
Opinion statement
The rise of immunotherapy is the greatest advance in oncology to occur over the last several years, but applications in gynecologic malignancies lag behind other tumors. The term “immunotherapy” envelops monoclonal antibodies as receptor mediators, including immune checkpoint inhibitors (ICPI), cancer vaccines, and adoptive immunotherapies alone or in combination with other therapeutic approaches. The purpose of this review is to summarize the status of immunotherapy trials in ovarian cancer and to specifically highlight data published in the last 1–2 years.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ovarian cancer is the fifth most common cause of cancer-related deaths in women, surpassing that of any other gynecological cancer. In the USA, an estimated 22,240 new cases of ovarian cancer will be diagnosed in 2018, and an estimated 14,070 women will succumb to the disease in this year alone [1]. In addition, there are more than 200,000 new cases of ovarian cancer and more than 150,000 deaths per year worldwide [2]. Initial treatment options for epithelial ovarian cancer (EOC) include cytoreductive surgery followed by platinum and taxane chemotherapy [3, 4] or neoadjuvant chemotherapy followed by interval debulking surgery [5, 6]. Due to the late stage at time of diagnosis (80%) [7], a high rate of recurrence (70–80%), and few treatment options for patients who develop resistance to frontline therapies, prognosis for ovarian cancer is poor, and the overall 5-year survival rate is only 47.4%. Thus, there is a great need to improve therapies available for ovarian cancer patients, particularly for patients with platinum-resistant disease.
Immunotherapy has shown promise in other cancers such as melanoma, bladder, lung, leukemia, and breast [8,9,10]. The tumor immune environment, such as the presence of CD3+ tumor-infiltrating T cells and intraepithelial CD8+ tumor-infiltrating lymphocytes (TILs), correlates with survival and progression in ovarian cancer [8, 11,12,14], indicating a role for modulation of the immune system in this disease site is possible. Current immunotherapeutic strategies for ovarian cancer consist mainly of (1) monoclonal antibodies as receptor mediators, including immune checkpoint inhibitors (ICPI); (2) cancer vaccines; or (3) adoptive immunotherapies alone or in combination with other approaches.
Immune checkpoint inhibitors for ovarian cancer
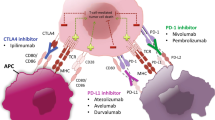
ICPIs impede the ability of the tumor to activate checkpoint proteins on the surface of T cells, thereby preventing the cancer from evading immune response and allowing the immune system to generate an antitumor response [15, 16]. Immune regulatory checkpoints are essential for cultivating peripheral tolerance and prevention of autoimmunity. Among the most well understood are cytotoxic lymphocyte-associated antigen 4 (CTLA-4) and programmed death receptor (PD-1). CTLA-4 abrogates autoreactive T cells in lymph nodes early in T cell activation, while PD-1 regulates previously activated T cells in non-lymphatic tissues later in the immune response and controls apoptosis of regulatory T cells [17, 18]. PD-1 expression occurs in instances of high T cell stimulation, such as cancer. Binding of PD-1 to its antigens PD-L1 and PD-L2 inhibits T cell proliferation/survival, via changes in interferon (IFN)-gamma, tumor necrosis factor-alpha, and IL-2, as well as phosphorylation downstream of the TCR, and increases in regulatory T cells [19]. Blocking these immune checkpoints with monoclonal antibodies is an attractive strategy [20].
A variety of ICPCs have gained FDA approval, yet the study of ICPIs in gynecologic cancers lags behind other disease sites. Nivolumab (Bristol-Meyers-Squibb) and pembrolizumab (Merck) target PD1. Both are IgG4 complexes, but differ subtly in binding site (N-loop versus CD-loop) and affinity (Kd = 3.06 pM versus 29 pM) [21]. Nivolumab has indications in melanoma, lung, renal cell, Hodgkin’s lymphoma, hepatocellular carcinoma, head and neck, urothelial, and MSI-high colorectal carcinomas. Pembrolizumab has indications in melanoma, lung, head and neck, Hodgkin’s lymphoma, PD-L1-expressing cervical, or gastric cancers and is notable as the first drug to gain site agnostic approval for tumors that exhibit microsatellite instability. Ipilimumab (Bristol-Meyers-Squibb; targets CTLA-4, with indications in melanoma, urothelial, and lung cancers. Atezolizumab (Genentech-Roche), darvulamab (Astra-Zeneca), and avelumab (EMD-Serono) target PD-L1 and are approved for urothelial/lung cancers, bladder/lung, and for lung/Merckel cell cancers, respectively.
