
Abstract
Human parainfluenza virus type 3 (hPIV3) is an important respiratory pathogen that causes the majority of viral pneumonia of infants and young children. hPIV3 can infect host cells through the synergistic action of hemagglutinin-neuraminidase (HN) protein and the homotypic fusion (F) protein on the viral surface. HN protein plays a variety of roles during the virus invasion process, such as promoting viral particles to bind to receptors, cleaving sialic acid, and activating the F protein. Crystal structure research shows that HN tetramer adopted a “heads-down” conformation, at least two heads dimmer on flank of the four-helix bundle stalk, which forms a symmetrical interaction interface. The stalk region determines interactions and activation of F protein in specificity, and the heads in down position statically shield these residues. In order to make further research on the function of these amino acids at the hPIV3 HN stalk/head interface, fifteen mutations (8 sites from stalk and 7 sites from head) were engineered into this interface by site-directed mutagenesis in this study. Alanine substitution in this region of hPIV3 HN had various effects on cell fusion promotion, receptor binding, and neuraminidase activity. Besides, L151A also affected surface protein expression efficiency. Moreover, I112A, D120A, and R122A mutations of the stalk region that were masked by global head in down position had influence on the interaction between F and HN proteins.
Similar content being viewed by others
Introduction
Viruses in the Paramyxoviridae are membrane-enveloped, whose genomes are organized on a single negative-sense strand of RNA that infect both humans and animals, causing significant threat to public health and safety [1, 2]. The Paramyxoviridae can be divided into two major subfamilies, Paramyxovirinae and Pneumovirinae. As representatives of Paramyxovirinae, according to different genetics and antigenicity, human parainfluenza viruses (hPIVs) can be divided into type 1 to 4, and further major subtypes of hPIV4 (4A and 4B) [3, 4]. Although the major structural and biological characteristics of hPIVs are similar, the clinical features and epidemiological characteristics of the various virus-induced diseases are distinct [5]. As a major cause of bronchiolitis and pneumonia in infants, young children, the chronically ill, the immunocompromised, and the elderly, human parainfluenza viruses 3 (hPIV3) is next to respiratory syncytial virus (RSV) in causing respiratory infections. Compared with other subtypes of hPIVs, hPIV3 has the highest infection rate, longer duration of infection, and significantly increasing hospitalization rate of the disease [3, 5,6,7]. Currently, there is still no effective vaccine or therapeutic drugs to battle against it [8].
The hPIV3 RNA genome encodes 6 proteins, which with a coding order from the 3’ end to the 5’ end are nucleocapsid protein (N), phosphate protein (P), matrix protein (M), fusion protein (F), hemagglutinin-neuraminidase protein (HN), and large protein (L), respectively [3]. Among them, HN protein and F protein are embedded in the lipid membrane [6]. For nearly all paramyxoviruses infection to proceed, the virion must fuse its envelope with host cell membrane, which is triggered at the plasma membrane in a pH-independent but receptor-dependent manner [9, 10]. For hPIV3, HN protein and F protein are necessary and critical to mediate cell penetration and infection [11, 12].
The HN protein is a type 2 homotetrameric integral membrane protein, with a short cytoplasmic tail at the N-terminal and transmembrane domains (TM) followed by a large ectodomain at the C-terminal region which is composed of a helical stalk and a globular head [12].
HN protein contributes at least three functions to the virus: (1) receptor recognition activity: they identify and bind sialic acid at the cell surface to initiate membrane fusion; (2) neuraminidase (NA) activity: neuraminidases hydrolyze sialic acid residues which destroys the receptor activity and finally results in releasing the progeny virus to infect additional cells; (3) fusion promotion activity: after binding to the receptor, HN proteins interact directly with the homology F protein and then activate them to undergo a series of irreversible conformational rearrangements which is coupled with membrane fusion [3, 9, 12, 13].
