
Abstract
Carotenoids in cassava storage roots play important roles in benefiting people’s health in the tropics because they provide essential nutrients and antioxidants. Although the related genes and loci associated with carotenoid metabolism in many species are well reported, in cassava they are poorly understood. In the present study, GWAS base on SLAF-seq was used in detecting the related genes and loci correlated to carotenoid contents in 98 accessions from a cassava F1 mapping population. The 98 accessions were divided into four subgroups. On the basis of general linear and compressed linear models, 144 genes were detected by selective sweep analysis, and 84 SNPs and 694 genes were detected by association mapping, in which Manes.04G164700 (XanDH) and Manes.11G105300 (AAO) were probably involved in the downstream pathway of carotenoid metabolism, and their expressions in six cassava genotypes were confirmed. Our results will be useful in yellow-root cassava variety improvement and provide the most effective and sustainable approach to maximize the nutritional and health benefits of carotenoid to a large number of populations.
Similar content being viewed by others


Introduction
Cassava (Manihot esculenta Crantz), originated in Latin America and is the fifth most important staple food crop in the world, particularly in sub-Saharan Africa. The storage root is rich in starch but poor in protein and micronutrients such as iron, zinc, and provitamin A (Welsch et al. 2010). The deficiency of vitamin A leads to loses of protection against eye diseases, such as macular degeneration, the leading cause of blindness in the elderly and young (Krinsky et al. 2003). Carotenoids are the synthesis precursor of provitamin A, which cannot be synthesized by animals including humans. In plants, carotenoids harvest light during photosynthesis and provide cleavage products (abscisic acid (ABA), strigolactones, β-cyclocitral) (Bouwmeester et al. 2007; Coesel et al. 2008; Gomez-Roldan et al. 2008; Umehara et al. 2008) that act as signal molecules for the regulation of physiological functions under abiotic stress modulating developmental processes and plant environmental responses (Ruiz-Solaa and Rodríguez-Concepción, 2012). They stabilize membrane lipids via antioxidant properties, and provide flowers and fruits with animal attracting colors that facilitate pollination and seed dispersal (Bouvier et al. 2005). Their formation is initiated by reactions catalyzed by carotenoid cleavage dioxygenases (CCDs) (Moise et al. 2005; Auldridge et al. 2006) in the pathway of carotenoid biosynthesis. Carotenoids are synthesized from isopentenyl diphosphate (IPP) and its isomer dimethylallyl diphosphate (DMAPP) which is mainly produced by the MEP pathway (Welsch et al. 2010). 20-carbon geranylgeranyl diphosphate (GGPP) was catalyzed by GGPP synthase from 10-carbon geranyl diphosphate (GPP) with a series of condensations (Deng et al. 2017). The key enzyme, phytoene synthase (PSY), is rate limiting in the carotenoid biosynthetic pathway of cassava roots (Welsch et al. 2010). It catalyzed GGPP in the carotenoid pathway which contains two pathway branches including α-carotene and ß-carotene (Cunningham and Gantt 1998, 2001; Isaacson et al. 2002). PSY1-3 is capable of enhancing the flux of carbon through carotenogenesis, and phytoene desaturase (PDS) may simply represent the subsequent rate limiting step in the pathway (Arango et al. 2010; Welsch et al. 2010). Interestingly, there was no correlation between root color and expression of major carotenoid biosynthesis genes (Welsch et al. 2010). Conventional breeding and genetic modification are the common strategy to improve total carotenoid content in cassava roots (Carvalho et al. 2016; Ceballos et al. 2013; Welsch et al. 2010). It was showed that fibrillin and Or proteins may induce carotenoid accumulation by initiating the synthesis of a carotenoid deposition sink in the form of the large carotenoid-sequestering sheets (Li et al. 2001; Carvalho et al. 2012). Deng et al. (2017) reported that in the mature period of storage root, the expression of PSY2 and LCYB involved in carotenoids biosynthesis in yellow cassava was significantly higher than that in swelling period, on the contrary, the expression of CCD1 and NCED3 associated with carotenoids degradation was significantly lower than that in swelling period. To further understand the pathway of carotenoid accumulation in cassava, genome-wide association studies (GWAS) based on specific-locus amplified fragment sequencing (SLAF-seq) was used to determine the related genes and the corresponding SNP in these researches.
