
Abstract
Pain is the most common symptom reported by patients with rheumatoid arthritis (RA) even after the resolution of chronic joint inflammation. It is believed that RA-associated pain is not solely due to inflammation, but could also be attributed to aberrant modifications to the central nervous system. The P2X4 receptor (P2X4R) is an ATP-activated purinergic receptor that plays a significant role in the transmission of information in the nervous system and pain. The involvement of P2X4R during the pathogenesis of chronic inflammatory pain and neuropathic pain is well-established. The attenuation of this receptor alleviates disease pathogenesis and related symptoms, including hyperalgesia and allodynia. Although some studies have revealed the contribution of P2X4R in promoting joint inflammation in RA, how it implicates pain associated with RA at peripheral and central nervous systems is still lacking. In this review, the possible contributions of P2X4R in the nervous system and how it implicates pain transmission and responses were examined.
Similar content being viewed by others


Introduction
Rheumatoid arthritis (RA) is one of the diseases resulting from prolonged chronic inflammation that predominantly affects joints. Recently, many advances have been made in the treatment of RA leading to better control of joint inflammation. However, pain resulting from RA is still a major problem reported by patients [1, 2]. It is believed that RA-related pain is not solely caused by inflammatory reactions. Lee and colleagues [3] revealed that 12–20% of patients reported prolonged pain without signs of inflammation. Moreover, Hewlett et al. [4] revealed that the severity of pain varies between patients and even RA patients with no indication of inflammation frequently experience pain. There is a weak association between the intensity of pain and the objective measures of inflammation [5]. Although the application of disease-modifying anti-rheumatic drugs (DMARDs) may resolve inflammation, 58% of early RA patients reported incomplete improvement in body pain after a year of treatment. Another 15% of patients reported no improvement of pain and 27% reported increased pain after one year compared to the baseline value [6]. These findings suggest that the pain associated with RA arises from non-inflammatory sources. Moreover, the nature of suffering from pain is multifactorial. Stress that resulted from sleep disturbance, depression, anxiety and catastrophising in RA patients may also influence the perception of pain. Meanwhile, RA is also believed to be derived from the presence of anti-citrullinated protein (ACPA), which is then developed into autoantibodies and may be present in the synovial fluid of the patients [7]. Enhanced pain hypersensitivity caused by ACPA was also reported by Wigerblad et al. [8] in mice model of collagen-induced arthritis administered with human or murine ACPA, which indicated that the ACPA-induced hypersensitivity is mediated by pathways depending on the IL-18 release. Although RA patients suffer from hyperalgesia as a result of lower pain threshold due to the pathogenesis of RA, the abnormalities may originate from the central nervous system (CNS), including the loss of conditioned pain modulation or central sensitisation, when there are the absent signs of chronic local inflammation or local tissue damage [1]. In a clinical trial, Christensen et al. [9] showed that 65% of RA patients experienced nociceptive pain, while the rest experienced non-nociceptive pain. As the consequence of the amplification of a nociceptive signal, neuropathic pain condition or central sensitisation is believed to be the culprit of the abnormal symptoms occurring in RA [1]. In this review, the aim was to discuss a comprehensive overview of P2X4 receptors (P2X4R) in the pathogenesis of RA and its possible mechanisms leading to RA-related pain.
The P2X4 receptor is ATP-gated purinergic signalling
Extracellular adenosine triphosphate (ATP) is a crucial signalling molecule regulating several biological processes involved in the nervous, endocrine, respiratory and cardiovascular systems. It is the energy substance in the body that is involved in the energy conversion of cells and tissues. The P2X4R activates channels in various neuronal, muscle, glandular, hair cells, microglia, astrocytes and B-lymphocytes. However, it is also heavily involved in the pathological progress of diseases such as inflammation, immune function, cancer, pain and ischaemia [10]. The effects of extracellular ATP activation implicate two types of P2 receptors cell surface; the G protein-coupled P2Y receptors and the P2X receptors, which have an integral cation channel that opens following the ATP binding process [11, 12]. They are known as ligand-gated cation-selective ion channels that bind to extracellular ATP [12, 13], mediating the inflow of sodium (Na+) and calcium ions (Ca2+) as well as the outflow of potassium ion (K+) [14]. These receptors belong to distinct structural types of Cys-loop and glutamate receptor families of ligand-gated cation channels. These channels are abundant in both excitable and non-excitable cells, including neurons, muscles, glands, hair, microglia, astrocytes and B lymphocyte cells [13]. Since ATP is a fast neurotransmitter at specific central synapses, it is also released in the periphery, which explains how pain transmission can be easily transmitted after ATP activation of P2X receptors [15].
Seven subtypes of P2X receptors designated as P2X1 through P2X7 that co-assemble as homotrimeric or heterotrimeric have been identified [16] ranging from 388 (P2X4) to 595 (P2X7) subunits [10, 11]. Each of the subunits contains two hydrophobic, putative membrane-spanning sections (TM1 and TM2) which are separated by an ectodomain containing 10 cysteine residues forming disulphide bonds [15]. These structures made them resemble the form of epithelial Na+ channels (ENaC) and acid-sensing channels (ASIC) [11]. It is believed that P2X receptors are closely involved in the development and transmission of pain and inflammatory-nociceptive signals [12]. P2X receptors are widely expressed in both neurons and glia in the nervous system, and P2X2, P2X4 and P2X6 receptors are most abundantly expressed [11, 15].
Similar to other P2X receptors, P2X4R is crucial in many physiological processes. It consists of three subunits consisting of the head, body and tail. The interactions between these subunits form the triple symmetry axis trimer channel protein [17]. It contains 388 amino acids comprising of an N-terminus (30 amino acids), a C-terminus (38 amino acids), two transmembrane domains (TM1 and TM2; 20 amino acids each) and a large extracellular domain (280 amino acids) [10] (Fig. 1). P2X4R is in fact, essential for the physiological importance of the body [10, 14] as it is involved in the normal regulation of inflammation, fibrogenesis, apoptosis and neuromodulation associated with pain sensitivity [18].

Structure of the P2X4 receptor. This receptor consists of N- and C-terminals, two transmembrane domains (TM1 and TM2) and a large extracellular domain (adapted from Zhang et al. [8])
During the physiological state, activation of P2X4R by binding of the extracellular ATP induces the opening of transmembrane pores and permits the influx of Ca2+, Na+ and K+ to modify their membrane voltage. This effect leads to the reduction of electrochemical gradients and activation of intracellular signalling components [19]. However, during tissue injury with the presence of noxious stimulation, especially in the case of chronic or neuropathic pain, the abundant ATP released from presynaptic nerves can bind to P2X4 receptors on microglia to produce distinctive, fast excitatory postsynaptic potentials (EPSPs) through the BDNF-TrkB receptor pathway [20, 21]. The inflammatory cascades lead to increased ATP-activated currents that change the voltage dependence of P2X4R and thus increasing the activation of this receptor [19]. This was demonstrated by the upregulation of P2X4R in the forelimb of the rat following complete Freund’s adjuvant (CFA) injection [19]. Since P2x4R has the highest permeability to Ca2+, the increased Ca2+ influx may activate related cells such as neurons, microglia and astrocytes. Consequently, these effects lead to the development of allodynia following nerve injury [20, 22]. These abnormal CNS changes result in the development of several neurological abnormalities including neuropathic pain [23], epilepsy [24, 25], anxiety [26], stroke [27] and Parkinson’s disease [28]. Furthermore, abnormal upregulation or deletion of P2X4R is also associated with the development of inflammation [29], auto-immune diseases [30], cardiovascular diseases [31] and endocrine disorders [32].
P2X4R alleviates inflammatory-induced pain responses
Arthritis-related pain is characterised by the occurrence of hyperalgesia and flare pain at the affected joints. Reduced pain thresholds and noxious hypersensitivity have been reported in RA patients [33, 34]. Patients with RA normally demonstrated reduced pressure pain thresholds and increased sensitivity to painful stimuli across several anatomical sites, including both inflamed joints and non-inflamed tissues [35, 36], suggesting central augmentation of pain signals. Patients may experience joint hyperalgesia when the affected joints are moved or when light pressure is exerted on the joints [37]. The essential mechanisms leading to RA-associated pain may be derived from modified central pain regulatory mechanisms such as abnormal pain modulation (central sensitisation), rather than prolonged stimulation of peripheral nociceptors [16]. It has also been reported that enhanced pain sensitivity was amplified in individuals that experienced a longer duration of RA [38].
