
Abstract
Porcine hemagglutinating encephalomyelitis virus (PHEV) is a member of the genus betacoronavirus within the family coronaviridae, which invades the central nervous system (CNS) via peripheral nervous system and causes encephalomyelitis or vomiting and wasting disease (VWD) in sucking piglets. Up to now, although few complete nucleotide sequences of PHEV have been reported, they are not annotated. This study aimed to illuminate genome characterization, phylogenesis and pathogenicity of the PHEV/2008 strain. The full length of the PHEV/2008 strain genome was 30,684 bp, with a G + C content of 37.27%. The genome included at a minimum of 11 predicted open reading frames (ORFs) flanked by 5′ and 3′ untranslated regions (UTR) of 211 and 289 nucleotides. The replicase polyproteins pp1a and pp1ab, which had 4382 and 7094 amino acid residues, respectively, were predicted to be cleaved into 16 subunits by two viral proteinases. Phylogenetic analysis based on the complete genome sequence revealed that PHEV/2008 strain was genetically different from other known PHEV types, which represented a novel genotype (GI-1). In addition, we found that PHEV/2008 was neurotropic and highly pathogenic to 4-week-old BALB/c mice. Taken together, this is the first detailed annotated, complete genomic sequence of a new genotype PHEV strain in China.
Similar content being viewed by others


Introduction
Coronaviruses are enveloped RNA viruses, which cause a great diversity of enteric, neurologic, and respiratory diseases in humans and other animals. The genomes of coronaviruses are about 30 kb, which are the largest genomes of all RNA viruses in nature, including RNA viruses contain segmented genomes [1]. According to the International Committee on Taxonomy of Viruses (ICTV), coronaviruses are divided into four genera: alphacoronavirus, betacoronavirus, gammacoronavirus, and deltacoronavirus [2]. In general, almost all alpha- and beta-coronaviruses have mammalian hosts, whereas gamma- and delta-coronaviruses mostly infect avian hosts.
Porcine hemagglutinating encephalomyelitis virus (PHEV), which is roughly spherical, enveloped virus with a linear, capped, polyadenylated, single-stranded, positive-polarity, non-segmented RNA genome belongs to genus betacoronavirus of the family coronaviridae, and order nidoviralesorder [3]. It was first isolated as early as the 1960s as the causative agents of encephalomyelitis and/or vomiting and wasting disease (VMD) in piglets [4, 5]. PHEV is a highly neurotropic virus that invades the CNS, causing neurological dysfunction in susceptible animals, including pigs, mice, and rats [6,7,8]. The clinical signs of PHEV infected pigs are greatly different, from subclinical symptoms to death. In young piglets, especially under 3 weeks of age, as well as other susceptible species, PHEV infection is often fatal because of the central nervous system disorders. The mortality rate is almost 100%. In contrast, older pigs generally have no obvious symptoms [9]. To date, there has been no vaccine or any treatment measures.
In previous studies, some regions, especially the structural protein genes of the PHEV genomes of different strains have been determined [10, 11]. Thus far, only few complete nucleotide sequences of PHEV have been publicly reported. In the present study, we determined the complete genomic sequence of the PHEV/2008 strain by molecular cloning and rapid amplification of cDNA ends techniques. Then, we discussed the data with respect to genome organization, expression strategy, phylogenetic relation, and pathogenicity of it.
Materials and methods
Virus and mice
PHEV/2008 and PHEV/CC14 were isolated from piglets in Changchun of Jilin province, China, and stored in our laboratory. The virus was propagated by passage in Neuro-2a (N2a) cells, purified using sucrose density gradient centrifugation [12], and stored in − 80 °C. Female BALB/c mice (4-week-old) were purchased and housed in the animal facility.
