
Abstract
Various metals are essential nutrients in humans, and metal shortages lead to a variety of deficiency diseases. Metal concentration abnormalities may cause metal deposition in the brain, and magnetic resonance imaging (MRI) is the most potent and sensitive technique now available for detecting metal deposition given the difficulties associated with performing brain tissue biopsy. However, the brain contains many kinds of metals that affect the signal intensity of MRI, which has led to numerous misunderstandings in the history of metal analysis. We reviewed the history of brain metal analysis with histologic findings. Typically, manganese overload causes high signal intensity on T1-weighted images (T1WI) in the globus pallidus, iron overload causes low signal intensity in the globus pallidus on T2-weighted images, and gadolinium deposition causes high signal intensity in the dentate nucleus, globus pallidus, and pulvinar of thalamus on T1WI. However, because nonparamagnetic materials and other coexisting metals also affect the signal intensity of brain MRI, the quantitative analysis of metal concentrations is difficult. Thus, when analyzing metal deposition using MRI, caution should be exercised when interpreting the validity and reliability of the obtained data.
Similar content being viewed by others

Introduction
Metals play an important role in diverse biochemical and physiological functions in humans, and various enzymes and vitamins contain metals. Several metals are essential nutrients in humans, and metal shortages lead to a variety of deficiency diseases or syndromes. On the other hand, excess amounts of such metals induce cellular and tissue damage, leading to a variety of adverse effects and diseases. The appropriate concentration of a metal lies within a very narrow range, and any excess or shortage easily causes toxic effects in vivo [1–3]. The method for quantifying the metal content in affected organs is important not only for diagnostic purposes but also for the assessment of disease progression and treatment effects. Magnetic resonance imaging (MRI) is the most potent and sensitive technique for detecting abnormal metal deposition; this technique involves evaluating small changes in relaxation times [4, 5]. Most substances in the body are diamagnetic, and thus barely influence magnetic fields. On the other hand, most metals are classified as paramagnetic, and show magnetization only in the presence of a surrounding magnetic field. Paramagnetism influences the T1 and T2 relaxation times due to the influence of unpaired electrons. Some metals, such as nonionic nickel, iron, and gadolinium (at temperatures below 18 °C), are ferromagnetic, and present very strong magnetization that causes metal artifacts in MRI. However, ferromagnetic metals do not exist in the body, except in surgical or traumatic implants [6]. Most metal deposition causes high signal intensity on T1-weighted images (T1WI) [7, 8] and low signal intensity on T2-weighted images (T2WI) [9], and these signal intensity changes are rather specific for metal deposition in vivo. This article focuses on the diseases associated with metal deposition in the brain and metal detectability on brain MRI.
Difficulties associated with metal analysis in the brain
Several factors make metal analysis uncertain. First, brain tissue from patients is not easily acquired. Many studies of metal deposition have based diagnoses on clinical or image analysis, while only a small number of studies have relied on histological analysis. However, except for brain tissue analysis, the cause of the abnormal signal intensity on MRI cannot be determined accurately [5]. Second, excess concentrations of multiple metals are often observed in the same subject. For example, patients with metal poisoning from exposure in mines frequently have excess deposition of multiple metals, since metal ores usually include multiple kinds of metals. In addition, abnormal metal storage in the human body occasionally affects the absorption of other metals. Iron deficiency induces enhanced absorption of iron mediated by greater expression of metal transporters of iron, as well as the increased absorption of other metals such as manganese, nickel, lead, and/or cadmium [10–13]. Third, the magnetism of a metal varies according to the oxidation state and the type of compound involved, and its signal intensity on MRI varies according to these parameters and the status of the metal. The oxidation state of a metal and the compound it is in frequently change in the body, so the MRI signal intensity of a metal is not constant [14, 15]. Below we review the effects of metals on brain MRIs in order of the atomic number of the metal.
Calcium
Calcium deposition in the brain occurs with Fahr disease, various metabolic diseases, and tumors [5]. Calcification is easily detected on CT, but it is difficult to detect on MRI because of the variety of signal intensities produced by calcified regions [16]. This variety of signal intensities from calcification is not caused by its paramagnetic effect but by a surface effect whereby the irregular surface of calcium restricts the motion of water close to the Larmor frequency, with the T1 relaxation time of water shortening as the protein content in the water increases. The surface effect varies according to the surface structure of the calcification, so the signal intensity from calcification on T1WI can vary even when there is the same concentration of calcium [17, 18]. On T1WI, the signal intensity of calcification is strongest at 30 % calcium concentration by weight, while on T2* the signal intensity and calcium concentration show a linear correlation [18, 19]. However, these results were obtained in vitro, and the true mechanism of the high signal intensity from the calcified region has not yet been determined because calcium tends to accumulate in the basal ganglia, where other paramagnetic metals such as iron and manganese also accumulate. Calcium causes a high signal intensity on T1WI due to the surface effect in vitro, but the cause of the high signal intensity from calcified regions in vivo is unclear.
Manganese
Manganese neurotoxicity was first reported by Couper [20] in 1837 in a case series of 5 manganese miners. Patients with manganese neurotoxicity suffer from Parkinson’s symptoms, including tremor in the extremities, gait disturbance, and whispering speech [20–22]. Manganese is highly paramagnetic, and MRI is expected to detect its presence in the brain. The first study used monkeys that were administered MnCl2 by intravenous injection or inhaled aerosol. Regardless of the administration route of MnCl2, an increasing signal intensity on MRI is observed in the caudate nucleus, globus pallidus, substantia nigra, ventromedial hypothalamus, and pituitary gland [23]. Even in man, a hyperintense globus pallidus was found in patients receiving long-term total parenteral nutrition therapy that included manganese [24] and in workers exposed to manganese [25–27], with these hyperintensities diminishing after cessation of the manganese exposure [25, 28, 29]. Intravenous administration or inhalation of manganese causes manganese deposition in the brain, but the oral intake of manganese rarely results in manganese deposition. Intellectual impairment in children after excess oral intake of manganese has been reported, albeit without histological proof [30]. Most orally ingested manganese does not remain in the human body, due to several mechanisms. Manganese is present in food and water, but the intestine adjusts manganese absorption and only 1–5 % of the manganese ingested is absorbed [31]. In addition, the absorbed excess manganese is promptly excreted into the bile from the liver and rarely causes manganese toxicity [22, 32]. Iron deficiency anemia promotes manganese absorption in the intestine and elevation of the manganese concentration in the blood, but the signal intensity of the globus pallidus on T1WI is not affected [33]. In this way, manganese neurotoxicity is caused by mine dust inhalation or intravenous administration but rarely by oral intake [34, 35]. The manganese metabolic pathway adapts to oral MRI contrast agents containing manganese, and the contrast effect of this oral contrast agent based on manganese is limited to liver and intestine [36, 37].
