
Abstract
Purpose
There is robust evidence that people with schizophrenia show elevated dopamine (DA) synthesis capacity in the striatum. This finding comes from positron emission tomography (PET) studies using radiolabelled l-3,4-dihydroxyphenylalanine (18F-DOPA). DA synthesis capacity also appears to be elevated in the midbrain of people with schizophrenia compared to healthy controls. We therefore aimed to optimise a method to quantify 18F-DOPA uptake in the midbrain of mice, and to utilise this method to quantify DA synthesis capacity in the midbrain of the sub-chronic ketamine model of schizophrenia-relevant hyperdopaminergia.
Procedures
Adult male C57Bl6 mice were treated daily with either ketamine (30 mg/kg, i.p.) or vehicle (saline) for 5 days. On day 7, animals were administered 18F-DOPA (i.p.) and scanned in an Inveon PET/CT scanner. Data from the saline-treated group were used to optimise an atlas-based template to position the midbrain region of interest and to determine the analysis parameters which resulted in the greatest intra-group consistency. These parameters were then used to compare midbrain DA synthesis capacity (KiMod) between ketamine- and saline-treated animals.
Results
Using an atlas-based template to position the 3.7 mm3 midbrain ROI with a T*–Tend window of 15–140 min to estimate KiMod resulted in the lowest intra-group variability and moderate test–retest agreement. Using these parameters, we found that KiMod was elevated in the midbrain of ketamine-treated animals in comparison to saline-treated animals (t(22) = 2.19, p = 0.048). A positive correlation between DA synthesis capacity in the striatum and the midbrain was also evident in the saline-treated animals (r2 = 0.59, p = 0.005) but was absent in ketamine-treated animals (r2 = 0.004, p = 0.83).
Conclusions
Using this optimised method for quantifying 18F-DOPA uptake in the midbrain, we found that elevated striatal DA synthesis capacity in the sub-chronic ketamine model extends to the midbrain. Interestingly, the dysconnectivity between the midbrain and striatum seen in this model is also evident in the clinical population. This model may therefore be ideal for assessing novel compounds which are designed to modulate pre-synaptic DA synthesis capacity.
Similar content being viewed by others

Introduction
Ketamine is an antagonist of the glutamatergic N-methyl-d-aspartate (NMDA) receptor. NMDA receptor hypofunction may contribute to the neurobiology of schizophrenia [1, 2], and ketamine can be used to generate a preclinical model with relevance to schizophrenia [3,4,5,6]. When given sub-chronically at sub-anaesthetic doses, rodents display behavioural and neurochemical phenotypes relevant to schizophrenia [3, 7], and we have previously shown that the sub-chronic ketamine model results in elevated [18F]-l-3,4-dihydroxyphenylalanine (18F-DOPA) uptake in the striatum in mice [6, 8]. This mimics the increase in 18F-DOPA uptake in the striatum which is evident in people with schizophrenia [9,10,11]. This finding likely reflects enhanced dopamine (DA) synthesis capacity in the striatum and demonstrates that the sub-chronic ketamine model replicates this key aspect of schizophrenia neurobiology.
Dopaminergic innervation to the striatum comes from neurons located in the midbrain, specifically in the substantia nigra pars compacta (SNpc) and the ventral tegmental area (VTA; [12]). Dopaminergic dysfunction in the striatum may therefore originate in the midbrain. Indeed, there is now neuroimaging and post-mortem evidence of dopaminergic dysfunction in the midbrain of people with schizophrenia [13,14,15]. For instance, a recent meta-analysis found an increase in neuromelanin in the midbrain of people with schizophrenia compared to healthy controls [14]. Neuromelanin is a product of excess cytosolic DA and its precursor 3,4-dihydroxyphenylalanine (DOPA) [16], and this is therefore a strong indication that DA synthesis in the midbrain is elevated in people with schizophrenia. Additionally, an 18F-DOPA PET study found that DA synthesis capacity was elevated in the nigra of people with schizophrenia compared to healthy controls, as well as in the striatum [13].
In the preclinical ketamine model, striatal DA dysfunction (and the hyperlocomotor phenotype) was abolished when midbrain DA neuron activity was inhibited chemogenetically [6]. This finding suggests that midbrain DA neuron function contributes to the neurochemical and behavioural phenotypes of this model. However, it is unknown whether ketamine directly affects DA synthesis capacity in the midbrain, or whether it acts via indirect mechanisms. Understanding this relationship between dopaminergic regions in this model with relevance to schizophrenia has implications for understanding the dopaminergic dysfunction in this disorder.