Genome cloning by RT-PCR
Total RNA from brain samples of infected mice were extracted using TriPure Isolation Reagent (Roche, USA) and quantified using a SmartSpec™ Plus spectrophotometer (BIO-RAD, USA). Reverse transcription was performed at 42 °C water bath for 2 h and 70 °C for 10 min using 1 µg of total RNA, 0.5 µL (200 U/µL) of Reverse Transcriptase M-MLV (RNase H-), 4 µL 5× Reverse Transcriptase M-MLV Buffer, 2 µL of Oligo(dT)18 primer, 0.5 µL(20U) Recombinant RNase Inhibitor (RRI), 2 µL (10 mM each) dNTP mixture (Takara, Dalian, China), and 9 µL sterile water was added into the mixture to reach a final volume of 20 µL. By reference to the genome sequence of PHEV strain VW572 (Genbank accession number: DQ011855), a set of fifteen pairs of specific primers (Table 1), which span the entire genome, were designed for gene-walking PCR. The PCR mixture contained 2 µL cDNA, 2 µL (2.5 mM) dNTP mixture, 1 µL forward and reverse primers (10 pmol), 0.5 µL PrimeSTAR HS DNA Polymerase (2.5 U/µl), 5 µL (5×) PrimeSTAR Buffer (Mg2+ plus) (Takara, Dalian, China),14.5 µL sterile water was added to the PCR mixture to reach a final volume of 25 µL. After a brief centrifugation, the PCR mixtures was amplified at 95 °C for 5 min, and then for 35 cycles of 95 °C for 50 s, 48–58 °C for 50 s (based on the Tm of every PCR fragment), 72 °C for 3 min, and then a final extension at 72 °C for 10 in an automated C1000™ thermal cycler (BIO-RAD, USA).
Cloning and sequencing of RT-PCR products
The PCR products of each gene fragment were cut from 0.8% agarose gel under UV light and then purified using the Ezgene™ Gel/PCR Extraction Kit (Biomiga). The purified PCR products were cloned and transformed using pEASY®-Blunt Zero Cloning Kit (TRANSGEN BIOTECH). At least four positive clones of selected amplification were sequenced at the Jilin Comate Bioscience Co., Ltd. China. Additionally, every PCR fragment was verified by direct sequencing of the PCR product. The 5′ and 3′ terminal sequences of the viral genome were determined by rapid amplification of cDNA ends (RACE), using a SMARTer® RACE 5′/3′ Kit (Clontech) according to the manufacturer’s instructions.
Sequence assembling and phylogenetic analysis
The sequences data were edited and assembled, and a final consensus full-length sequence of the viral genome was determined and analyzed using the Clustal W method available in Bioeditd software package v7.0.0. (http://www.mbio.ncsu.edu/bioedit/bioedit). The potential open reading frames (ORFs) were predicted using the ORF finder program in the website of the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/orffinder/). The homologous analysis of PHEV/2008 and other coronaviruses available in the GenBank database was performed using the Basic local alignment search tool (BLAST) program [13]. Phylogenetic trees derived from nucleotide sequences were constructed using the neighbor-joining method of MEGA 6. Prediction of cleavage sites of polyproteins was performed by using ZCURVE_CoV [14]. Protein family was analyzed by using PFAM [15]. Signal peptides and their cleavage sites were predicted by using SignalP 4.1 [16]. Transmembrane domains were predicted by using TMHMM and verified by using TMpred.
Infection in BALB/c mice with PHEV/2008
To investigate the pathogenicity of PHEV/2008, twenty female BALB/c mice of 4-week-old were randomly divided into two groups. One group of 10 mice were intranasally (i.n.) inoculated with 0.1 ml PHEV (the TCID50 was 10−4.45 0.1 ml−1). The other group of 10 mice mock infected i.n. with 0.1 ml DMEM supplemented with 2% fetal bovine serum was used as control. Clinical signs were examined daily for 7 days, brains were harvest on 5th day post infection and fixed in 10% buffered formalin for histological examination.
Histopathology and immunohistochemistry analysis
Histopathology and immunohistochemistry (IHC) were conducted as previously [17]. Briefly, the tissue samples were embedded in paraffin, sectioned, and stained with hematoxylin and eosin (H&E) for histopathology. For IHC, the sections were deparaffinized in xylene and were rehydrated in ethanol. H2O2 was used to block endogenous peroxidase, and the antigen was retrieved in citrate buffer (10 mM, pH 6.0). The sections were incubated with rabbit anti-PHEV N protein polyclonal antibody (diluted at 1:100) overnight at 4 °C and then incubated with biotinylated secondary antibody. An avidin–biotin-peroxidase complex was used to detect antigen–antibody reaction. Diamino benzidine (DAB) was used as a peroxidase substrate (Maixin Biotechnology Company, Fujian, China). A negative control was performed by replacing primary antibody with normal rabbit serum.
Nucleotide sequence accession number
The complete genome sequences of PHEV/2008 and PHEV/CC14 have been submitted to the GenBank database with accession number KY994645 and MF083115, respectively.