Pain responses in animal models of arthritis are well-established. Several researchers investigating the involvement of P2X4R in the pathogenesis of chronic arthritis and inflammation have identified positive contributions of this receptor in pain behaviour responses. For instance, a rat model of collagen-induced arthritis and CFA-induced arthritis mimicked RA via several main characteristics such as systemic effects on joints, preferential injury to peripheral extremity joints, the participation of rheumatoid factors, greater severity in females and histological discoveries including synovial hyperplasia and infiltration of inflammatory cells [39, 40].
In a P2X4R-knockout mice model, Tsuda and colleagues [41] reported significant suppression of pain hypersensitivity to innocuous mechanical stimuli (tactile allodynia) and hind paw swelling following the intraplantar injection of CFA. Similar attenuation of mechanical allodynia was shown in a rat model of chronic constriction injury following the inhibition of P2X4R activation [42]. Furthermore, the inhibition of P2X4R via the intrathecal route significantly attenuated its upregulation in the spinal cord, thereby suppressing the development of mechanical allodynia in a mice model of herpetic pain [22]. Meanwhile, a reduced tail pressure pain threshold was reported in the adjuvant-induced arthritic rat [43]. A similar result was also reported by previous studies [44,45,46], with the polyarthritic rat showing a reduction in tail-flick latency, indicating reduced spinal reflex starting from day 7 to 22 following arthritic induction [44]. Other than that, the arthritic rats demonstrated the development of hyperalgesia as determined by the reduction in latency to cold [45, 47, 48] and hot stimuli [49]. In the paw pressure test, the CFA-injected arthritic rat demonstrated a significantly reduced threshold compared to the non-arthritic rat [50]. It was reported that the absence of P2X4R produced no effects in the mice with acute physiological pain or pain induced by tissue injury. The study highlighted P2X4R’s critical role in the development of chronic pain rather than acute pain [41, 51]. Therefore, it is essential to understand the underlying pathophysiology of modified central pain regulation which may be the leading cause of RA-associated pain.
Involvement of the P2X4 receptor in the peripheral nervous system
Since the symptoms of RA include peripheral neuropathy due to neurogenic lesions [52], similar pathomechanisms of neuropathic pain may be partially shared in RA. Previous studies revealed an extensive distribution of P2X4R in the dorsal root ganglia (DRG), autonomic and sensory ganglia [13, 14, 53]. The expression of peripheral P2X4R has been reported in the DRG or macrophages during inflammatory pain [54,55,56], but possibly not during neuropathic pain [56]. In the DRG, immunofluorescence staining demonstrated that P2X4R expression was mainly in satellite glial cells [57].
Following peripheral nerve lesion or inflammation, the enhanced peripheral nerve signals drive ɣ-aminobutyric acidergic (GABAergic) neurons to increase presynaptic ɣ-aminobutyric acid A (GABAA) receptors, leading to depolarisation of central terminals of the sensory neuron (i.e. primary afferent depolarisation), which in turn develops an impulse train that travels peripherally. These effects, therefore, trigger the release of inflammatory peptides from peripheral terminals of C-fibres that lead to the activation of inflammatory cascades in the affected tissues [58]. ATP released from injured nerves may bind and activate P2X4R on the related cells [53]. However, the inflammatory mediators can also stimulate P2X4R activation expressed on macrophages, which in turn leads to the production of prostaglandins (PGs) via the p38 mitogen-activated protein kinase (p38 MAPK) activation pathway [54].
This mechanism may also lead to the increased release of brain-derived neurotrophic factor (BDNF) from monocytes, B- and T-cells as shown in the culture of human peripheral blood mononuclear cells [59]. In the synovial fluid culture of RA patients, Klein et al. [60] reported that increased BDNF release in the synovial fluid was not mediated by the indirect action of PGs, but possibly via the actions of pro-inflammatory cytokines such as tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6) and interleukin-17 (IL-17) as demonstrated in the culture of monocytes [61] and microglia in the central nervous system [62] (Fig. 2). In microglial cell cultures, the activation of P2X4R on their cell surfaces leads to an increased Ca2+ influx into cells that further activates p38 MAPK, resulting in increased synthesis and release of BDNF via soluble N-ethylmaleimide-sensitive factor attachment protein (SNARE)-dependent exocytosis [63]. The production of BDNF is secreted by both fibroblast cells and non-fibroblast cells, including endothelial cells and macrophages, at the later stage of osteoarthritis [9].

Mechanism of P2X4R at peripheral region involving peripheral nerve lesion and inflammatory cells. Following the injury of a peripheral nerve due to inflammatory reactions in synovial fluid, the released ATP and PGs from the damaged nerve activate P2X4R expressed on the macrophage. Subsequently, the highly Ca2+ influx into the macrophage cell may activate p38 MAPK leading to the release of BDNF and PGE2 into the synovial fluid and surrounding nerves. The high influx of Ca2+ also indirectly stimulates P2X7R to allow the production of pro-inflammatory factors from macrophage, which further enhances the BDNF release by monocytes, B- and T-cells. Moreover, the released pro-inflammatory cytokines can further stimulate NLRP1 inflammasome on the macrophage. The synergistic effects between injured peripheral nerves and inflammatory cells lead to the leukocyte infiltration in joints and synovial hyperplasia. The prolonged synovial inflammation may further lead to bone erosion and cartilage destruction (osteoarthritis) and therefore transmit the nociceptive signals to a central level that develop thermal hyperalgesia and mechanical allodynia; the common symptoms of RA-associated chronic pain
Li and colleagues [64] reported that the inhibition of P2X4R resulted in decreased serum interleukin-1β (IL-1β), TNF-α, IL-6 and IL-17 levels in a mouse model of collagen-induced arthritis. Synovial inflammation and joint damage indicated by prominent leukocyte infiltration, synovial hyperplasia, bone erosion and evident cartilage damage were attenuated following the administration of a P2X4R antisense nucleotide; a P2X4R gene suppressor. In addition, the activated NLR family, pyrin domain containing 1 (NLRP1), PYD- and CARD-domain containing (ASC) and caspase-1 (NLRP inflammasome) were inhibited following the suppression of P2X4R in this mouse model and synovial cells of RA patients. This study highlighted the importance of NRLP1 involvement in the inflammasome signalling pathway in RA pathogenesis following P2X4R activation.
Furthermore, Lalisse and colleagues [65] demonstrated that P2X4R regulates Ca2+ influx in DRG neurons in a mouse model for chronic inflammation. The activation of P2X4R may also implicate other peripheral signalling pathways. In an in vitro study of mouse macrophage, PX4R was found to inhibit P2X7R-mediated autophagy [66]. Kawano et al. [66] revealed that P2X4R modulates several mechanisms of P2X7R-mediated inflammation, including the production of IL-1β that targets macrophages, causing their death. These pathological events can lead to an abnormal discharge of primary sensory neurons, enhanced nociceptive input transmission and enhanced synaptic plasticity of the spinal cord, as revealed in an in-vitro study using superficial spinal dorsal horn slices [67]. Through the above studies, it was demonstrated that P2X4R activation may damage nervous systems. It is important to attenuate its activation following chronic pain and inflammatory conditions. Minimising P2X4R overexpression may become a potential target for the treatment of RA and other nervous system-related diseases.
Involvement of the P2X4 receptor during central pain transmission
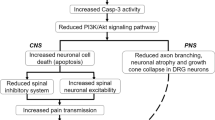
In the CNS, P2X4R is extensively distributed in the brain and cerebral cortex, basal ganglia, diencephalon, dorsal thalamus and spinal cord, but more densely found in the pons and medulla of the brain [13, 14, 53]. This receptor is specifically found in excitatory synapses in the brain, together with P2X2 receptors [11]. In pathological conditions such as neuropathic pain or epilepsy, the upregulation of P2X4R expression was detected in the neurons and microglia of the hippocampus and spinal cord [25, 64]. The activation of P2X4R in these cells is triggered by episodic tetanus stimuli contributing to synaptic plasticity in the hippocampal pyramidal neurons [67]. Moreover, the activation of P2X4R increases Ca2+ signalling to allow gene expression, increase the release of neurotransmitters to augment the transmission of sensory signals and to enhance sensory neuronal hyperexcitability and central sensitisation that subsequently develop prolonged inflammatory responses and pain [10, 65].
