
Abstract
Probabilistic models over strings have played a key role in developing methods that take into consideration indels as phylogenetically informative events. There is an extensive literature on using automata and transducers on phylogenies to do inference on these probabilistic models, in which an important theoretical question is the complexity of computing the normalization of a class of string-valued graphical models. This question has been investigated using tools from combinatorics, dynamic programming, and graph theory, and has practical applications in Bayesian phylogenetics. In this work, we revisit this theoretical question from a different point of view, based on linear algebra. The main contribution is a set of results based on this linear algebra view that facilitate the analysis and design of inference algorithms on string-valued graphical models. As an illustration, we use this method to give a new elementary proof of a known result on the complexity of inference on the “TKF91” model, a well-known probabilistic model over strings. Compared to previous work, our proving method is easier to extend to other models, since it relies on a novel weak condition, triangular transducers, which is easy to establish in practice. The linear algebra view provides a concise way of describing transducer algorithms and their compositions, opens the possibility of transferring fast linear algebra libraries (for example, based on GPUs), as well as low rank matrix approximation methods, to string-valued inference problems.










Similar content being viewed by others


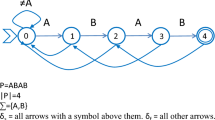
Notes
In reversible models such as the TKF91 model, an arbitrary root can be selected without changing the likelihood. In this case, the root is used for computational convenience and has no special phylogenetic meaning.
Note that we use the convention of using \(\hat{\sigma}\) when the character is an element of the extended set of characters \(\hat{\varSigma}\).
In technical terms, we use the Mealy model.
The inverse problem, learning the values for this map is also of interest (Holmes and Rubin 2002), but here we focus on the forward problem.
The rate matrix Q comes from a substitution model (we will assume the Jukes–Cantor model for simplicity, but all our results carry over to the General Time Reversible models (Felsenstein 2003)).
Note that we avoided the standard tensor product notation ⊗ because of a notation conflict with the automaton and transducer literature, in which ⊗ denotes multiplication in an abstract semiring (the generalization of normal multiplication, ⋅ used here). The operator ⊗ is also often overloaded to mean the product or concatenation of automata or transducers, which is not the same as the pointwise product as defined here.
We omit the factor at the root since its effect on the running time is a constant independent of L and N.
References
Airoldi, E. M. (2007). Getting started in probabilistic graphical models. PLoS Comput. Biol., 3(12).
Bishop, C. M. (2006). Pattern recognition and machine learning (pp. 359–422). Berlin: Springer. Chap. 8.
Bouchard-Côté, A., & Jordan, M. I. (2012). Evolutionary inference via the Poisson indel process. Proc. Nat. Acad. Sci. USA. doi:10.1073/pnas.1220450110.
Bouchard-Côté, A., Jordan, M. I., & Klein, D. (2009). Efficient inference in phylogenetic InDel trees. In Advances in neural information processing systems (Vol. 21).
Bouchard-Côté, A., Sankararaman, S., & Jordan, M. I. (2012). Phylogenetic inference via sequential Monte Carlo. Syst. Biol., 61, 579–593.
Bradley, R. K., & Holmes, I. (2007). Transducers: an emerging probabilistic framework for modeling indels on trees. Bioinformatics, 23(23), 3258–3262.
Daskalakis, C., & Roch, S. (2012). Alignment-free phylogenetic reconstruction: Sample complexity via a branching process analysis. Ann. Appl. Probab.
Dreyer, M., Smith, J. R., & Eisner, J. (2008). Latent-variable modeling of string transductions with finite-state methods. In Proceedings of EMNLP 2008.
Droste, M., & Kuich, W. (2009). Handbook of weighted automata. Monographs in theoretical computer science. Berlin: Springer. Chap. 1.
Eilenberg, S. (1974). Automata, languages and machines (Vol. A). San Diego: Academic Press.
Felsenstein, J. (1981). Evolutionary trees from DNA sequences: a maximum likelihood approach. J. Mol. Evol., 17, 368–376.
Felsenstein, J. (2003). Inferring phylogenies. Sunderland: Sinauer Associates.
Fernandez, P., Plateau, B., & Stewart, W. J. (1998). Optimizing tensor product computations in stochastic automata networks. RAIRO. Rech. Opér., 32(3), 325–351.
Görür, D., & Teh, Y. W. (2008). An efficient sequential Monte-Carlo algorithm for coalescent clustering. In Advances in neural information processing (pp. 521–528). Red Hook: Curran Associates.
Hein, J. (1990). A unified approach to phylogenies and alignments. Methods Enzymol., 183, 625–944.
Hein, J. (2000). A generalisation of the Thorne–Kishino–Felsenstein model of statistical alignment to k sequences related by a binary tree. In Pac. symp. biocomput. (pp. 179–190).
Hein, J. (2001). An algorithm for statistical alignment of sequences related by a binary tree. In Pac. symp. biocomput. (pp. 179–190).
Hein, J., Jensen, J., & Pedersen, C. (2003). Recursions for statistical multiple alignment. Proc. Natl. Acad. Sci. USA, 100(25), 14960–14965.
Higdon, D. M. (1998). Auxiliary variable methods for Markov Chain Monte Carlo with applications. J. Am. Stat. Assoc., 93(442), 585–595.
Holmes, I. (2003). Using guide trees to construct multiple-sequence evolutionary hmms. Bioinformatics, 19(1), 147–157.
Holmes, I. (2007). Phylocomposer and phylodirector: analysis and visualization of transducer indel models. Bioinformatics, 23(23), 3263–3264.
Holmes, I., & Bruno, W. J. (2001). Evolutionary HMM: a Bayesian approach to multiple alignment. Bioinformatics, 17, 803–820.
