
Abstract
Fatty acid desaturases (FADs) catalyze the introduction of a double bond into acyl chains. Two FAD groups have been identified in plants: acyl-acyl carrier proteins (ACPs) and acyl-lipid or membrane-bound FAD. The former catalyze the conversion of 18:0 to 18:1 and to date have only been identified in plants. The latter are found in eukaryotes and bacteria and are responsible for multiple desaturations. In this study, we identified 82 desaturase gene and protein sequences from 10 grass species deposited in GenBank that were analyzed using bioinformatic approaches. Subcellular localization predictions of desaturase family revealed their localization in plasma membranes, chloroplasts, endoplasmic reticula, and mitochondria. The in silico mapping showed multiple chromosomal locations in most species. Furthermore, the presence of the characteristic histidine domains, the predicted motifs, and the finding of transmembrane regions strongly support the protein functionality. The identification of putative regulatory sites in the promotor and the expression profiles revealed the wide range of pathways in which fatty acid desaturases are involved. This study is an updated survey on desaturases of grasses that provides a comprehensive insight into diversity and evolution. This characterization is a necessary first step before considering these genes as candidates for new biotechnological approaches.
Similar content being viewed by others


Introduction
Polyunsaturated fatty acids (PUFAs) are important components of plant cell membranes, particularly chloroplast and endoplasmic reticulum (ER) membranes. Variations in their composition affect cellular and physiological processes such as cold adaptation, ion channel modifications, pathogen defense, pollen formation, and chloroplast development [1]. Desaturases are the key enzymes in the synthesis of PUFA. Their main role is the activation of the oxygen destined to subsequent modifications of C–H bonds in saturated or monounsaturated carbons. Desaturases are classified as delta or omega according to whether the position of the double bond is numbered relative to the carboxyl or methyl ends, respectively.
Two main fatty acid desaturase (FAD) groups have been described in plants. The first group is represented by soluble acyl–acyl carrier protein (acyl-ACP) desaturases found so far only in plants [2]. Acyl-ACP desaturases introduce the first double bond into the saturated acyl chain; they localize exclusively in plastids where they introduce the double bond into fatty acids that are linked to an acyl carrier protein. In this reaction, the NADPH/ferredoxin system acts as the electron donor. Members of this class are specific for a particular substrate chain length and for specific carbon atoms of the target fatty acid. As an example, delta 9 stearoyl-ACP specifically adds a double bond between carbons 9 and 10 of the stearic acid-ACP form to yield oleic acid. Solubility properties of this class of desaturases have allowed the access to their crystal structure [3]. The second and larger group of plant desaturases, acyl-lipid desaturases, comprises membrane-bound proteins. Most acyl-lipid desaturases consist of 300–350 amino acid residues, being hydrophobic proteins that appear to span membranes four times [4]. No crystal structure is currently available. Membrane-bound FADs are present in the chloroplast and in the ER. In chloroplasts, the formation of the double bond uses the NADPH/ferredoxin system as electron donor, whereas in the ER, this role is accomplished by the NADH/cytochrome b5 system. The acyl-lipid group includes the omega desaturase subfamily that introduces a double bond between an existing one and the methyl end. For instance, omega-3 FAD creates the double bond between the third and fourth carbon from the methyl end of the oleic acid to form linoleic acid. A second subfamily, front-end desaturases (FEDs), is present in the ER and introduces double bonds between a pre-existing double bond and the carboxyl end of unsaturated fatty acids. FEDs predominantly contain an N-terminal cytochrome b5 fused to the main desaturation domain [5]. A third subfamily is constituted by sphingolipid desaturases that also use cytochrome b5 hemoprotein as the electron donor [6].
Fatty acid desaturation is an important step in oil biochemistry and therefore determinant of the quality of vegetable oils [7], thus explaining why the focus of most desaturase studies has long been on oleaginous species such as olive [8] and sunflower [9]. However, changes of the agricultural frontiers together with an increasing demand of crop varieties with enhanced tolerance to environmental stresses have brought renewed interest in desaturases and their role in stress tolerance [10, 11]. It is known that cell membranes are a primary site for cold-induced injury and that cold acclimation includes the activation of mechanisms that protect membrane fluidity. The ability to adjust membrane lipid fluidity by changing the levels of unsaturated fatty acids is a feature of cold-responsive species, and is mainly provided by the regulated activity of FADs [12].
This study presents a complete survey of the information about FAD in grasses available in the GenBank database. Bioinformatic tools were used to characterize sequences and structures of the different FAD groups and to predict expression patterns.
Materials and Methods
Sequence Sources
Genomic and complementary DNA (cDNA) sequences and the corresponding deduced protein of desaturases were retrieved from GenBank (http://www.ncbi.nlm.nih.gov/genbank/) using “desaturase” as keyword in all databases available at this site. The resulting proteins were used as queries in a second round of BLAST search in order to identify additional complete sequences that might have been overlooked in the first search (e-value ≤e−80).
Moreover, Gramene database (http://www.gramene.org/) was interrogated since grass genomes deposited in this platform could provide additional putative desaturases.