Multiple early-phase clinical trials are ongoing to examine ICPIs in ovarian cancer (Table 1). Preliminary results from the JAVELIN Solid Tumor [22••] clinical trial of avelumab (anti-PD-L1) in 124 patients with refractory/recurrent ovarian cancer found an overall response rate (ORR) of 9.7%; stable disease (SD) was observed in 55 patients (44.4%), yielding a DCR of 54.0%. Only 6.5% of patients experienced a grade 3/4 treatment-related adverse event (AE). PD-L1 expression was present in 77% of patients; ORR was 12.3% in PDL-1+ versus 5.9% in PD-L1- patients. Preliminary results from a trial of pembrolizumab in 16 patients with PD-L1+ advanced ovarian cancer reported an ORR of 11.5%, with 1 patient with a complete response (CR), 2 with partial responses (PR), and 6 with SD. Progression-free survival (PFS) was reported as 1.9 months and overall survival (OS) as 13.1 months, with 73.1% of patients experiencing an AE but only 1 patient with a grade 3 treatment-related AE. A phase II study of ipilimumab in 40 patients with recurrent platinum-sensitive ovarian cancer reported a best overall response rate (BORR) of 10.3% by RECIST 1.1 (n = 39); however, 50% (20/40) of the patients experienced a grade 3 or higher treatment-related AE [23•]. Toxicities related to ICPIs most commonly include skin (rash, pruritus), gastrointestinal tract (diarrhea), infusion-related reactions, and fatigue, which may limit the clinical effectiveness of this class of drugs [24, 25].
There is immense interest in ICPIs in combination with poly-ADP-ribose polymerase inhibitors (PARPi) or vascular endothelial growth factor receptor (VEGF) inhibitors, including both monoclonal antibodies (e.g., bevacizumab) and small molecule inhibitors (e.g., cedirinib). There are currently numerous ongoing early-phase clinical trials focused on ICPIs in combination with these agents [26,26,27,28,30] (Table 1). A phase I study of durvalumab in combination with olaparib or cediranib in 26 patients with women’s cancers found an 83% disease control rate (DCR) in patients who received durvalumab plus olaparib and a 75% DCR and 50% response rate in patients who received durvalumab plus cediranib [31]. IMagyn050 is an ongoing phase III clinical trial of atezolizumab (anti-PD-L1) versus placebo with standard frontline chemotherapeutics and bevacizumab on yet untreated patient with advanced EOC and fallopian or primary peritoneal cancers (NCT03038100) [32].
Biomarkers to predict response to ICPI in ovarian cancer must be validated. The definition of PD-L1 “positivity” varies greatly. Microsatellite instability (MSI) leading to high mutational burden was first shown to be predictive of response to ICPIs in colorectal carcinomas [33]. Rates of MSI may vary by histologic subtype of ovarian cancer. One group identified a subset of ovarian clear cell carcinomas, typically characterized by worse prognosis compared to other histological subtypes, associated with increased MSI and increased tumor immunogenicity [34]. BRCA1/2 mutations may also be associated with increased mutation load, immunogenicity, and greater expression of PD-1/PD-L1, indicating that these patients may respond well to ICPIs [35]. Others have suggested that whole exome sequencing may also identify disruptions of the PD-L1 gene, signifying that genetic characteristics may, one day, be used to identify ICPI responsiveness [36]. Such observations underscore the need for further study of the effects of genomic instability, homologous recombination status, or BRCA 1/2 mutations on ICPI outcome [29, 37, 38].
Vaccination strategies for ovarian cancer
The first attempt to use vaccination to stimulate immune function occurred in 1891 when William Coley injected Streptococcus pyogenes and Serratia marcescens intra-tumorally, after observing regression of sarcoma in a patient with erysipelas [39]. Modern cancer vaccines consist of autologous whole cells, dendritic cells loaded with autologous tumor-specific antigens, or plasmids that result in antigen expression and immune activation after repeated exposure. Each vaccination strategy may be further augmented by a variety of immune-modulatory agents.
Engineered autologous tumor cell immunotherapy represents an elegant approach to restoration of T cell effector function. This may involve transfection of autologous irradiated tumor cells with a plasmid encoding for the upregulation of immunostimulants and downregulation of immunosuppressants. One notable example is Vigil® (Gradalis, Carrollton, TX), containing a proprietary plasmid leading to up regulation of granulocyte-monocyte colony stimulating factor (GM-CSF) and silencing of furin, an enzyme that activates transforming growth factor (TGF)-ß1/2. Expression of GM-CSF stimulates antigen presentation; inhibition of TGF-ß1/2 allows antigen-presenting cells to recognize tumor antigens. Early studies showed that response to vaccination (as manifested by positivity in gamma-interferon) using ≥ 107 cells with the construct for up to 12 monthly injections in solid tumors correlated with survival (25.7 versus 11.6 months) [40]. In 2017, similar immune results were found using a 1-log lower-dose formulation in a study of 15 patients, including 40% with heavily pre-treated ovarian cancer [41], which suggests smaller tumor samples are required for initial engineering. Additional studies of this technology as maintenance therapy remain in progress (NCT02346747, NCT01309230) [42, 43].