Results
A new lineage of PHEV was identified in China
Phylogenetic tree was constructed using nucleotide sequence of ORF1ab (Fig. 1a) of PHEV/2008 and other 22 representative coronaviruses. The results showed that the PHEV/2008 strain and reference strain PHEV-VW572 were clearly clustered within lineage A of the genus betacoronavirus, relatively close to BCoV (U00735) and HCoV-OC43 (KU131570). In order to genotype and further analyze the genetic diversity both PHEV/2008 and other PHEV strains, we constructed phylogenetic tree with 12 PHEV reference strains and PHEV/2008 based on the complete genome sequences. The result indicated 2 different clusters, referred to as genotype I (GI-1 to GI-2) and genotype II (GII-1 to GII-3). Our two isolates were clustered together and clearly set apart from PHEV-VW572 strain (Fig. 1b) in the phylogenetic tree. Hence, we proposed our two isolates as a new lineage of PHEV in genotype I (GI-1).

Phylogenetic trees for PHEV/2008 and other selected coronavirus. Unrooted neighbor-joining phylogenies based on the nucleotide sequences of full-length ORF1ab a and complete genome sequence b of virus strains of 4 coronavirus genera (alphacoronavirus, betacoronavirus, gammacoronavirus, and deltacoronavirus). Our two PHEV isolates in this study are indicated with triangle. Reference sequences obtained from GenBank are indicated by strain name and accession number. Bootstrap analysis was performed with 1000 trials. The scale bar beneath the tree indicates the number of nucleotide substitution per site
Genomic characterization of the new PHEV GI-1 genome
Using gene walking RT-PCR and RACE methods, we obtained seventeen overlapping cDNA clones, which spans the whole genomes of two PHEV strains. The final sequences of these clones revealed that the genomes of PHEV/2008 and PHEV/CC14 contain 30,684 and 30,682 nucleotides, respectively, including the 5′- and 3′-UTRs and excluding the poly (A) tract. The G + C content is 37.27 and 37.29%, which has high similarity to values reported for other coronaviruses (37–42%) (Table 2) [18]. The complete genome sequences of the two PHEV isolates showed a typical coronavirus gene order 5′-UTR, replicase, HE, S, E, M, N, and UTR-3′.
Further analysis revealed that our two isolates contain deletions/insertions in different genes. Compared with PHEV-VW572, two strains contain a 3-nt deletion (GTT) in the ORF 1a at positions 325–327, leading to a single amino acid deletion (V, Val). The NS2 genes of two strains are 702 nt in length, encoding two 203 amino acid proteins, which are 9 aa longer than that of VW-572. In the case of NS4.9 genes, 12 nt deletion were identified in the two isolates, which was reported in 2 Canadian PHEV strains (IAF-404 and 67N) previously. Overall, genome characterization and phylogenetic analysis revealed that our two isolates are novel PHEV variants.
Coding features and expression strategies of predicted proteins
The protein coding potential of the 30,684 bp PHEV/2008 genome is depicted (Fig. 2). The genome of PHEV/2008 contains two large and partially overlapping ORFs (ORF1a and ORF1b) in the 5′-proximal two-thirds of the genome, which encode replicase polyproteins (pp1a/pp1ab). In addition, there are at least nine ORFs in the downstream of ORF1b.

Map of the predicted ORFs in the PHEV/2008 genome sequence. A schematic of the complete genome organization of PHEV/2008 is shown. The replicase gen consists of two ORFs, replicase 1a and replicase 1b, which are expressed by a ribosomal frameshifting mechanism. The expand region below shows the downstream ORFs of the genome. The sizes and positions of accessory genes (NS2, NS4.9, NS12.7, and N2) are indicated, relative to the basic genes HE, S, E, M, and N
The ORF1a (base pairs 211–13,338) and ORF1b (base pairs 13,338–21,494) occupy about 21.3 kb of the viral genome. They are translated from the genomic mRNA through ribosomal frame shifting to generate polyproteins pp1a (4382 amino acid residues) and pp1ab (7094 amino acid residues), which are cleaved into 16 nonstructural proteins (nsp1 to nsp16) by two viral proteinases, papain-like proteinase and 3C-like cysteine proteinase. As compared with other coronaviruses, we predicted the putative pp1a/pp1ab cleavage sites (Fig. 3a). There are two elements that participate in ribosomal frame shifting. The first is a conserved “slippery sequence” which is located in the 13,332–13,338 nt (5′-UUUAAAC-3′) of PHEV/2008 genome. The second is a three-stemmed pseudoknots located a short distance downstream of the slippery sequence (Fig. 3b). Additionally, a 190 nt packaging signal (PS) domain contains a 98 nt hairpin structure have also been identified, which may act as a starting point during viral nucleocapsid assembly. The PS domain is located in the nsp15 of pp1ab in the PHEV/2008 genome, and the hairpin is consisted of four copies of a repeating structural subunit that composes conserved AGC/GUAAU motifs at 10 bp intervals (Fig. 3c).

