corby S et al (2011) Knocking down of the KCC2 in rat hippocampal neurons increases intracellular chloride concentration and compromises neuronal survival. J Physiol 589:2475-2496
Watanabe M, Fukuda A (2015) Development and regulation of chloride homeostasis in the central nervous system. Front Cell Neurosci 9:371
Price GD, Trussell LO (2006) Estiimate of the chloride concentration in a central glutamatergic terminal: a gramicidin perforated-patch study on the Calyx of Held. J Neurosci 26:11432-11436
Hori T, Takahashi T (2012) Kinetics of synaptic vesicle refilling with neurotransmitter glutamate. Neuron 76:511-517
Zhu L, Polley N, Matthews GC, Delpire E (2008) NKCC1 and KCC2 prevent hyperexcitability in the mouse hippocampus. Epilepsy Res 79:201-212
Sato SS, Artoni P, Landi S, Cozzolino O, Parra R, Pracucci E, Trovato F, Szczurkowska J et al (2017) Simultaneous two-photon imaging of intracellular chloride concentration and pH in mouse pyramidal neurons in vivo. Proc Natl Acad Sci 201702861. https://doi.org/10.1073/pnas.1702861114
Hassel B, Brathe A (2000) Neuronal pyruvate carboxylation supports formation of transmitter glutamate. J Neurosci 20:1342-1347
Bak LK, Schousboe A, Sonnewald U, Waagepetersen HS (2006) Glucose is necessary to maintain neurotransmitter homeostasis during synaptic activity in cultured glutamatergic neurons. J Cereb Blood Flow Metab 26:1285-1297
Takeda K, Ishida A, Takahashi K, Ueda T (2012) Synaptic vesicles are capable of synthesizing the VGLUT substrate glutamate from a-ketoglutarate for vesicle loading. J Neurochem 121:184-196
Takeda K, Ueda T (2017) Effective mechanism for synthesis of neurotransmitter glutamate and its loading into synaptic vesicles. Neurochem Res 42:64-76
Chen W, Mahadomrongkul V, Berger UV, Bassan M, DeSilva T, Tanaka K, Irwin N, Aoki C et al (2004) The glutamate transproter GLT1a is expressed in excitatory axon terminals of mature hippocampal neurons. J Neurosci 24:1136-1148
Waagepetersen HS, Qu H, Sonnewald U, Shimamoto K, Schousboe A (2005) Role of glutamine and neuronal glutamate uptake in glutamate homeostasis and synthesis during vesicular release in cultured glutamatergic neurons. Neurochem Int 47:92-102
Danbolt NC, Furness DN, Zhou Y (2016) Neuronal vs glial glutamate uptake: Resolving the conundrum. Neurochem Int 98:29-45
Ishikawa T, Sahara Y, Takahashi T (2002) A single packet of transmitter does not saturate postsynaptic glutamate receptors. Neuron 34:613-621
Yamashita T, Ishikawa T, Takahashi T (2003) Developmental increase in vesicular glutamate content does not cause saturation of AMPA receptors at the calyx of Held synapse. J Neurosci 23:3633-3638
Wu XS (2007) The origin of quantal size variation: vesicular glutamate concentration plays a significant role. J Neurosci 27:3046-3056
Yamashita T, Kanda T, Eguchi K, Takahashi T (2009) Vesicular glutamate filling and AMPA receptor occupancy at the calyx of Held synapse of immature rats. J Physiol 587:2327-2339. https://doi.org/10.1113/jphysiol.2008.167759
Liu G, Choi S, Tsien RW (1999) Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron 22:395-409
Rosenmund C, Clements JD, Westbrook GL (1993) Nonuniform probability of glutamate release at a hippocampal synapse. Science 262:754-757
McAllister AK, Stevens CF (2000) Nonsaturation of AMPA and NMDA receptors at hippocampal synapses. Proc Natl Acad Sci 97:6173-6178
Bramham CR, Torp R, Zhang N, Storm-Mathisen J, Ottersen OP (1990) Distribution of glutamate-like immunoreactivity in excitatory hippocampal pathways: a semiquantitative electron microscopic study in rats. Neuroscience 39(2):405-417