Spinal cord
Several studies revealed that blocking P2X4R activation by ATP binding may reduce the occurrence of tactile allodynia in the spinal cord following intrathecal antisense oligonucleotides [20]. This may occur due to the inhibition of microglial activation since the binding of ATP to P2X receptors expressed on microglia may lead to its activation. Extracellular ATP stimulates P2X4R, triggering a transient increase in Ca2+ influx in microglia. In fact, microglial activation relies on high concentrations of Ca2+ influx [68]. Ulmann and colleagues [25] revealed the upregulation of P2X4R in activated spinal microglia at the level of transcription, translation and post-translation following peripheral nerve injury. Although P2X4R is capable of high Ca2+ concentration fluxes comparable to that of N-methyl-d-aspartate receptor (NMDAR) subtypes, it cannot implicate NMDAR because this receptor is blocked by Mg2+. Therefore, P2X4R potentially matches the regulatory function of the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) subtype of glutamate receptors to develop synaptic plasticity [69].
The activation of P2X4R activation also implicates BDNF-tyrosine kinase B (BDNF-TrKB) signalling pathway, resulting in the augmentation of BDNF production, c-fos in microglia, p38 level and stimulation of the trigeminal neuralgia [70]. In the spinal cord of P2X4R-deficient mouse, it was discovered that increased BDNF signalling, extracellular signal-regulated kinase 1/2 (ERK ½) phosphorylation of NMDAR GluN1 subtype and KCC2 downregulation following peripheral inflammation were impaired [65], suggesting that P2X4R played a significant role leading to chronic inflammatory pain. In another study, certain signalling mediators such as fibronectin, Lyn tyrosine kinase and chemokine ligand 2 (CCL2) can stimulate the upregulation of P2X4R on microglia or induce its trafficking to the cell surface of microglia following peripheral nerve injury [42]. The activated P2X4R may lead to the activation of the phosphatidylinositol 3-kinases-protein kinase B (P13K/Akt) pathway, which further leads to microglial activation and promoting its migration [47].
The increased activation of P2X4R is also linked to the enhanced level of catestatin (CST) and subsequently leads to pain aggravation [71]. Although the development of CST has yet to be investigated during the central pathogenesis of RA, it may have some effects on causing further aberrant stimulation of P2X4R on the surface of microglia. Deng et al. [71] revealed that following nerve injury, phosphorylation of ERK 1/2 in microglia leads to the increased expression of CST which further enhances the activated P2X4R. CST can also chemotactically accumulate monocytes and mediate neurogenic inflammation [72]. Moreover, the activation of ERK 1/2 can lead to the release of pro-inflammatory mediators in the spinal dorsal horn region [71]. Likewise, activated T-helper cell 1 (Th1 lymphocytes) from peripheral blood can infiltrate the blood-brain barrier and communicate with spinal microglia via the release of pro-inflammatory mediators, including interferon-ɣ (IFN-ɣ), to activate P2X4R [42, 73]. Consequently, these mechanisms lead to the stimulation of microglia and astrocytes productions in the spinal cord, produce multiple neuroinflammatory signalling mediators (i.e. IL-6, IL-10, IL-1β, IL-6 and IL-18), stimulate a variety of cyclooxygenase-2 ((COX-2), cysteine protease-1, nitric oxide synthase 2, carboxypeptidase, metalloproteinase and chymotrypsin) and promote neuroinflammatory responses [74,75,76] which can be deleterious, or facilitate synaptic plasticity and cell death. The highly activated microglia can further enhance the expression levels of P2X4R and, therefore, prolong the occurrence of hyperalgesia. The simplified proposed mechanism following P2X4R activation in the spinal cord is illustrated in Fig. 3.

Central mechanisms after P2X4R activation in the spinal cord. The affected neuron secretes adenosine triphosphate (ATP), chemokine ligand 2 (CCL2) and fibronectin that upregulate microglia P2X4 receptor (P2X4R) and promote its trafficking to cell surface prior to peripheral nerve injury due to rheumatoid arthritis (RA). Helper T-cells from peripheral blood vessel may pass through the blood-brain barrier (BBB) and interact with microglia via the released pro-inflammatory cytokines including interferon-ɣ (IFN-ɣ). As a consequence of ATP binding to P2X4R, several pathways are activated including phosphorylation of extracellular signal-regulated kinase 1/2 (ERK 1/2) and p38 mitogen-activated protein kinase (p38 MAPK) and phosphatidylinositol 3-kinases-protein kinase B (PI3K/Akt) that leads to the release of brain-derived neurotrophic factor (BDNF) and pro-inflammatory mediators (IL-1β, TNF-α, IL-6 and prostaglandins) that contributes to neurogenic inflammation. Secreted BDNF may then bind to tropomyosin receptor kinase B (TrkB) receptors on nociceptive neurons that further downregulate potassium chloride cotransporter (KCC2) resulting in the accumulation of chloride anions (Cl-) in the neurons. Consequently, the subsequent binding to ɣ-aminobutyric acid A receptor (GABAA receptor) that is normally inhibitory, shifts to the disinhibition of inhibitory neurons. Meanwhile, the production of cathestatin (CST) via the phosphorylation of extracellular signal-regulated kinase 1/2 (ERK 1/2) in microglia may further augment the P2X4R activation on the activated microglia. The hyperexcitability resulted from GABA depolarisation together with the release of pro-inflammatory cytokines from microglia and lesioned nerve, as well as N-methyl-d-aspartate receptors (NMDARs) activation, leads to sensitisation of spinal circuits and therefore, chronic pain
Brain
A significant number of studies have shown prominent expression of P2X4R in different neuronal populations in the hippocampus, cerebellum and cortex [65]. P2X4R expression has been identified in neurons throughout CNS, mediating synaptic transmission, and maintaining biological responses in tissues. In the pyramidal neurons of the hippocampus, activation of P2X4R is triggered by the occurrence of electrical activity contributed by tetanus stimuli, leading to the production of synaptic plasticity [68]. In the rat brain, dense distribution of P2X4R was identified in granule cells of the dentate gyrus, CA1/CA3 pyramidal cells, Purkinje cells of the cerebellar cortex and neurons pontine nucleus [77, 78]. The localisation of P2X4R was also found at postsynaptic membranes as opposed to terminals of Schaffer collaterals in CA1 pyramidal cells, densely located at the peripheral region of parallel fibre postsynaptic specialisation and Schaffer collateral synapses [77]. Although electron microscopy immunostaining analysis revealed that the P2X4R was preferentially localised to post-synaptic terminals, its localisation was also found at both presynaptic and postsynaptic terminals in various brain regions [77, 79].
The mechanisms of ATP activation involving microglia activation together with neuronal interactions in the brain are also similar in the spinal cord since microglia is ubiquitously distributed throughout CNS [80]. In neurodegenerative diseases, P2X4R expression is upregulated in GABAergic interneurons and GABAergic spiny neurons of the striatum and substantia nigra as reported in a rat model of Parkinson’s disease [81]. In addition, the abundant expression of P2X4R in the hypothalamus and anterior pituitary glands indicates that P2X4R plays a significant role in hypothalamic-pituitary mechanisms in the CNS [82]. Furthermore, the reduced synaptic facilitation and long-term potentiation (LTP) in P2X4R-knockout mice indicate that P2X4R plays a crucial role in the development of central sensitisation [26]. However, there is still limited discovery regarding the role of P2X4R in a brain region associated with RA.
Previous studies have shown that, as one of the ATP-activated P2 receptors, P2X4R can play a role in the reward circuit in a particular brain region by regulating glutamate [83, 84] or dopamine release [85]. P2X4R is also involved in the development of long-term potentiation (LTP) and long-term depression (LTD) [80]. It is highly probable that the unstable emotion and depression reported by RA patients involves the activation of P2X4R via certain specific pathways. Therefore, more research is required to explore the role of P2X4R within a particular brain region associated with pain transmission so that the specific pathway can be modulated to combat chronic pain and negative emotions associated with RA.