In previous studies of hPIV3, on the basis of cryotomography researchers have been suggested a model of HN tetramer in a “4-heads-down” conformation whereby at least two of the heads form a symmetrical interaction on the two sides of the stalk while other two of four heads away from the stalk without any contacts [11]. Eventually the stalk forms a parallel tetrameric coiled-coil bundle [four-helix bundle (4HB)] [14]. It has been proposed that HNs in the “heads-down” position are responsible for hiding the residues that are associated with the interaction and activation of F protein on the stalk region. It was shown that the HN-F interaction has occurred before receptor engagement, but it is not sufficient to activate F until the heads of HN transition from down to up position binding receptor thereby inducing HN head to covey the activation signal to stalk. In the end, stalk interacts with the F protein and then triggers the fusion process [11, 15].
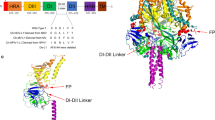
It is reported that both NDV and PIV5 HN heads in the down position form a similar interaction with the 4HB stalk, indicating that the formation of this interaction is common to HN proteins of other paramyxoviruses [14, 15]. In this report, based on primary sequence alignment of PIV5, NDV, and hPIV3, we selected 7 residues from the global head and 8 residues from 4HB stalk that form the stalk/head interface of hPIV3, and then mutated these sites to alanine. The homology structural model of the hPIV3 HN is based on NDV HN structure (PDB ID:3TIE) to locate the stalk/head interface on it and then each mutant is identified in the structure model. By means of the vaccinia virus-T7 RNA polymerase expression system, the wild-type or mutated HN proteins can be expressed. The ability to form syncytia, neuraminidase activity, receptor binding ability, and protein surface expression was detected.
Materials and methods
Cells, viruses, and vectors
BHK-21 cells were kindly provided by the Shandong Center for Disease Control and Prevention, China, cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal calf serum (FCS) and 1% penicillin–streptomycin. Both wild-type (wt) vaccinia virus and recombinant vaccinia virus vTF7-3 cultured in BHK-21 cells were generous gifts from Dr. Bernard Moss, aimed to quantify cell fusion and provide T7 RNA polymerase in the vaccinia-T7 RNA polymerase expression system, respectively [16]. The recombinant plasmid vectors, given by Professor Ronald M. Iorio, mainly refer to pBSK-HN and pBSK-F, which are made in the BamH I site of pBluescriptSK(+) (pBSK+) by inserting hPIV3 HN and F genes, respectively [17].
Transient expression system
Recombinant vaccinia virus vTF7-3 was made by inserting the T7 RNA polymerase gene into vaccinia virus, and then the polymerase gene integrated within the vaccinia virus genome, which made the virus vTF7-3 infectious and could stably express T7 RNA polymerase in BHK-21 cells. T7 RNA polymerase has high catalytic activity and strict specificities for both promoter and terminator [16]. There were T7 promoter and terminator in pBSK plasmid, therefore mutants from pBSK-HN plasmid also had T7 promoter and terminator. The strong T7 promoters were completely controlled by T7 RNA polymerase. When BHK-21 cells were infected with the recombinant vaccinia virus and transfected with plasmids containing the target genes, the latter were expressed at higher levels. Wild-type (wt) HN and all mutated HN proteins were used in the vaccinia virus-T7 RNA polymerase expression system to be expressed in BHK-21 cells which were seeded in six-well plates with density of 4 × 105 cells per well 8 h before transfection. Monolayer cells were infected by the recombinant vaccinia virus at a multiplicity of infection (MOI) of 10 PFU, cultured in a 5% CO2 incubator for 1 h at 37 °C, and then washed with serum-free DMEM. TurboFect™ Transfection Reagent (Thermo Fisher Scientific) transfections were performed using 1 μg of each plasmid for transfection [18].
Construction of single-point mutations at the stalk/head interface
Plasmid pBSK-HN was used as the template in each PCR mutagenesis to generate target amino acid substitution. The primers are shown in Table 1 (mutated sites are underlined in the primer sequences).