GWAS have become one of common and useful tools for identifying genes for many traits in animals and plants (Wang et al. 2015; Iqbal et al. 2015; Lee et al. 2015; Zhao et al. 2015; Han et al. 2015a, b; Huang et al. 2010). SLAF-seq has been developed, and is more efficient for large-scale genotyping. SLAF-seq has advantages such as deep sequencing to ensure genotyping accuracy, reduced representation strategy to reduce sequencing costs, pre-designed reduced representation scheme to optimize marker efficiency and a double barcoding system to examine large populations (Sun et al. 2013). It has significant advantages for GWAS, which has been successfully applied to soybean (Zhao et al. 2015) and cucumber (Zhang et al. 2015). In the present study, the SLAF-seq technology was used to perform a GWAS for a segregating F1 population derived from a cross of South China 9 (SC9, ♀), a yellow-root cassava, with other white-root SC varieties (♂) to identify the relative SNPs and determine the related functional genes of carotenoid metabolism pathway. It will provide a clue to increase carotenoid content in cassava varieties and benefit human health in the tropics.
Materials and methods
Material preparation and carotenoid detection
The segregating F1 population derived from the cross of SC9 (♀) with other white-root SC varieties (♂) was developed in the National Cassava Germplasm Bank, Tropical Crops Genetic Resources Institute, Chinese Academy of Tropical Agricultural Sciences. The F1 populations were numbered from No. 1 to No. 377. Ten months after planting, cassava storage roots were harvested washed, and a 2.0 g sample of flesh was transferred into a cooled mortar. The flesh was ground with a small amount of acetone until the color of the residual flesh was white, and the extracting solution acetone was colorless. The extracting solution and residual were diluted with 25 mL acetone and placed in a centrifuge at 3000 rpm for 5 min. The supernatant was collected and the absorbance measured using a spectrophotometer at a wavelength of 454 nm. Three biological replications were used in the present study. The content of β-carotene CA = A454nm × 0.5 (where, CA is the content of β-carotene; A454nm is absorbance; the unit is mg/hg) (Zhang and Fang 2006; Rodriguez-Amaya 2001).
Sample preparation for SLAF and SNP calling
The genome DNA was extracted using the method described in Wu et al. (2010) for each accession. Genomic DNA was isolated from the fresh leaves and analyzed according to SLAF-seq (Sun et al. 2013). Two restriction enzymes (Hpy166II and HaeIII) were applied, and 100 bp pair end reads were sequenced by HiSeq™2500 platform. More than 100,000 fragments with insertion length range from 360 to 420 bp were obtained. The high quality paired-end reads were mapped on to the cassava genome using the Burrows–Wheeler Aligner (Li and Durbin 2009; Li et al. 2009). These fragments were evenly distributed in the reference of cassava genome which was downloaded from JGI (https://phytozome.jgi.doe.gov/pz/portal.html). The reads were mapped to the same genome loci that were used as a SLAF fragment. SNPs were obtained using GATK and SAM tools (McKenna et al. 2010; Li et al. 2009). SNPs with minor allele frequency (MAF) < 5% and integrity < 0.8 were excluded from the genotype data sets of all the accessions.
Construction of phylogenetic tree
The high quality SNPs were used to construct the phylogenetic tree using the neighbor-joining method in MEGA5 (Koichiro et al. 2011). A bootstrap consensus tree was obtained from 1000 replicates (Saitou and Nei 1987).
Identification of putative selective sweep regions
To investigate further whether specific regions in the genome of cassava have been under positive selection, a selective sweep analysis was performed on the 98 accessions’ genome sequence. The genetic differentiation coefficient (Fst) and πratio of the two groups were calculated by the 500-kb sliding windows method (Brown 1970; Tajima 1983). Genomic regions at which both Fst and πratio value reach the critical value, corresponding to a 5% significant level, were taken as significantly differentiated genomic regions.