Sex differences in microglia-P2X4 receptor signalling
Although the occurrence of RA is more worsening in female than male patients [86] with a higher prevalence rate (3:1 ratio) [87], the specific molecular mechanisms leading to this complication is less extensively studied. It is reported that the pro-inflammatory immune responses to tissue damage in females are higher than in males, therefore, directly results in more pain. It is demonstrated in an in vitro study that the female sex hormone, oestrogen, is found to inhibit the release of pro-inflammatory cytokines such as IL-1α, IL-1β, TNF-α and apoptosis from microglia following the exposure of lipopolysaccharide (LPS) [88]. Other than that, a recent study by Vacca et al. [89] discovered that the oestradiol-treated female mice showed decreased number of astrocyte after the induction of nerve injury, explaining the possible significant role of hormones in the development of pain responses. However, there is very limited report regarding the differences in P2X4R activation across the gender in RA, in which, need to be discovered.
However, the recent researches by Sorge et al. [90] and Mapplebeck et al. [91] have revealed the major differences in gender in regards to the microglial and P2X4R signalling of chronic neuropathic pain. These novel discoveries justify that the P2X4R signalling in microglia is not the underlying key leading to chronic pain hypersensitivity in the females. In contrast, the specific upregulation of P2X4R is consistently found in male variant across the species and pain models. In the mice model of peripheral nerve injury (PNI), Sorge et al. [90] reported that the P2X4 transcript was increased in male but not in the female mice as shown in the quantitative reverse-transcriptase polymerase chain reaction (qRT-PCR) analysis, although both gender showed the similar levels of allodynia. Furthermore, the similar findings have been shown by Mapplebeck et al. [91] in the similar neuropathic pain model that the P2X4R from the fluorescent-activated cell sorting-isolated CD11b + (microglia) cells in the spinal cord was upregulated in males but not female rat. In the same study, the authors also demonstrated that the stimulation of ATP alone contributes to the increase of P2X4R level in the microglia primary cell culture of the male rat following a chronic pain-inducing PNI, but not in the female rat microglia cell culture. These novel findings showed that the P2X4R is increased merely in the males following the chronic neuropathic pain, therefore justifies the sex differences in the activation of microglia-P2X4R signalling. It is possible that these findings are associated with the higher affinity of interferon regulatory factor-5 (IRF5) binding to the P2X4 promoter region in male compared to female-derived primary microglia [91]. Moreover, it is possible that the hormones may influence the microglia-P2X4R signalling since the injection of P2X4R antagonist to the testosterone-treated female mice following PNI results in the reduced allodynia compared to the castrated male mice [90]. As the dimorphism is shown in the activation of P2X4R across the sexes, it is possible that the mechanism of P2X4R during the pathogenesis of RA also differ between the gender, which needs further investigations.
Potent P2X4 receptor inhibitors
In the treatment of RA-related pain, there have only been a limited number of drugs tested to demonstrate antagonism towards P2X4R. Several studies identified potential agents to block P2X4R in the neuropathic pain models. However, there is a possibility that tested drugs could be beneficial in the treatment of RA pain. The use of a general broad-range purinergic or general P2X receptor antagonist in several diseases produced unsatisfactory outcomes. In an in vitro study, pyridoxal phosphate-6-azo (benzene-2,4-disulfonic acid (PPADS)), a general non-selective P2 receptor antagonist [83, 92], demonstrated no mark effect on basal adenosine diphosphate (ADP)-directed and morphine-induced migration of microglia [47]. Furthermore, in an in vitro study using HEK293 cells transfected with the P2X4R, the responses of this receptor to ATP were found to be insensitive to the blockade of PPADS although being administered at a very high concentration [93]. Meanwhile, TNP-ATP has been shown to produce beneficial effects as a P2X4R antagonist [47]. Nevertheless, this compound implicates other types of P2X receptors with high potency [94]. It is suggested that the use of drugs specifically targeting P2X4R is best applied for the treatment of related diseases.
Interestingly, certain drugs that strongly antagonise P2X4R and prevent Ca2+ influx such as paroxetine, an analogue of monoamine uptake blockers, also produce potent antidepressant effects [95]. In a rat model for chronic constriction injury (CCI), paroxetine showed potent inhibitory effects against P2X4R and attenuated allodynia and thermal hyperalgesia [96]. A phenoxazine derivative, PSB-12602, inhibits P2X4R via an allosteric mechanism [97]. Other than that, 1-(2,6-dibromo-4-isopropyl-phenyl)-3-(3-pyridyl) urea or BX430 has recently been identified as possessing strong antagonist properties against human P2X4R-mediated Ca2+ uptake [98]. However, these drugs have yet to be tested on animal models.
Several studies have discovered 5-(3-bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-one (5-BDBD) to have strong anti-P2X4R activity in various animal models, such as chronic migraine [99], inflamed airways [42] and central sensitisation [94]. This drug specifically discriminates against P2X4R and exerts no effects on other P2X receptors when co-expressed in the same tissue [94]. Coddou and colleagues [94] revealed that 5-BDBD inhibited LTP via dissociation of PSD-95 multi-protein complex interactions that further downregulate NMDAR activity. However, this drug dissolves poorly in water and requires the best medium to dissolve in order to produce a good P2X4R antagonistic effect.
Increasing attention has been given to (5-[3-(5-thioxo-4H-[1,2,4]oxadiazol-3-yl)phenyl]-1H-naphtho[1, 2-b][1,4]diazepine-2,4(3H,5H)-dione), also known as NP-1815-PX, which has recently discovered as a novel P2X4R antagonist. In a murine model for herpetic pain, the selective antagonistic effect of NP-1815-PX against P2X4R alleviates allodynia without implicating acute nociceptive pain and motor function. However, it was found to be effective when administered intrathecally but produced no significant effects when given orally [22]. Another drug, PSB-12054, inhibits P2X4R via allosteric mechanisms, with a 30-fold selectivity towards P2X4R compared to P2X1 receptor and a 50-fold selectivity compared to other types of P2X receptors [14].
In an in vitro study using human monocyte and macrophage cell cultures, PSB-12062 strongly attenuated thapsigargin-resistant components [100]. It positively modulated the ATP-mediated activation of transforming growth factor β2 (TGF-β2) gene expression while attenuating the activation of ATP-mediated C-X-C motif chemokine-5 (CXCL5) gene expression [100]. Another novel P2X4R antagonist, PSB-15417, also showed strong attenuation of mechanical allodynia in a diabetic neuropathy rat model and inhibited P2X4R activation that was mainly expressed on satellite glial cells of DRG neurons [57].
Tricarbonyldichlororuthenium (II) dimer (CORM-2) has been identified as a potent P2X4R antagonist in a study by Jurga et al. [101] that attenuated hyperalgesia and boosted the antihyperalgesic and antiallodynic effects of morphine and buprenorphine in a CCI rat model. It was further discovered that antagonising P2X4R by CORM-2 suppressed microglial and astrocyte activations decreased the expressions of matrix metallopeptidase 9 (MMP-9), IL-1β, IL-18 and IL-6 and reduced nociceptive responses, possibly through the inhibition of p38 MAPK, ERK 1/2 and PI3K/Akt signalling pathways [102]. However, there was a contradiction by Hervera et al. [103] asserting that P2X4R was potentially not the main target of CORM-2, as the modulation of HO-1/CO pathways by CORM-2 was implicated by the inhibition of NO produced by the NOS1 expression from activated neurons. A small number of novel P2X4R antagonists have been tested on chronic pain associated with RA, and the potential drugs available need to be tested to assess their efficiency in managing RA-related pain.
Conclusion and future research
Flare pain in RA is not solely caused joint inflammatory reactions, but also through several possible mechanisms resulting from other abnormalities. There is currently no optimum treatment method available to manage the chronic pain associated with RA. The discovery of P2X4R has provided a new avenue for understanding the molecular mechanisms underlying chronic pain associated with RA and other related diseases. The understanding of the role of P2X4R activation in the pathophysiology of chronic pain related to RA is still at an early stage. However, a collection of literature currently available provides hope that the flaring pain experienced by RA patients can be minimised when P2X4R is attenuated. Hence, identifying and exploring the relevant pathological mechanisms involving P2X4R in RA could be a novel approach to manage pain resulting from RA.