In Silico Mapping
Desaturase genes from Oryza sativa, Setaria italica, Bachypodium distachyon, Oryza brachyanta, Zea mays, Hordeum vulgare, and Sorghum bicolor were in silico mapped using the comparative grass genome database available in Gramene (http://ensembl.gramene.org/genome_browser/index.html). Sequences from Triticum aestivum (genomes A, B, and D) were mapped using the Unité de Recherche Génomique Info (URGI) (https://urgi.versailles.inra.fr/blast/blast.php). Sequences from Triticum urartu and Aegilops tauschii were also mapped using URGI considering the ancestry homology with the corresponding A and D genomes of T. aestivum, respectively [13, 14].
Searching for Cis-Regulatory Elements in the T. aestivum Genome
When available, the 1500-bp sequence upstream of the transcription start site of desaturase genes was obtained from the International Wheat Genome Sequencing Consortium (IWGSC) database. In total, seven 5′ regions were retrieved using the cDNA sequences corresponding to ADV59911.1 and ADN59912.1 from T. aestivum and ACG41542.1, NP_001149122.1, XP_008652067.1, NP_001266540.1, and XP_008665998.1 from Z. mays as queries (e-value ≤3e−93) (Supplementary Table 1). These upstream regions were analyzed using the web-based PlantPAN system (http://plantpan.mbc.nctu.edu.tw) that identifies transcription factor (TF) binding sites.
Transcript Profiles
The wheat expression database http://wheat.pw.usda.gov/WheatExp/ allowed us to compare desaturase transcript profiles across a range of tissue samples. This database compiles publicly available RNA-Seq datasets, namely, developmental time course in five tissues (spike, root, leaf, grain, and stem) [15], senescing leaf time course at heading date, 12 and 22 days after anthesis [16], and drought and heat stress (seedlings exposed to 40 °C and 20% PEG-6000 for1 and 6 h) [17]. The WheatExp database was queried by BLAST using the three desaturase sequences HQ589252.1, HQ589253.1, and D84678.1 from T. aestivum (Supplementary Table 1). Matches with the highest identity scores were considered (e-value 0.0). All databases available in WheatExp have been generated from hexaploid wheat (A, B, and D genomes), except for the leaf senescence database that was generated from durum tetraploid wheat (A and B genomes). In the case of the developmental time course data available for each of the five organs, a mean level of expression was calculated over the three progressive stages available. An ad hoc scale was developed based on the number of plus (+) symbols to allow the rapid visualization of differences in levels of expression among tissues and treatments. The expression level was arranged in four categories as follows: <10, 10–20, 20–60, and >100 FPKM/RPKM.
Protein Analyses and Domain Identification
Physicochemical data comprising number of amino acids, molecular weight, and isoelectric point (pI) were obtained with the ProtParam tool (http://web.expasy.org/protparam/) [18]. Then, a bounded set of sequences containing a relatively homogeneous number of proteins from the different desaturase groups was defined. In this subset, protein domains and conserved motifs were searched using Multiple Expectation Maximization for Motif Elicitation (MEME-Suite) 4.11.1 software (http://meme-suite.org) [19] with the following parameters: distribution of motifs: 0 or 1 per sequence, maximum number of motifs: 6, minimum number of sites: 2, minimum motif width: 5, and maximum motif width: 50. The identified motifs were analyzed in their predicted functionality using Pfam server (http://pfam.xfam.org). The transmembrane helix motifs in the acyl-lipid desaturase sequences were predicted using two platforms: the TMHMM 2.0 server (http://www.cbs.dtu.dk/services/TMHMM) based on a hidden Markov model [20] and the SOSUI system (http://harrier.nagahama-i-bio.ac.jp/sosui/) [21]. The subcellular localization of desaturase proteins was estimated with CELLO v.2.5 (subCELlular Localization predictor) server http://cello.life.nctu.edu.tw/ [22].
Molecular Modeling of Acyl-ACP-Desaturases from Wheat, Brachypodium distachyon, and Maize
The protein structural modeling of three acyl-ACP desaturases from T. aestivum (ADV59911.1), B. distachyon (XP_003573524.1), and maize (ACG49052.1) was performed using the interface SwissModel (swissmodel.expasy.org/interactive) with the template (2xZ1.A), a crystal form of Ricinus communis [3] deposited in Protein Data Bank (PDB). Ramachandran plot analysis for the best predicted model was obtained by RAMPAGE server (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php) [23].
Phylogenetic Analysis of Grass Desaturase Proteins
A phylogenetic tree of 20 desaturase proteins was built from a p-distance matrix using the neighbor-joining method available in software MEGA 6.0.6 [24]. The model was tested by the bootstrap method (1000 replications). Positions with gaps or missing data were eliminated by selecting the pairwise deletion option. A zeta-carotene desaturase (T. aestivum ACI04664.1) was included as an outgroup sequence in the phylogenetic tree since it is involved in a different biosynthesis pathway that converts zeta-carotene to lycopene through two consecutive desaturation steps.
Results
Eighty-two DNA FAD sequences of grasses were found in GenBank. They corresponded to A. tauschii (2), B. distachyon (12), H. vulgare (7), Oryza brachyantha (5), O. sativa (7), S. italica (5), S. bicolor (10), T. aestivum (6), T. urartu (2), and Z. mays (26) (Supplementary Table 1). Both soluble acyl-ACP and acyl-lipid desaturases were represented albeit most of them belonged to the second group. In Gramene database, 121 FAD deduced from grass genomes were available. Both groups of desaturases were represented in the following species: A. tauschii (15), B. distachyon (4), H. vulgare (5), O. brachyantha (6), O. barthii (4), O. glaberrima (3), O. glumaepatula (3), O. nivara (3), O. punctata (2), O. sativa Indica (8), O. sativa Japonica (20), S. italica (4), S. bicolor (3), T. aestivum (9), T. urartu (20), and Z. mays (12).