Patients with chronic hepatic failure also show a hyperintense globus pallidus and to a lesser extent putamen and red nucleus on T1WI (Fig. 1) [38, 39]. The pattern of MRI abnormalities resembles that of manganese overload. Patients with liver cirrhosis and high signal intensity in the globus pallidus on T1WI have higher blood manganese concentrations than healthy volunteers [40]. The hyperintense globus pallidus on T1WI seen in patients with chronic hepatic failure due to Alagille syndrome diminished 3 months after hepatic transplantation. A high manganese concentration in the blood and symptomatic manganese toxicity were reported to improve following hepatic transplantation [41]. Manganese is considered to be the cause of hyperintense globus pallidus in patients with chronic hepatic failure, but copper and iron as well as manganese concentrations are elevated in the globus pallidus of autopsy cases [42]. The hyperintense globus pallidus seen in hepatic failure occurs due to an elevated concentration of manganese in the blood, which passes through the portal-systemic collateral vessels without migrating through the liver [43]. The hyperintense globus pallidus observed in schistosomiasis [44], Osler–Weber–Rendu disease [45], and post-transjugular intrahepatic portosystemic shunt placement [46] is also considered to be related to portal-systemic shunting. On the other hand, a hyperintense globus pallidus sometimes develops in patients with chronic liver failure in the absence of portal-systemic shunting [47]. This is because liver dysfunction associated with insufficient biliary excretion of manganese leads to an increased manganese concentration in the blood and hyperintensity of the globus pallidus [48].

A 57-year-old man with liver cirrhosis. A hyperintense lesion is present in the globus pallidus (white arrow) on sagittal T1WI due to manganese deposition
Transient hyperintensity of the striatum on T1WI after an ischemic event is also associated with the manganese concentration. In a study on brief focal ischemia in rats, transient hyperintensity of the striatum was observed from 5 days to 4 weeks after the ischemia, as was a synchronous increase in the manganese concentration in the striatum. The increased manganese was due to increases in mitochondrial manganese superoxide dismutase and glutamine synthetase, which are manganese-containing metalloproteins. Endogenous manganese also can affect the signal intensity from the brain [49].
Iron
The iron distribution in the human brain was first uncovered by histochemical analysis (Prussian blue or Perls’ stain). However, more recently, biochemical analytical techniques such as atomic absorption spectrophotometry (AAS), inductively coupled plasma mass spectrometry (ICP-MS), or X-ray fluorescence spectrometry (XRF) has been used to evaluate the iron concentration in brain tissue owing to the precise and quantitative nature of the data obtained [4, 50]. The iron in the brain is divided into heme iron and non-heme iron. Heme iron is present in hemoglobin, whereas the non-heme iron includes low-molecular-weight complexes, metalloproteins such as transferrin, storage proteins such as ferritin and hemosiderin, and ionic iron. Ferritin and hemosiderin are the most important sources of iron-related signal changes in cerebral MRI, since ferritin and hemosiderin contain 33–88 % of the non-heme iron found in the brain [4, 50]. Ferritin, which is the main chemical form of iron in the brain, has a T2-shortening effect and shows a low signal intensity on T2WI. More than 0.01 mg/ml of iron can be detected on MRI in the laboratory [51], and the iron concentration in the human brain is up to 0.2 (mg/g tissue wet weight) in the globus pallidus, which is theoretically sufficient to evaluate the iron concentration with T2WI [50]. The iron concentration in the basal ganglia in the brain increases until 30 years of age on MRI [52] (Fig. 2). However, the iron concentration estimated via T2WI is not well correlated with the results of biochemical analysis [53, 54]. There are several reasons that the iron content estimated with T2WI is not accurate. One is that the regional T2 signal intensity is affected not only by iron but also by the water and myelin contents [4]. The second is that iron transforms various chemical structures in vivo, causing different paramagnetic effects. For example, oxyhemoglobin and deoxyhemoglobin both contain an Fe2+ species, but because of differences in their electronic structures, oxyhemoglobin is diamagnetic while deoxyhemoglobin is paramagnetic. However, apart from ferritin, which has been extensively studied, little is known about the magnetic properties of other forms of iron and their contributions to the MRI contrast [4]. The third reason is the clustering effect of iron particles. Even in the same chemical structures, the heterogeneous distribution of iron particles causes a T2-shortening effect (clustering effect) and a low signal intensity on T2WI [55–58]. As the correlation between the iron content and T2WI is questionable, T2*-weighted imaging [59], susceptibility-weighted imaging [60], and quantitative susceptibility mapping [61, 62] have been tested as methods of estimating the iron content. While the iron content can indeed by estimated more accurately with MRI using these techniques, the paramagnetic effects caused by other elements still affect such measurements. In addition, hemosiderin shows the best image contrast on T2*-weighted images and susceptibility-weighted images [63, 64], but ferritin shows more prominent image contrast on T2-weighted images obtained by conventional spin-echo images than on T2*-weighted ones [63].