Abbreviations
- AMPA:
-
α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor
- ASC:
-
PYD- and CARD-domain containing caspase-1
- ASIC:
-
Acid-sensing channel
- ADP:
-
Adenosine diphosphate
- ATP:
-
Adenosine triphosphate
- BDNF:
-
Brain-derived neurotrophin factor
- BDNF-TrKB:
-
BDNF-tyrosine kinase B
- BX430:
-
1-(2,6-Dibromo-4-isopropyl-phenyl)-3-(3-pyridyl)urea
- Ca2+ :
-
Calcium ion
- CCI:
-
Chronic constriction injury
- CCL2:
-
Chemokine ligand 2
- CFA:
-
Complete Freund’s adjuvant
- CNS:
-
Central nervous system
- CORM-2:
-
Tricarbonyldichlororuthenium (II) dimer
- COX-2:
-
Cyclooxygenase-2
- CST:
-
Catestatin
- CXCL25:
-
ATP-mediated C-X-C motif chemokine-5
- DMARDs:
-
Disease-modifying anti-rheumatic drugs
- DRG:
-
Dorsal root ganglion
- ENaC:
-
Epithelial Na+ channel
- EPSPs:
-
Excitatory postsynaptic potentials
- ERK 1/2 :
-
Extracellular signal-regulated kinase 1/2
- GABAA :
-
ɣ-Aminobutyric acid A receptor
- GABAergic:
-
ɣ-Aminobutyric acidergic
- IFN-ɣ:
-
Interferon-ɣ
- IL-6:
-
Interleukin-6
- IL-17:
-
Interleukin-17
- IL-1β:
-
Interleukin-1β
- IRF5:
-
Interferon regulatory factor-5
- KCC2:
-
Potassium-chloride cotransporter
- K+ :
-
Potassium ion
- LTD:
-
Long-term depression
- LTP:
-
Long-term potentiation
- MMP-9:
-
Matrix metallopeptidase-9
- Na+ :
-
Sodium ion
- NLRP1:
-
Pyrin domain containing 1 of activated NLR family
- NMDAR:
-
N-Methyl-d-aspartate receptor
- NP-1815-PX:
-
(5-[3-(5-Thioxo-4H-[1,2,4]oxadiazol-3-yl)phenyl]-1H-naphtho[1, 2-b][1,4]diazepine-2,4(3H,5H)-dione)
- PGs:
-
Prostaglandins
- PNI:
-
Peripheral nerve injury
- PPADs:
-
Pyridoxal phosphate-6-azo(benzene-2,4-disulfonic acid)
- P2X4R:
-
P2X4 receptor
- P2X7R:
-
P2X7 receptor
- P13K/Akt:
-
Phosphatidylinositol 3-kinase-protein kinase B
- p38 MAPK:
-
p38 Mitogen-activated protein kinase
- RA:
-
Rheumatoid arthritis
- TGF-β2:
-
Transforming growth factor-β2
- Th1:
-
T-helper cell 1
- TNF-α:
-
Tumor necrosis factor-α
- 5-BDBD:
-
5-(3-Bromophenyl)-1,3-dihydro-2H-benzofuro[3,2-e]-1,4-diazepin-2-one
References
Ten Klooster P, De Graaf N, Vonkeman H (2019) Association between pain phenotype and disease activity in rheumatoid arthritis patients: a non-interventional, longitudinal cohort study. Arthritis Res Ther 21:257
Walsh DA, McWilliams DF (2014) Mechanisms, impact and management of pain in rheumatoid arthritis. Nat Rev Rheumatol 10:581–592. https://doi.org/10.1038/nrrheum.2014.64
Lee YC, Cui J, Lu B, Frits ML, Iannacone CK, Shadick NA, Weinblatt ME, Solomon DH (2011) Pain persists in DAS28 rheumatoid arthritis remission but not in ACR/EULAR remission: a longitudinal observational study. Arhritis Res Ther 13:R83
Hewlett ST, Sanderson T, May J, Alten R, Bingham CO III, Cross M, March L, Pohl C, Woodworth T, Bartlett SJ (2012) I’m hurting, I want to kill myself’: rheumatoid arthritis flare is more than a high joint count—an international patient perspective on flare where medical help is sought. Rheumatology 51(1):69–76. https://doi.org/10.1093/rheumatology/keq455
Courvoisier N, Dougados M, Cantagrel A, Goupille P, Meyer O, Sibilia J, Daures JP, Combe B (2008) Prognostic factors of 10-year radiographic outcome in early rheumatoid arthritis: a prospective study. Arthritis Res Ther 10:R106. https://doi.org/10.1186/ar2498
McWilliams DF, Zhang W, Mansell JS, Kiely PD, Young A, Walsh DA (2012) Predictors of change in bodily pain in early rheumatoid arthritis: an inception cohort study. Arthritis Care Res 64:1505–1513. https://doi.org/10.1002/acr.21723
Umeda N, Matsumoto I, Sumida T (2017) The pathogenic role of ACPA in rheumatoid arthritis. Nihon Rinsho Meneki Gakkai Kaishi 40:391–395. https://doi.org/10.2177/jsci.40.391
Wigerbald G, Bas DU, Fernandes-Cerqueira C, Krishanmurthy A, Nandakumar KS, Rogoz S, Kato J, Sandor K, Su J, Lundlberg K, Holmdahl R, Jakobsson P-J, Malmström V, Catrina AI, Klareskorg L, Svensson CI (2016) Autoantibodies to citrullinated proteins may induce joint pain independent of inflammation. Ann Rheum Dis 75:730–865. https://doi.org/10.1136/annrheumdis-2015-208094
Christensen AW, Rifbjerg-Madsen S, Christensen R, Dreyer L, Tillingsøe H, Seven S, Boesen M, Ellegard K, Bliddal H, Danneskiold-Samsøe B (2016) Non-nociceptive pain in rheumatoid arthritis is frequent and affects disease activity estimation: cross-sectional data from the FRAME study. Scand J Rheumatol 45:461–469. https://doi.org/10.3109/03009742.2016.1139174
Zhang W-J, Zhu Z-M, Liu Z-X (2020a) The role of P2X4 receptor in neuropathic pain and its pharmacological properties. Pharmacol Res 158:104875. https://doi.org/10.1016/j.phrs.2020.104875
Khakh BS, North RA (2006) P2X receptors as cell-surface ATP sensors in health and disease. Nature 442:527–532. https://doi.org/10.1038/nature04886
Murrel-Lagnado RD, Qureshi OS (2008) Assembly and trafficking of P2X purinergic receptors. Mol Membr Biol 25:321–331. https://doi.org/10.1080/09687680802050385
Collo G, North RA, Kawashima E, Merlo-Pich E, Neidhart S, Suprenant A, Buell G (1996) Cloning of P2X5 and P2X6 receptors and the distribution and properties of an extended family of ATP-gated ion channels. J Neurosci 16:2495–2507. https://doi.org/10.1523/JNEUROSCI.16-08-02495.1996
Zhang W-J, Luo H-L, Zhu Z-M (2020b) The role of P2X4 receptors in chronic pain: a potential pharmacological target. Biomed Pharmacother 129:110447. https://doi.org/10.1016/j.biopha.2020.110447
North RA (2002) Molecular physiology of P2X receptors. Physiol Rev 82:1013–1067. https://doi.org/10.1152/physrev.00015.2002
Boyden SD, Hossain IN, Wohlfahrt A, Lee YC (2016) Non-inflammatory causes of pain in patients with rheumatoid arthritis. Curr Rheumatol Rep 18:30. https://doi.org/10.1007/s11926-016-0581-0
Dal Ben D, Buccioni M, Lambertucci C, Marucci G, Thomas A, Volpini R (2015) Purinergic P2X receptors: structural models and analysis of ligand-target interaction. Eur J Med Chem 89:561–580. https://doi.org/10.1016/j.ejmech.2014.10.071
Asif A, Khalid M, Ahmad H, Rehman AU (2019) Role of purinergic receptors in hepatobiliary carcinoma in Pakistani population: an approach towards proinflammatory role of P2X4 and P2X7 receptors. Purinergic Signal 15:367–374. https://doi.org/10.1007/s11302-019-09675-0
Wang Y, Chen Z, Liu C, Lu X, Yang C, Qiu S (2019) Distributive differences of P2Xs between the forelimb and hind limb of adjuvant arthritis rats and intervention by Notopterygh rhizoma et radix. Pharm Biol 57:81–88. https://doi.org/10.1080/13880209.2018.1561730
Tsuda M, Shigemoto-Mogami Y, Koizumi S, Mizokoshi A, Kohsaka S, Salter MW, Inoue K (2003) P2X4 receptors induced in spinal microglia gate tactile allodynia after nerve injury. Nature 424:778–783. https://doi.org/10.1038/nature01786
Harkat M (2017) Etude moléculaire de la dilatation des récepteurs P2X. Doctoral dissertation, Université de Strasbourg