An antigen of recent interest in gynecologic malignancies is folate receptor (FR)-α (also known as folate-binding protein, FBP), as it is highly overexpressed in ovarian and type 2 endometrial cancers, coinciding with increased DNA production and turnover. Jackson and colleagues (2017) recently published results of a phase I/IIa trial employing E39 (GALE 301), an HLA-A2 restricted, FRα peptide in women without evidence of disease following treatment of initial disease (n = 24 with 16 controls) or recurrence (n = 5 with 6 controls). A total of 6 monthly injections (100, 500, or 1000 μg) with granulocyte macrophage-colony stimulating factor (GM-CSF) were provided. Delayed-type hypersensitivity increased in a dose-dependent fashion (13.3% vs 55% CG, p = 0.01). Estimated 2-year disease-free survival (DFS) was 85.7% in the 1000 μg group vs 33.6% in controls (p = 0.021). A subsequent phase Ib study of breast and ovarian cancer patients evaluated six serial immunizations using E39 and an attenuated variant (E39’) (E39 × 6, versus E39 × 3 then E39’ × 3, E39’ × 3 then E39 × 3). This study suggested benefit to incorporation of attenuated vaccine with no new safety signals [44]. Immune response was successfully generated without significant AEs in 90% of patients in a related phase I trial of 22 patients with a history of ovarian cancer or breast cancer and no evidence of disease who were treated with metronomic cyclophosphamide then vaccinated 6 times monthly [45••]. While this provides biologic plausibility, there is presently no estimate on clinical efficacy of this approach.
Of note, FRα is also the target for mirvetuximab soravtansine (IMGN853), a humanized monoclonal antibody attached via a disulfide-containing linker to the cytotoxic maytansinoid, DM4. Once released within the target cell, DM4 acts as an anti-mitotic agent that inhibits tubulin polymerization and microtubule assembly, resulting in cell cycle arrest and apoptosis. A phase III study of mirvetuximab soravtansine versus investigator’s choice chemotherapy (paclitaxel, pegylated liposomal doxorubicin, or topotecan) in platinum-resistant ovarian cancer is currently ongoing (FORWARD1, NCT02631876) [46]. Mirvetuximab soravtansine is not independently cytotoxic to FRα-negative endometrial cells in vitro; however, bystander killing on FRα-negative cells in co-culture with FRα-positive cells does occur [47]. This effect is presumably secondary to diffusion of metabolites to adjacent cells following lysosomal processing post-internalization [48]. Thus, antibody-drug conjugation may offer prominent advantages relative to other FRα-targeting strategies including farletuzumab (MORAb-003; Morphotek, Inc., Exton, PA), which relies only upon antibody-dependent cellular and complement-dependent cytotoxicity.
Other tumor-associated antigens (TAA) of interest in ovarian cancer belong to cancer-testis family and p53, the latter of which is nearly universally mutated in ovarian cancers [49]. Vaccination strategies targeting cancer-testis antigen (e.g., MAGE, NY-ESO) are reviewed extensively elsewhere [50]. Despite much early excitement surrounding various vaccination strategies targeting various TAAs, phase III trials have historically been disappointing. One notable example is vaccination with anti-idiotypic antibodies against glycoprotein CA-125, an FDA-approved serum biomarker to monitor clinical response to therapy in ovarian cancer, which failed at the phase III level for reasons still not fully understood, but which remain under active investigation [51]. Recently, a phase I trial of a modified vaccinia ankara vaccine delivering wild-type human p53 (p53MVA) in combination with gemcitabine chemotherapy in patients with platinum-resistant ovarian cancer showed longer PFS in patients with p53 reactive CD4+/CD8+ T cells but unacceptable toxicity with chemotherapy administration [52•]. These studies serve as a humbling reminder of the complexity of the immune response in disease.
Adoptive immunotherapy in ovarian cancer
Adoptive immunotherapy is based on the infusion of autologous or allogeneic neoplastic targeting immune cells that have been expanded and/or activated ex vivo [53, 54]. Adoptive immunotherapy can be based on antigen-independent (innate immunity, e.g., natural killer (NK) and cytokine-induced killer (CIK) cells) or antigen-dependent (adaptive immunity, e.g., TILs, chimeric antigen receptor (CAR) T cells) strategies [53].
NK cells can kill tumor cells without prior sensitization or need for MHC expression and have an important role in tumor immunosurveillance. Individuals with impaired NK cell function display an increased risk of cancer development [55, 56]. Initial clinical trials involving NK cells focused on response modifiers and cytokine therapy to potentiate autologous antitumor response in vivo and demonstrated conflicting results [55].
Most of the clinical trials using NK cells remain in early-phase development; however, data thus far suggest that administration of large numbers of donor NK cells is safe, feasible, and efficacious in patients with leukemia and solid tumors (NCT01212341) [57, 58]. Administration of lymphodepleting chemotherapy and T cell suppression with allogenic NK cells has been shown to increase TTP from 52 days with fludarabine, cyclophosphamide, cyclosporine, NK cells, IL-2 (Arm 1) to 98 and 100 days using this regimen plus 10 mg methylprednisolone (Arm 2) or 1 mg methylprednisolone (Arm 3), respectively (NCT01105650). Arm 2 experienced a decrease in disease progression (33%, 1/3) compared to Arm 1 (67%, 2/3) and Arm 3 (71%, 5/7) suggesting benefit from higher doses of methylprednisolone. Both Arm 2 (1/3) and Arm 3 (3/7) treatments resulted in improved OS at 1 year compared to Arm 1 (0/3). CAR-NK cells, chimeric antigen receptor-engineered NK cells, have also been explored in vitro and in hematologic and MUC1-positive solid tumors, but have yet to be tested clinically in ovarian cancer [59, 60]. While data from many NK cell clinical trials demonstrates that this treatment is safe, one trial (NCT00652899) was terminated early due to toxicity.