Replicase gene and protein products of PHEV/2008. a Polyprotein pp1a and pp1b processing scheme for PHEV/2008. Rounds and triangles represent sites in the replicase polyproteins pp1a and pp1b that predicted to be cleaved by papain-like proteinases (yellow) or the 3C-like cysteine proteinase (blue), respectively. Cleavage products are numbered nsp1 to nsp16, nsp11 is an oligopeptide generated when ribosomal frameshifting does not occur. b Two ribosomal frameshifting elements of the PHEV/2008 replicase gene. The first element is the 5′-UUUAAAC-3′ heptanucletide slippery sequence, the second element is an extensively characterized RNA pseudoknot structure. c Highly conserved secondary structure of the RNA packaging signal of PHEV/2008. The repeat units of AGC/GUAAU motifs are boxed. (Color figure online)
ORF 2 (base pairs 21,504–22,205) encodes a putative protein (NS2) with 233 amino acids, which is a group 2-specific protein. PFAM analysis revealed that the putative protein belongs to the Coronavirus NS2A protein family with accession number PF05213. Previous studies demonstrated that NS2 protein is dispensable for MHV, but deleted by reverse genetics results in a significant attenuation of the virulence in mice [19]. It was reported that the deletion of NS2 protein of PHEV may lead to viral evolution and confer respiratory tropism [20].
ORF 3 (base pairs 22,354–23,628) encodes the putative hemagglutinin-esterase (HE) glycoprotein, which contains 424 amino acids. HE presents in only a subset of betacoronaviruses and influenza C virus. The HE protein, which may be functionally similar to the S protein, has high homology to that of influenza C virus [21]. It was supposed that the ancestors of the betacoronaviruses acquired HE by the means of gene recombination when infected a same host cell with influenza virus. Homologies of the deduced amino acid sequences of HE proteins between PHEV/2008 and other betacoronaviruses have 41–90% amino acid identities (Table 2). The results of PFAM analysis revealed that amino acid residues 2–378 of HE protein form a member of the HE family with an accession number PF03996. Prediction of signal peptide in the N-terminal 70 amino acids was performed using SignalP, revealing that a signal peptide with a cleavage site between positions 18 and 19 is present (D = 0.654; D-cutoff = 0.450). TMHMM analysis revealed that a transmembrane domain is located in 392–414. The putative receptor-binding (aa 140–283) and receptor-destroying sialate O-acetylesterase (acetylesterase; aa 25–139 and 284–341) domains of PHEV/2008 HE protein were identified by multi-alignment with other group 2a coronaviruses (Fig. 4). Previous experiments have been demonstrated that, in BCoV, HE protein is required for viral replication, however, it is not necessary and sufficient for viral infection [22].

Schematic diagrams of structural proteins of PHEV/2008. Models of the five structural proteins, spike (S), hemagglutinin-esterase (HE), envelop (E), membrane (M), and nucleocapsid (N) of PHEV/2008 showing domains identified in this study. Numbering indicates amino acid residues
ORF 4 (base pairs 23,643–27,692) encode the putative S glycoprotein with 1349 amino acids in length (PFAM accession no. PF01601). The amino acid identities of S glycoprotein between PHEV/2008 and other betacoronaviruses are 27–81%, however, less than 23% with non-betacoronaviruses (Table 2). Percentage identities between the deduced amino acid sequences of the PHEV/2008 and BCoV main structural proteins revealed that the S protein to be less conserved (81.13%) than the HE (90.09%), E (97.62%), M (93.48%), and N (95.32%) protein. It could be hypothesized that divergences of the S protein are related to tissue tropism. SignalP analysis showed that a signal peptide is present with a cleavage site between amino acid residues 14 and 15 (D = 0.769 D-cutoff = 0.450). TMHMM analysis of this predicted protein showed only one transmembrane domain at positions 1294–1316. On this basis, we concluded that most of the S protein (residues 1–1293) is located outside of the virus. The S protein is cleaved into two subunits (S1 and S2) by host-cell proteases at a potential site located after LRQRR between residues 754 and 755 by comparing with S proteins of other group 2a coronaviruses. The S1 subunit is expected to contain a receptor-binding domain which plays a crucial role in the stage of virus attachment to host cell surface receptors. The S2 subunit is predicted to mediate fusion of the viral and cellular membranes. Two heptad repeats (HR), which is located at residues 977–1088 (HR1) and 1245–1290 (HR2), were identified in the PHEV S2 subunit (Fig. 4).