Osen KK, Storm-Mathisen J, Ottersen OP, Dihle B (1995) Glutamate is concentrated in and released from parallel fiber terminals in the dorsal cochlear nucleus: a quantitative immunocytochemical analysis in guinea pig. J Comp Neurol 357(3):482-500. https://doi.org/10.1002/cne.903570311
Ottersen OP, Storm-Mathisen J, Bramham C, Torp R, Laake J, Gundersen V (1990) A quantitative electron microscopic immunocytochemical study of the distribution and synaptic handling of glutamate in rat hippocampus. Prog Brain Res 83:99-114
Ottersen OP, Zhang N, Walberg F (1992) Metabolic compartmentation of glutamate and glutamine: morphological evidence obtained by quantitative immunocytochemistry in rat cerebellum. Neuroscience 46(3):519-534
Shupliakov O, Brodin L, Cullheim S, Ottersen OP, Storm-Mathisen J (1992) Immunogold quantification of glutamate in two types of excitatory synapse with different firing patterns. J Neurosci 12:3789-3803
Varoqui H, Erickson JD (1997) Vesicular neurotransmitter transporters. Potential sites for the regulation of synaptic function. Mol Neurobiol 15:165-191
Van der Kloot W, Molgo J, Cameron R, Colasante C (2002) Vesicle size and transmitter release at the frog neuromuscular junction when quantal acetylcholine content is increased or decreased. J Physiol 541:385-393
Pothos E, Desmond M, Sulzer D (1996) L-3,4-Dihydroxyphenylalanine increases the quantal size of exocytotic dopamine release in vitro. J Neurochem 66:629-636
Marx M-C, Billups D, Billups B (2015) Maintaining the presynaptic glutamate supply for excitatory neurotransmission. J Neurosci Res 93:1031-1044
Martinez-Hernandez A, Bell KP, Norenberg MD (1977) Glutamine synthetase: glial localization in brain. Science 195:1356-1358
Hamberger AC, Chiang GH, Nylen ES, Scheff SW, Cotman CW (1978) Stimulus evoked increase in the biosynthesis of the putative neurotransmitter glutamate in the hippocampus. Brain Res 143:549-555
Hamberger AC, Chiang GH, Nylen ES, Scheff SW, Cotman CW (1979) Glutamate as a CNS transmitter. I. Evaluation of glucose and glutamine as precursors for the synthesis of preferentially released glutamate. Brain Res 168:513-530
Hamberger AC, Chiang GH, Sandoval E, Cotman CW (1979) Glutamate as a CNS transmitter. II. Regulation of synthesis in the releasable pool. Brain Res 168:531-541
Laake JH, Slyngstad TA, Haug FM, Ottersen OP (1995) Glutamine from glial cells is essential for the maintenance of the nerve terminal pool of glutamate: immunogold evidence from hippocampal slice cultures. J Neurochem 65:871-881
Schousboe A, Bak LK, Waagepetersen HS (2013) Astrocytic control of biosynthesis and turnover of the neurotransmitters glutamate and GABA. Front Endocrinol. https://doi.org/10.3389/fendo.2013.00102
Tani H, Dulla CG, Farzampour Z, Taylor-Weiner A, Huguenard JR, Reimer RJ (2014) A local glutamate-glutamine cycle sustains synaptic excitatory transmitter release. Neuron 81:888-900
Leke R, Schousboe A (2016) The glutamine transporters and their role in the glutamate/GABA-glutamine cycle. Adv Neurobiol 13:223-257
Kam K, Nicoll R (2007) Excitatory synaptic transmission persists independently of the glutamate-glutamine cycle. J Neurosci 27:9192-9200
Masson J, Darmon M, Conjard A, Chuhma N, Ropert N, Thoby-Brisson M, Foutz AS, Parrot S et al (2006) Mice lacking brain/kidney phosphate-activated glutaminase have impaired glutamatergic synaptic transmission, altered breathing, disorganized goal-directed behavior and die shortly after birth. J Neurosci 26:4660-4671
Kvamme E, Roberg B, Torgner IA (2000) Phosphate-activated glutaminase and mitrochondrial glutamine transport in the brain. Neurochem Res 25:1407-1419
Eid T, Hammer J, Runden-Pran E, Roberg B, Thomas MJ, Osen K, Davanger S, Laake P et al (2007) Increased expression of phosphate-activated glutaminase in hippocampal neurons in human mesial temporal lobe epilepsy. Acta Neuropathol 113:137-152