Matsumura Y, Yamashita T, Sasaki A, Nakata A, Kohno K, Masuda T, Tozaki-Saitoh H, Imai T, Kuraishi Y, Tsuda M (2016) A novel P2X4 receptor-selective antagonist produces anti-allodynic effect in a mouse model of herpetic pain. Sci Rep 6:32461. https://doi.org/10.1038/srep32461
Zhang W, Liu Y, Yang B, Liu Z, Yu Q (2019) Microencapsulated olfactory ensheathing-cell transplantation reduces pain in rats by inhibiting P2X4 receptor overexpression in the dorsal root ganglion. Neuro Report 30:120–126. https://doi.org/10.1097/WNR.0000000000001170
Engel T, Alves M, Sheedy C, Henshall DC (2016) ATPergic signalling during seizures and epilepsy. Neuropharmacology 104:140–153. https://doi.org/10.1016/j.neuropharm.2015.11.001
Ulmann L, Levavasseur F, Avignone E, Peyroutou R, Hirbec H, Audinat E, Rassendren F (2013) Involvement of P2X4 receptors in hippocampal microglial activation after status epilepticus. Glia 61:1306–1319. https://doi.org/10.1002/glia.22516
Bertin E, Deluc T, Pilch KS, Martinez A, Pougnet J-T, Doudnikoff E, Allain A-E, Bergmann P, Rousseau M, Toulmé E (2020) Increased surface P2X4 receptor regulates anxiety and memory in P2X4 internalization-defective knock-in mice. Mol Psychiatry. https://doi.org/10.1038/s41380-019-0641-8
Srivastava P, Cronin CG, Scranton VL, Jacobson KA, Liang BT, Verma R (2020) Neuroprotective and neuro-rehabilitative effects of acute purinergic receptor P2X4 (P2X4R) blockade after ischemic stroke. Exp Neurol 329:113308. https://doi.org/10.1016/j.expneurol.2020.113308
Tóth A, Antal Z, Bereczeki D, Sperlágh B (2019) Purinergic signalling in Parkinson’s disease: a multi-target system to combat neurodegeneration. Neurochem Res 44:2413–2422. https://doi.org/10.1007/s11064-019-02798-1
Wilkermann VE, Thompson KE, Neuland K, Jaramillo AM, Fois G, Schmidt H, Wittekindt OH, Han W, Tuvim MJ, Dickey BF (2019) Inflammation-induced upregulation of P2X4 expression augments mucin secretion in airway epithelia. Am J Physiol-Lung C 316:L58–L70. https://doi.org/10.1152/ajplung.00157.2018
Zabala A, Vazquez-Villoldo N, Rissiek B, Gejo J, Martin A, Palomino A, Perez-Samartín A, Pulagam KR, Lukowiak M, Capetillo-Zarate E (2018) P2X4 receptor controls microglia activation and favors remyelination in autoimmune encephalitis. EMBO Mol Med 10:e8743. https://doi.org/10.15252/emmm.201708743
Verma R, Cronin CG, Hudobenko J, Venna VR, McCullough LD, Liang BT (2017) Deletion of the P2X4 receptor is neuroprotective acutely, but induces a depressive phenotype during recovery from ischemic stroke. Brain Behav Immun 66:302–312. https://doi.org/10.1016/j.bbi.2017.07.155
Rana I, Badoer E, Alahmadi E, Leo C, Woodman O, Stebbing M (2014) Microglia are selectively activated in endocrine and cardiovascular control centres in streptozotocin-induced diabetic rats. J Neuroendocrinol 26:413–425. https://doi.org/10.1111/jne.12161
Dhont TWL, Verbruggen A, Oostendorp RAB, Duquet W (1999) Pain threshold in patients with rheumatoid arthritis and effect of manual oscillations. Scand J Rheumatol 28:88–93. https://doi.org/10.1080/030097499442540
Lee YC, Lu B, Edwards RR, Wasan AD, Nassikas NJ, Clauw DJ, Solomon DH, Karlson EW (2013) The role of sleep problems in central pain processing in rheumatoid arthritis. Arthritis Rheum 65:59–68. https://doi.org/10.1002/art.37733
Wendler J, Hummel T, Reissinger M, Manger B, Pauli E, Kalden JR, Kobal G (2001) Patients with rheumatoid arthritis adapt differently to repetitive painful stimuli compared to healthy controls. J Clin Neurosci 8:272–277. https://doi.org/10.1054/jocn.1999.0775
Gerecz-Simon EM, Tunks ER, Heale JA, Kean WF, Buchanan WW (1989) Measurement of pain threshold in patients with rheumatoid arthritis, ankylosing spondylitis, and healthy controls. Clin Rheumatol 8:467–474. https://doi.org/10.1007/BF02032098
Schaible H-G, Ebersberger A, Von Banchet GS (2002) Mechanisms of pain in arthritis. Ann N Y Acad Sci 966:343–354. https://doi.org/10.1111/j.1749-6632.2002.tb04234.x
Leffler A-S, Kosek E, Lerndal T, Nordmark B, Hansson P (2002) Somatosensory perception and function of diffuse noxious inhibitory controls (DNIC) in patients suffering from rheumatoid arthritis. Eur J Pain 6:161–176. https://doi.org/10.1053/eujp.2001.0313
Duris FH, Fava RA, Noelle RJ (1994) Collagen-induced arthritis as a model of rheumatoid arthritis. J Clin Immunol 73:11–18. https://doi.org/10.1006/clin.1994.1164
Billiau A, Matthys P (2001) Modes of action of Freund’s adjuvants in experimental models of autoimmune diseases. J Leukoc Biol 70:849–860. https://doi.org/10.1189/jlb.70.6.849
Tsuda M, Kuboyama K, Inoue T, Nagata K, Tozaki-Saitoh H, Inoue K (2009) Behavioral phenotypes of mice lacking purinergic P2X4 receptors in acute and chronic pain assays. Mol Pain 5(1744-8069):5–28. https://doi.org/10.1186/1744-8069-5-28
Chen X-M, Xu J, Song J-G, Zheng B-J, Wang X-R (2015) Electroacupuncture inhibits excessive interferon-γ evoked up-regulation of P2X4 receptor in spinal microglia in a CCI rat model for neuropathic pain. Br J Anaesth 114:150–157. https://doi.org/10.1093/bja/aeu199
Lyness WH, Smith FL, Heavner JE, Iacono CU, Garvin RD (1989) Morphine self-administration in the rat during adjuvant-induced arthritis. Life Sci 45:2217–2224. https://doi.org/10.1016/0024-3205(89)90062-3
Park EH, Kahng JH (1999) Suppressive effects of propolis in rat adjuvant arthritis. Arch Pharm Res 22:554–558. https://doi.org/10.1007/BF02975325
Liu Y-L, Lin H-M, Rong Z, Wu J-C, Han R, Raymond LN, Reid RF, Qin Z-H (2009) Suppression of complete Freund’s adjuvant-induced adjuvant arthritis by cobratoxin. Acta Pharmacol Sin 30:219–227. https://doi.org/10.1038/aps.2008.20
Mahdi HJ, Khan NAK, Asmawi MZ, Mahmud R, Vikneswaran A, Murugaiyah L (2018) In vivo anti-arthritic and anti-nociceptive effects of ethanol extract of Moringa oleifera leaves on complete Freund’s adjuvant (CFA)-induced arthritis in rats. Integr Med Res 7:85–94. https://doi.org/10.1016/j.imr.2017.11.002
Horvath RJ, De Leo JA (2009) Morphine enhances microglial migration through modulation of P2X4 receptor signaling. J Neurosci 29:998–1005. https://doi.org/10.1523/JNEUROSCI.4595-08.2009
Dewangan AK, Perumal Y, Pavurala N, Chopra K, Mazumder S (2017) Preparation, characterization and anti-inflammatory effects of curcumin loaded carboxymethyl cellulose acetate butyrate nanoparticles on adjuvant induced arthritis in rats. J Drug Deliv Sci Technol 41:269–279. https://doi.org/10.1016/j.jddst.2017.07.022
Cook CD, Moore KI (2006) Effects of sex, hindpaw injection site and stimulus modality on nociceptive sensitivity in arthritic rats. Physiol Behav 87:552–562. https://doi.org/10.1016/j.physbeh.2005.12.005
Smith FL, Ken F, Lowe J, Welch SP (1998) Characterization of Δ9-tetrahydrocannabinol and anandamide antinociception in nonarthritic and arthritic rats. Pharmacol Biochem Behav 60:183–191. https://doi.org/10.1016/S0091-3057(97)00583-2