ORF 5 (base pairs 27,682–27,744), which partially overlaps with S protein, encodes a predicted protein with 20 amino acids in length and named NS4.9. It was failed to identify any matching sequence using PFAM analysis. TMHMM analysis of this putative protein did not reveal any transmembrane helix either.
ORF 6 (base pairs 27,760–28,089), which overlaps with ORF 5, encoding a putative protein with 109 amino acids in length and named NS12.7. PFAM analysis revealed that this putative protein belongs to Coronavirus non-structural protein NS2 family with PFAM accession number PF04753. SignalP and TMHMM showed neither signal peptide nor transmembrane helix.
ORF 7 (base pairs 28,076–28,330) encoding the small envelop (E) protein yields a putative protein of 84 amino acids in length. The amino acid identity of E proteins between PHEV/2008 and BCoV is as high as 98%, however, less than 23% with non-betacoronaviruses (Table 2). According to PFAM analysis, the predicted E protein belongs to Non-structural protein NS3/Small envelop (NS3_envE) family with PFAM accession number PF02723. There is absence of signal peptide and cleavage site (D = 0.139, D-cutoff = 0.500). TMHMM analysis revealed that a transmembrane domain is present, which is located in positions 15–37 (Fig. 4).
ORF 8 (base pairs 28,345–29,037) encodes the putative membrane (M) glycoprotein, which contains 230 amino acids. The amino acid identities of M proteins between PHEV/2008 and other betacoronaviruses are 40–93%, while less than 37% with non-betacoronaviruses (Table 2). PFAM analysis revealed that the predicted M protein belongs to Coronavirus M matrix/glycoprotein family with PFAM accession number PF01635. The result of SignalP analysis showed absence of any signal peptide (D = 0.101 D-cutoff = 0.500). Based on TMHMM analysis, three transmembrane domains are located in positions 25–44, 49–71, 81–103, leaving the C-terminal 125-amino acid hydrophilic domain is positioned inside of the virus (Fig. 4).
ORF 9a (base pairs 29,047–30,396) encodes the nucleocapsid (N) protein and produces a putative protein of 449 amino acids in length (PFAM accession number, PF00937). The amino acid identities between the N proteins of PHEV/2008 and those of other betacoronaviruses are 30–95%, however, less than 22% amino acid identities with non-betacoronaviruses (Table 2). Neither signal peptide nor transmembrane helix was identified by using SignalP and TMHMM. The N protein resides in the inside of the virion and interacts with the RNA genome in a beads-on-a-string configuration to form a viral nucleocapsid. The RNA binging domain is located at two ends of the N protein, designated the N-terminal domain (NTD) and the C-terminal domain (CTD). Flanking the RNA-binding domains are three spacer segments, domain N1a at the hydroxy terminus, spacer B and domain N3 at the carboxy terminus, and domain N2a which contains a serine- and arginine-rich region (SR) at the center of the N molecule (Fig. 4). Besides, the N protein also binds to the M protein to promote assembly and budding, and plays a part in coronaviruses replication.
ORF 9b (base pairs 29,108–29,731), which completely overlaps with ORF9a, encoding a predicted nonstructural protein (N2) of 207 amino acids in length. Based on PFAM analysis, the N2 protein belongs to Coronavirus nucleocapsid I protein family with PFAM accession number PF03187.
Characterization of untranslated regions of the PHEV/2008 genome sequence
Untranslated regions were detected at the 5′ (210 nt) and 3′ (289 nt) ends of the genome, which were similar to the cases with other coronaviruses genomes. Additionally, there was only one evident untranslated region in the PHEV/2008 genome that was larger than 50 nucleotides and located in the intergenic region between ORF2 and ORF3 (nucleotide position 22,206–22,353).
At the 5′ and 3′ ends of the coronaviruses genomes, cis-acting sequences and structure elements are present, which function in RNA synthesis and decay, gene expression, and virion assembly [23]. The 5′ end of the PHEV/2008 genome includes a predicted leader sequence and a coronavirus transcription regulatory sequence motif, 5′-CUAAAC-3′. There are two highly conserved RNA elements at the 3′ end of the PHEV/2008 genome, which also found in MHV and BCoV. One is a bulged stem-loop and the other is an adjacent pseudoknot [24]. Furthermore, downstream of these elements is a hypervariable region which makes up most of the rest of the 3′ UTR [25]. Nucleotide sequences are less well conserved in this region among coronaviruses; however, we also found two conserved structures. The first is an octanucleotide motif, 5′-GGGAGAGC-3′, which is nearly conserved in all coronaviruses. The other structure is a minimal signal that is required for initiation of negative-strand RNA synthesis, for MHV, it is located within the final 55 nucleotides (nt) of the 3′ UTR [26].