Holmes, I., & Rubin, G. M. (2002). An expectation maximization algorithm for training hidden substitution models. J. Mol. Biol.
Jensen, J., & Hein, J. (2002). Gibbs sampler for statistical multiple alignment (Technical report). Dept of Theor Stat, University of Aarhus.
Jordan, M. I. (2004). Graphical models. Stat. Sci., 19, 140–155.
Kawakita, A., Sota, T., Ascher, J. S., Ito, M., Tanaka, H., & Kato, M. (2003). Evolution and phylogenetic utility of alignment gaps within intron sequences of three nuclear genes in bumble bees (Bombus). Mol. Biol. Evol., 20(1), 87–92.
Knudsen, B., & Miyamoto, M. (2003). Sequence alignments and pair hidden Markov models using evolutionary history. J. Mol. Biol., 333, 453–460.
Langville, A. N., & Stewart, W. J. (2004). The Kronecker product and stochastic automata networks. J. Comput. Appl. Math., 167(2), 429–447.
Lunter, G., Miklós, I., Drummond, A., Jensen, J., & Hein, J. (2005). Bayesian coestimation of phylogeny and sequence alignment. BMC Bioinform., 6(1), 83.
Metzler, D., Fleissner, R., Wakolbinger, A., & von Haeseler, A. (2001). Assessing variability by joint sampling of alignments and mutation rates. J. Mol. Biol..
Miklós, I., & Toroczkai, Z. (2001). An improved model for statistical alignment. In First workshop on algorithms in bioinformatics, Berlin: Springer.
Miklós, I., Drummond, A., Lunter, G., & Hein, J. (2003a). Bayesian phylogenetic inference under a statistical insertion–deletion model. In Algorithms in bioinformatics, Berlin: Springer.
Miklós, I., Song, Y. S., Lunter, G. A., & Hein, J. (2003b). An efficient algorithm for statistical multiple alignment on arbitrary phylogenetic trees. J. Comput. Biol., 10, 869–889.
Miklós, I., Lunter, G. A., & Holmes, I. (2004). A long indel model for evolutionary sequence alignment. Mol. Biol. Evol., 21(3), 529–540.
Mingming, S. (2012). Gpumatrix library.
Mohri, M. (2002). Generic epsilon-removal and input epsilon-normalization algorithms for weighted transducers. Int. J. Found. Comput. Sci., 13(1), 129–143.
Mohri, M. (2009). Handbook of weighted automata. Monographs in theoretical computer science. Berlin: Springer. Chap. 6.
Novák, Á., Miklós, I., Lyngsoe, R., & Hein, J. (2008). Statalign: an extendable software package for joint Bayesian estimation of alignments and evolutionary trees. Bioinformatics, 24, 2403–2404.
Redelings, B. D., & Suchard, M. A. (2005). Joint Bayesian estimation of alignment and phylogeny. Syst. Biol., 54(3), 401–418.
Redelings, B. D., & Suchard, M. A. (2007). Incorporating indel information into phylogeny estimation for rapidly emerging pathogens. BMC Evol. Biol., 7(40).
Rivas, E. (2005). Evolutionary models for insertions and deletions in a probabilistic modeling framework. BMC Bioinform., 6(1), 63.
Satija, R., Pachter, L., & Hein, J. (2008). Combining statistical alignment and phylogenetic footprinting to detect regulatory elements. Bioinformatics, 24, 1236–1242.
Schützenberger, M. P. (1961). On the definition of a family of automata. Inf. Control, 4, 245–270.
Song, Y. S. (2006). A sufficient condition for reducing recursions in hidden Markov models. Bull. Math. Biol., 68, 361–384.
Steel, M., & Hein, J. (2001). Applying the Thorne–Kishino–Felsenstein model to sequence evolution on a star-shaped tree. Appl. Math. Lett., 14, 679–684.
Teh, Y. W., Daume, H. III, & Roy, D. M. (2008). Bayesian agglomerative clustering with coalescents. In Advances in neural information processing (pp. 1473–1480). Cambridge: MIT Press.
Thorne, J. L., Kishino, H., & Felsenstein, J. (1991). An evolutionary model for maximum likelihood alignment of DNA sequences. J. Mol. Evol., 33, 114–124.
Thorne, J. L., Kishino, H., & Felsenstein, J. (1992). Inching toward reality: an improved likelihood model of sequence evolution. J. Mol. Evol., 34, 3–16.
Westesson, O., Lunter, G., Paten, B., & Holmes, I. (2011). Phylogenetic automata, pruning, and multiple alignment. Preprint, arXiv:1103.4347.
Westesson, O., Lunter, G., Paten, B., & Holmes, I. (2012). Accurate reconstruction of insertion-deletion histories by statistical phylogenetics. PLoS ONE, 7(4), e34572.
Whaley, R.C., Petitet, A., & Dongarra, J. J. (2001). Automated empirical optimization of software and the ATLAS project. Parallel Comput., 27(1–2), 3–35.
Williams, V. V. (2012). Multiplying matrices faster than Coppersmith–Winograd. In STOC.
Wong, K. M., Suchard, M. A., & Huelsenbeck, J. P. (2008). Alignment uncertainty and genomic analysis. Science, 319(5862), 473–476.
Acknowledgements
I would like to thank Ian Holmes, Bonnie Kirkpatrick and the anonymous reviewers for their comments. This work was partially funded by a NSERC Discovery Grant and a Google Faculty Award, and computing was supported by WestGrid.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bouchard-Côté, A. A Note on Probabilistic Models over Strings: The Linear Algebra Approach. Bull Math Biol 75, 2529–2550 (2013). https://doi.org/10.1007/s11538-013-9906-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11538-013-9906-6