Further analyses were performed using only those sequences from GenBank for which either messenger RNA (mRNA) or cDNA were available. Therefore, sequences automatically deduced from genomic DNA were excluded, excepting seven desaturases that can be distinguished by an ID number that begins with an XP_ format (Supplementary Table 1).
Some incomplete sequences were detected. As an example, maize FAD8 (BAA22440) lacked the first 49 amino acids, and was hence excluded from the analysis. Duplicated DNA or protein sequences bearing different accession numbers were also detected, as was the case of maize delta-12 FAD (BAE93382.1) and FAD2 (ABF50053.1). Similarly, rice FAD8 accession numbers AAW32557.1 and BAE79784.1 corresponded to the same gene sequence that was, respectively, induced by cold or repressed after pathogen infection. In the analysis that followed, duplicated sequences were included only once.
Mapping of Desaturase Genes
In silico mapping of desaturase sequences identified multiple chromosomal sites in ten grass species (Supplementary Table 1). When more than one locus was found in a species, they were spread in several different linkage groups. For instance, corn desaturases mapped in all ten chromosomes of its haploid complement.
In polyploid wheat, desaturase homologous genes could be assigned to specific A, B, or D genomes only when the alignment between the query and the corresponding contig hit was complete (four out of six cases; Supplementary Table 1).
Analysis of Regulatory Gene Regions
The 5′ regulatory region of seven desaturase genes from T. aestivum was analyzed with PlantPAN. In total, 34 different TF binding motifs were detected. The most represented TF families were TCP, AP2, NF-YB, B3, AT-Hook, bZIP, GATA, and MYB. Twelve (35%) of these TF families were found to be involved in abiotic stress response (Plant TFDB; http://planttfdb.cbi.pku.edu.cn) [25] (Supplementary Fig. 1).
Results of PlantPAN scanning analyzed by enzymes showed that bHLH was the most abundant stress-inducible TF binding site in acyl-ACP NP_001151082.1 and acyl-lipid omega-3 desaturases, whereas AP2/ERF was the most abundant one in acyl-ACP ADV59911.0 and ADV59912.1 (stearoyl), and acyl-lipid omega-6 desaturases.
Transcription Analysis
Table 1 summarizes the expression levels of three desaturase genes from common wheat resulting from BLAST against the WheatExp pipeline built on RNA-Seq experiments of T. aestivum and T. turgidum. Acyl-ACP desaturases HQ589252.1 (SAD1) and HQ589253.1 (SAD2) expressed rather evenly in different plant tissues, whereas acyl-lipid omega-3 presented a low expression level in grain samples. Regarding stress conditions, drought and heat treatments did not increase the level of desaturase transcripts but rather kept constant or diminished after stress. Moreover, transcriptional profiles of flag leaf desaturases were not modified during senescence after anthesis. All the homologous copies of HQ589252.1, HQ589253.1, and D84678.1 genes showed similar levels of expression among the A, B, and D genomes, except for the HQ589253.1 gene at genome B that presented a relatively low level of expression in all of the RNA-Seq experiments available in WheatExp.
Sequence Analysis and Subcellular Localization
Amino acid sequences deduced from mRNA sequences of 23 desaturases from T. aestivum, Z. mays, O. sativa, and S. bicolor were obtained. The predicted molecular weight of acyl-ACP desaturases varied between 42.25 and 45.39 kDa (both from Z. mays), while that of acyl-lipid desaturases ranged from 43.68 kDa (T. aestivum) to 51.37 kDa (O. sativa) (Table 2). Nineteen out of 23 theoretical pI values were above 8, suggesting a general basic character for grass desaturases; four relatively acid proteins presented pI values below 7 (Table 2).
Cello prediction tool showed that all acyl-ACP desaturases were localized in the chloroplast, even though SAD1 (ADV59911.1) was also located in mitochondria, with a reliability predictor value of 1.867 (Table 2). Acyl-lipid desaturases were mostly found in the ER, except for FAD7 (BAA22441.1) from Z. mays and FAD7 (BAE79783.1) and FAD8 (AAW32557.1) from O. sativa that were localized in chloroplasts, and cytochrome b5 desaturases, whose predicted localization was in plasma membranes (Table 2). In our study, all desaturases with predicted ER localization contained the motif responsible for retrieving these proteins from other compartments to the ER at the carboxi termini [26], with the exception of ACG28208.1 from Z. mays (Table 2).
Transmembrane Domains in Acyl-Lipid Desaturases
Sequences from a set of the omega desaturase subfamily (16 in total) presented the expected transmembrane domains, although differences in the predicted number were observed depending on the program that was used. Thus, the number of transmembrane domains varied between 1 and 5 with the TMHMM method and between 3 and 6 with the SOSUI method (Supplementary Fig. 2a, b). Most of the proteins presented three transmembrane domains with TMHMM and four to five helices with SOUSI. In total, TMHMM predicted 46 transmembrane helices while SOSUI predicted 66.