Pathogenicity of PHEV/2008
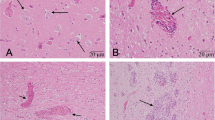
Mice body weight and temperature were determined daily throughout the experiment as indicators of the disease progression. Body weight increased slightly during the first 2 days, after day 3, it showed a sharp decrease. At the end of experiment, the body weight decreased to approximately 85% of their origination (Fig. 5a). Similarly, the temperature of infected mice decreased after day 3 (data are not shown). Decreases in body weight and temperature were in accord with the clinical symptoms. In the control group, no obvious clinical signs were observed and no mice died throughout the experimental period. However, mild to moderate neurological signs, such as bristle coat, hunched back, swing arms and paralysis were observed between 3 and 7 days in mice infected with PHEV/2008. All PHEV-infected mice died in the end of the experiment, implicating that PHEV/2008 is highly pathogenic in mice (Fig. 5b). In addition, pathological changes were observed in brain of the PHEV-infected mice on 5th day. A large number of mononuclear cells gathered in perivascular and formed perivascular cuffing in the cerebral parenchyma (Fig. 5d). A vascular oedema was observed and the peripheral space had become enlarged (Fig. 5e). Hemorrhage was also found and red blood cells infiltrated into the brain parenchyma (Fig. 5f). Activated microglia encircled degenerating neurons and formed microglial nodules (Fig. 5g). Immunohistochemistry of cerebellar cortex showed that PHEV N protein was mostly found in neurons (Fig. 5h–j).

Clinical signs and pathology features of PHEV/2008 infection in mice. Body weight changes (a) and survival curves (b) of BALB/c mice. Brain histopathological changes of PHEV-infected mice with clinical signs were examined by hematoxylin and eosin (H&E) staining at 5 days post infection, ×400; c negative control; d perivascular cuff; e vascular oedema; f hemorrhage; g microglia nodule. Immunohistochemistry staining of neuron infection (h–j). No PHEV-positive labeling of neurons in the negative control group;×400 (h); Brains from an affected mouse showing PHEV-positive labeling in the cytoplasm of nerve cells, ×100 (i) and ×400 (j)
Discussion
Since it was first isolated in Canada in 1962, PHEV has been reported in many countries all over the word, including Japan, America, Belgium, Argentina, China, Korea, Thailand, Netherlands, and so on [7]. In 2007, PHE-like outbreaks occurred in two individual pig farms in Changchun and Siping in Jilin province, the mortality rate is 47.6% (30/63) and 100% (38/38), respectively. Since then,the disease occurs almost every year in Jilin province, especially during the early spring season. Thus far, there is no effective vaccine and treatment measures to protect farmers from economic losses. Besides its economic importance, PHEV has proven to be an excellent new trans-synaptic tracer for analyzing neuroanatomical connections in the CNS in the field of neuroscience [9].
In this study, we report the full length genome sequence and characteristic features of PHEV/2008 strain. The results of genomic analysis revealed that PHEV/2008 is a lineage A of the genus betacoronavirus, with the typical gene order 5′-UTR, replicase, HE, S, E, M, N, and UTR-3′. The first ORF (ORF1a/b) encodes replicase proteins, usually referred to as pp1a and pp1ab, respectively, and comprises approximately two-thirds of the genome. The translation of ORF1ab involves − 1 ribosomal frame shifting in order to synthesis polyproteins pp1ab. During or after synthesis, these polyproteins are cleaved at different specific enzyme sites by virus-encoded proteinases into 16 proteins. That is, nsp1 to nsp11 are encoded in ORF1a, nsp12 to nsp16 are encoded in ORF1b, and nsp11 is generated only when ribosomal frameshifting does not occur [27]. The ORF3, ORF4, ORF7, ORF8, and ORF9a are predicted to encode the five main structure proteins, HE, S, E, M, and N proteins, respectively. Translation of these genes is different from ORF1ab which translated from genomic mRNA, while these main structure protein genes are translated from subgenomic mRNAs (sgRNAs). The sgRNAs was consist of common 5′ leader sequences and variable “body” parts. It is assumed that the leader-to-body fusion event involves in the mechanism of discontinuous negative-strand RNA synthesis [28]. Both genomic RNA and sgRNAs are synthesized through negative-strand intermediates which possess 5′ oligouridylate tracts and 3′ antileaders.