Population structure and principal component analysis (PCA) analysis
Admixture was used for population structure analysis; the predefined genetic clusters of K values from 1 to 10 were used to evaluate the convergence of individuals. The PCA was inferred by cluster based on the total of SNPs (Alexander et al. 2009; De Hoon et al. 2004; Dunteman 1989).
Genome-wide association analyses
The total of the high-integrity SNPs was used to implement association analyses with compressed mixed linear models (MLM) and general linear models (GLM) in TASSEL (software for association mapping of complex traits in diverse samples) (Bradbury et al. 2007). The population structure inferred by Admixture (Alexander et al. 2009), and SPAGeDi (Hardy and Vekemans 2002) was used to calculate the relative kinship matrix (K) between individuals. The Bonferroni method (Hochberg 1988) at α < 0.1 and α < 0.01 (p ≤ 1.0 × 10−6 and p ≤ 1.0 × 10−7, respectively) was used to calibrate the p value at the threshold to determine whether a significant association existed. Candidate genes located within the 100-kb region upstream or downstream of peak SNPs were identified.
RNA isolation and QRT-PCR
Total RNA isolation from six cassava genotypes included high, medium and low carotenoid contents in storage roots was conducted following the instruction of Plant RNA extraction kit (TIANGEN Co., Ltd, China). Reverse transcription (RT) reaction was followed the Reverse first strand cDNA Synthesis kit, and the quantitative real-time (QRT)-PCR reaction was performed according to a Super Real Pre Mix kit (TARATA Co., Ltd, China), respectively. The amplification protocol was as follows: 10 min denaturization at 95 °C, then 40 cycles of 15 s denaturing at 95 °C, 30 s annealing at 60 °C, and 30 s extending at 72 °C. The relative expression level was normalized against selected housekeeping genes such as ubiquitin. Quantification of the gene expression was carried out based on the relative quantitative analysis method (2−\({{\Delta \Delta C_{\text{T}} }}\)).
Results
Classification of cassava samples
To classify cassava samples, the β-carotene contents of 98 accessions were determined; 48 accessions with high carotenoid content varied from 2.5 to 14.8 mg/hg and 50 accessions with low carotenoid content arranged from 0.25 to 2.5 mg/hg from F1 population were selected to use in the present study. The frequency of the carotenoid content for 98 accessions is shown in Fig. 1.

The frequency of carotenoid content among 98 accessions. 98 accessions were divided into 17 groups. The X-axis is standard for different carotenoid content groups. The Y-axis is standard for frequency of different carotenoid content groups
SNP calling
For all 98 accessions, we obtained 167.10M single end reads with average genome depth of 8.31X. After filtering low integrity and minor allele frequency SNPs, a total of 104,059 high quality SNPs were used for further analysis. All 104,059 SNPs were distributed along 18 chromosomes and unanchored scaffolds. There were 6584 SNPs in Chromosome 1 which is the most and 4159 SNPs in Chromosome 16 which is the least; 10,772 SNPs were located in unanchored scaffolds (Fig. 2).

Genome-wide SNP distributions. The Y-axis is standard for the numbers of SNP in each chromosome
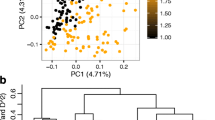
The phylogenetic tree
As shown in the phylogenetic tree (Fig. 3), 98 accessions were classified into four subgroups. The high carotenoid accessions and low carotenoid accessions were evenly distributed among four subgroups showing that these samples are suitable for association analysis.

The phylogenetic tree of 98 accessions. The red branch presented the accession with low carotenoids content, and the blue one is the accession with high carotenoids content. Each branch represents one accession, and the genetic distance scale bar is shown in the left side
Putative selective sweep region
Regions in the genome under strong positive selection after the divergence of these two populations are expected to share fewer derived alleles. Using stringent criteria genomic region with Fst > 0.02 (95%) and πratio < 0.84 (5%) were defined as potential selective sweep regions (Fig. 4). These regions, distributed in chromosomes 2, 4, 5, 7 and 14, harbor 144 annotated protein-coding genes. Of particular interest is the selective sweep region observed at gene, such as Manes.04G164700, which is annotated as xanthoxin dehydrogenase. Xanthoxin dehydrogenase is involved in the pathway of carotenoid metabolism. The X-axis represents πratio (high carotenoid group/low carotenoid group) and Y-axis represents Fst. The blue and red denote the differential regions with πratio > 95% or < 5% and Fst > 95%.