Motif Analysis
Six conserved motifs were detected in 18 desaturase protein sequences (e-value ≤1.1e−095; Fig. 1) using de novo motif discovery of MEME-Suite tools. Motifs 1, 2, and 5 were common for all the desaturases analyzed. In acyl-lipid desaturases (Supplementary Table 1), these domains corresponded to the three histidine boxes known to form part of the active site [4] (Fig. 2). The first histidine domain was included in motif 5, the second in motif 2, and the third in motif 1. In the omega-3 group, the first, second, and third histidine boxes were HDCGH, HGWRYSHRTHH, and HVIHH, respectively. In the omega-6/delta-12 group, the first, second, and third histidine boxes were HECGH, HRRHH, and HVXHH, respectively. Finally, in the FED group, the first, second, and third histidine boxes were HDSGH, HNTHH, and QYEHH, respectively.

Motif analysis performed with MEME server in acyl-lipid and acyl-ACP desaturases according to Supplementary Table 1

Logo representation of four desaturase motifs based on 18 protein sequences. Motif 1 includes the third histidine box, motif 2 corresponds to the second histidine box, and motif 5 includes the first histidine box, all of them reported for acyl-lipid desaturases. Motif 4 is present exclusively in acyl-ACP desaturases
Motifs 3 and 6 were only present in the acyl-lipid omega subfamily, whereas motif 4 was exclusive to acyl-ACP desaturases. This last motif contained the box sequence EENRH reported as part of the active site of acyl-ACP desaturases [27]. The other motif previously described for acyl-ACP desaturases with consensus D/EEX2H [27] was not detected by MEME, but it could be recognized by visual inspection and consisted in DEKRH residues in all of the grass acyl-ACP desaturases analyzed. Motif 4 included the EEX2H sequence reported as part of the active site of acyl-ACP.
When motifs 1 and 2 were submitted to the Pfam database, hits with PF00487 were found, whereas motif 4 matched with PF03405, conserved domains associated with FAD-1 and 2, respectively. Motif 6 corresponded to a domain with unknown function (DUF). No Pfam-A matches were found for motifs 3 and 5. The acyl-lipid group information obtained with MEME motif analysis and TMHMM transmembrane domains was integrated. The resulting protein topology corresponded to a Cin-Nin structure (Fig. 3).

Homology Modeling
The molecular model of the three acyl-ACP desaturases from T. aestivum, B. distachyon, and Z. mays consisted equally of a domain of 11 α-helices (Fig. 4). In model validation, the Ramachandran plot analysis by using the RAMPAGE server revealed that >90% were in favored regions, indicating that the models were in fairly good quality.

Homology modeling of T. aestivum stearoyl-ACP desaturase (ADV59911.1) using the template of Ricinus communis performed with the SwissModel server. Sequence boxes associated with the active site, the amino, and carboxi termini are indicated
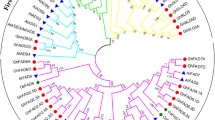
Phylogenetic Analysis
To evaluate the relationships among FAD proteins in different grass species, a phylogenetic tree was constructed according to the alignments of 20 full-length protein sequences belonging to acyl-ACP and acyl-lipid desaturases of seven grass species (Supplementary Fig. 3).
According to the tree diagram, four main groups (I–IV) with high bootstrap values, mostly 100, could be recognized. The first, second, and third main groups contained acyl-lipid desaturases and the fourth group included acyl-ACP desaturases. Group I included subgroups Ia and Ib, corresponding to omega-3 and omega-6/delta-12 desaturases, respectively. Group II included the sphingolipid class, and group III was composed of FEDs. Zeta-carotene desaturase was external and close to the acyl-ACP group.
Discussion
GenBank interrogation retrieved information on desaturases from a total of ten species belonging to the grass family Poaceae. Of these, Z. mays was the most represented species, most likely because of the importance of corn as an oleaginous crop. More recent large-scale genome sequencing has provided new information about many proteins and is available in another public database, Gramene.
The description of their structural conformation and the mechanisms by which gene expression is controlled constitute necessary information to hypothesize about potential roles and functions. All known desaturases are characterized by the presence of histidine domains localized at highly conserved positions that are believed to be responsible for di-iron binding at the catalytic center [5, 27].
The conserved occurrence of histidine domains in grass desaturases plays an essential role in enzyme functionality. The results obtained with MEME confirmed the presence of three histidine domains in acyl-lipid desaturases [2, 4] in motifs 1, 2, and 5. In the FED group in particular, the third consensus histidine domain has been defined as H/QX2-3HH [28]. In all of the FEDs analyzed in this study, the first residue was a glutamine Q instead of H, which is crucial for enzyme activity. In relation to the acyl-ACP group, we found two consensus-binding boxes differing from those found in acyl-lipid desaturases that have been associated to the active site [27]. Although motifs 1, 2, and 5 were detected by MEME in both acyl-lipid and acyl-ACP desaturases, the latter lacked the typical histidine clusters, suggesting that one domain detected by MEME across a group of structurally heterogeneous proteins could even contain important differences in sequences. Thus, the equivalence of motifs should be considered with caution.