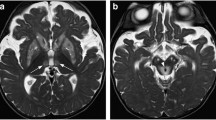
A 15-year-old female (a) and 43-year-old female (b), both without any past history of severe disease. Due to iron deposition, the signal intensity of the globus pallidus (white arrows) is lower in the former than in the latter on T2WI
Although various factors other than iron content also affect the estimated iron concentration in the brain on MRI, MRI is used to diagnose many kinds of iron-storage diseases, such as neurodegeneration with brain iron accumulation (NBIA) [65–68], Alzheimer’s disease [69, 70], Parkinson’s disease [71, 72], multiple sclerosis [73, 74], Friedreich’s ataxia [75, 76], and Huntington’s disease [53, 77]. NBIA is a group of neurodegenerative diseases characterized by iron accumulation in the basal ganglia. The diagnosis of NBIA has advanced with the development of MRI, gene analysis, and postmortem histological analysis. Ten disease-associated genes have been identified so far, but 20 % of NBIA cases remain genetically undefined. Only two of these NBIA genes are associated with iron homeostasis; the others are associated with mitochondria or fatty acid metabolism [66]. All of the NBIA disorders feature iron deposition in the globus pallidus but differ in their other findings. They are discriminated based on clinical movement disorder and neurodegenerative course [64]. In other iron-storage diseases, the true mechanism underlying the iron deposition is not known.
Iron has a T1-shortening effect, and its usefulness as a positive contrast agent for brain MRI [ultrasmall superparamagnetic iron oxides (USPIO)] has been examined. The signal intensity of iron on T1WI is influenced by the size of the iron particles [78], the clustering effect [55, 56], and T1WI parameters [79]. However, compared with gadolinium, the pattern of iron enhancement is heterogeneous and variable, so iron-based contrast agents cannot be used instead of gadolinium-based contrast agents (GBCAs) [80, 81]. The T1-shortening effect of iron is seldom used for the quantitative analysis of iron; instead, it is used to qualitatively check for the presence of iron. Subacute hemorrhage and melanin are typically identified by the presence of hyperintensity on T1WI [8]. The hyperintensity seen in subacute hemorrhage on T1WI is due to methemoglobin, which contains Fe3+. Even in artificial cerebral spinal fluid, hemoglobin changes to methemoglobin after incubation for 84 h at 38 ° C, just like the chemical change observed for NaNO2. When hemoglobin changes to methemoglobin, Fe2+ changes to Fe3+, leading to hyperintensity on T1WI [82, 83].
Melanin is a metal-binding protein that binds various metals in vivo. The signal intensity of melanin is related to the binding of melanin to paramagnetic metals such as Cu2+, Fe3+, Zn2+, and Mn2+; however, it shows a particularly strong affinity for Fe3+ which affects the signal intensity of melanin on T1WI [84, 85]. Melanin appears in the brain as neuromelanin [86] and in the presence of metastatic melanoma, primary diffuse meningeal melanomatosis, neurocutaneous melanosis, and Aspergillus infection [8]. The location and extent of a region of high signal intensity on T1WI are useful diagnostic findings. Neuromelanin is present mainly in the substantia nigra and locus coeruleus within the pons, and shows high signal intensity on T1WI in normal human brain. A loss of high signal intensity in these areas on T1WI indicates a decrease in the amount of neuromelanin, which can lead to a diagnosis of Parkinson’s disease [86].
Copper
Copper is another essential trace element, i.e., an important cofactor for many enzymes. Copper is obtained orally from the daily diet and is excreted via the biliary system. Copper metabolic abnormalities include Wilson’s disease, which is a condition associated with excessive copper accumulation, and Menkes disease, which is a condition related to copper deficiency [87–90]. Wilson’s disease is caused by a mutation in the gene for ATP7B, which binds copper to ceruloplasmin and facilitates the excretion of the copper–ceruloplasmin complex in the bile; the mutation leads to the gradual accumulation of copper in the body [89–91]. It is known that copper has a relatively small paramagnetic effect on MRI [92, 93] and that, on its own, copper accumulation does not directly cause signal intensity changes [91]. Brain MRI abnormalities in Wilson’s disease patients include brain atrophy, symmetric regional hyperintensity on T2WI, hypointensity in basal ganglia on T2WI, and a hyperintense globus pallidus on T1WI [94–96]. The “face of giant panda” sign characterized by hypointensity in the red nucleus and substantia nigra surrounded by hyperintensity in the midbrain is known to be characteristic of patients with Wilson’s disease, although it is only rarely seen [96, 97]. Hyperintensity on T2WI is observed in putamen (72 %), caudate (61 %), thalami (58 %), midbrain (49 %), pons (20 %), cerebral white matter (25 %), cortex (9 %), medulla (12 %), and cerebellum (10 %) [96], but these signal changes are the opposite to those produced by the paramagnetic effect of copper. The cause of this hyperintensity on T2WI is unclear, but edema, gliosis, neuronal necrosis, and cystic degeneration due to copper toxicity have all been proposed. Hyperintense regions on T2WI occasionally diminish after therapy, and this change may reflect edema and gliosis [98–100]. Hyperintensity of the globus pallidus on T1WI was initially attributed to copper accumulation [101], but manganese deposition due to liver dysfunction has more recently been implicated [91, 100]. Hypointensity of the substantia nigra, striatum, and globus pallidus on T2WI is believed to be due to increased accumulation of both copper and iron (Fig. 3) [91, 96, 102]. Ceruloplasmin, which transports not only copper but also iron, is decreased in Wilson’s disease, and this enhances iron accumulation in the body [89, 90]. Indeed, in autopsy studies, both copper and iron accumulation were found to be higher in brains with Wilson’s disease than in control brains [103, 104]. In addition, a PET study with radioactively labeled iron in human subjects showed increased iron uptake by the brain in Wilson’s disease patients compared to healthy volunteers [105]. In Wilson’s disease, the paramagnetic effects of manganese and iron and the neurotoxicity of copper greatly affect the abnormality of brain MRI.