While clinical trials involving cytokine-induced killer (CIK) cells are numerous for other cancers, data are much more limited for ovarian cancer. In one study, stage II ovarian cancer patients receive radiofrequency ablation (RFA) with and without autologous transfer of CIKs (Table 2). Several studies have been published analyzing the efficacy of CIK cells in the treatment of solid tumors including ovarian cancer [61,61,63]. Liu et al. in a phase II study tested the role of CIK cells in maintenance therapy after first-line treatment in advanced epithelial ovarian cancer [61]. Ninety-two patients underwent cytoreductive surgery followed by 6–8 courses of carboplatin and paclitaxel chemotherapy and 1 month after the last course half of the patients received monthly infusions of autologous CIK cells with the remaining patients receiving no treatment [53, 61]. In patients treated with CIK therapy, an increase in median PFS was observed (37.7 vs 22.2 months, p = 0.004) while OS differences did not reach statistical significance. Zhang et al. demonstrated that pre-treating CIK cells with an immunological adjuvant PA-MSHA (Pseudomonas aeruginosa-mannose-sensitive hemagglutinin) led to improved function of CIK cells; patients treated with PA-MSHA-stimulated CIK cells plus chemotherapy achieved PR or SD (with only one patient exhibiting PD), whereas no patients in the CIK plus chemotherapy group achieved partial remission [63].
Ovarian cancer is not generally considered an immunogenic cancer, however, evidence of immune evasion, clinical response to immunotherapy, and the correlation of CD8+ CD3+ T cells with improved survival suggest otherwise [64]. TILs (tumor-infiltrating lymphocytes) are endogenous autologous T cells derived from resected tumors or the peripheral blood of naïve or vaccinated patients and expanded ex vivo [64]. Non-reactive autologous T cells can be stimulated by tumor antigen-specific stimulation in vitro or by genetic engineering to express either an exogenous tumor antigen-specific T cell receptor (TCR) or chimeric antigen receptor (CAR).
TILs have been explored in clinical trials since the 1990s with early human trials showing mixed results [65]. Prospective studies demonstrate spontaneous recognition of tumor neo-epitopes in immunotherapy naïve ovarian cancer patients validating the resurgence of TIL clinical trials [66•, 67]. Results for a pilot study determined that TIL treatment for platinum-resistant metastatic ovarian cancer with a decrescendo IL-2 was feasible and tolerable. All patients had SD for a minimum of 3 months (NCT02482090) [68]. TIL therapy in combination with checkpoint inhibitors or new methods for TIL expansion could results in improved clinical responses in ovarian cancer.
Numerous clinical trials using engineered T cells, TCR, and CAR-T cells to target antigens such as HER2, NY-ESO-1, FR-alpha, MSLN, MUC16, and p53 are currently recruiting. These TAAs are also targets in cancer vaccine development [64]. ALT-801 is a biologic compound composed of IL-2 genetically fused to a humanized soluble T cell receptor directed against p53-derived antigen which recognized an epitope displayed on cancer cells in the context of HLA-A*0201 [69]. In patients with progressive metastatic malignancies, this regimen was well tolerated and elicited a clinical antitumor response at the three doses analyzed (0.015 mg/kg, 0.040 mg/kg, and 0.080 mg/kg, the 0.040 mg/kg demonstrated the highest production of IFN-gamma and immunogenicity titer (NCT00496860).
A variant of adoptive T cell immunotherapy, CAR-T cell immunotherapy allows for the combination of antigen specificity through conjugation of a specific antibody with T cell activating properties in a single fusion molecule [70]. CARs bypass the immune escape mechanism of cancer cells because they endow T lymphocytes with cytotoxic effector features in an MHC-unrestricted manner. The current CAR-T cell trials mainly target antigens mesothelin and HER2. In a trial of CAR-T cells directed against mesothelin (CART-meso) (NCT02159716) [71], six recurrent serous ovarian cancer patients were treated with either a single dose (3 × 10^7 cells/m2) of CART-meso or CART-meso (3 × 10^8 cells/m2) with or without lymphodepletion. This therapy was found to be safe and feasible with patients showing SD after 1 month of treatment. Preclinical data demonstrated that established and primary ovarian cancer cells were recognized and reacted against by anti-HER2-CAR-T cells with little to no reactivity to normal ovarian surface epithelium, thus showing feasibility for the current HER2 clinical trials [72].