Fatty acids in plants are synthesized in plastids and desaturated in plastids or in the ER, with acyl-ACP proteins associated to the chloroplast and acyl-lipid desaturases associated to both ER [29] and chloroplasts [30]. Our results are in line with these previous studies, although additional locations were predicted for one acyl-ACP desaturase in mitochondria and two acyl-lipid FEDs in plasma membranes. Although the presence of desaturases in mitochondria has been suggested in animal cells [31], to our knowledge, there is no previous report about plant desaturases in the mitochondrial compartment. Meanwhile, some acyl-lipid desaturases have been found associated to plasma membranes in addition to their typical localization in the reticulum [32].
Acyl-lipid desaturases from reticulum present characteristic sequence motifs at their carboxi termini which are involved in the retrieval of the escaped membrane proteins back to the ER [33] (Table 3). Two different types of motifs have been described, one rich in aromatic amino acids with a general FWYNN/S/KKF structure and a more variable motif with two conserved lysines described as dilysine ER targeting signals [26]. Within the set of desaturases localized in the ER, we found that the enzymes responsible for adding a double bond at the C6-C7 site relative to the methyl end (i.e., omega-6) had the former motif, whereas those involved in C3-C4 site desaturation (i.e., omega-3) possessed the dilysine motif. One exception was ACG28208.1 from Z. mays, an omega-3 for which Cello predictions included both ER and chloroplast compartments although they lacked the typical ER retrieval motif. This feature could be some evidence that the chloroplast was the most likely location for ACG28208.1.
The in silico mapping of grass desaturase genes showed a number of hit sites in chromosomes ranging from one in Aegilops tauchii to ten in Z. mays, but it should be noted that this range in site numbers is directly affected by the number of sequences we were able to retrieve from NCBI for each species. Our results are in accordance with those observed in diploid Gossypium raimondii, where the loci of 19 acyl-lipid desaturases spread to 11 of their 13 chromosomes in the haploid complement [34].
The localization of FAD2 (BAE93382.1) from Z. mays was analyzed in more detail because the outcome of BLAST against Gramene consisted in two positions on chromosome 5 (77,169 bp apart) and an additional position on chromosome 4, all three with high and confident alignment scores and identity percentage. The two contiguous positions in chromosome 5 were interpreted as the products of known mechanisms operating in genome evolution such as tandem direct duplications often mediated by transposons [35]. On the other hand, more than one copy of a given gene map in different chromosomes has been frequently reported for Z. mays [36] and attributed to an ancient duplication polyploidy event with subsequent evolution during which the extra copies of each gene were unevenly retained or lost.
Regarding the protein secondary structure, the availability of helix predictors takes on special relevance in proteins associated with membranes, since their crystal structure is less frequently available due to technical difficulties to obtain large quantities of purified proteins. We observed that the number of predicted transmembrane helices varied depending on the method considered [20, 21], with a tendency to detect more helices with SOSUI than with TMHMM (Supplementary Material Fig. 2). This difference may be due to the fact that SOSUI predicts transmembrane helices assuming the presence of hydrophobic primary transmembrane helices and hydrophilic secondary ones, whereas TMHMM also considers the alternation of cytoplasmic and non-cytoplasmic loops. Although the number of transmembrane helices based on hydrophobicity in each residue is variable [37], the most frequent topology assumes that the enzyme spans the membrane four times [3].
In silico analysis of expression using RNA-Seq data revealed differences among desaturase genes and tissue or treatment (Table 1). For instance, when the two acyl-ACP desaturases are compared, HQ58952.1 gene maintained a higher level of transcription than HQ58953.1 under stress conditions and during leaf senescence. Further, the expression of the former was relatively stable across a wide range of organs and growing conditions, reflecting a constitutive mode of gene expression. We detected some differences among homologous copies, in particular, a lower expression of HQ589253.1 located in genome B relative to those in genomes A and D. In contrast, D84678.1 showed a similar level of expression in all the three homologous copies. The relative expression levels of homologous genes in polyploid wheat frequently differ, causing genome-specific transcription profiles [38]. This differential gene regulation has been associated with mechanisms of dosage compensation operating in the allopolyploid species and with intergenomic interactions [39].
Most desaturase transcripts obtained from the WheatExp database did not show induction under drought and heat stress conditions. Contrarily, a decreased expression was observed in four out of nine cases when comparing stress treatments with the control (Table 1), in agreement with the observed decrease in unsaturation activity in plants at elevated temperatures [12]. It is worth mentioning that translational regulation can also operate to modulate enzyme accumulation during the response to temperature changes [40].
Although the expression patterns of desaturases did not change during leaf senescence, differences were observed among enzyme genes. Within acyl-ACP desaturases, SAD1 constantly expressed at high levels, whereas SAD2 showed a minimum expression. These observations may reflect different roles of these two enzymes in the chloroplast stroma. Both are constitutively involved in stearic acid desaturation to yield oleic acid, but some degree of subfunctionality could be expected between SAD1 and SAD2 based on differences in acidity character and subcellular localization (Table 2). To our knowledge, the role of desaturases in grass senescence has not been largely evaluated. In this sense, a delta 9 acyl-lipid desaturase of rose flowers has been identified as a senescence-inducible gene [41].