Within the rodent brain, the striata show the strongest 18F-DOPA signal, and the experimental parameters for estimation of DA synthesis capacity in this region are published elsewhere [6, 8, 17]. However, the considerable differences in size and physiology between the striatum and the midbrain—including an elevated ratio of DA to its metabolite 3,4-dihydroxyphenylacetic acid (DOPAC) in the midbrain compared to the striatum [18]—mean that we cannot presume to use the same analytical parameters for 18F-DOPA quantification in both compartments. This work therefore has two aims. The first is to establish a method to place the midbrain region of interest accurately based on the acquired CT structural scan and to define the optimal analysis parameters which allow for robust, reliable quantification of 18F-DOPA signal in the mouse midbrain. The second aim of this work is to use this method to determine whether the sub-chronic ketamine model displays elevated DA synthesis capacity in the midbrain alongside the striatum.
Methods
Subjects
The analysis of 18F-DOPA uptake in the striatum has already been published for this dataset (for both the saline- and ketamine-treated animals and the test–retest cohort) [8]. These scans were re-analysed here to assess 18F-DOPA uptake in the midbrain, and therefore no new animals were used for this study. The details below refer to the original cohorts of animals. Wild-type male C57BL/6J mice were acquired from Charles River (Kent, UK). Animals were housed in groups of up to 4 and provided with ad libitum food and water. Animals were acclimatised to the facility for at least 1 week prior to any experimental procedures, which were undertaken in adult animals (8–12 weeks old). For animal weights, see Table 1. All animal experimental procedures were performed in accordance with the UK Animals (Scientific Procedures) Act 1986 and EU directive 2010/63/EU, and protocols were approved by the Imperial College Animal Welfare and Ethical Review Body and conducted under the licence PE0206466.
Experimental Cohorts
Saline-Treated Cohort
Animals were administered saline intraperitoneally (i.p.; 0.9%) once daily for 5 consecutive days, as they formed the control cohort for the ketamine treatment (see below). Animals then underwent the PET scan on day 7. Animals were euthanized immediately after the scan. Although 14 animals were originally treated with saline, 2 animals were excluded from analysis due to issues with the scan data, resulting in a final n = 12 for this group. This cohort was used to establish the midbrain template and the analysis parameters which resulted in the best-fitting data and lowest intra-group variability when estimating 18F-DOPA uptake in the midbrain.
Ketamine-Treated Cohort
To generate the sub-chronic ketamine model, ketamine hydrochloride (Sigma-Aldrich, K2753) was dissolved in 0.9% saline solution and administered to mice (n = 12) at a dose of 30 mg/kg (i.p) once daily for 5 consecutive days. On day 7, after 2 days of washout, animals underwent an 18F-DOPA preclinical PET scan. Animals were euthanized immediately after the scan.
Test-Retest Cohort
A separate cohort of untreated mice (n = 6) was scanned twice to assess reproducibility of results. Animals underwent the initial scan (“test”) and were then recovered from the anaesthesia. They were re-scanned (“retest”) approximately 48 h later, after which they were immediately euthanized.
PET Acquisition
18F-DOPA was synthesised as described previously [19]. One hour prior to the administration of 18F-DOPA, mice were anaesthetised with isoflurane (4%), which was maintained at 1–2% for the duration of the scan (O2 1 L/min). Respiration rate and body temperature were continuously monitored (BioVet, m2m Imaging Corp, OH, USA). To prevent peripheral metabolism of 18F-DOPA, inhibitors of catechol-O-methyl-transferase (COMT) and aromatic amino acid decarboxylase (AADC)—entacapone (40 mg/kg; Sigma-Aldrich, SML0654) and benserazide hydrochloride (10 mg/kg; Sigma-Aldrich, B7283)—were given i.p. 45 min and 30 min prior to the administration of 18F-DOPA, respectively [6, 17, 20]. To avoid competition with the uptake of other neutral amino acids in the diet, animals were fasted for 45 min before being anaesthetised [6, 21]. An i.p. cannula used to deliver the radiotracer was inserted 30 min prior to the scan. The mice were then placed into an Inveon PET/CT scanner (Siemens, Surrey, UK) and underwent a 10-min CT scan to allow for attenuation correction of the PET signal. A dynamic PET scan was then started concomitantly with the delivery of 18F-DOPA via the i.p. cannula, and data were collected for 140 min. The test–retest datasets were only acquired up to 120 min, due to changes in the scanning parameters between cohorts. The average 18F-DOPA doses delivered for each experimental cohort are provided in Table 1.