Cytokine release syndrome is a potentially severe systemic toxicity seen after adoptive T cell therapy caused by T cell activation and proliferation. It is associated with elevated levels of circulating cytokines and has been previously described as a systemic response in hematologic malignancies. Tanyi et al. (2017) described a new phenomenon, compartmental cytokine release syndrome, in a patient with advanced recurrent serous ovarian cancer treated with autologous mesothelin-redirected CAR-T cells [73]. This was thought to arise from antigen-specific T cell activation and innate immune activation targeted to the site of tumor in the pleural cavity rather than other compartments that harbored tumor or compartments in which CART-meso cells were detected. The unique nature of ovarian cancer growth and spread could therefore lead to unexpected responses to CAR-T cell therapy compared to other solid tumors.
Conclusions
Immunotherapeutic strategies for the treatment of ovarian cancer must be explored and refined to improve morbidity and mortality of this disease and render therapies available to these patients which are on par with those available to other tumor sites.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Ovarian cancer—cancer stat facts [Internet]. [cited 2018 May 21]. Available from: https://seer.cancer.gov/statfacts/html/ovary.html
Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–86.
Vargas-Hernández VM, Moreno-Eutimio MA, Acosta-Altamirano G, Vargas-Aguilar VM. Management of recurrent epithelial ovarian cancer. Gland Surg. 2014;3(3):198–202.
Marth C, Reimer D, Zeimet AG. Front-line therapy of advanced epithelial ovarian cancer: standard treatment. Ann Oncol. 2017;28(suppl_8):viii36–9.
Vergote I, Tropé CG, Amant F, Kristensen GB, Ehlen T, Johnson N, et al. Neoadjuvant chemotherapy or primary surgery in stage IIIC or IV ovarian cancer. N Engl J Med. 2010;363(10):943–53.
National Comprehensive Cancer Network. Ovarian Cancer v 2.2018.
National Institutes of Health. Surveillance, epidemiology, end results: ovarian cancer.
Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia | NEJM [Internet]. 2011 [cited 2018 May 22]. Available from: https://www.nejm.org/doi/full/10.1056/nejmoa1103849
Forde PM, Chaft JE, Smith KN, Anagnostou V, Cottrell TR, Hellmann MD, et al. Neoadjuvant PD-1 blockade in resectable lung cancer. N Engl J Med. 2018 [cited 2018 May 22]; Available from: https://www.nejm.org/doi/10.1056/NEJMoa1716078?url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org&rfr_dat=cr_pub%3Dwww.ncbi.nlm.nih.gov
Trastuzumab after adjuvant chemotherapy in HER2-Positive breast cancer | NEJM 2005 [Internet]. [cited 2018 May 22]. Available from: https://www.nejm.org/doi/full/10.1056/NEJMoa052306
Clarke B, Tinker AV, Lee C-H, Subramanian S, van de Rijn M, Turbin D, et al. Intraepithelial T cells and prognosis in ovarian carcinoma: novel associations with stage, tumor type, and BRCA1 loss. Mod Pathol. 2009;22(3):393–402.
Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer | NEJM [Internet]. 2003 [cited 2018 May 22]. Available from: https://www.nejm.org/doi/10.1056/NEJMoa020177?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dwww.ncbi.nlm.nih.gov
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102(51):18538–43.
Hwang W-T, Adams SF, Tahirovic E, Hagemann IS, Coukos G. Prognostic significance of tumor-infiltrating T cells in ovarian cancer: a meta-analysis. Gynecol Oncol. 2012;124(2):192–8.
Intlekofer AM, Thompson CB. At the bench: preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. J Leukoc Biol. 2013;94(1):25–39.
Mocellin S, Benna C, Pilati P. Coinhibitory molecules in cancer biology and therapy. Cytokine Growth Factor Rev. 2013;24(2):147–61.
Fife BT, Bluestone JA. Control of peripheral T cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–82.
Krummel MF, Allison JP. Pillars article: CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. The Journal of Experimental Medicine. 1995. 182: 459–465. J Immunol. 2011;187(7):3459–65.
Buchbinder EI, Desai A. CTLA-4 and PD-1 pathways: similarities, differences, and implications of their inhibition. Am J Clin Oncol. 2016;39(1):98–106.
Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12(4):252–64.
Fessas P, Lee H, Ikemizu S, Janowitz T. A molecular and preclinical comparison of the PD-1-targeted T-cell checkpoint inhibitors nivolumab and pembrolizumab. Semin Oncol. 2017;44(2):136–40.
•• Disis ML, Patel MR, Pant S, Hamilton EP, Lockhart AC, Kelly K, et al. Avelumab (MSB0010718C; anti-PD-L1) in patients with recurrent/refractory ovarian cancer from the JAVELIN Solid Tumor phase Ib trial: safety and clinical activity. JCO. 2016;34(15_suppl):5533 Available from: http://ascopubs.org/doi/abs/10.1200/JCO.2016.34.15_suppl.5533. A preliminary analysis for trial of avelumab in patients with recurrent or refractory ovarian cancer. Responses for patients that were PD-L1+ were compared with PD-L1- expression.