Miller KE, Hoffman EM, Sutharshan M, Schechter R (2011) Glutamate pharmacology and metabolism in peripheral primary aferents: Physiological and pathophysiological mechanisms. Pharmacol Ther 130:283-309
Zhou Y, Dhaher R, Parent M, Hu QX, Hassel B, Yee SP, Hyder F, Gruenbaum SE et al (2019) Selective deletion of glutamine synthetase in the mouse cerebral cortex includes glial dysfunction and vascular impairment that precede epilepsy and neurodegeneration. Neurochem Int 123:22-33
Maragakis NJ, Rothstein JD (2001) Glutamate transporters in neurologic disease. Arch Neurol 58:365-370
Sheldon AL, Robinson MB (2007) The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int 51:333-355
Bacci A, Sancini G, Verderio C, Armano S, Pravettoni E, Fesce R, Franceschetti S, Matteoli M (2002) Block of glutamate-glutamine cycle between astrocytes and neurons inhibits epileptiform activity in hippocampus. J Neurophysiol 88:2302-2310
Tani H, Bandrowski AE, Parada I, Wynn M, Huguenard JR, Prince DA, Reimer RJ (2007) Modulation of epileptiform activity by glutamine and system A transport in a model of post-traumatic epilepsy. Neurobiol Dis 25:230-238
Tani H, Dulla CG, Huguenard JR, Reimer RJ (2010) Glutamine is required for persistent epileptiform activity in the disinhibited neocortical brain slice. J Neurosci 30:1288-1300
Kanamori K, Ross BD (2013) Electrographic seizures are significantly reduced by in vivo inhibition of neuronal uptake of extracellular glutamine in rat hippocampus. Epilepsy Res 107:20-36
Mackenzie B, Erickson JD (2004) Sodium-coupled neutral amino acid (System N/A) transporters of the SLC38 gene family. Pflugers Arch 447:784-795
Conti F, Melone M (2006) The glutamine commute: lost in the tube? Neurochem Int 48:459-464
Erickson JD (2017) Functional identification of activity-regulated, high-affinity glutamine transport in hippocampal neurons inhibited by riluzole. J Neurochem 142:29-40
Neale SA, Copeland CS, Salt TE (2014) Effect of VGLUT inhibitors on glutamatergic synaptic transmission in the rodent hippocampus and prefrontal cortex. Neurochem Int 73:159-165. https://doi.org/10.1016/j.neuint.2013.10.001
Neale SA, Copeland CS, Uebele VN, Thomson FJ, Salt TE (2013) Modulation of hppocampal synaptic transmission by the kynurenine pathway member xanthurenic acid and other VGLUT inhibitors. Neuropsychopharmacology 38(6):1060-1067. https://doi.org/10.1038/npp.2013.4
Thompson CM, Davis E, Carrigan CN, Cox HD, Bridges RJ, Gerdes JM (2005) Inhibitors of the glutamate vesicular transporter (VGLUT). Curr Med Chem 12(18):2041-2056
Winter HC, Ueda T (1993) Glutamate uptake system in the presynaptic vesicle: glutamic acid analogs as inhibitors and alternate substrates. Neurochem Res 18:79-85
Winter HC, Ueda T (2007) The glutamate uptake system in presynaptic vesicles: further characterization of structural requirements for inhibitors and substrates. Neurochem Res 33(2):223-231. https://doi.org/10.1007/s11064-007-9493-8
Schoepp DD, True RA (1992) 1S,3R-ACPD-sensitive (metabotropic) [3H]glutamate receptor binding in membranes. Neurosci Lett 145:100-104
Bartlett RD, Esslinger CS, Thompson CM, Bridges RJ (1998) Substituted quinolines as inhibitors of L-glutamate transport into synaptic vesicles. Neuropharmacology 37(7):839-846
Shigeri Y, Seal RP, Shimamoto K (2004) Molecular pharmacology of glutamate transporters, EAATs and VGLUTs. Brain Res Rev 45(3):250-265
Carrigan CN, Bartlett RD, Esslinger CS, Cybulski KA, Tongcharoensirikul P, Bridges RJ, Thompson CM (2002) Synthesis and in vitro pharmacology of substituted quinoline-2,4-dicarboxylic acids as inhibitors of vesicular glutamate transport. J Med Chem 45(11):2260-2276