A 43-year-old female with Wilson’s disease. The globus pallidus (white arrows), putamen (blue arrows), and pulvinar of thalamus (yellow arrows) are hypointense compared with a normal age-matched female (Fig. 2b). The hypointensity is probably caused by iron deposition due to low ceruloplasmin
Gadolinium
Gadolinium is used in MRI contrast media owing to the strong paramagnetic effect of the gadolinium ion. Because gadolinium is toxic to humans, it is injected along with chelating compounds that can prevent its toxicity by promoting its quick excretion from the body [106]. However, increased signal intensity in the dentate nucleus, globus pallidus, and pulvinar of thalamus on T1WI in patients subjected to repeated GBCA administration was recently reported for the first time. The administered gadolinium remains in the human body for a long time and causes abnormal signal intensity on T1WI (Fig. 4) [106–108]. Because of the toxicity of gadolinium, residual gadolinium has been attracting increasing attention [109–116]. These regions show increased signal with the administration of linear-type GBCAs but not macrocyclic-type GBCAs; because linear-type GBCAs are more unstable than macrocyclic-type GBCAs, it is strongly suspected that the change in signal is caused by the release of free gadolinium ion from the chelating compound in the GBCA [116–118]. In autopsy studies, gadolinium is detected in the brain tissue from patients with a history of GBCA administration using ICP-MS [119–121]. The signal intensity on T1WI and the residual gadolinium in these brain regions are both correlated with the total dose of GBCA administered, and the gadolinium deposition is clearly due to past GBCA administration [106]. Animal studies have also confirmed that the signal from the dentate nucleus on T1WI is affected by previous gadolinium administration, and that gadolinium is deposited [122, 123], but none of these clinical phenomena have been reported in connection with gadolinium deposition in the brain in the presence of normal renal function [106].

A 63-year-old man with a history of multiple linear-type GBCA administration. Hyperintense lesions are present on the globus pallidus (white arrow) and dentate nucleus (red arrow) on sagittal T1WI due to gadolinium deposition
Mercury
Mercury occurs naturally in three major forms: elemental mercury, inorganic compounds, and organic compounds. Elemental mercury can cross the blood–brain barrier and cause neurotoxicity. The first outbreak of methylmercury poisoning occurred in Japan around Minamata Bay. The mercury was discharged by an industrial plant, and biological accumulation occurred in the fish and shellfish in this bay. The patients developed a progressive neurologic illness (Minamata disease) caused by the ingestion of methylmercury-contaminated seafood. In the acute phase, high signal intensity regions appear in brain white matter on T2WI [124, 125]. Organic mercury poisoning causes central nerve and peripheral nerve toxicity. In the chronic phase of organic mercury poisoning, atrophy of the calcarine area, cerebellum, and postcentral gyri is observed [125, 126]. However, no specific signal intensity due to histologically proven mercury deposition has been reported.
References
Tchounwou PB, Yedjou CG, Patlolla AK, Sutton DJ. Heavy metal toxicity and the environment. EXS. 2012;101:133–64.
Jomova K, Valko M. Advances in metal-induced oxidative stress and human disease. Toxicology. 2011;283:65–87.
Jaishankar M, Tseten T, Anbalagan N, Mathew BB, Beeregowda KN. Toxicity, mechanism and health effects of some heavy metals. Interdiscip Toxicol. 2014;7:60–72.
Dusek P, Dezortova M, Wuerfel J. Imaging of iron. Int Rev Neurobiol. 2013;110:195–239.
Valdés Hernández Mdel C, Maconick LC, Tan EM, Wardlaw JM. Identification of mineral deposits in the brain on radiological images: a systematic review. Eur Radiol. 2012;22:2371–81.
Wood ML, Hardy PA. Proton relaxation enhancement. J Magn Reson Imaging. 1993;3:149–56.
Zimny A, Zińska L, Bladowska J, Neska-Matuszewska M, Sąsiadek M. Intracranial lesions with high signal intensity on T1-weighted MR images—review of pathologies. Pol J Radiol. 2013;78:36–46.
Ginat DT, Meyers SP. Intracranial lesions with high signal intensity on T1-weighted MR images: differential diagnosis. Radiographics. 2012;32:499–516.
Zimny A, Neska-Matuszewska M, Bladowska J, Sąsiadek MJ. Intracranial lesions with low signal intensity on T2-weighted MR images—review of pathologies. Pol J Radiol. 2015;80:40–50.
Bressler JP, Olivi L, Cheong JH, Kim Y, Maerten A, Bannon D. Metal transporters in intestine and brain: their involvement in metal-associated neurotoxicities. Hum Exp Toxicol. 2007;26:221–9.
Knöpfel M, Zhao L, Garrick MD. Transport of divalent transition-metal ions is lost in small-intestinal tissue of b/b Belgrade rats. Biochemistry. 2005;44:3454–65.
Park JD, Cherrington NJ, Klaassen CD. Intestinal absorption of cadmium is associated with divalent metal transporter 1 in rats. Toxicol Sci. 2002;68:288–94.
Ragan HA. Effects of iron deficiency on the absorption and distribution of lead and cadmium in rats. J Lab Clin Med. 1977;90:700–6.
Westbrook C, Roth CK, Talbot J. MRI in Practice. In: Westbrook C, Roth CK, Talbot J, editors. MRI in practice. 4th ed. Philadelphia: Wiley-Blackwell; 2011. p. 372–95.
Runge VM, Clanton JA, Lukehart CM, Partain CL, James AE Jr. Paramagnetic agents for contrast-enhanced NMR imaging: a review. AJR Am J Roentgenol. 1983;141:1209–15.
Tsuruda JS, Bradley WG. MR detection of intracranial calcification: a phantom study. AJNR Am J Neuroradiol. 1987;8:1049–55.
Saito M, Matsumoto K, Sakamoto M, Iikubo M, Sugawara Y, Sasano T. Experimental study on MR signal intensity changes with the degree of calcification. Dent Radiol. 2004;44:104–9.
Henkelman RM, Watts JF, Kucharczyk W. High signal intensity in MR images of calcified brain tissue. Radiology. 1991;179:199–206.
Kucharczyk W, Henkelman RM. Visibility of calcium on MR and CT: can MR show calcium that CT cannot? AJNR Am J Neuroradiol. 1994;15:1145–8.