It is believed that all PHEV strains spread via the peripheral nervous system to the CNS, casing VMD and/or encephalomyelitis in suckling piglets. The clinical symptoms of VMD include sneezing, coughing, inappetence, and vomition. On the other hand, the clinical signs of encephalomyelitis are characterized by neurological disorder such as muscle tremor, hyperesthesia, incoordination, paddling, or paralysis [29]. Upper respiratory tract and pulmonary lesions have rarely been reported; however, acute outbreaks of influenza-like illness (ILI) was noted in exhibition swine at agricultural fairs in Michigan, USA, in the summer of 2015 [21]. The atypical clinical presentation reflects that PHEV may cause respiratory disease in older swine.
In the cases of other coronaviruses, the nucleocapsid gene has been successfully used for phylogenetic analysis. Hence, a phylogenetic tree was constructed based on the nucleocapsid gene to analyze the evolutionary pattern of our two isolates and other PHEV strains (data are not shown). According to the phylogenetic tree, our two isolates are clustered together and closely related to Chinese type (JT06, FJ009234) and two North American type (67N, AY078417; IAF-404, AF481863), suggesting that PHEV strains currently circulating in China could be originate from a similar ancestral strain with North American type PHEV strains.
This is the first annotated, full-length genome sequence of an individual PHEV strain in china. The availability of the PHEV/2008 nucleotide sequence will promote the development of future comparative genomic studies and numerous PCR-based diagnostic assays for the virus. We expect that the usability of this full-length, detailed-annotation virus genome sequence will be useful for the development of antivirus research.
References
van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA (2012) Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3(6):e00473–e00412
Adams MJ, Lefkowitz EJ, King AM, Harrach B, Harrison RL, Knowles N, Kropinski AM, Krupovic M, Kuhn JH, Mushegian AR, Nibert M, Sabanadzovic S, Sanfacon H, Siddell SG, Simmonds P, Varsani A, Zerbini FM, Gorbalenya AE, Davison AJ (2016) Ratification vote on taxonomic proposals to the international committee on taxonomy of viruses. Arch Virol 161(10):2921–2949
Li Z, Zhao K, Lan Y, Lv X, Hu S, Guan J, Lu H, Zhang J, Shi J, Yang Y, Song D, Gao F, He W (2017) Porcine hemagglutinating encephalomyelitis virus enters neuro-2a cells via clathrin-mediated endocytosis in a Rab5-, cholesterol-, and pH-dependent manner. J Virol 91(23):01083–01017
Roe CK, Alexander TJ (1958) A disease of nursing pigs previously unreported in Ontario. Can J Comp Med Vet Sci 22(9):305–307
Li Z, He W, Lan Y, Zhao K, Lv X, Lu H, Ding N, Zhang J, Shi J, Shan C, Gao F (2016) The evidence of porcine hemagglutinating encephalomyelitis virus induced nonsuppurative encephalitis as the cause of death in piglets. PeerJ 4:e2443
Quiroga MA, Cappuccio J, Pineyro P, Basso W, More G, Kienast M, Schonfeld S, Cancer JL, Arauz S, Pintos ME, Nanni M, Machuca M, Hirano N, Perfumo CJ (2008) Hemagglutinating encephalomyelitis coronavirus infection in pigs, Argentina. Emerg Inf Dis 14(3):484–486
Li Z, Lan Y, Zhao K, Lv X, Ding N, Lu H, Zhang J, Yue H, Shi J, Song D, Gao F, He W (2017) miR-142-5p disrupts neuronal morphogenesis underlying porcine hemagglutinating encephalomyelitis virus infection by targeting Ulk1. Front Cell Infect Microbiol 7:155
Hirano N, Haga S, Sada Y, Tohyama K (2001) Susceptibility of rats of different ages to inoculation with swine haemagglutinating encephalomyelitis virus (a coronavirus) by various routes. J Comp Pathol 125(1):8–14
Hirano N (2004) Neurotropism of swine haemagglutinating encephalomyelitis virus (coronavirus) in mice depending upon host age and route of infection. J Comp Pathol 130(1):58–65
Goebel SJ, Taylor J, Masters PS (2004) The 3′ cis-acting genomic replication element of the severe acute respiratory syndrome coronavirus can function in the murine coronavirus genome. J Virol 78(14):7846–7851
Gao W, Zhao K, Zhao C, Du C, Ren W, Song D, Lu H, Chen K, Li Z, Lan Y, Xie S, He W, Gao F (2011) Vomiting and wasting disease associated with hemagglutinating encephalomyelitis viruses infection in piglets in Jilin, China. Virol J 8:130