Carrigan CN, Patel SA, Cox HD, Bolstad ES, Gerdes JM, Smith WE, Bridges RJ, Thompson CM (2014) The development of benzo- and naphtho-fused quinoline-2,4-dicarboxylic acids as vesicular glutamate transporter (VGLUT) inhibitors reveals a possible role for neuroactive steroid. Bioorg Med Chem Lett 24(3):850-854. https://doi.org/10.1016/j.bmcl.2013.12.086
Carrigan CN, Esslinger CS, Bartlett RD, Bridges RJ, Thompson CM (1999) Quinoline-2,4-dicarboxylic acids: synthesis and evaluation as inhibitors of the glutamate vesicular transport system. Bioorg Med Chem Lett 9(17):2607-2612
Tamura Y, Ogita K, Ueda T (2013) A new VGLUT-specific potent inhibitor: Pharmacophore of brilliant yellow. Neurochem Res 39:117-128. https://doi.org/10.1007/s11064-013-1196-8
Favre-Besse FC, Poirel O, Bersot T, Kim-Grellier E, Daumas S, El Mestikawy S, Acher FC, Pietrancosta N (2014) Design, synthesis and biological evaluation of small-azo-dyes as potent vesicular glutamate transporters inhibitors. Eur J Med Chem 78:236-247. https://doi.org/10.1016/j.ejmech.2014.03.056
Roseth S, Fykse EM, Fonnum F (1995) Uptake of L-glutamate into rat brain synaptic vesicles: effect of inhibitors that bind specifically to the glutamate transporter. J Neurochem 65(1):96-103
Yaffe D, Vergara-Jaque A, Forrest LR, Schuldiner S (2016) Emulating proton-induced conformational changes in the vesicular monoamine transporter VMAT2 by mutagenesis. Proc Natl Acad Sci 113:E7390-E7398
Muller C, Viry S, Miehe M, Andriamampandry C, Aunis D, Maitre M (2002) Evidence for a gamma-hydroxybutyrate (GHB) uptake by rat brain synaptic vesicles. J Neurochem 80:899-904
Cao Q, Zhao K, Zhong XZ, Zou Y, Yu H, Huang P, Xu T-L, Dong X-P (2014) SLC17A9 protein functions as a lysosomal ATP transporter and regulates cell viability. J Biol Chem 289:23189-23199
Goldim MPS, Della Giustina A, Petronilho F (2019) Using evans blue dye to determine blood-brain barrier integrity in rodents. Curr Protoc Immunol 126(1):e83. https://doi.org/10.1002/cpim.83
Pietrancosta N, Kessler A, Favre-Besse FC, Triballeau N, Quentin T, Giros B, El Mestikawy S, Acher FC (2010) Rose Bengal analogs and vesicular glutamate transporters (VGLUTs). Bioorg Med Chem 18(18):6922-6933. https://doi.org/10.1016/j.bmc.2010.06.069
Ogita K, Hirata K, Bole DG, Yoshida S, Tamura Y, Leckenby AM, Ueda T (2001) Inhibition of vesicular glutamate storage and exocytotic release by Rose Bengal. J Neurochem 77:34-42
Bole DG, Ueda T (2005) Inhibition of vesicular glutamate uptake by Rose Bengal-related compounds: Structure-activity relationship. Neurochem Res 30(3):363-369
Acknowledgments
We would like to thank Dr. Heike Wulff (UCDavis) for reading this article and her helpful suggestions.
Funding
The present work was supported by grants from the Fond de Recherche Santé Québec (FRQS 30582), the Natural Sciences and Engineering Research Council (RGPIN/386431-2012 and RGPIN/04682-2017), the Agence Nationale de la Recherche (ANR, ANR-13-SAMA-0005-01), the Centre National de la Recherche Scientifique, the Institut National de la Santé et de la Recherche Médicale and Sorbonne Université and to JDE by the National Institute of Neurological Disorders and Stroke (NS109668 and NS113955).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
This article does not contain any studies with animals performed by any of the authors. Informed consent was obtained from all participants included in the study.
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Pietrancosta, N., Djibo, M., Daumas, S. et al. Molecular, Structural, Functional, and Pharmacological Sites for Vesicular Glutamate Transporter Regulation. Mol Neurobiol 57, 3118–3142 (2020). https://doi.org/10.1007/s12035-020-01912-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-01912-7