Phase II study of ipilimumab monotherapy in recurrent platinum-sensitive ovarian cancer—study results - ClinicalTrials.gov [Internet]. [cited 2018 Mar 28]. Available from: https://clinicaltrials.gov/ct2/show/results/NCT01611558. A trial to investigate the overall response and adverse events related to ipilimumab as a monotherapy for recurrent ovarian cancer.
Brahmer JR, Lacchetti C, Schneider BJ, Atkins MB, Brassil KJ, Caterino JM, et al. Management of immune-related adverse events in patients treated with immune checkpoint inhibitor therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2018;14:JCO2017776385.
Safety and activity of anti-PD-L1 antibody in patients with advanced cancer | NEJM [Internet]. 2012 [cited 2018 May 25]. Available from: https://www.nejm.org/doi/full/10.1056/nejmoa1200694
Niraparib in combination with pembrolizumab in patients with triple-negative breast cancer or ovarian cancer - full text view - ClinicalTrials.gov [Internet]. [cited 2018 Mar 26]. Available from: https://clinicaltrials.gov/ct2/show/NCT02657889
PEMBRO with chemo in neo adj treatment of ovarian cancer - full text view - ClinicalTrials.gov [Internet]. [cited 2018 Jun 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT03275506
A phase II study of nivolumab/bevacizumab - full text view - ClinicalTrials.gov [Internet]. [cited 2018 Jun 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT02873962
A study in ovarian cancer patients evaluating rucaparib and nivolumab as maintenance treatment following response to front-line platinum-based chemotherapy - full text view - ClinicalTrials.gov [Internet]. [cited 2018 Jun 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT03522246
ATALANTE: atezolizumab vs placebo phase III study in late relapse ovarian cancer treated with chemotherapy + bevacizumab - full text view - ClinicalTrials.gov [Internet]. [cited 2018 Jun 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT02891824
Lee J-M, Cimino-Mathews A, Peer CJ, Zimmer A, Lipkowitz S, Annunziata CM, et al. Safety and clinical activity of the programmed death-ligand 1 inhibitor durvalumab in combination with poly (ADP-ribose) polymerase inhibitor olaparib or vascular endothelial growth factor receptor 1–3 inhibitor cediranib in women’s cancers: a dose-escalation, phase I study. JCO. 2017;35(19):2193–202.
A Study of atezolizumab versus placebo in combination with paclitaxel, carboplatin, and bevacizumab in participants with newly-diagnosed stage iii or stage iv ovarian, fallopian tube, or primary peritoneal cancer - full text view - ClinicalTrials.gov [Internet]. [cited 2018 Mar 26]. Available from: https://clinicaltrials.gov/ct2/show/NCT03038100
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20.
Howitt BE, Strickland KC, Sholl LM, Rodig S, Ritterhouse LL, Chowdhury D, et al. Clear cell ovarian cancers with microsatellite instability: a unique subset of ovarian cancers with increased tumor-infiltrating lymphocytes and PD-1/PD-L1 expression. Oncoimmunology [Internet]. 2017 Jan 6 [cited 2018 Apr 2];6(2). Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5353914/
Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, et al. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016;7(12):13587–98.
Bellone S, Buza N, Choi J, Zammataro L, Gay L, Elvin J, et al. Exceptional response to pembrolizumab in a metastatic, chemotherapy/radiation-resistant ovarian cancer patient harboring a PD-L1-genetic rearrangement. Clinical Cancer Research [Internet]. 2018 Jan 19 [cited 2018 Jun 6]; Available from: http://clincancerres.aacrjournals.org/lookup/doi/10.1158/1078-0432.CCR-17-1805
Olaparib, durvalumab, and tremelimumab in treating patients with recurrent or refractory ovarian, fallopian tube or primary peritoneal cancer with BRCA1 or BRCA2 mutation - full text view - ClinicalTrials.gov [Internet]. [cited 2018 Jun 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT02953457
Pembrolizumab in treating participants with metastatic, recurrent or locally advanced cancer and genomic instability - full text view - ClinicalTrials.gov [Internet]. [cited 2018 Jun 12]. Available from: https://clinicaltrials.gov/ct2/show/NCT03428802
Guo C, Manjili MH, Subjeck JR, Sarkar D, Fisher PB, Wang X-Y. Therapeutic cancer vaccines: past, present, and future. Adv Cancer Res. 2013;119:421–75.
Senzer N, Barve M, Kuhn J, Melnyk A, Beitsch P, Lazar M, et al. Phase I trial of “bi-shRNAifurin/GMCSF DNA/autologous tumor cell” vaccine (FANG) in advanced cancer. Mol Ther. 2012;20(3):679–86 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3293620/.
Manning L, Barve M, Wallraven G, Kumar P, Taquet N, Bognar E, et al. Assessment of low dose Vigil® engineered autologous tumor cell (EATC) immunotherapy in patients with advanced solid tumors. Clin Oncol. 2017;2:4.