PET Image Processing
Following acquisition, data were histogrammed into 45 frames (10 × 3 s, 6 × 5 s, 8 × 30 s, 5 × 60 s, 6 × 300 s, 8 × 600s for data acquired up to 120 min). For data acquired up to 140 min, an additional 2 × 600 s frames were added. Data were reconstructed using filtered back projection with CT attenuation correction, adjusting for random noise, scatter, and radiotracer decay. Image processing was carried out using the Inveon Research Workspace software (Siemens, USA). The CT image was manually co-registered to the reconstructed PET image using the outline of the skull as a reference. For each subject, the percentage-injected dose was normalised for body weight and injected activity to provide standardised uptake values (SUV). The striatal (0.70 mm3) and cerebellar (the reference region; rectangular; 1.1 mm3) regions of interest (ROI) were placed manually on summation radioactivity images guided by CT and mouse brain anatomy based on previous studies [6, 8, 17].
Developing the Midbrain Template
Based on stereotaxic coordinates from the Paxinos Mouse Brain Atlas, the volume of the midbrain region which incorporates the bilateral SNpc and VTA is approximately 3.2 mm3 [22] (Supp. Figure S1a). Therefore, we established a rectangular midbrain ROI of 3.7 mm3 (16 voxels, Supp. Table S1). A template was developed to standardise the placement of this ROI in the brain; a representative CT image at the midline sagittal viewpoint was aligned with a mask based on the sagittal P56 Allen Mouse Brain Atlas, image 21 (mouse.brain-map.org/static/atlas [23]) to identify the location of the midbrain (Fig. 1). The resulting template was applied to the CT images of each animal to place the midbrain ROI. Supplementary figure S1b shows the placements for all 12 saline-treated animals.

Developing the midbrain template. a An atlas image of the mousebrain sagittal midline (from the Allen Mouse Brain Atlas) was used to identify the midbrain region, and a mask based on this atlas image (green) was aligned to an ROI drawn within the outline of the skull from a representative mouse CT scan. The cerebellum (yellow) was placed in the posterior part of the brain. This template was then used to place the 3.7-mm3 midbrain ROI (blue), seen from the b coronal and c sagittal perspective. The 18F-DOPA signal (yellow-red spectrum) can be seen in the d coronal and e sagittal planes, along with the placement of the midbrain ROI
Modified Patlak Analysis
The kinetic analysis to provide an estimate of the rate of 18F-DOPA uptake was performed using a MATLAB pipeline developed in-house (version R2022a; MathWorks, MA, USA). A modified Patlak graphical analysis was used to determine the influx rate constant KiMod (min−1). KiMod is a modified version of the influx rate constant KiStd (min−1) and takes into account an estimation of kloss (min−1), the rate at which signal is removed from the system due to metabolism of 18F-dopamine, as described previously [20, 24,25,26]. To establish the optimal time window for estimating KiMod in the midbrain, data from the saline-treated animals were processed through the modified Patlak analysis pipeline using varying start (T*) and end times (Tend). The analysis pipeline also quantifies the coefficient of variation (CV%) for the Patlak modelling, which gives an indication of how well the fitting has worked. These numbers were also compared for each T*–Tend window, with the lowest CV% indicating the best fit of the data to the graphical model. In some instances, the Patlak model can fail due to excessive noise in the data, or too few usable data points. This requires these datasets to be excluded, and therefore the number of animals included for each T*–Tend window (as a fraction of the whole group) is also noted.
Statistical Analysis
Statistical tests were carried out using GraphPad Prism v9 (GraphPad Software, La Jolla, California, USA). Data were tested for statistical outliers (Grubbs, α = 0.05) and normality (Kolmogorov-Smirnov test). Significance was defined as α < 0.05. Data is always presented as mean ± standard deviation (SD). SD of the saline-treated cohort was compared for each T*–Tend time windows to determine the parameters which produced the least within-group variability. T-test was used to compare mean differences between groups, with Welch’s correction used where variance was significantly different between groups. Consistency between the test–retest scans was analysed using an intra-class correlation coefficient (ICC; using a two-way mixed model with absolute agreement) and absolute percentage variance (%). Pearson’s correlations coefficients were calculated to assess significant linear correlations.