Syncytium formation experiment
BHK-21 cells were plated on 12-well plates 6 h earlier before transfection. Co-transfections with wt or mutated HN and pBSK-F plasmids were performed as soon as monolayer cells were full of wells. The pBSK-F plasmid was transfected separately as a negative control. At 24 h post-transfection, the cells were washed with PBS and then fixed with methanol for 10 min. Then the monolayer cells were stained with Giemsa Stain solution (Solarbio) for syncytium observation. An inverted microscope was used to capture syncytia where three or more nuclei fuse together [19].
Content mixing assay
Monolayer of BHK-21 cells was plated in 12-well plates and 60-mm culture dish. On the one hand, cells in the 12-well plates were infected with recombinant vaccinia virus, 1 h later at 37 °C, and then were co-transfected with wt or mutated HN and pBSK-F plasmids. On the other hand, at the same time, monolayer cells in the dish were already infected with wild-type vaccinia virus (MOI = 10) and then cultured for an hour at 37 °C and finally transfected with 1 μg of plasmid pG1NT7β-gal. After incubation for 24 h, equal numbers (1 × 105) of the two population cells were combined in duplicate wells of a 96-well dish. After being incubated for 5 more hours at 37 °C, cell lysates were pelleted by centrifugation (12,000 rpm for 2 min), and were assayed for β-galactosidase activity according to the procedures of the High Sensitivity β-Galactosidase Assay Kit (Agilent Stratagene). The extent of fusion was quantified by the absorbance of each well which was measured at a wavelength of 570 nm. The well with pBSK+ empty vector acts as a negative control [20].
Dye transfer assay
Human red blood cells (RBCs) were washed three times with cold PBS-CM(containing 0.1 mM CaCl2 and 1 mM MgCl2), and resuspended in cold PBS-CM (1% hematocrit) and then incubated with 15 μl octadecyl rhodamine B (R18; Invitrogen) (1 mg/ml in ethanol) at room temperature in the dark for 30 min. Then unbound R18 was washed with cold PBS. Monolayer cells co-expressing wt or mutated HN and F proteins were overlaid with R18-labeled RBCs at 4 °C for 30 min. The cells were washed and then incubated for 60 min at 37 °C and finally recorded with a fluorescence microscope [20].
Hemadsorption assay
The receptor binding activity of each mutant was detected by the ability to adsorb human red blood cells (RBCs). After transfection for 24 h, the monolayers that expressed wt or mutated HN protein were incubated at 4 °C to be appropriated for receptor binding (but not fusion) with a 2% suspension of RBCs in PBS–CM (0.1 mM CaCl2 and 1 mM MgCl2). Unbound RBCs were washed away with ice-cold PBS and the results of RBCs adsorption were observed under inverted microscope. Then the cells were treated with 50 ml NH4Cl at 4 °C until all bound RBCs were lysed and the lysate was cleared by centrifugation (12,000 rpm for 2 min). The absorbance was read at 540 nm on the UV spectrophotometer (Shimadzu, Kyoto, Japan) [21].
Neuraminidase assay
Transfection was performed as previously described. Following 22 h incubation at 37 °C, the cells were detached from the plates using 0.53 mM EDTA in PBS, the cells were centrifuged at 2000 rpm for 3 min. The lysates were assayed for neuraminidase activity according to the procedures of Neuraminidase Assay Kit (Beyotime Biotechnology). Fluorescence was measured with excitation and emission wavelengths of 360 and 460 nm, respectively [17].
Flow cytometry
Monoclonal antibody specific for the hPIV3 HN protein was discontinued, so every wt or mutated HN was tagged at its C-terminus with the 6*His tag.
Cell surface expression efficiency was quantified as the mean fluorescence intensity per cell. Monolayer BHK-21 cells were transfected separately with hPIV3 wt or mutated HN plasmids. After transfection for 36 h, the cells were removed from the wells by PBS-EDTA (0.53 mM EDTA), detached, pelleted, washed twice with PBSA (PBS containing 5% BSA), and then incubated in a 1:100 dilution of anti-6*His tag monoclonal antibody at room temperature for 2 h. The unbounded primary antibody was washed away twice with PBSA and followed by incubation with 1:100 dilution of fluorescein isothiocyanate (FITC)-conjugated goat anti-mouse IgG. Finally, the cells were fixed in PBS containing 4% paraformaldehyde at room temperature for 10 min. 0.4 ml PBSA was used to resuspend the cells and subjected to flow cytometry in a FACS Calibur flow cytometer (Becton–Dickinson Biosciences). Cells transfected with pBSK+ empty vector separately were used as a negative control [22].