Couper J. On the effects of black oxide of manganese when inhaled into the lungs. Br Ann Med Pharm. 1837;1:41.
Mena I, Marin O, Fuenzalida S, Cotzias GC. Chronic manganese poisoning. Clinical picture and manganese turnover. Neurology. 1967;17:128–36.
Tuschl K, Mills PB, Clayton PT. Manganese and the brain. Int Rev Neurobiol. 2013;110:277–312.
Newland MC, Ceckler TL, Kordower JH, Weiss B. Visualizing manganese in the primate basal ganglia with magnetic resonance imaging. Exp Neurol. 1989;106:251–8.
Mirowitz SA, Westrich TJ, Hirsch JD. Hyperintense basal ganglia on T1-weighted MR images in patients receiving parenteral nutrition. Radiology. 1991;181:117–20.
Nelson K, Golnick J, Korn T, Angle C. Manganese encephalopathy: utility of early magnetic resonance imaging. Br J Ind Med. 1993;50:510–3.
Kim Y, Kim KS, Yang JS, Park IJ, Kim E, Jin Y, et al. Increase in signal intensities on T1-weighted magnetic resonance images in asymptomatic manganese-exposed workers. Neurotoxicology. 1999;20:901–7.
Shin YC, Kim E, Cheong HK, Cho S, Sakong J, Kim KS, et al. High signal intensity on magnetic resonance imaging as a predictor of neurobehavioral performance of workers exposed to manganese. Neurotoxicology. 2007;28:257–62.
Mirowitz SA, Westrich TJ. Basal ganglial signal intensity alterations: reversal after discontinuation of parenteral manganese administration. Radiology. 1992;185:535–6.
Ejima A, Imamura T, Nakamura S, Saito H, Matsumoto K, Momono S. Manganese intoxication during total parenteral nutrition. Lancet. 1992;339:426.
Bouchard MF, Sauvé S, Barbeau B, Legrand M, Brodeur MÈ, Bouffard T, et al. Intellectual impairment in school-age children exposed to manganese from drinking water. Environ Health Perspect. 2011;119:138–43.
Davidsson L, Cederblad A, Lönnerdal B, Sandström B. The effect of individual dietary components on manganese absorption in humans. Am J Clin Nutr. 1991;54:1065–70.
Roth JA. Homeostatic and toxic mechanisms regulating manganese uptake, retention, and elimination. Biol Res. 2006;39:45–57.
Kim Y, Park JK, Choi Y, Yoo CI, Lee CR, Lee H, et al. Blood manganese concentration is elevated in iron deficiency anemia patients, whereas globus pallidus signal intensity is minimally affected. Neurotoxicology. 2005;26:107–11.
Rivera-Mancía S, Ríos C, Montes S. Manganese accumulation in the CNS and associated pathologies. Biometals. 2011;24:811–25.
Roels H, Meiers G, Delos M, Ortega I, Lauwerys R, Buchet JP, et al. Influence of the route of administration and the chemical form (MnCl2, MnO2) on the absorption and cerebral distribution of manganese in rats. Arch Toxicol. 1997;71:223–30.
Thomsen HS, Loegager V, Noergaard H, Chabanova E, Moller JM, Sonne J. Oral manganese for liver imaging at three different field strengths. Acad Radiol. 2004;11:630–6.
Chabanova E, Thomsen HS, Løgager V, Moller JM, Brage K, Fogh K, et al. Effect of new manganese contrast agent on tissue intensities in human volunteers: comparison of 0.23, 0.6 and 1.5 T MRI, a part of a phase I trial. MAGMA. 2004;17:28–35.
Brunberg JA, Kanal E, Hirsch W, Van Thiel DH. Chronic acquired hepatic failure: MR imaging of the brain at 1.5 T. AJNR Am J Neuroradiol. 1991;12:909–14.
Zeneroli ML, Cioni G, Crisi G, Vezzelli C, Ventura E. Globus pallidus alterations and brain atrophy in liver cirrhosis patients with encephalopathy: an MR imaging study. Magn Reson Imaging. 1991;9:295–302.
Hauser RA, Zesiewicz TA, Rosemurgy AS, Martinez C, Olanow CW. Manganese intoxication and chronic liver failure. Ann Neurol. 1994;36:871–5.
Barron TF, Devenyi AG, Mamourian AC. Symptomatic manganese neurotoxicity in a patient with chronic liver disease: correlation of clinical symptoms with MRI findings. Pediatr Neurol. 1994;10:145–8.
Maeda H, Sato M, Yoshikawa A, Kimura M, Sonomura T, Terada M, Kishi K. Brain MR imaging in patients with hepatic cirrhosis: relationship between high intensity signal in basal ganglia on T1-weighted images and elemental concentrations in brain. Neuroradiology. 1997;39(8):546–50.
Inoue E, Hori S, Narumi Y, Fujita M, Kuriyama K, Kadota T, et al. Portal-systemic encephalopathy: presence of basal ganglia lesions with high signal intensity on MR images. Radiology. 1991;179:551–5.
Li Y, Qiang JW, Ju S. Brain MR imaging changes in patients with hepatic schistosomiasis japonicum without liver dysfunction. Neurotoxicology. 2013;35:101–5.
Oikonomou A, Chatzistefanou A, Zezos P, Mintzopoulou P, Vadikolias K, Prassopoulos P. Basal ganglia hyperintensity on T1-weighted MRI in Rendu–Osler–Weber disease. J Magn Reson Imaging. 2012;35:426–30.
Krieger S, Jauss M, Jansen O, Stiehl A, Sauer P, Geissler M, et al. MRI findings in chronic hepatic encephalopathy depend on portosystemic shunt: results of a controlled prospective clinical investigation. J Hepatol. 1997;27:121–6.
Skehan S, Norris S, Hegarty J, Owens A, MacErlaine D. Brain MRI changes in chronic liver disease. Eur Radiol. 1997;7:905–9.