Trial of Adjuvant FANG™ vaccine for high risk stage III/IV ovarian cancer - full text view - ClinicalTrials.gov [Internet]. [cited 2018 Jun 25]. Available from: https://clinicaltrials.gov/ct2/show/NCT01309230
Phase 2 trial of maintenance vigil for high risk stage IIIb-IV ovarian cancer - full text view - ClinicalTrials.gov [Internet]. [cited 2018 Jun 25]. Available from: https://clinicaltrials.gov/ct2/show/NCT02346747
Vreeland TJ, Litton JK, Qiao N, Philips AV, Alatrash G, Hale DF, et al. Phase Ib trial of folate binding protein (FBP)-derived peptide vaccines, E39 and an attenuated version, E39’: an analysis of safety and immune response. Clin Immunol. 2018; Available from: http://www.sciencedirect.com/science/article/pii/S1521661617308653.
Kalli KR, Block MS, Kasi PM, Erskine CL, Hobday TJ, Dietz A, et al. Folate receptor alpha peptide vaccine generates immunity in breast and ovarian cancer patients. Clin Cancer Res. 2018. A phase I study that demonstrates that generation of an immune response to folate receptor vaccination occurs in a large number of patients in clinical remission and remains detectable at 1 year.
PH3 study of mirvetuximab soravtansine vs investigator’s choice of chemotherapy in women with Fra+ Adv. EOC, primary peritoneal or fallopian tube cancer - full text view - ClinicalTrials.gov [Internet]. [cited 2018 Jun 25]. Available from: https://clinicaltrials.gov/ct2/show/NCT02631876
Altwerger G, Bonazzoli E, Bellone S. In Vitro and in vivo activity of IMGN853, an antibody–drug conjugate targeting folate receptor alpha linked to DM4, in biologically aggressive endometrial cancers. Mol Cancer Ther. 2018; Available from: http://mct.aacrjournals.org/content/17/5/1003.long.
Ab O, Whiteman KR, Bartle L. IMGN853, a folate receptor-α (FRα)–targeting antibody–drug conjugate, exhibits potent targeted antitumor activity against FRα-expressing tumors. Mol Cancer Ther. 2015; Available from: http://mct.aacrjournals.org/content/14/7/1605.long.
Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474(7353):609–15.
Odunsi K. Immunotherapy in ovarian cancer. Ann Oncol. 2017;28(suppl_8):viii1–7 Available from: https://academic.oup.com/annonc/article/28/suppl_8/viii1/4693810.
Battaglia A, Fossati M, Buzzonetti A, Scambia G, Fattorossi A. A robust immune system conditions the response to abagovomab (anti-idiotypic monoclonal antibody mimicking the CA125 protein) vaccination in ovarian cancer patients. Immunol Lett. 2017;191:35–9.
• Hardwick NR, Frankel P, Ruel C, Kilpatrick J, Tsai W, Kos F, et al. p53-reactive T cells are associated with clinical benefit in patients with platinum-resistant epithelial ovarian cancer after treatment with a p53 vaccine and gemcitabine chemotherapy. Clin Cancer Res. 2018;24(6):1315–25 Available from: http://clincancerres.aacrjournals.org.proxy-hs.researchport.umd.edu/content/24/6/1315. A phase I trial to demonstrate that response to p53 vaccination in conjunction with cytotoxic chemotherapy correlates with longer PFS.
Mittica G, Capellero S, Genta S, Cagnazzo C, Aglietta M, Sangiolo D, et al. Adoptive immunotherapy against ovarian cancer. J Ovarian Res. 2016;9 Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4869278/.
Krishnan V, Berek JS, Dorigo O. Immunotherapy in ovarian cancer. Curr Probl Cancer. 2017;41(1):48–63.
Uppendahl LD, Dahl CM, Miller JS, Felices M, Geller MA. Natural killer cell-based immunotherapy in gynecologic malignancy: a review. Front Immunol. 2018;8 Available from: https://www.frontiersin.org/articles/10.3389/fimmu.2017.01825/full.
Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. 2000;356(9244):1795–9.
Yang Y, Lim O, Kim TM, Ahn Y-O, Choi H, Chung H, et al. Phase I study of random healthy donor-derived allogeneic natural killer cell therapy in patients with malignant lymphoma or advanced solid tumors. Cancer Immunol Res. 2016;4(3):215–24.
Eguizabal C, Zenarruzabeitia O, Monge J, Santos S, Vesga MA, Maruri N, et al. Natural killer cells for cancer immunotherapy: pluripotent stem cells-derived NK cells as an immunotherapeutic perspective. Front Immunol. 2014;5:439.
Klapdor R, Wang S, Hacker U, Büning H, Morgan M, Dörk T, et al. Improved Killing of ovarian cancer stem cells by combining a novel chimeric antigen receptor-based immunotherapy and chemotherapy. Hum Gene Ther. 2017;28(10):886–96.
Martín-Antonio B, Suñe G, Perez-Amill L, Castella M, Urbano-Ispizua A. Natural killer cells: angels and devils for immunotherapy. Int J Mol Sci. 2017;29:18(9).