Gene expression patterns can be predicted by finding specific motifs in promoters. PlantPAN scanning of the desaturase 5′ region in wheat genome detected binding sites for diverse TF families, confirming that desaturases are able to respond to different environmental and physiological factors. The most abundant binding sites corresponded to the TEOSINTE-BRANCHED1/CYCLOIDEA/PCF (TCP) TF family, varying from 235 in wheat omega-6 desaturase homologous to NP_001149122.1 from Z. mays to 97 in wheat omega-3 desaturase homologous to XP_008665998.1 from Z. mays. This TF family is exclusive to higher plants and plays versatile functions in multiple aspects of plant growth and development [42].
It has been reported [43, 44] that when plants are exposed to low temperatures, there are changes in desaturase expression that correlate with cold tolerance. In agreement with this, we identified numerous TF binding sites at the desaturase 5′ regions related with the response to low temperatures, such as families MYB, NAC, and AP2/ERF [45,46,47]. Desaturase expression is involved in several other forms of abiotic stress. Among the TF associated with abiotic stress response, we found that bHLH, a group of regulatory proteins modulated by salt or drought, was another well-represented family [48].
Motif analysis in promoter sequences suggested that desaturase gene expression could be regulated under osmotic stress by at least 12 different TF families. Computational methods able to detect TF binding sites at the regulatory regions of a gene have provided essential information to functional analysis and to the building of models for transcriptional regulatory networks [49].
The evolutionary pathway of FADs could be inferred by combining the biochemical role with the taxonomical position in species-wide samples [4, 37]. Based on the amino acid sequences, we confirmed that acyl-ACP and acyl-lipid proteins constitute two groups of unrelated desaturases despite some similarities regarding cofactor use and spatial arrangement during hydrogen removal [27].
Within acyl-lipid or membrane proteins, a common ancestor has been suggested for omega-3 and omega-6/delta-12 desaturases (Supplementary Fig. 3, group Ia and Ib) after analyzing almost 400 desaturases from different eukaryotic species [39], a point that is also evidenced by the tree topology of our study. Sphingolipid desaturases (group II) and FEDs (group III) appear more distantly positioned despite the fact that all the enzymes included in groups I, II, and III share the functional role of adding a double bond in a pre-existing unsaturated chain. FED clustering can be explained by common characteristics, such as the presence of N-terminal cytochrome b5, an intronless gene structure [6] and a glutamine as the first residue at the third histidine box. All these shared features support a common origin of grass FEDs.
The wide availability of bioinformatic tools and public sequence databases open the possibility to plan experiments and to make statistically supported predictions of the structure and function of genes and proteins. However, it is essential to have an integrated insight between the in silico predictions and the biological context as presented in this study. The dynamic structure of the genomes could also be interpreted through the in silico mapping. The identification of structural and functional patterns, as well as the variations, is an essential starting point to identify targets for gene manipulation in future experiments.
Conclusion
Currently, it is estimated that more than 10,000 grass species (Poaceae) exist, distributed in 700 genera grouped in 30 tribes. In this study, sequences of desaturase genes from eight different genera representative of six tribes were included, compiled, and described. Protein genes were assigned to specific functional groups that distinguish the enzymes according to the position and number of double-bond insertions on the acyl chain and subcellular localization. The patterns of expression of some wheat desaturases could be retrieved, and putative regulatory TF binding sites were identified. Consensus motifs in the active site previously obtained from a wide range of taxa could be specifically defined for grass genes. The characterization of desaturases takes on relevance due to their role in the response to different environmental conditions and in oil quality or nutritional level. This characterization is a necessary first step before considering these genes as candidate for new biotechnological approaches.
References
Wallis, J., Watts, J., & Browse, J. (2002). Polyunsaturated fatty acid synthesis: what will they think of next? Trends in Biochemical Sciences, 27, 467–473.
Murata, N., & Wada, H. (1995). Acyl-lipid desaturases and their importance in the tolerance and acclimatization to cold of cyanobacteria. The Biochemical Journal, 308, 1–8.
Lindqvist, Y., Huang, W., Schneider, G., & Shanklin, J. (1996). Crystal structure of Δ9 stearoyl-acyl carrier protein desaturase from castor seed and its relationship to other di-iron proteins. The EMBO Journal, 15, 4081–4092.
Los, D., & Murata, N. (1998). Structure and expression of fatty acid desaturases. Biochimica et Biophysica Acta, 1394, 3–15.
López Alonso, D., García-Maroto, F., Rodríguez-Ruiz, J., Garrido, J., & Vilches, M. (2003). Evolution of the membrane-bound fatty acid desaturases. Biochemical Systematics and Ecology, 31, 1111–1124.
Sperling, P., Zahringer, U., & Heinz, E. (1998). A sphingolipid desaturase from higher plants. Identification of a new cytochrome b5 fusion protein. The Journal of Biological Chemistry, 273, 28,590–28,596.
Mikkilineni, V., & Rocheford, T. (2003). Sequence variation and genomic organization of fatty acid desaturase-2 (fad2) and fatty acid desaturase-6 (fad6) cDNAs in maize. Theoretical and Applied Genetics, 106, 1326–1332.
Hernández, M., Mancha, M., & Martínez-Rivas, J. M. (2005). Molecular cloning and characterization of genes encoding two microsomal oleate desaturases (FAD2) from olive. Phytochemistry, 66, 1417–1426.
Rousselin, P., Hahne, G., Prieto-Dapena, P., & Jordano, J. (2002). Modification of sunflower oil quality by seed-specific expression of a heterologous Δ9-stearoyl-(acyl carrier protein) desaturase gene. Plant Breeding, 121, 108–116.