Rose C, Butterworth RF, Zayed J, Normandin L, Todd K, Michalak A, et al. Manganese deposition in basal ganglia structures results from both portal-systemic shunting and liver dysfunction. Gastroenterology. 1999;117:640–4.
Fujioka M, Taoka T, Matsuo Y, Mishima K, Ogoshi K, Kondo Y, et al. Magnetic resonance imaging shows delayed ischemic striatal neurodegeneration. Ann Neurol. 2003;54:732–47.
Haacke EM, Cheng NY, House MJ, Liu Q, Neelavalli J, Ogg RJ, et al. Imaging iron stores in the brain using magnetic resonance imaging. Magn Reson Imaging. 2005;23:1–25.
Reimer P, Allkemper T, Schuierer G, Peters PE. Brain imaging: reduced sensitivity of RARE-derived techniques to susceptibility effects. J Comput Assist Tomogr. 1996;20:201–5.
Aoki S, Okada Y, Nishimura K, Barkovich AJ, Kjos BO, Brasch RC, et al. Normal deposition of brain iron in childhood and adolescence: MR imaging at 1.5 T. Radiology. 1989;172:381–5.
Chen JC, Hardy PA, Kucharczyk W, Clauberg M, Joshi JG, Vourlas A, et al. MR of human postmortem brain tissue: correlative study between T2 and assays of iron and ferritin in Parkinson and Huntington disease. AJNR Am J Neuroradiol. 1993;14:275–81.
Chen JC, Hardy PA, Clauberg M, Joshi JG, Parravano J, Deck JH, et al. T2 values in the human brain: comparison with quantitative assays of iron and ferritin. Radiology. 1989;173:521–6.
Tanimoto A, Pouliquen D, Kreft BP, Stark DD. Effects of spatial distribution on proton relaxation enhancement by particulate iron oxide. J Magn Reson Imaging. 1994;4:653–7.
Tanimoto A, Oshio K, Suematsu M, Pouliquen D, Stark DD. Relaxation effects of clustered particles. J Magn Reson Imaging. 2001;14:72–7.
Gossuin Y, Gillis P, Muller RN, Hocq A. Relaxation by clustered ferritin: a model for ferritin-induced relaxation in vivo. NMR Biomed. 2007;20:749–56.
Gossuin Y, Muller RN, Gillis P. Relaxation induced by ferritin: a better understanding for an improved MRI iron quantification. NMR Biomed. 2004;17:427–32.
Yao B, Li TQ, Gelderen Pv, Shmueli K, de de Zwart JA, Duyn JH. Susceptibility contrast in high field MRI of human brain as a function of tissue iron content. Neuroimage. 2009;44:1259–66.
Langkammer C, Krebs N, Goessler W, Scheurer E, Ebner F, Yen K, Fazekas F, Ropele S. Quantitative MR imaging of brain iron: a postmortem validation study. Radiology. 2010;257:455–62.
Hopp K, Popescu BF, McCrea RP, Harder SL, Robinson CA, Haacke ME, et al. Brain iron detected by SWI high pass filtered phase calibrated with synchrotron X-ray fluorescence. J Magn Reson Imaging. 2010;31:1346–54.
Zheng W, Nichol H, Liu S, Cheng YC, Haacke EM. Measuring iron in the brain using quantitative susceptibility mapping and X-ray fluorescence imaging. Neuroimage. 2013;78:68–74.
Haque TL, Miki Y, Kanagaki M, Takahashi T, Yamamoto A, Konishi J, et al. MR contrast of ferritin and hemosiderin in the brain: comparison among gradient-echo, conventional spin-echo and fast spin-echo sequences. Eur J Radiol. 2003;48:230–6.
Mori N, et al. Microbleeds in moyamoya disease: susceptibility-weighted imaging versus T2*-weighted imaging at 3 Tesla. Investig Radiol. 2008;43:574–9.
Kruer MC, Boddaert N, Schneider SA, Houlden H, Bhatia KP, Gregory A, et al. Neuroimaging features of neurodegeneration with brain iron accumulation. AJNR Am J Neuroradiol. 2012;33:407–14.
Colombelli C, Aoun M, Tiranti V. Defective lipid metabolism in neurodegeneration with brain iron accumulation (NBIA) syndromes: not only a matter of iron. J Inherit Metab Dis. 2015;38:123–36.
Levi S, Finazzi D. Neurodegeneration with brain iron accumulation: update on pathogenic mechanisms. Front Pharmacol. 2014;5:99.
Arber CE, Li A, Houlden H, Wray S. Insights into molecular mechanisms of disease in neurodegeneration with brain iron accumulation: unifying theories. Neuropathol Appl Neurobiol. 2015;. doi:10.1111/nan.12242.
Zhu WZ, Zhong WD, Wang W, Zhan CJ, Wang CY, Qi JP, et al. Quantitative MR phase-corrected imaging to investigate increased brain iron deposition of patients with Alzheimer disease. Radiology. 2009;253:497–504.
van Rooden S, Doan NT, Versluis MJ, Goos JD, Webb AG, Oleksik AM, et al. 7T T2*-weighted magnetic resonance imaging reveals cortical phase differences between early- and late-onset Alzheimer’s disease. Neurobiol Aging. 2015;36:20–6.
Drayer BP, Olanow W, Burger P, Johnson GA, Herfkens R, Riederer S. Parkinson plus syndrome: diagnosis using high field MR imaging of brain iron. Radiology. 1986;159:493–8.
Griffiths PD, Dobson BR, Jones GR, Clarke DT. Iron in the basal ganglia in Parkinson’s disease. An in vitro study using extended X-ray absorption fine structure and cryo-electron microscopy. Brain. 1999;122:667–73.
Craelius W, Migdal MW, Luessenhop CP, Sugar A, Mihalakis I. Iron deposits surrounding multiple sclerosis plaques. Arch Pathol Lab Med. 1982;106:397–9.
Stankiewicz JM, Neema M, Ceccarelli A. Iron and multiple sclerosis. Neurobiol Aging. 2014;35:S51–8.