Results
Optimising a Method for Quantification of 18F-DOPA Uptake in the Midbrain
Based on the time activity curves (TAC; change in SUV across the scan period), the midbrain shows a similar pattern of 18F-DOPA uptake compared to the striatum, indicating that the radiotracer behaves in a similar way between these regions (Supp. Figure S2). For instance, the average time at which the TAC peaked (SUVpeak) was not significantly different between the striatum (100 ± 22.3 min) and midbrain (95 ± 22.3 min; t(22) = 0.45, p = 0.65; Supp. Figure S2a,b). The slope of the SUVratio (the cerebellar SUV relative to the SUV of the ROI) was significantly different between the midbrain and striatum (F = 240.9, p < 0.0001); however, both were linear (striatum r2 =0.88, midbrain r2 = 0.61; Supp. Figure S2c).
By comparing different T*–Tend windows to calculate KiMod, we found that the lowest CV% values were acquired when the T*–Tend window omitted the first 5 min of the scan (Table 2). SD values generally decreased with an earlier T* time; however, the lowest SD for midbrain KiMod which also permitted the inclusion of 12/12 animals were acquired using T* times from 10 min upwards. Although data up to 120 mins showed the lowest CV%, the modelling was successful for more individuals (12/12 animals) when data was included up to 140 min. As T* increased (with a Tend of 140 min), CV% decreased while SD increased. The SD of the kloss parameter was also lowest when data was included up to 140 min, and again was lowest using T* times from 10 min upwards (Supp. Table S2).
Using the Test–Retest Cohort to Assess Consistency for Estimating Midbrain Ki Mod
The ICC coefficient values for midbrain KiMod from the test–retest cohort were compared for a subset of T*–Tend windows (Fig. 2). The ICC coefficient for the 10–120 min T*–Tend window was 0.53 and was 0.74 for both the 15–120 and 20–120 min T*–Tend windows. However, the absolute variability between test and retest values was lowest for the 15–120 minute T*–Tend window (43.7% ± 50.1%), compared to the 20–120 minute T*–Tend window (48.5 ± 55.0%) and the 10–120 min T*–Tend window (55.2 ± 53.1%).

Comparison of KiMod values for the test-retest cohort, for a subset of T*–Tend time windows. N = 6
Summary of Parameter Testing and Comparison to Analysis in the Striatum
Based on the above results, we determined that a T*–Tend window of 15–140 min was optimal for quantification of 18F-DOPA signal in the midbrain. This resulted in one outlier for the analysis of the midbrain, which was removed for analysis. We next compared outputs for the midbrain with the striatum. Previous work has determined that the optimal T*–Tend window for analysis of KiMod in the striatum with 18F-DOPA is 20–140 min [8]. KiMod in the midbrain (using a 15–140 min T*–Tend window) showed significantly decreased variability (based on SD) compared to the striatum (using a 20–140 min T*–Tend; F(11) = 4.1, p = 0.03; Fig. 3). Additionally, the test–retest analysis resulted in a lower ICC coefficient for the striatum (0.56) compared to the midbrain (0.74). However, KiMod was significantly lower in the midbrain compared to the striatum (striatum KiMod 0.019 ± 0.006, midbrain KiMod 0.005 ± 0.003; t(16.3) = 7.4, p < 0.0001; Fig. 3). There was also a significant decrease in how well the data from the midbrain fitted to the modified Patlak model compared to the striatum (striatum CV% 5.3 ± 1.93%, midbrain CV% 8.4 ± 4.4%; t(21) = 2.2, p = 0.037; Supp. Figure S3), suggesting that the signal from the midbrain may be noisier than in the striatum. The midbrain also showed significantly decreased kloss in comparison to the striatum (t(21) = 2.8, p = 0.01; Supp. Figure S4).

KiMod in the striatum and midbrain. KiMod was significantly decreased in the midbrain (using the 15–140 min T*–Tend window) compared to the striatum (using the 20–140 min T*-Tend window). ****p < 0.0001. Mean ± SD. n(striatum) = 12, n(midbrain) = 11
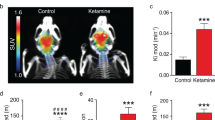
Midbrain DA Synthesis Capacity is Increased in the Sub-Chronic Ketamine Model
We next applied this midbrain method to the sub-chronic ketamine model. There was no significant difference in animal weight, or the 18F-DOPA dose injected between the saline- and ketamine-treated groups (Table 1). There was a significant increase in KiMod in the striatum of the ketamine-treated animals compared to those treated with saline (t(22) = 2.48, p = 0.02; Supp. Figure S5). This replicates the previous analysis of this dataset [8]. We found elevated KiMod in the midbrain for animals treated with ketamine compared to those treated with saline (t(21) = 2.18, p = 0.04; Fig. 4a). There was also a significant positive correlation between KiMod in the striatal and midbrain compartments in saline-treated animals (r2 = 0.59, p = 0.005; Fig. 4b). However, this relationship was absent in animals which had been treated with ketamine (r2 = 0.004, p = 0.83). To assess the magnitude of change in the striatum relative to the midbrain, individual KiMod values from ketamine-treated animals were compared to the mean values for the control animals in each region. Compared to the mean of the saline-treated group, ketamine-treated animals showed an increased KiMod of 26% in the striatum (± 20.1%), and 55% in the midbrain (± 65.3%; Supp. Figure S6). Although these mean values were not statistically different (t(11.6) = 1.36, p = 0.19, with Welch’s correction), the change in KiMod in the midbrain as a result of ketamine treatment was significantly more variable compared to the striatum (F(10) = 10.8, p = 0.0005).