Selective sweep detection and the plot of Fst and πratio. The X-axis is π and Y-axis is Fst. The red and blue are differential area by selecting π < 0.05 and π > 0.95 and Fst > 0.95
Population structure and PCA analysis
Admixture was applied for population structure analysis. The lowest number of subgroups was four (K = 4, ∆K) for the 98 accessions and this was consistent with the phylogenetic tree. The first subgroup includes 21 accessions with 13 high carotenoid accessions and 8 low carotenoid accessions; the second subgroup includes 19 accessions with 8 high carotenoid and 11 low carotenoid accessions; the third subgroup includes 40 accessions with 21 high carotenoid and 19 low carotenoid accessions; the fourth subgroup includes 18 accession with 6 high carotenoid and 12 low carotenoid accessions (Fig. 5). For all the subgroups, both high and low carotenoid accessions are distributed. The result of PCA analysis was shown in Fig. 6. PCA1 denotes the first primary component, PCA2 denotes the second primary component, and PCA3 denotes the third primary component. Each point represents one sample and each color represents one subgroup. The PCA analysis showed that whose genetic relationships of the accessions were relative close and those accessions were distant, and these data can be able to explain 4.9% of the genetic variation.

The clustering plot of 98 accessions. a shows the relationship between ∆K values and K values. ∆K get the lowest value while K equals to 4. b The ten different population structures with different K values. Each color denotes one subgroup in each strap. For example, the first strap shows the population structure while K = 1

The three-dimensional plot of PCA analysis of 98 accessions. 98 accessions were gathered into three dimensions through PCA analysis. PCA1 denotes the first primary component, PCA2 denotes the second primary component, and PCA3 denotes the third primary component. Each point represents one sample and each color represents one subgroup
Genome-wide association analysis
GLM and compressed MLM in TASSEL were used for association analysis. For GLM model, Q population structure information was applied. While both Q population structure and affiliation information was used for MLM. Based on GLM, eight SNP loci were significantly associated with p value < 1.0 × 10−7 distributed in chromosome 2, 4, 10 and 11. While p value < 1.0 × 10−6, 84 SNPs were significant associated, which are distributed in all chromosome except chromosome 5. For each SNP loci, the 100 kb region from upstream and downstream was regarded as significantly associated regions. 694 genes are distributed in these regions, and their ID and annotation were listed in Table S1. Based on MLM model, no SNP loci were significantly associated with p value < 1.0 × 10−7, while 3 SNPs and 25 genes were found with p value < 1.0 × 10−6. The ID of these genes and annotation was listed in Table S2. In total, 84 SNPs and 694 genes are associated with carotenoid trait. For example, among these genes, Manes.11G105300 annotated as abscisic aldehyde oxidase is probably involved in the pathway of carotenoid metabolism. The carotenoid associate analysis based on both GLM and MLM was shown in Fig. 7.

Genome-wide association study (GWAS) of traits associated with carotenoid in Manihot esculenta Crantz. a QQ-plot based on the MLM model. b QQ-plot based on the GLM model. The real line is the predicted threshold, and SNP and the solid line are jointed consistent in the front-end, only upwarped at the end, which means SNP is low false-positive and the correlation results are reliable. c Manhattan figure based on the MLM model for correlation analysis. d Manhattan figure based on the GLM model for correlation analysis. The X-axis is chromosome and Y-axis is − lg of associated value. A dot represents an SNP site. The blue dashed line is a significant threshold for multiple hypothesis tests (FDR), higher than the threshold point for significant correlation. The red dotted line is the negative log of the total number of 0.1/SNP
Gene expression levels in cassava storage roots
The expression of candidate genes was confirmed at the storage roots of six cassava genotypes included high (Nos. 327, 74), media (Nos. 261, 292) and low carotenoid (Nos. 54, 333) contents. Among the related genes associated with carotenoid trait, the gene expression levels of Manes.11G105300 (AAO) in the storage roots of six cassava genotypes (Nos. 54, 74, 261, 292, 327 and 333) were shown in Fig. 8. The highest expression was No. 292, following by Nos. 261, 333, 327, 54 and 74. The protein xanthoxin dehydrogenase related with carotenoid metabolism is shown in Fig. 9; its gene Manes.04G164700 (XanDH) expression in the storage roots of six cassava genotypes (Nos. 54, 74, 261, 292, 327and 333) indicated that No. 292 is the highest, following by Nos. 333, 327, 261, 54 and 74.