Guo L-H, Trautmann K, Schluesener HJ (2005) Expression of P2X4 receptor by lesional activated microglia during formalin-induced inflammatory pain. J Neuroimmunol 163:120–127. https://doi.org/10.1016/j.jneuroim.2005.03.007
Majdal HM, Sulaiman SM, Sulaiman ME (2012) Nerve conduction and electromyography in rheumatoid arthritis patients: a case-control study. Ann Coll Med Mosul 38:44–51
Bo X, Kim M, Nori SL, Schoepfer R, Burnstock G, North RA (2003) Tissue distribution of P2X 4 receptors studied with an ectodomain antibody. Cell Tissue Res 313:159–165. https://doi.org/10.1007/s00441-003-0758-5
Ulmann L, Hirbec H, Rassendren F (2010) P2X4 receptors mediate PGE2 release by tissue-resident macrophages and initiate inflammatory pain. EMBO J 29:2290–2300. https://doi.org/10.1038/emboj.2010.126
Ying M, Liu H, Zhang T, Jiang C, Gong Y, Wu B, Zou L, Yi Z, Rao S, Li G (2017) Effect of artemisinin on neuropathic pain mediated by P2X4 receptor in dorsal root ganglia. Neurochem Int 108:27–33. https://doi.org/10.1016/j.neuint.2017.02.004
Williams WA, Linley JE, Jones CA, Shibata Y, Snijder A, Button J, Hatcher JP, Huang L, Taddese B, Thornton P (2019) Antibodies binding the head domain of P2X4 inhibit channel function and reverse neuropathic pain. Pain 160:1989–2003. https://doi.org/10.1097/j.pain.0000000000001587
Teixeira JM, Dos Santos GG, Neves AF, Athie MCP, Bonet IJM, Nishijima CM, Farias FH, Figueiredo JG, Hernandez-Olmos V, Alshaibani S (2019) Diabetes-induced neuropathic mechanical hyperalgesia depends on P2X4 receptor activation in dorsal root ganglia. Neuroscience 398:158–170. https://doi.org/10.1016/j.neuroscience.2018.12.003
Pan B, Zhang Z, Chao D, Hogan QH (2018) Dorsal root ganglion field stimulation prevents inflammation and joint damage in a rat model of rheumatoid arthritis. Neuromodulation. 21:247–253. https://doi.org/10.1111/ner.12648
Kerschensteiner M, Gallmeier E, Behrens L, Leal VV, Hoppe E, Oropeza-Wekerle RL, Bartke I, Stadelmann C, Lassmann H, Wekerle H, Hohlfeld (1999) Activated human T cells, and monocytes produce brain-derived neurotrophic factor in vitro and in inflammatory brain lessions: a neuroprotective rol of inflammation? J Exp Med 189:865–870. https://doi.org/10.1084/jem.189.5.865
Klein K, Aeschlimann A, Jordan S, Gay R, Gay S, Sprott H (2012) ATP induced brain-derived neurotrophic factor expression and release from osteoarthritis synovial fibroblasts is mediated by purinergic receptor P2X4. PLoS One 7:e36693. https://doi.org/10.1371/journal.pone.0036693
Schulte-Herbrüggen O, Nassenstein C, Lommatzsch M, Quarcoo D, Renz H, Braun A (2005) Tumor necrosis factor-α and interleukin-6 regulate secretion of brain-derived neurotrophic factor in human monocytes. J Neuroimmunol 160:204–209. https://doi.org/10.1016/j.jneuroim.2004.10.026
Kawanokuchi J, Shimizu K, Nitta A, Yamada K, Mizuno T, Takeuchi H, Suzumura A (2008) Production and functions of IL-17 in microglia. J Neuroimmunol 194:54–61. https://doi.org/10.1016/j.jneuroim.2007.11.006
Trang T, Beggs S, Wan X, Salter MW (2009) P2X4-receptor-mediated synthesis and release of brain-derived neurotrophic factor in microglia is dependent on calcium and p38-mitogen-activated protein kinase activation. J Neuro-Oncol 29:3518–3528. https://doi.org/10.1523/JNEUROSCI.5714-08.2009
Li F, Guo N, Ma Y, Ning B, Wang Y, Kou L (2014) Inhibition of P2X4 suppresses joint inflammation and damage in collagen-induced arthritis. Inflammation 37:146–153. https://doi.org/10.1007/s10753-013-9723-y
Lalisse S, Hua J, Lenoir M, Linkc N, Rassendren F, Ulmann L (2018) Sensory neuronal P2RX4 receptors controls BDNF signaling in inflammatory pain. Sci Rep 8:1–12. https://doi.org/10.1038/s41598-018-19301-5
Kawano A, Tsukimoto M, Mori D, Noguchi T, Harada H, Takenouchi T, Kitani H, Kojima S (2012) Regulation of P2X7-dependent inflammatory functions by P2X4 receptor in mouse macrophages. Biochem Biophys Res Commun 420:102–107. https://doi.org/10.1016/j.bbrc.2012.02.122
Liu S, Liu Y-P, Huang Y-K, Zhang Y-K, Song AA, Ma P-C, Song X-J (2015) Wnt/Ryk signaling contributes to neuropathic pain by regulating sensory neuron excitability and spinal synaptic plasticity in rats. Pain 156:2572–2584. https://doi.org/10.1097/j.pain.0000000000000366
Sim JA, Chaumont S, Jo J, Ulmann L, Young MT, Cho K, Buell G, North RA, Rassendren F (2006) Altered hippocampal synaptic potentiation in P2X4 knock-out mice. J Neurosci 26:9006–9009. https://doi.org/10.1523/JNEUROSCI.2370-06.2006
Chun BJ, Stewart BD, Vaughan DD, Bachstetter AD, Kekenes-Huskey PM (2019) Simulation of P2X-mediated calcium signalling in microglia. J Physiol 597:799–818. https://doi.org/10.1113/JP277377
Liu C, Zhang Y, Liu Q, Jiang L, Li M, Wang S, Long T, He W, Kong X, Qin G (2018) P2X4-receptor participates in EAAT3 regulation via BDNF-TrkB signaling in a model of trigeminal allodynia. Mol Pain 14:1744806918795930. https://doi.org/10.1177/1744806918795930
Deng Z, Li C, Liu C, Du E, Xu C (2018) Catestatin is involved in neuropathic pain mediated by purinergic receptor P2X4 in the spinal microglia of rats. Brain Res Bull 142:138–146. https://doi.org/10.1016/j.brainresbull.2018.07.003
Egger M, Beer AGE, Theurl M, Schgoer W, Hotter B, Tatarczyk T, Vasiljevic D, Frauscher S, Marksteiner J, Patsch JR (2008) Monocyte migration: a novel effect and signaling pathways of catestatin. Eur J Pharmacol 598:104–111. https://doi.org/10.1016/j.ejphar.2008.09.016
Sweitzer SM, Hickey WF, Rutkowski MD, Pahl JL, DeLeo JA (2002) Focal peripheral nerve injury induces leukocyte trafficking into the central nervous system: potential relationship to neuropathic pain. Pain 100:163–170. https://doi.org/10.1016/S0304-3959(02)00257-9
Ji R-R, Berta T, Nedergaard M (2013) Glia and pain: is chronic pain a gliopathy? Pain 154:S10–S28. https://doi.org/10.1016/j.pain.2013.06.022
Chu Y-X, Zhang Y-Q, Zhao Z-Q (2012) Involvement of microglia and interleukin-18 in the induction of long-term potentiation of spinal nociceptive responses induced by tetanic sciatic stimulation. Neurosci Bull 28:49–60
Sommer C, Schäfers M, Marziniak M, Toyka KV (2008) Etanercept reduces hyperalgesia in experimental painful neuropathy. J Peripher Nerv Syst 6:67–72. https://doi.org/10.1111/j.1529-8027.2001.01010.x
Rubio ME, Soto F (2001) Distinct localization of P2X receptors at excitatory postsynaptic specializations. J Neurosci 21:641–653. https://doi.org/10.1523/JNEUROSCI.21-02-00641.2001
Soto F, Garcia-Guzman M, Gomez-Hernandez JM, Holmann M, Karschin C, Stühmer W (1996) P2X4: an ATP-activated ionotropic receptor cloned from rat brain. Proc Natl Acad Sci U S A 93:3684–3688. https://doi.org/10.1073/pnas.93.8.3684
Franklin KM, Asatryan L, Jakowec MW, Trudell JR, Bell RL, Davies DL (2014) P2X4 receptors (P2X4Rs) represent a novel target for the development of drugs to prevent and/or treat alcohol use disorders. Front Neurosci 8. https://doi.org/10.3389/fnins.2014.00176