The effect of sub-chronic ketamine treatment on KiMod in the midbrain. a KiMod was elevated in the midbrain of ketamine-treated animals compared to saline-treated animals (p = 0.04). b There was a significant positive correlation between KiMod values for the striatum and midbrain for each animal in the saline-treated group (p = 0.005). This was absent in animals which had been treated with ketamine for 5 days (p = 0.83). Each data-point represents an individual animal. *p < 0.05. Mean ± SD. n(saline) = 11, n(ketamine) = 12
Discussion
We found that it is possible to estimate 18F-DOPA uptake in the mouse midbrain with moderate test–retest agreement, using an atlas-based template and a T*–Tend window of 15–140 min. DA synthesis capacity in the mouse midbrain was approximately a quarter of that in the striatum. This is consistent with ex vivo work which has shown that levels of the tyrosine hydroxylase (TH) protein are lower in the midbrain compared to the dorsal striatum [18]. TH is the rate-limiting enzyme in the synthesis of DA, and this is therefore a likely explanation for this regional difference in the rate of 18F-DOPA uptake we observed. This relative difference in DA synthesis capacity between these regions has also been found in human studies using 18F-DOPA PET [13, 27]. The study by Howes and colleagues also found that healthy people demonstrated a positive correlation between nigral and striatal DA synthesis capacity [13], which was evident in the saline-treated mice. While not conclusive, this concordance between human and mouse studies of nigral and striatal 18F-DOPA uptake supports the validity of the preclinical method developed here.
Sub-chronic administration of ketamine has been shown to increase DA synthesis capacity in the striatum [6]. Here, we show that this model also results in an elevated DA synthesis capacity in the midbrain. Although we were not able to conduct an ex vivo analysis of the cohort which underwent PET scans here, another study in which ketamine (10 mg/kg) was delivered to mice daily for 7 days found increased TH protein levels in the VTA [28]. A separate study found that repeated ketamine exposure increased the firing activity of DA neurons in the VTA [29]. These studies provide indirect support for our finding of elevated DA synthesis capacity in the midbrain following sub-chronic ketamine administration.
This finding in the sub-chronic ketamine model of schizophrenia also appears to replicate the findings in the clinical population; an 18F-DOPA PET study found that DA synthesis capacity was elevated in both the nigra and striatum of people with schizophrenia, compared to healthy controls [13]. However, it should be noted that another study found no difference between 18F-DOPA in the nigra for patients with schizophrenia and healthy controls, although this study may have been underpowered to detect a difference in this region [19]. Another study which assessed dopaminergic activity in the nigra found blunted amphetamine-induced DA release in this region [30] which would suggest diminished DA synthesis capacity. However, this study used a radiotracer which binds to dopamine 2/3 receptors, and these results therefore cannot be directly compared to those acquired with 18F-DOPA.
There is additional indirect evidence that dysfunctional dopaminergic transmission is present in the nigra of people with schizophrenia. Although a recent review of post-mortem studies found no clear difference in levels of midbrain TH in the patient population [15], it is possible to use magnetic resonance imaging to assess levels of neuromelanin—an accumulated product of cytosolic DA and DOPA—in the living brain [31]. A recent meta-analysis of these studies found that neuromelanin levels in the nigra were elevated in people with schizophrenia, compared to healthy controls [14]. The current work therefore supports the theory of hyperdopaminergia in the midbrain as a component of schizophrenia neurobiology.
The exact mechanism by which midbrain DA is elevated in this sub-chronic ketamine model is still unclear. Previous work in the sub-chronic ketamine model has found decreased parvalbumin (PV) interneuron number in the prelimbic cortex and in a subregion of the hippocampus, and that the activation of PV interneurons could ameliorate the behavioural and neurochemical deficits in this model [6]. This reduction in PV interneuron number may contribute to the disinhibition of excitatory glutamatergic input to the midbrain [32]. This in turn may result in elevated firing rates of DA neurons in the midbrain. A decrease in mRNA levels of parvalbumin has also been found in cortical post-mortem tissue from people with schizophrenia [33]. This work is therefore congruent with the hypothesis that striatal hyperdopaminergia in schizophrenia may stem from disinhibition of midbrain DA neurons [34, 35].