The expression level of AAO in the storage roots of six cassava genotypes (Nos. 54, 333, 261, 292, 327 and 74). Nos. 327, 74: high carotenoid contents; Nos. 261, 292: media carotenoid contents; Nos. 54, 333: low carotenoid contents

The expression level of XanDH in the storage roots of six cassava genotypes (Nos. 54, 333, 261, 292, 327 and 74). Nos. 327, 74: high carotenoid contents; Nos. 261, 292: media carotenoid contents; Nos. 54, 333: low carotenoid contents
Discussion
Genome-wide association studies that take full advantage of ancient recombination events to identify the genetic loci underlying traits is becoming a powerful tool for detecting natural variation underlying complex traits in crops (Rafalski 2010). Chen et al. (2013) developed multi-state tagging in rice based on SLAF-seq technology, and obtain 518 chromosome specificity sequence of 7E and developed 89 specific molecular markers in Thinopyrum elongatum, and the success rate was 65.9%. These specific markers were verified in other materials such as wheat varieties, polyploidy T. elongatum, and the results show that the research and development of chromosome specific molecular markers in T. elongatum has high repeatability, stability and specificity, which can be applied to the resistant breeding of wheat. At early stage, scientists developed SNP tags through EST sequencing, which were used in detecting the species affinity and evolution and founded there existed multiple domestication or gene multiple transfer phenomenon between cultivar and wild type (Kanazin et al. 2002). SNP tags as a high stability and polymorphism abundant molecular marker have been widely used in species identification, origin of species and ties of consanguinity. It can be used on the crop breeding through establishing the genealogy of genetic relations by determining the genetic distance between the species. Julia et al. (2013) used SLAF-seq technology to develop 149,362 SNP tags with 26 Xiphophorus fish, and constructed the phylogenetic tree, described the history of the evolution of physiologically, and found that the species had an interesting evolutionary story: at least two species could be formed through cross breeding, and speculated that these may be the father of the hybrid origin. As a result, SNP tags developed using SLAF-seq technology can also answer uncertain systems, especially, those sibling species. In the present study, GWAS base on SLAF-seq was used to detect the related genes and the corresponding SNP in 98 accessions from a cassava F1 mapping population. 104,059 SNPs were found to be distributed along 18 chromosomes and unanchored scaffolds, in which 84 SNPs and 694 genes are determined to associate with carotenoid metabolism. These corresponding SNPs and related genes will be helpful to understand the molecular mechanism of carotenoid accumulation and increase the progress towards cassava variety improvement and benefit to human health. The 98 cassava accessions, divided into high and low carotenoids contents, were analyzed based on the phylogeny, selective sweep, population structure, kinship and genome-wide association. PCA analysis showed that the genotype data is able to explain 4.9% of the genetic variation. We speculated there are a lot of factors affected on the growth of cassava such as typhoon and rainstorm and border effect of cultivation and β-carotenoid. 