Gordon N. Friedreich’s ataxia and iron metabolism. Brain Dev. 2000;22:465–8.
Waldvogel D, van Gelderen P, Hallett M. Increased iron in the dentate nucleus of patients with Friedrich’s ataxia. Ann Neurol. 1999;46(1):123–5.
Rosas HD, Chen YI, Doros G, Salat DH, Chen NK, Kwong KK, et al. Alterations in brain transition metals in Huntington disease: an evolving and intricate story. Arch Neurol. 2012;69:887–93.
Varallyay P, Nesbit G, Muldoon LL, Nixon RR, Delashaw J, Cohen JI, et al. Comparison of two superparamagnetic viral-sized iron oxide particles ferumoxides and ferumoxtran-10 with a gadolinium chelate in imaging intracranial tumors. AJNR Am J Neuroradiol. 2002;23:510–9.
Takahama K, Amano Y, Hayashi H, Kumazaki T. T1-weighted magnetic resonance imaging sequence appropriate for the evaluation of the longitudinal relaxation effect of superparamagnetic iron oxide: a phantom study. J Nippon Med Sch. 2002;69:571–6.
Taschner CA, Wetzel SG, Tolnay M, Froehlich J, Merlo A, Radue EW. Characteristics of ultrasmall superparamagnetic iron oxides in patients with brain tumors. AJR Am J Roentgenol. 2005;185:1477–86.
Neuwelt EA, Várallyay P, Bagó AG, Muldoon LL, Nesbit G, Nixon R. Imaging of iron oxide nanoparticles by MR and light microscopy in patients with malignant brain tumours. Neuropathol Appl Neurobiol. 2004;30:456–71.
Bradley WG Jr, Schmidt PG. Effect of methemoglobin formation on the MR appearance of subarachnoid hemorrhage. Radiology. 1985;156:99–103.
Gomori JM, Grossman RI, Goldberg HI, Zimmerman RA, Bilaniuk LT. Intracranial hematomas: imaging by high-field MR. Radiology. 1985;157:87–93.
Enochs WS, Hyslop WB, Bennett HF, Brown RD 3rd, Koenig SH, Swartz HM. Sources of the increased longitudinal relaxation rates observed in melanotic melanoma. An in vitro study of synthetic melanins. Investig Radiol. 1989;24:794–804.
Enochs WS, Petherick P, Bogdanova A, Mohr U, Weissleder R. Paramagnetic metal scavenging by melanin: MR imaging. Radiology. 1997;204:417–23.
Nakamura K, Sugaya K. Neuromelanin-sensitive magnetic resonance imaging: a promising technique for depicting tissue characteristics containing neuromelanin. Neural Regen Res. 2014;9:759–60.
Seshadri R, Bindu PS, Gupta AK. Teaching NeuroImages: Menkes kinky hair syndrome. Neurology. 2013;81:e12–3.
Tümer Z. An overview and update of ATP7A mutations leading to Menkes disease and occipital horn syndrome. Hum Mutat. 2013;34:417–29.
Patil M, Sheth KA, Krishnamurthy AC, Devarbhavi H. A review and current perspective on Wilson disease. J Clin Exp Hepatol. 2013;3:321–36.
Hordyjewska A, Popiołek Ł, Kocot J. The many “faces” of copper in medicine and treatment. Biometals. 2014;27:611–21.
Dusek P, Roos PM, Litwin T, Schneider SA, Flaten TP, Aaseth J. The neurotoxicity of iron, copper and manganese in Parkinson’s and Wilson’s diseases. J Trace Elem Med Biol. 2015;31:193–203.
Runge VM, Stewart RG, Clanton JA, Jones MM, Lukehart CM, Partain CL, et al. Work in progress: potential oral and intravenous paramagnetic NMR contrast agents. Radiology. 1983;147:789–91.
Vymazal J, Bulte JW, Frank JA, Di Chiro G, Brooks RA. Frequency dependence of MR relaxation times. I. Paramagnetic ions. J Magn Reson Imaging. 1993;3:637–40.
Starosta-Rubinstein S, Young AB, Kluin K, Hill G, Aisen AM, Gabrielsen T, et al. Clinical assessment of 31 patients with Wilson’s disease. Correlations with structural changes on magnetic resonance imaging. Arch Neurol. 1987;44:365–70.
Aisen AM, Martel W, Gabrielsen TO, Glazer GM, Brewer G, Young AB, et al. Wilson disease of the brain: MR imaging. Radiology. 1985;157:137–41.
Sinha S, Taly AB, Ravishankar S, Prashanth LK, Venugopal KS, Arunodaya GR, Vasudev MK, et al. Wilson’s disease: cranial MRI observations and clinical correlation. Neuroradiology. 2006;48:613–21.
Hitoshi S, Iwata M, Yoshikawa K. Mid-brain pathology of Wilson’s disease: MRI analysis of three cases. J Neurol Neurosurg Psychiatry. 1991;54:624–6.
Roh JK, Lee TG, Wie BA, Lee SB, Park SH, Chang KH. Initial and follow-up brain MRI findings and correlation with the clinical course in Wilson’s disease. Neurology. 1994;44:1064–8.
Sinha S, Taly AB, Prashanth LK, Ravishankar S, Arunodaya GR, Vasudev MK. Sequential MRI changes in Wilson’s disease with de-coppering therapy: a study of 50 patients. Br J Radiol. 2007;80:744–9.
Kim TJ, Kim IO, Kim WS, Cheon JE, Moon SG, Kwon JW, et al. MR imaging of the brain in Wilson disease of childhood: findings before and after treatment with clinical correlation. AJNR Am J Neuroradiol. 2006;27:1373–8.
Mochizuki H, Kamakura K, Masaki T, Okano M, Nagata N, Inui A, et al. Atypical MRI features of Wilson’s disease: high signal in globus pallidus on T1-weighted images. Neuroradiology. 1997;39:171–4.
Lee JH, Yang TI, Cho M, Yoon KT, Baik SK, Han YH. Widespread cerebral cortical mineralization in Wilson’s disease detected by susceptibility-weighted imaging. J Neurol Sci. 2012;313:54–6.