Sub-chronic ketamine treatment also disrupted the positive correlation between DA synthesis capacity in the midbrain and nigra which was evident in saline-treated animals. Although there was no significant difference in the magnitude of change induced by ketamine in each region, the change induced by sub-chronic ketamine was more variable in the midbrain compared to the striatum, with some ketamine-treated animals showing a midbrain KiMod below the mean of the saline-treated animals. Interestingly, this finding of a disrupted relationship between DA synthesis capacity in these two compartments has also been found in people with schizophrenia [13]. This suggests that the ketamine model can replicate this decoupling between the dopaminergic compartments which is seen in the patient population. This model may therefore be extremely useful for trialling treatments designed to rescue dopaminergic dysfunction throughout the nigro-striatal circuit.
Ketamine is also utilised as an antidepressant [36], and there is some evidence that the positive effects of ketamine are due to increased midbrain DA neuron firing and activity [29, 37]. Our findings provide support for this hypothesis by indicating that sub-chronic ketamine can alter DA synthesis capacity in this region. Future studies could clarify the role of ketamine-induced midbrain hyperdopaminergia on the antidepressant actions of this drug by correlating midbrain DA synthesis capacity with behavioural outputs relevant to depression.
Limitations
The nigra is situated very close to the midline, and this method therefore uses an ROI which encompassed the bilateral midbrain. As a result, this technique does not allow for studies of lateralization, including the use of unilateral models. The location of the nigra also means that the midbrain ROI may incorporate other monoaminergic regions including the raphe nucleus (serotonergic) and the locus coeruleus (noradrenergic). 18F-DOPA signal from these monoaminergic regions may contribute to the findings seen here [27]. Determining the contribution of these regions to this signal may be possible by assessing 18F-DOPA signal in the midbrain following a DA-specific lesion. The small size of the nigra also means that partial volume effects may contribute to the KiMod values acquired for the midbrain. Additionally, this study uses only male animals. There are known sex-specific effects on the outcomes for ketamine administration [5, 38, 39], and therefore, a comparable study using female animals would be extremely valuable. Finally, performing ex vivo confirmation of increased DA synthesis capacity in a cohort of ketamine-treated animals which also underwent 18F-DOPA PET scans would directly link the results from this neuroimaging method with a biological phenotype.
Conclusion
This study developed an atlas-based template to quantify 18F-DOPA uptake in the mouse midbrain. This methodological advance will permit the interrogation of the nigro-striatal pathway both in healthy animals and in preclinical models of relevance to schizophrenia and other dopaminergic disorders. We have applied this method to show that the sub-chronic ketamine model results in elevated DA synthesis capacity throughout the nigro-striatal pathway. As a result, we can now utilise the sub-chronic ketamine model to trial novel therapeutics which may act to normalise the midbrain DA function.
Data Availability
The datasets generated and/or analysed in this study, as well as all codes used in the present study, are available from the corresponding author upon reasonable request.