Matteucci, M., D'Angeli, S., Errico, S., Lamanna, R., Perrotta, G., & Altamura, M. M. (2011). Cold affects the transcription of fatty acid desaturases and oil quality in the fruit of Olea europaea L. genotypes with different cold hardiness. Journal of Experimental Botany, 62, 3403–3420.
Karabudak, T., Bor, M., Ozdemir, F., & Turkan, I. (2014). Glycine betaine protects tomato (Solanum lycopersicum) plants at low temperature by inducing fatty acid desaturase7 and lipoxygenase gene expression. Molecular Biology Reports, 41, 1401–1410.
Upchurch, R. (2008). Fatty acid unsaturation, mobilization, and regulation in the response of plants to stress. Biotechnology Letters, 30, 967–977.
Ling, H., Zhao, S., Liu, D., Wang, J., Sun, H., Zhang, C., et al. (2013). Draft genome of the wheat A-genome progenitor Triticum urartu. Nature, 496, 87–90.
Jia, J., Zhao, S., Kong, X., Li, Y., Zhao, G., He, W., et al. (2013). Aegilops tauschii draft genome sequence reveals a gene repertoire for wheat adaptation. Nature, 496, 91–95.
Choulet, F., Alberti, A., Theil, S., Glover, N., Barbe, V., Daron, J., Pingault, L., et al. (2014). Structural and functional partitioning of bread wheat chromosome 3B. Science, 345, 1249721.
Pearce, S., Tabbita, F., Cantu, D., Buffalo, V., Avni, R., Vazquez-Gross, H., Zhao, R., Conley, C., Distelfeld, A., & Dubcovksy, J. (2014). Regulation of Zn and Fe transporters by the GPC1 gene during early wheat monocarpic senescence. BMC Plant Biology, 14, 368. doi:10.1186/s12870-014-0368-2.
Liu, Z., Xin, M., Qin, J., Peng, H., Ni, Z., Yao, Y., & Sun, Q. (2015). Temporal transcriptome profiling reveals expression partitioning of homologous genes contributing to heat and drought acclimation in wheat (Triticum aestivum L.) BMC Plant Biology, 15, 152. doi:10.1186/s12870-015-0511-8.
Gasteiger, E., Hoogland, C., Gattiker, A., Duvaud, S., Wilkins, M., Appel, R., & Bairoch A. (2005). Protein identification and analysis tools on the ExPASy server. In: Walker J. M. (ed.), The proteomics protocols handbook (pp. 571–607). Humana.
Bailey, T., Williams, N., Misleh, C., & Li, W. (2006). MEME: discovering and analyzing DNA and protein sequence motifs. Nucleic Acids Research, 34(Web Server issue), W369–W373. doi:10.1093/nar/gkl198.
Krogh, A., von Heijne, B. L., & Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Journal of Molecular Biology, 305, 567–580.
Hirokawa, T., Boon-Chieng, S., & Mitaku, S. (1998). SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics, 14, 378–379.
Yu, C., Chen, Y., Lu, C., & Hwang, J. (2006). Prediction of protein subcellular localization. Proteins, 64, 643–651.
Lovell, S., Davis, I., Arendall, W., Bakker, P., Word, J., Prisant, M., Richardson, J., & Richardson, D. C. (2003). Structure Validation by Cα Geometry: φ, ψ and Cβ Deviation. Proteins: Structure, Function, and Genetics, 50, 437–450.
Tamura, K., Stecher, G., Peterson, D., Filipski, A., & Kumar, S. (2013). MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Molecular Biology and Evolution, 30, 2725–2729.
Jin, J., Tian, F., Yang, D. C., Meng, Y. Q., Kong, L., Luo, J. C., & Gao, G. (2016). PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Research. doi:10.1093/nar/gkw982.
Mc Cartney, A., Dyer, J., Dhanoa, P., Kim, P., Andrews, D., McNew, J., & Mullen, R. (2004). Membrane-bound fatty acid desaturase are inserted co-translationally into the ER and contain different ER retrieval motif at the carboxi termini. The Plant Journal, 37, 156–173.
Shanklin, J., & Cahoon, E. (1998). Desaturation and related modifications of fatty acids. Annual Review of Plant Physiology and Plant Molecular Biology, 49, 611–641.
Sayanova, O., Smith, M. A., Lapinskas, P., Stobart, A. K., Dobson, G., Christie, W. W., Shewry, P. R., & Napier, J. A. (1997). Expression of aborage desaturase cDNA containing an N-terminal cytochromeb5 domain results in the accumulation of high levels of delta6-desaturated fatty acids in transgenic tobacco. Proceedings of the National Academy of Sciences of the United States of America, 94, 4211–4216.
Dyer, J. M., & Mullen, R. T. (2001). Immunocytological localization of two plant fatty acid desaturases in the endoplasmic reticulum. FEBS Letters, 494, 44–47.
Ferro, M., Salvi, D., Brugiere, S., Miras, S., & Kowalski, S. (2003). Proteomics of the chloroplast envelope membranes from Arabidopsis thaliana. Molecular & Cellular Proteomics, 2, 325–345.