144 genes were detected to distribute on chromosomes 2, 4, 5, 7 and 14. Based on the GLM and MLM, we detected 84 SNPs and 694 genes associated with the carotenoid trait. Among these genes, Manes.04G164700 was annotated as xanthoxin dehydrogenase and as well as Manes.11G105300 as abscisic aldehyde oxidase which is concerned with plant response to drought stress as it controls the closing of stomata to minimize water loss and also senescence. Both proteins probably participated in the downstream pathways of carotenoid metabolism. There are many previous reports that focus on root-nutrient improvement and selection for high carotenoid content for cassava (Ceballos et al. 2013; Carvalho et al. 2016), while there are few reports involved in the relative genes and loci associated with carotenoid metabolism. XanDH and AAO in a flacca Tomato Mutant with Deficient Abscisic Acid and Wilty Phenotype, which suggests that XanDH and AAO are key enzyme genes in ABA synthesis (Sagi et al. 1999). It was also reported that XanDH played an important role in ABA biosynthesis in rice (Endo et al. 2014). In the present study, the result of expressions level of XanDH and AAO genes with carotenoid content in six cassava F1 genotypes suggests these genes were probably associated with the reduction of carotenoids and the increase of ABA, however, it will need further confirmation to validate the biological functions in cassava carotenoid metabolism for both proteins. So far, the traditional breeding was the common and effective approaches for selecting cassava genotypes with high carotenoid contents (Carvalho et al. 2016; Ceballos et al. 2013; Welsch et al. 2010). In the previous reports, the proportion of β-carotene/total carotenoid content was from 22.0 to 62.3% in 23 cassava landraces (Carvalho et al. 2016), indicating β-carotene is the main component in the carotenoids of the cassava storage roots. Our results showed that the highest content of β-carotene is 14.8 mg/hg in the cassava F1 population; it was much more than that of its parents SC9 and white-root genotypes located at the group of low carotenoid contents.
Author contribution statement
SC made the major contributions to this study in the conception, design, drafting part of manuscript, and final revision. XL contributed to data analysis and drafting manuscript. KL contributed to conception and design of manuscript. KIT and LJCBC contributed to parts of conception, and critical revision of manuscript. All authors read and approved the final manuscript.
References
Alexander DH, Novembre J, Lange K (2009) Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19:1655–1664
Arango J, Wust F, Beyer P, Welsch R (2010) Characterization of phytoene synthases from cassava and their involvement in abiotic stress-mediated responses. Planta 232:1251–1262
Auldridge ME, Mc Carty DR, Klee HJ (2006) Plant carotenoid cleavage oxygenases and their apocarotenoid products. Curr Opin Plant Biol 9:315–321
Bouvier F, Isner JC, Dogbo O, Camara B (2005) Oxidative tailoring of carotenoid: a prospect towards novel functions in plants. Trends Plant Sci 10:187–194
Bouwmeester HJ, Roux C, Lopez-Raez JA, Bécard G (2007) Rhizosphere communication of plants, parasitic plants and AM fungi. Trends Plant Sci 12:224–230
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Brown AH (1970) The estimation of Wright’s fixation index from genotypic frequencies. Genetica 41:399–406
Carvalho LJCB, Lippolis J, Chen S, de Souza CRB, Vieira EA (2012) Anderson JV (2012) Characterization of carotenoid-protein complexes and gene expression analysis associated with carotenoid sequestration in pigmented cassava (Manihot esculenta Crantz) storage root. Open Biochem J 6:116–130
Carvalho LJCB, Agustini MAV, Anderson JV, Vieira EA, de Souza CRB, Chen S, Schaal BA, Silva JP (2016) Natural variation in expression of genes associated with carotenoid biosynthesis and accumulation in cassava (Manihot esculenta Crantz) storage root. BMC Plant Biol 16:133
Ceballos H, Morante N, Sánchez T, Ortiz D, Aragón I, Chávez AL, Pizarro M, Calle F, Dufour D (2013) Rapid cycling recurrent selection for increased carotenoid content in cassava roots. Crop Sci 53:1–10