Change history
03 April 2024
A Correction to this paper has been published: https://doi.org/10.1007/s11307-024-01916-y
Abbreviations
- AADC:
-
amino acid decarboxylase
- COMT:
-
catechol-o-methyltransferase
- CV:
-
coefficient of variation
- DA:
-
dopamine
- DOPA:
-
l-3,4-dihydroxyphenylalanine
- DOPAC:
-
3,4-Dihydroxyphenylacetic acid
- 18F-DOPA:
-
fluorinated l-3,4-dihydroxyphenylalanine
- IP:
-
intraperitoneal
- Ki:
-
influx constant
- MBq:
-
megabecquerel
- NMDA:
-
N-methyl-d-aspartate
- PET:
-
positron emission tomography
- PV:
-
parvalbumin
- ROI:
-
region of interest
- SD:
-
standard deviation
- SNpc:
-
substantia nigra pars compacta
- SUV:
-
standardised uptake values
- TAAR1:
-
trace amine-associated receptor 1
- TAC:
-
time activity curve
- TH:
-
tyrosine hydroxylase
- VTA:
-
ventral tegmental area
References
Howes OD, Shatalina E (2022) Integrating the neurodevelopmental and dopamine hypotheses of schizophrenia and the role of cortical excitation-inhibition balance. Biol Psychiatry 92(6):501–513
Beck K et al (2020) Association of ketamine with psychiatric symptoms and implications for its therapeutic use and for understanding schizophrenia: a systematic review and meta-analysis. JAMA Netw Open 3(5):e204693
Becker A et al (2003) Ketamine-induced changes in rat behaviour: a possible animal model of schizophrenia. Prog Neuro-Psychopharmacol Biol Psychiatry 27(4):687–700
Bates MLS, Trujillo KA (2019) Long-lasting effects of repeated ketamine administration in adult and adolescent rats. Behav Brain Res 369:111928
Strong CE et al (2017) Locomotor sensitization to intermittent ketamine administration is associated with nucleus accumbens plasticity in male and female rats. Neuropharmacology 121:195–203
Kokkinou M et al (2021) Reproducing the dopamine pathophysiology of schizophrenia and approaches to ameliorate it: a translational imaging study with ketamine. Mol Psychiatry 26(6):2562–2576
Chatterjee M et al (2012) Neurochemical and molecular characterization of ketamine-induced experimental psychosis model in mice. Neuropharmacology 63(6):1161–1171
Halff EF et al (2022) Evaluation of Intraperitoneal [(18)F]-FDOPA Administration for micro-PET imaging in mice and assessment of the effect of subchronic ketamine dosing on dopamine synthesis capacity. Mol Imaging 2022:4419221
Brugger SP et al (2020) Heterogeneity of striatal dopamine function in schizophrenia: meta-analysis of variance. Biol Psychiatry 87(3):215–224
McCutcheon R et al (2018) Defining the locus of dopaminergic dysfunction in schizophrenia: a meta-analysis and test of the mesolimbic hypothesis. Schizophr Bull 44(6):1301–1311
McCutcheon RA, Abi-Dargham A, Howes OD (2019) Schizophrenia, dopamine and the striatum: from biology to symptoms. Trends Neurosci 42(3):205–220
Bjorklund A, Dunnett SB (2007) Dopamine neuron systems in the brain: an update. Trends Neurosci 30(5):194–202
Howes OD et al (2013) Midbrain dopamine function in schizophrenia and depression: a post-mortem and positron emission tomographic imaging study. Brain 136(Pt 11):3242–3251
Ueno F et al (2022) Neuromelanin accumulation in patients with schizophrenia: a systematic review and meta-analysis. Neurosci Biobehav Rev 132:1205–1213
van Hooijdonk CFM et al (2023) The substantia nigra in the pathology of schizophrenia: a review on post-mortem and molecular imaging findings. Eur Neuropsychopharmacol 68:57–77
Sulzer D et al (2000) Neuromelanin biosynthesis is driven by excess cytosolic catecholamines not accumulated by synaptic vesicles. Proc Natl Acad Sci U S A 97(22):11869–11874
Bonsall DR et al (2017) Single cocaine exposure does not alter striatal pre-synaptic dopamine function in mice: an [(18) F]-FDOPA PET study. J Neurochem 143(5):551–560
Salvatore MF, Pruett BS (2012) Dichotomy of tyrosine hydroxylase and dopamine regulation between somatodendritic and terminal field areas of nigrostriatal and mesoaccumbens pathways. PLoS One 7(1):e29867
Jauhar S et al (2017) A Test of the Transdiagnostic dopamine hypothesis of psychosis using positron emission tomographic imaging in bipolar affective disorder and schizophrenia. JAMA Psychiatry 74(12):1206–1213
Walker MD et al (2013) In-vivo measurement of LDOPA uptake, dopamine reserve and turnover in the rat brain using [18F]FDOPA PET. J Cereb Blood Flow Metab 33(1):59–66
Camargo SM et al (2014) The molecular mechanism of intestinal levodopa absorption and its possible implications for the treatment of Parkinson’s disease. J Pharmacol Exp Ther 351(1):114–123
Keith BJ, Franklin M, Paxinos G (2008) The mouse brain in stereotaxic coordinates, compact: the coronal plates and diagrams. Elsevier Science
Allen Reference Atlas. Mouse brain [brain atlas]. Available from atlas.brain-map.org [cited 2023].