Beauchamp, E., Tekpli, X., Marteil, G., Lagadic-Gossmann, D., Legrand, P., & Rioux, V. (2009). N-myristoylation targets dihydroceramide delta4-desaturase 1 to mitochondria: partial involvement in the apoptotic effect of myristic acid. Biochimie, 91, 1411–1419.
Todorova, R. (2008). Expression and localization of FAD2 desaturase from spinach in tobacco cells. Russian Journal of Plant Physiology, 55, 513–520.
Pagny, S., Lerouge, P., Faye, L., & Gomord, V. (1999). Signals and mechanisms for protein retention in the endoplasmic reticulum. Journal of Experimental Botany, 50, 157–164.
Liu, W., Li, W., He, Q., Khan Daud, M., Chen, J., & Zhu, S. (2015). Characterization of 19 genes encoding membrane-bound fatty acid desaturases and their expression profiles in Gossypium raimondii under low temperature. PloS One. doi:10.1371/journal.pone.0123281.
Zhang, J., Zuo, T., & Peterson, T. (2013). Generation of tandem direct duplications by reversed ends transposition of maize ac elements. PLoS Genetics. doi:10.1371/journal.pgen.1003691.
Schnable, J. C. (2015). Genome evolution in maize: from genomes back to genes. Annual Review of Plant Biology, 66, 329–343.
Sperling, P., Ternes, P., Zank, T. K., & Heinz, E. (2003). The evolution of desaturases. Prostaglandins, Leukotrienes and Essential Fatty Acids, 68, 73–95.
Basualdo, J., Díaz, M. L., Cuppari, S., Cardone, S., Soresi, D., Pérez, C. G., & Carrera, A. (2015). Allelic variation and differential expression of VRN-A1 in durum wheat genotypes varying in the vernalization response. Plant Breeding, 134, 520–528.
Feldman, M., & Levy, A. A. (2012). Genome evolution due to allopolyploidization in wheat. Genetics, 192, 763–774.
Horiguchi, G., Horiguchi, T., Takuichi, F., Naoto, K., & Hiroaki, K. (2000). Temperature-dependent translational regulation of the ER w-3 fatty acid desaturase gene in wheat root tips. The Plant Journal, 24, 805–813.
Fukuchi-Mizutani, M., Savin, K., Cornish, E., Tanaka, Y., Ashikari, T., Kusumi, T., & Murata, N. (1995). Senescence-induced expression of a homologue of delta 9 desaturase in rose petals. Plant Molecular Biology, 29, 627–635.
Ma, J., Liu, F., Wang, Q., Wang, K., Jones, D., & Zhang, B. (2016). Comprehensive analysis of TCP transcription factors and their expression during cotton (Gossypium arboreum) fiber early development. Scientific Reports. doi:10.1038/srep21535.
Domínguez, T., Hernández, L., Pennycooke, J., Jiménez, P., Martínez Riva, J. M., Sanz, C., Stockinger, E., Sánchez-Serrano, J., & Sanmartín, M. (2010). Increasing ω-3 desaturase expression in tomato results in altered aroma profile and enhanced resistance to cold stress. Plant Physiology, 153, 655–665.
Shi, J., Cao, Y., Fan, X., Li, M., Wang, Y., & Ming, F. (2012). A rice microsomal delta-12 fatty acid desaturase can enhance resistance to cold stress in yeast and Oryza sativa. Molecular Breeding, 29, 743–757.
Agarwal, M., Hao, Y., Kapoor, A., Dong, C. H., Fuji, H., Zheng, X., & Zhu, J. K. (2006). A R2R3 type MYB transcription factor is involved in the cold regulation of CBF genes and in acquired freezing tolerance. Journal of Biological Chemistry, 281, 37,636–37,645.
Xia, N., Zhang, G., Liu, X.-Y., Deng, L., Cai, G.-L., Zhang, Y., Wang, X.-J., Zhao, J., Huang, L.-L., & Kang, Z. S. (2010). Characterization of a novel wheat NAC transcription factor gene involved in defense response against stripe rust pathogen infection and abiotic stresses. Molecular Biology Reports, 37, 3703–3712.
Liu, Q., Kasuga, M., Sakuma, Y., Abe, H., Miura, S., Goda, H., Shimada, Y., Yoshida, S., Shinozaki, K., & Yamaguchi-Shinozaki, K. (1998). Two transcription factors, DREB1 and DREB2, with an EREBP/AP2 DNA binding domain separate two cellular signal transduction pathways in drought- and low-temperature-responsive gene expression, respectively, in Arabidopsis. The Plant Cell, 10, 391–406.
Abe, H., Urao, T., Ito, T., Seki, M., Shinozaki, K., & Yamaguchi-Shinozakia, K. (2003). Arabidopsis AtMYC2 (bHLH) and AtMYB2 (MYB) function as transcriptional activators in abscisic acid signaling. The Plant Cell, 15, 63–78.
Blais, A., & Dynlacht, B. (2005). Constructing transcriptional regulatory networks. Genes & Development, 19, 1499–1511.
Author information
Authors and Affiliations
Corresponding author



Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Díaz, M.L., Cuppari, S., Soresi, D. et al. In Silico Analysis of Fatty Acid Desaturase Genes and Proteins in Grasses. Appl Biochem Biotechnol 184, 484–499 (2018). https://doi.org/10.1007/s12010-017-2556-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-017-2556-7