Liu J, Li H, Cao S, Zhang X, Yu J, Qi J, et al. Maintenance therapy with autologous cytokine-induced killer cells in patients with advanced epithelial ovarian cancer after first-line treatment. J Immunother. 2014;37(2):115–22.
Zhang Z, Wang L, Luo Z, Zhao X, Huang J, Li H, et al. Efficacy and safety of cord blood-derived cytokine-induced killer cells in treatment of patients with malignancies. Cytotherapy. 2015;17(8):1130–8.
Zhang C, Zhang Z, Wang L, Han J, Li F, Shen C, et al. Pseudomonas aeruginosa-mannose sensitive hemagglutinin injection treated cytokine-induced killer cells combined with chemotherapy in the treatment of malignancies. Int Immunopharmacol. 2017;51:57–65.
Rodriguez-Garcia A, Minutolo NG, Robinson JM, Powell DJ. T cell target antigens across major gynecologic cancers. Gynecol Oncol. 2017;145(3):426–35.
Andersen R, Donia M, Westergaard MCW, Pedersen M, Hansen M, Svane IM. Tumor infiltrating lymphocyte therapy for ovarian cancer and renal cell carcinoma. Hum Vaccin Immunother. 2015;11(12):2790–5.
Deniger DC, Pasetto A, Robbins PF, Gartner JJ, Prickett TD, Paria BC, et al. T-cell responses to TP53 “hotspot” mutations and unique neoantigens expressed by human ovarian cancers. Clin Cancer Res. 2018. In this paper, TILs from ovarian cancer patients were found to have specificity to mutated neoantigens and that these T cells could be used for adoptive cell therapy. TP53 “hotspot” reactive T cells were also found and these cells could recognize a broad range of tumor types in unrelated individuals.
Bobisse S, Genolet R, Roberti A, Tanyi JL, Racle J, Stevenson BJ, et al. Sensitive and frequent identification of high avidity neo-epitope specific CD8+ T cells in immunotherapy-naive ovarian cancer. Nat Commun. 2018. 1092;9(1):15.
Pedersen M, Westergaard M, Nielsen M, Borch TH, Poulsen LG, Hendel H, et al. 1145PDAdoptive cell therapy with tumor-infiltrating lymphocytes for patients with metastatic ovarian cancer: a pilot study. Ann Oncol. 2017;28(suppl_5) Available from: https://academic.oup.com/annonc/article/28/suppl_5/mdx376.010/4109224.
Fishman MN, Thompson JA, Pennock GK, Gonzalez R, Diez LM, Daud AI, et al. Phase I trial of ALT-801, an interleukin-2/T cell receptor fusion protein targeting p53 (aa264-272)/HLA-A*0201 complex, in patients with advanced malignancies. Clin Cancer Res. 2011;17(24):7765–75.
Zhu X, Cai H, Zhao L, Ning L, Lang J, Zhu X, et al. CAR-T cell therapy in ovarian cancer: from the bench to the bedside. Oncotarget. 2017;8(38):64607–21.
Tanyi JL, Haas AR, Beatty GL, Stashwick CJ, O’Hara MH, Morgan MA, et al. Anti-mesothelin chimeric antigen receptor T cells in patients with epithelial ovarian cancer. JCO. 2016;34(15_suppl):5511.
Lanitis E, Dangaj D, Hagemann IS, Song D-G, Best A, Sandaltzopoulos R, et al. Primary human ovarian epithelial cancer cells broadly express HER2 at immunologically-detectable levels. PLoS ONE. 2012;7(11):e49829.
Tanyi JL, Stashwick C, Plesa G, Morgan MA, Porter D, Maus MV, et al. Possible compartmental cytokine release syndrome in a patient with recurrent ovarian cancer after treatment with mesothelin-targeted CAR-T cells. J Immunother. 2017;40(3):104–7.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Gynecologic Cancers
Rights and permissions
About this article

Cite this article
Fan, C.(., Reader, J. & Roque, D.M. Review of Immune Therapies Targeting Ovarian Cancer. Curr. Treat. Options in Oncol. 19, 74 (2018). https://doi.org/10.1007/s11864-018-0584-3
Published:
DOI: https://doi.org/10.1007/s11864-018-0584-3
Keywords
- Ovarian cancer
- Immunotherapy
- Immune checkpoint inhibitors
- Cancer vaccine
- Adoptive immunotherapy
- PD1
- PD-L1
- PD-L2
- CTLA-4
- Nivolumab
- Ipilimumab
- Pembrolizumab
- Avelumab
- Atezolizumab
- PARP inhibitors
- BRCA 1/2 mutation
- Microsatellite instability
- Bevacizumab
- Anti-angiogenic
- NY-ESO
- MAGE
- p53
- Mirvetuximab soravtansine
- Folate receptor alpha
- Folate-binding protein
- TILS
- CAR-T cells
- NK cells
- T cells
- HER2
- Mesothelin