Tsuda M, Masuda T, Tozaki-Saitoh H, Inoue K (2013) P2X4 Receptors and neuropathic pain. front cell neurosci. https://doi.org/10.3389/fncel.2013.00191
Amadio S, Montilli C, Picconi B, Calabresi P, Volonté C (2007) Mapping P2X and P2Y receptor proteins in striatum and substantia nigra: an immunohistological study. Purinergic Signal 3:389–398. https://doi.org/10.1007/s11302-007-9069-8
Stojilkovic SS (2009) Purinergic regulation of hypothalamopituitary functions. Trends Endocrinol Metab 20:460–468. https://doi.org/10.1016/j.tem.2009.05.005
Kru gel U, Spies O, Regenthal R, Illes P, Kittner H (2004) P2 receptors are involved in the mediation of motivation-related behaviour. Purinergic Signal 1:21–29. https://doi.org/10.1007/s11302-004-4745-4
Kru gel U, Kittner H, Franke H, Illes P (2003) Purinergic modulation of neuronal activity in the mesolimbic dopaminergic system in vivo. Synapse 47:134–142. https://doi.org/10.1002/syn.10162
Khoja S, Shah V, Garcia D, Asatryan L, Jakowec MW, Davies DL (2016) Role of purinergic P2X4 receptors in regulating striatal dopamine homeostasis and dependent behaviours. J Neurochem 139:134–148. https://doi.org/10.1111/jnc.13734
Chen G, Zhang Y-Q, Qadri YJ, Serhan CN, Ji R-R (2018) Microglia in pain: detrimental and protective roles in pathogenesis and resolution of pain. Neuron 100:1292–1311. https://doi.org/10.1016/j.neuron.2018.11.009
Linos A, Worthington JW, O’Fallon WM, Kurland LT (1980) The epidemiology of rheumatoid arthritis in Rochester, Minnesota: a study of incidence, prevalence and mortality. Am J Epidemiol 111:87–98. https://doi.org/10.1093/oxfordjournals.aje.a112878
Smith JA, Das A, Butler JT, Ray SK, Banik NL (2011) Estrogen or estrogen receptor agonist inhibits lipopolysaccharide-induced microglial activation and death. Neurochem Res 36:1587–1593. https://doi.org/10.1007/s11064-010-0336-7
Vacca V, Marinelli S, Pieroni L, Urbani A, Luvisetto S, Pavone F (2016) 17β-estradiol counteracts neuropathic pain: a behavioural, immunohistochemical and proteomic investigation on sex-related differences in mice. Sci Rep 6:18980. https://doi.org/10.1038/srep18980
Sorge RE, Mapplebeck JC, Rosen S, Beggs S, Taves S, Alexander JK, Martin LJ, Austin JS, Sotocinal SG, Chen D, Yang M, Shi XQ, Huang H, Pillon NJ, Bilan PJ, Tu Y, Klip A, Ji RR, Zhang J, Salter MW, Mogil JS (2015) Different immune cells mediate mechanical pain hypersensitivity in male and female mice. Nat Neurosci 18:1081–1083. https://doi.org/10.1038/nn.4053
Mapplebeck JCS, Dalgarno R, Tu Y, Moriarty O, Beggs S, Kwok CHT, Halievski K, Assi S, Mogil JS, Trang T, Salter MW (2018) Microglial P2X4R-evoked pain hypersensitivity is sexually dimorphic in rats. Pain 159:1752–1763. https://doi.org/10.1097/j.pain.0000000000001265
Lambrecht G (2000) Agonists and antagonists acting at P2X receptors: selectivity profiles and functional implications. Naunyn Schmiedeberg's Arch Pharmacol 362:340–350. https://doi.org/10.1007/s002100000312
Buell G, Lewis C, Collo G, North RA, Surprenant A (1996) An antagonist-insensitive P2X receptor expressed in epithelia and brain. EMBO J 15:55–62. https://doi.org/10.1002/j.1460-2075.1996.tb00333.x
Coddou C, Yan Z, Obsil T, Huidobro-Toro JP, Stojilkovic S (2011) Activation and regulation of purinergic P2X receptor channels. Pharmacol Rev 63:641–683. https://doi.org/10.1124/pr.110.003129
Nagata K, Imai T, Yamashita T, Tsuda M, Tozaki-Saitoh H, Inoue K (2009) Antidepressants inhibit P2X4 receptor function: a possible involvement in neuropathic pain relief. Mol Pain 5(1744-8069):5–20. https://doi.org/10.1186/1744-8069-5-20
Zarei M, Sabetkasei M, Zanjani TM (2014) Paroxetine attenuates the development and existing pain in a rat model of neurophatic pain. Iran Biomed J 18:94–100. https://doi.org/10.6091/ibj.1282.2013
Hernandez-Olmos V, Abdelrahman A, El-Tayeb A, Freudendahl D, Weinhausen S, Müller CE (2012) N-substituted phenoxazine and acridone derivatives: structure–activity relationships of potent P2X4 receptor antagonists. J Med Chem 55:9576–9588. https://doi.org/10.1021/jm300845v
Ase AR, Honson NS, Zaghdane H, Pfeifer TA, Séguéla P (2015) Identification and characterization of a selective allosteric antagonist of human P2X4 receptor channels. Mol Pharmacol 87:606–616. https://doi.org/10.1124/mol.114.096222
Long T, He W, Pan Q, Zhang S, Zhang D, Qin G, Chen L, Zhou J (2020) Microglia P2X4R-BDNF signalling contributes to central sensitization in a recurrent nitroglycerin-induced chronic migraine model. J Headache Pain 21:1–17. https://doi.org/10.1186/s10194-019-1070-4
Layhadi JA, Turner J, Crossman D, Fountain SJ (2018) ATP evokes Ca2+ responses and CXCL5 secretion via P2X4 receptor activation in human monocyte-derived macrophages. J Immunol 200:1159–1168. https://doi.org/10.4049/jimmunol.1700965
Jurga AM, Piotrowska A, Starnowska J, Rojewska E, Makuch W, Mika J (2016) Treatment with a carbon monoxide-releasing molecule (CORM-2) inhibits neuropathic pain and enhances opioid effectiveness in rats. Pharmacol Rep 68:206–213. https://doi.org/10.1016/j.pharep.2015.08.016
Jurga AM, Piotrowska A, Makuch W, Przewlocka B, Mika J (2017) Blockade of P2X4 receptors inhibits neuropathic pain-related behavior by preventing MMP-9 activation and, consequently, pronociceptive interleukin release in a rat model. Front Pharmacol. https://doi.org/10.3389/fphar.2017.00048
Hervera A, Lea’nez S, Negrete R, Motterlini R, Pol O (2012) Carbon monoxide reduced neuropathic pain and spinal microglial actuvation by inhibiting nitric oxide synthesis in mice. PLoS One 7:1–10. https://doi.org/10.1186/1744-8069-9-16
Funding
This study was funded by the Ministry of Higher Education, Malaysia and Universiti Sains Malaysia under Fundamental Research Grant Scheme (203.PPSP.6171238) and Short Term Grant (304/PPSP/6315333).
Author information
Authors and Affiliations
Contributions
Khir NAM and Ismail CAN developed the contents and wrote the manuscript, Noh ASM prepared illustrations and did the search strings and Shafin N proofread and ensured the relevant flow of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
Nurul Ajilah Mohamed Khir declares that she has no conflict of interest. Ain’ Sabreena Mohd Noh declares that she has no conflict of interest. Nazlahshaniza Shafin declares that she has no conflict of interest. Che Aishah Nazariah Ismail declares that she has no conflict of interest.
Ethical approval
Not applicable
Consent for publication
Not applicable.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Khir, N.A.M., Noh, A.S.M., Shafin, N. et al. Contribution of P2X4 receptor in pain associated with rheumatoid arthritis: a review. Purinergic Signalling 17, 201–213 (2021). https://doi.org/10.1007/s11302-021-09764-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11302-021-09764-z