Chen S, Huang Z, Dai Y, Qin S, Gao Y, Zhang L, Gao Y, Chen J (2013) The development of 7E chromosome-specific molecular markers for Thinopyrum elongatum based on SLAF-seq technology. PLoS One 8:e65122
Coesel S, Oborník M, Varela J, Falciatore A, Bowler C (2008) Evolutionary origins and functions of the carotenoid biosynthetic pathway in marine diatoms. PLoS One 3:e2896
Cunningham FX Jr, Gantt E (1998) Genes and enzymes of carotenoid biosynthetic pathway in plants. Annu Rev Plant Physiol Mol Biol 49:557–583
Cunningham FX Jr, Gantt E (2001) One ring or two? Determination of ring number in carotenoid by lycopene ε-cyclases. Proc Natl Acad Sci USA 98:2905–2910
De Hoon Michiel J L, Imoto S, Miyano S (2004) Open source clustering software. Bioinformatics 20:1453–1454
Deng C, Yao H, An F, Li K, Chen S (2017) Analysis of genes and enzymes associated with carotenoid metabolism in cassava. J Plant Genet Resour 18:556–563
Dunteman GH (1989) Principal components analysis. Sage, Newbury Park
Endo A, Nelson KM, Thoms K, Abrams SR, Nambara E, Sato Y (2014) Functional characterization of xanthoxin dehydrogenase in rice. J Plant Physiol 171:1231–1240
Gomez-Roldan V, Soraya F, Philip B, Puech-Pagès V, Dun Elizabeth A, Pillot JP, Letisse F, Matusova R, Danoun S, Portais JC, Bouwmeester H, Bécard G, Beveridge CA, Rameau C, Rochange SF (2008) Strigolactone inhibition of shoot-branching. Nature 455:189–194
Han Y, Zhao X, Cao G, Wang Y, Li Y, Liu D, Teng W, Zhang Z, Li D, Qiu L, Zheng H, Li W (2015a) Genetic characteristics of soybean resistance to HG type 0 and HG type1.2.3.5.7 of the cyst nematode analyzed by genome-wide association mapping. BMC Genom 16:598. https://doi.org/10.1186/s12864-015-1800-1
Han Y, Zhao X, Liu D, Li Y, Lightfoot DA, Yang Z, Zhao L, Zhou G, Wang Z, Huang L, Zhang Z, Qiu L, Zheng H, Li W (2015b) Domestication footprints anchor genomic regions of agronomic importance in soybeans. New Phytol. https://doi.org/10.1111/nph.13626
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hochberg Y (1988) A sharper Bonferroni procedure for multiple tests of significance. Biometrika 75:800–802
Huang X, Wei X, Sang T, Zhao Q, Feng Q, Zhao Y, Li C, Zhu C, Lu T, Zhang Z, Li M, Fan D, Guo Y, Wang A, Wang L, Deng L, Li W, Lu Y, Weng Q, Liu K, Huang T, Zhou T, Jing Y, Li W, Lin Z, Buckler ES, Qian Q, Zhang QF, Li J, Han B (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 11:961–969
Iqbal A, Kim YS, Kang JM, Lee YM, Rai R, Jung JH, Oh DY, Nam KC, Lee HK, Kim JJ (2015) Genome-wide association study to identify quantitative trait loci for meat and carcass quality traits in Berkshire. Asian Australas J Anim Sci 28:1537–1544
Isaacson RG, Zamir D, Hirsehberg J (2002) Cloning of tangerine from tomato reveals a carotenoid isomerase essential for the production of β-carotene and xanthophylls in plants. Plant Cell 14:333–342
Julia CJ, Shaohua F, Paolo F, Manfred S, Axel M (2013) The evolutionary history of Xiphophorus fish and their sexually selected sword: a genome-wide approach using restriction site-associated DNA sequencing. Mol Ecol 22:2986–3001
Kanazin V, Talbert H, See D, DeCamp P, Nevo E, Blake (2002) Discovery and assay of single-nucleotide polymorphisms in barley (Hordeum vulgare). Plant Mol Biol 48:529–537
Koichiro T, Daniel P, Glen S, Masatoshi N, Sudhir K (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Krinsky NI, Landrum JT, Bone RA (2003) Biologic mechanisms of the protective role of lutein and zeaxanthin in the eye. Annu Rev Nutr 23:171–201
Lee YS, Jeong H, Taye M, Kim HJ, Ka S, Ryu YC, Cho S (2015) Genome-wide association study (GWAS) and its application for improving the genomic estimated breeding values (GEBV) of the Berkshire pork quality traits. Asian Australas J Anim Sci 28:1551–1557