Chen K, Zhao K, He W, Gao W, Zhao C, Wang L, Pan W, Song D, Wang C, Gao F (2012) Comparative evaluation of two hemagglutinating encephalomyelitis coronavirus vaccine candidates in mice. Clin Vaccine Immunol 19(7):1102–1109
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410
Gao F, Ou HY, Chen LL, Zheng WX, Zhang CT (2003) Prediction of proteinase cleavage sites in polyproteins of coronaviruses and its applications in analyzing SARS-CoV genomes. FEBS Lett 553(3):451–456
Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Griffiths-Jones S, Khanna A, Marshall M, Moxon S, Sonnhammer EL, Studholme DJ, Yeats C, Eddy SR (2004) The Pfam protein families database. Nucleic Acids Res 32(Database issue):D138–D141
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8(10):785–786
Zhao K, Song D, He W, Lu H, Zhang B, Li C, Chen K, Gao F (2010) Identification and phylogenetic analysis of an Orf virus isolated from an outbreak in sheep in the Jilin province of China. Vet Microbiol 142(3–4):408–415
Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK, Poon LL, Wong SS, Guan Y, Peiris JS, Yuen KY (2005) Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J Virol 79(2):884–895
de Haan CA, Masters PS, Shen X, Weiss S, Rottier PJ (2002) The group-specific murine coronavirus genes are not essential, but their deletion, by reverse genetics, is attenuating in the natural host. Virology 296(1):177–189
Lorbach JN, Wang L, Nolting JM, Benjamin MG, Killian ML, Zhang Y, Bowman AS (2017) Porcine hemagglutinating encephalomyelitis virus and respiratory disease in exhibition Swine, Michigan, USA, 2015. Emerg Inf Dis 23(7):1168–1171
Vlasak R, Luytjes W, Spaan W, Palese P (1988) Human and bovine coronaviruses recognize sialic acid-containing receptors similar to those of influenza C viruses. Proc Natl Acad Sci USA 85(12):4526–4529
Popova R, Zhang X (2002) The spike but not the hemagglutinin/esterase protein of bovine coronavirus is necessary and sufficient for viral infection. Virology 294(1):222–236
Sola I, Almazan F, Zuniga S, Enjuanes L (2015) Continuous and discontinuous RNA synthesis in coronaviruses. Annu Rev Virol 2(1):265–288
Hsue B, Hartshorne T, Masters PS (2000) Characterization of an essential RNA secondary structure in the 3’ untranslated region of the murine coronavirus genome. J Virol 74(15):6911–6921
Liu Q, Johnson RF, Leibowitz JL (2001) Secondary structural elements within the 3’ untranslated region of mouse hepatitis virus strain JHM genomic RNA. J Virol 75(24):12105–12113
Lin YJ, Liao CL, Lai MM (1994) Identification of the cis-acting signal for minus-strand RNA synthesis of a murine coronavirus: implications for the role of minus-strand RNA in RNA replication and transcription. J Virol 68(12):8131–8140
Perlman S, Netland J (2009) Coronaviruses post-SARS: update on replication and pathogenesis. Nat Rev Microbiol 7(6):439–450
Sawicki SG, Sawicki DL, Siddell SG (2007) A contemporary view of coronavirus transcription. J Virol 81(1):20–29
Rho S, Moon HJ, Park SJ, Kim HK, Keum HO, Han JY, Van Nguyen G, Park BK (2011) Detection and genetic analysis of porcine hemagglutinating encephalomyelitis virus in South Korea. Virus Genes 42(1):90–96
Acknowledgements
This study was supported by the National Key Research and Development Program of China (Grants 2016YFD0500102, 2016YFD0500707), the National Natural Science Foundation of China (Grants 31772704, 31672519, and 31602018), the Scientific and Technological Project of Jilin Province (Grants 20180101270JC, 20170204033NY, and 20160520033JH).
Author information
Authors and Affiliations
Contributions
JS and FG conceived the study and participated in its design and coordination. JS, KZ, HL, ZL, XL performed the research. YL, JG, WH analyzed and interpreted data. JS and KZ wrote the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This article does not contain any studies with human participants performed by any of the authors. All animal experiments were in accordance with the Animal Welfare Ethical Committee of the College of Veterinary Medicine, Jilin University.
Additional information
Edited by Juergen A Richt.
Rights and permissions
About this article

Cite this article
Shi, J., Zhao, K., Lu, H. et al. Genomic characterization and pathogenicity of a porcine hemagglutinating encephalomyelitis virus strain isolated in China. Virus Genes 54, 672–683 (2018). https://doi.org/10.1007/s11262-018-1591-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-018-1591-y