Patlak CS, Blasberg RG (1985) Graphical evaluation of blood-to-brain transfer constants from multiple-time uptake data. Generalizations. J Cereb Blood Flow Metab 5(4):584–590
Holden JE et al (1997) Graphical analysis of 6-fluoro-L-dopa trapping: effect of inhibition of catechol-O-methyltransferase. J Nucl Med 38(10):1568–1574
Sossi V, Doudet DJ, Holden JE (2001) A reversible tracer analysis approach to the study of effective dopamine turnover. J Cereb Blood Flow Metab 21(4):469–476
Moore RY et al (2003) Monoamine neuron innervation of the normal human brain: an 18F-DOPA PET study. Brain Res 982(2):137–145
Abiero A et al (2021) 1-Phenylcyclohexan-1-amine hydrochloride (PCA HCl) alters mesolimbic dopamine system accompanied by neuroplastic changes: a neuropsychopharmacological evaluation in rodents. Neurochem Int 144:104962
Iro CM et al (2021) Repeated but not single administration of ketamine prolongs increases of the firing activity of norepinephrine and dopamine neurons. Int J Neuropsychopharmacol 24(7):570–579
Slifstein M et al (2015) Deficits in prefrontal cortical and extrastriatal dopamine release in schizophrenia: a positron emission tomographic functional magnetic resonance imaging study. JAMA Psychiatry 72(4):316–324
Zucca FA et al (2014) Neuromelanin of the human substantia nigra: an update. Neurotox Res 25(1):13–23
Homayoun H, Moghaddam B (2007) NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci 27(43):11496–11500
Dienel SJ, Lewis DA (2019) Alterations in cortical interneurons and cognitive function in schizophrenia. Neurobiol Dis 131:104208
Sonnenschein SF, Gomes FV, Grace AA (2020) Dysregulation of midbrain dopamine system and the pathophysiology of schizophrenia. Front Psychiatry 11:613
Grace AA, Gomes FV (2019) The circuitry of dopamine system regulation and its disruption in schizophrenia: insights into treatment and prevention. Schizophr Bull 45(1):148–157
Meshkat S et al (2023) Oral ketamine for depression: an updated systematic review. World J Biol Psychiatry 24(7):545–557
Parise EM et al (2021) The resilient phenotype induced by prophylactic ketamine exposure during adolescence is mediated by the ventral tegmental area-nucleus accumbens pathway. Biol Psychiatry 90(7):482–493
Garcia-Carachure I et al (2020) Enduring effects of adolescent ketamine exposure on cocaine- and sucrose-induced reward in male and female C57BL/6 mice. Neuropsychopharmacology 45(9):1536–1544
Schoepfer KJ et al (2019) Sex- and dose-dependent abuse liability of repeated subanesthetic ketamine in rats. Physiol Behav 203:60–69
Acknowledgements
We would like to acknowledge Sharon Ashworth (Biological Operations Manager, Invicro) and Nicholas Keat (PET-CT Physicist, Invicro) for their support provided to the study.
Funding
This work was supported by grants from the Medical Research Council (MRC) UK (MC_A656_5QD30_2135 to OH and MC-A654-5QB40 to DJW) and the Wellcome Trust (094849/Z/10/Z to OH). MV is supported by the National Institute for Health Research Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 101028661.
Author information
Authors and Affiliations
Contributions
Experiments were designed by OH, SN, DJW, EH and MK. The original data was collected by AG-H, EH, MK and SN with assistance from EI. Data analysis and interpretation was conducted by AP with support from LW, DB, S-PT and MV. The manuscript was written by AP, and all other authors provided feedback and editing of the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
LW is an associate editor of Molecular Imaging and Biology (MIBI; unpaid). LW is on the leadership committee of Women in Molecular Imaging, which is part of the World Molecular Imaging Society (unpaid). LW has stock with Glaxosmithkline, and is a fulltime employee of Invicro LLC. MV and OH are named inventors for the patent “Dopaminergic Imaging to Predict Treatment Response in Mental Illness” ref: US20230024712A1. OH is also part-time employee and stock-holder of H Lundbeck A/s. OH has received investigator-initiated research funding from and/or participated in advisory/speaker meetings organised by Angellini, Autifony, Biogen, Boehringer-Ingelheim, Eli Lilly, Heptares, Global Medical Education, Invicro, Jansenn, Karuna, Lundbeck, Neurocrine, Ontrack/Pangea, Otsuka, Sunovion, Recordati, Roche and Viatris/Mylan. No other authors have competing interests to declare.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 5.57 MB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Petty, A., Garcia-Hidalgo, A., Halff, E.F. et al. Sub-Chronic Ketamine Administration Increases Dopamine Synthesis Capacity in the Mouse Midbrain: a Preclinical In Vivo PET Study. Mol Imaging Biol 25, 1054–1062 (2023). https://doi.org/10.1007/s11307-023-01865-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11307-023-01865-y