Li H, Durbin R (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25:1754–1760
Li L, Paolillo DJ, Parthasarathy MV, DiMuzio EM, Garvin DF (2001) A novel gene mutation that confers abnormal patterns of b-carotene accumulation in cauliflower (Brassica oleracea var. botrytis). Plant J 26:59–67
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA (2010) The Genome Analysis Toolkit: a map reduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
Moise AR, von Lintig J, Palczewski K (2005) Related enzymes solve evolutionarily recurrent problems in the metabolism of carotenoid. Trends Plant Sci 10:178–186
Rafalski JA (2010) Association genetics in crop improvement. Curr Opin Plant Biol 13:174–180
Rodriguez-Amaya DB (2001) A guide to carotenoid analysis in foods. ILSI Human Nutrition Institute, One Thomas Circle
Ruiz-Solaa MÁ, Rodríguez-Concepción M (2012) Carotenoid biosynthesis in Arabidopsis: a colorful pathway. Arabidopsis Book. https://doi.org/10.1199/tab.0158
Sagi M, Fluhr R, Lips SH (1999) Aldehyde oxidase and xanthine dehydrogenase in a flacca tomato mutant with deficient abscisic acid and wilty phenotype. Plant Physiol 120:571–578
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Sun X, Liu D, Zhang X, Li W, Liu H, Hong W, Jiang C, Guan N, Ma C, Zeng H, Xu C, Song J, Huang L, Wang C, Shi J, Wang R, Zheng X, Lu C, Wang X, Zheng H (2013) SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS One 8:e58700
Tajima F (1983) Evolutionary relationship of DNA sequence in finite populations. Genetics 105:437–460
Umehara M, Hanada A, Yoshida S, Akiyama K, Arite T, Takeda-Kamiya N, Magome H, Kamiya Y, Shirasu K, Yoneyama K, Kyozuka J, Yamaguchi S (2008) Inhibition of shoot branching by new terpenoid plant hormones. Nature 455:195–200
Wang W, Zhang T, Zhang G, Wang J, Han K, Wang Y, Zhang Y (2015) Genome-wide association study of antibody level response to NDV and IBV in Jinghai yellow chicken based on SLAF-seq technology. J Appl Genet 56:365–373
Welsch R, Arango J, Bär C, Salazar B, Al-Babili S, Beltrán J, Chavarriaga P, Ceballos H, Tohme J, Beyera P (2010) Provitamin A accumulation in cassava (Manihot esculenta) roots driven by a single nucleotide polymorphism in a phytoene synthase gene. Plant Cell 22:3348–3356
Wu X, Ren C, Joshi T, Vuong T, Xu D, Nguyen H (2010) SNP discovery by high-throughput sequencing in soybean. BMC Genome 11:469
Zhang YG, Fang BP (2006) Descriptors and data standard for sweet potato [Ipomoea batatas (L.) Lam.]. China Agriculture Press, Beijing (in Chinese)
Zhang P, Zhu Y, Wang L, Chen L, Zhou S (2015) Mining candidate genes associated with powdery mildew resistance in cucumber via super-BSA by specific length amplified fragment (SLAF) sequencing. BMC Genom 16:1
Zhao X, Han Y, Li Y, Liu D, Sun M, Zhao Y, Lv C, Li D, Yang Z, Huang L, Teng W, Qiu L, Zheng H, Li W (2015) Loci and candidate gene identification for resistance to Sclerotinia sclerotiorum in soybean (Glycine max L. Merr.) via association and linkage maps. Plant J 82:245–255
Acknowledgements
This work was supported by NSFC (No. 31271776), NSFC-CGIAR International (Regional) Cooperation and Exchange Programs (No. 31361140366), Open Fund Project of Ministry of Agriculture Key Laboratory (1630032014053), National Science and Technology Infrastructure Program (2015BAD15B01), and the Initial Fund of High-level Creative Talents in Hainan Province.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by J.-H. Liu.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Luo, X., Tomlins, K.I., Carvalho, L.J.C.B. et al. The analysis of candidate genes and loci involved with carotenoid metabolism in cassava (Manihot esculenta Crantz) using SLAF-seq. Acta Physiol Plant 40, 66 (2018). https://doi.org/10.1007/s11738-018-2634-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-018-2634-7