Cumings JN. The copper and iron content of brain and liver in the normal and in hepato-lenticular degeneration. Brain. 1948;71:410–5.
Litwin T, Gromadzka G, Szpak GM, Jabłonka-Salach K, Bulska E, Członkowska A. Brain metal accumulation in Wilson’s disease. J Neurol Sci. 2013;329:55–8.
Bruehlmeier M, Leenders KL, Vontobel P, Calonder C, Antonini A, Weindl A. Increased cerebral iron uptake in Wilson’s disease: a 52Fe-citrate PET study. J Nucl Med. 2000;41:781–7.
Kanda T, Oba H, Toyoda K, Kitajima K, Furui S. Brain gadolinium deposition after administration of gadolinium-based contrast agents. Jpn J Radiol. 2016;34:3–9.
Kanda T, Ishii K, Kawaguchi H, Kitajima K, Takenaka D. High signal intensity in the dentate nucleus and globus pallidus on unenhanced T1-weighted MR images: relationship with increasing cumulative dose of a gadolinium-based contrast material. Radiology. 2014;270:834–41.
Errante Y, Cirimele V, Mallio CA, Di Lazzaro V, Zobel BB, Quattrocchi CC. Progressive increase of T1 signal intensity of the dentate nucleus on unenhanced magnetic resonance images is associated with cumulative doses of intravenously administered gadodiamide in patients with normal renal function, suggesting dechelation. Investig Radiol. 2014;49:685–90.
Kanal E, Tweedle MF. Residual or retained gadolinium: practical implications for radiologists and our patients. Radiology. 2015;275:630–4.
Ramalho J, Semelka RC, Ramalho M, Nunes RH, AlObaidy M, Castillo M. Gadolinium-based contrast agent accumulation and toxicity: an update. AJNR Am J Neuroradiol. 2015;. doi:10.3174/ajnr.A4615.
Huckle JE, Altun E, Jay M, Semelka RC. Gadolinium deposition in humans: when did we learn that gadolinium was deposited in vivo? Investig Radiol. 2015;. doi:10.1097/RLI.0000000000000228.
Beomonte Zobel B, Quattrocchi CC, Errante Y, Grasso RF. Gadolinium-based contrast agents: did we miss something in the last 25 years? Radiol Med. 2015;. doi:10.1007/s11547-015-0614-1.
Montagne A, Toga AW, Zlokovic BV. Blood–brain barrier permeability and gadolinium: benefits and potential pitfalls in research. JAMA Neurol. 2016;73:13–4.
Malayeri AA, Brooks KM, Bryant LH, Evers R, Kumar P, Reich DS, et al. National Institutes of Health perspective on reports of gadolinium deposition in the brain. J Am Coll Radiol. 2016;. doi:10.1016/j.jacr.2015.11.009.
Guermazi A, Kressel HY. Getting published in radiology: a deputy editor’s perspective. Jpn J Radiol. 2015;33:678–85.
Kanda T, Osawa M, Oba H, Toyoda K, Kotoku J, Haruyama T, et al. High signal intensity in dentate nucleus on unenhanced T1-weighted MR images: association with linear versus macrocyclic gadolinium chelate administration. Radiology. 2015;275:803–9.
Radbruch A, Weberling LD, Kieslich PJ, Eidel O, Burth S, Kickingereder P, et al. Gadolinium retention in the dentate nucleus and globus pallidus is dependent on the class of contrast agent. Radiology. 2015;275:783–91.
Cao Y, Huang DQ, Shih G, Prince MR. Signal change in the dentate nucleus on T1-weighted mr images after multiple administrations of gadopentetate dimeglumine versus gadobutrol. AJR Am J Roentgenol. 2016;206:414–9.
McDonald RJ, McDonald JS, Kallmes DF, Jentoft ME, Murray DL, Thielen KR, et al. Intracranial gadolinium deposition after contrast-enhanced MR imaging. Radiology. 2015;275:772–82.
Kanda T, Fukusato T, Matsuda M, Toyoda K, Oba H, Kotoku J, et al. Gadolinium-based contrast agent accumulates in the brain even in subjects without severe renal dysfunction: evaluation of autopsy brain specimens with inductively coupled plasma mass spectroscopy. Radiology. 2015;276:228–32.
Murata N, Gonzalez-Cuyar LF, Murata K, Fligner C, Dills R, Hippe D, et al. Macrocyclic and other non-group 1 gadolinium contrast agents deposit low levels of gadolinium in brain and bone tissue: preliminary results from 9 patients with normal renal function. Investig Radiol. 2016;. doi:10.1097/RLI.0000000000000252.
Robert P, Violas X, Grand S, Lehericy S, Idée JM, Ballet S, Corot C. Linear gadolinium-based contrast agents are associated with brain gadolinium retention in healthy rats. Investig Radiol. 2016;51:73–82.
Jost G, Lenhard DC, Sieber MA, Lohrke J, Frenzel T, Pietsch H. Signal increase on unenhanced T1-weighted images in the rat brain after repeated, extended doses of gadolinium-based contrast agents: comparison of linear and macrocyclic agents. Investig Radiol. 2016;51:83–9.
Taber KH, Hurley RA. Mercury exposure: effects across the lifespan. J Neuropsychiatry Clin Neurosci. 2008;20:384–9.
Korogi Y, Takahashi M, Shinzato J, Okajima T. MR findings in seven patients with organic mercury poisoning (Minamata disease). AJNR Am J Neuroradiol. 1994;15:1575–8.
Korogi Y, Takahashi M, Okajima T, Eto K. MR findings of Minamata disease—organic mercury poisoning. J Magn Reson Imaging. 1998;8:308–16.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
About this article

Cite this article
Kanda, T., Nakai, Y., Aoki, S. et al. Contribution of metals to brain MR signal intensity: review articles. Jpn J Radiol 34, 258–266 (2016). https://doi.org/10.1007/s11604-016-0532-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11604-016-0532-8