Statistical analysis
All results were expressed as mean ± SD of at least 3 independent experiments. Statistical analysis was performed using Student’s T test, and P < 0.05 was statistically considered as significant.
Results
Point mutations at the stalk/head interface
This homology model was generated based on the NDV HN crystal structure (PDB ID:3TIE) (Fig. 1). The localization of selected amino acids at the hPIV3 stalk/head interface is shown in Fig. 1, with 8 residues from stalk helices (I112, Q116, M118, D120, R122, K123, S126, E127) and 7 residues from the head (R141, I142, D145, G147, I148, L151, N152). All of these sites were individually mutated to alanine in succession. Alanine was introduced because it has minimal disruptive impact on protein structure because of a short side chain.

The location of mutations at the stalk/head interface. The homology modeling of the hPIV3 HN protein was generated based on the NDV HN crystal structure (PDB ID: 3TIE) in the “4-heads-down” conformation. The residues that comprise this stalk/head interface in hPIV3 and NDV HN align are based on primary sequence alignment. The mutant residues are shown in space-filling mode in this interface. The figure was made by PyMOL 2.0
Fusion promotion ability of individual mutants
The ability of each mutant to promote cell fusion was evaluated with Giemsa staining, a content mixing assay, and a lipid-mixing assay. To examine the effect of mutants on syncytium fusion, monolayer BHK cells were transfected with plasmids to express wt HN or HN mutants and F proteins. The cells that were transfected only with pBSK+ plasmid were used as negative control. As shown in Fig. 2a, b, compared with the wt HN, four single mutants (I112A, R122A, I148A, and L151A) almost lost the ability of forming syncytium. But as for R141A, I142A, and G147A, the number of syncytia was more than that in wt HN and the size of syncytia was also bigger. Syncytia caused by other mutants were not only smaller but also fewer.

Fusogenic properties of stalk/head interface mutants. BHK-21 cells were transfected with wt or mutated HN and hPIV3 F, while pBSK-F plasmid was expressed separately as a negative control. Formation of syncytia was observed at 20 h, and the cells were stained with Giemsa. a Syncytia caused by HN stalk region mutants (arrows). b Syncytia caused by HN head region mutants (arrows). c Quantification of content mixing measured by a beta-galactosidase reporter gene assay, which indicates the membrane fusion activity of the mutants. Value of content mixing detected in cells co-transfected with wt F and HN proteins serves as a positive control. The other values are expressed as percentages of this positive control. The experimental data are the average of three independent replicates (*P < 0.05; **P < 0.01; ***P < 0.001; otherwise, P > 0.05)
To quantify the membrane fusion activity of the mutants more accurately, we performed a β-galactosidase reporter gene assay as described in materials and methods to measure the levels of content mixing. When cells co-expressing with wt or mutated HN and F proteins were mixed with the cells transfected with pGINT7 β-gal plasmid separately, the pGINT7 β-gal gene is activated to produce galactosidase. The reaction of the enzyme with the substrate can produce a color change. While the expression level of pGINT7 β-gal is related to the amount of T7 RNA polymerase that was also identical with the degree of cell fusion. The results are reported in Fig. 2c. The fusion ability of I112A, R122A, I148A, and L151A was significantly reduced, and other six mutants (Q116A, M118A, K123A, S126A, D145A, N152A) reduced the fusion activity to around 50% of that of wt HN. There was no difference in cell fusion ability between E127A and wt HN. It is noteworthy that R141A, I142A, and G147A had a significantly increased fusion activity, especially for R141A residue, with an equivalent percentage 178.0% as much as that of the wt HN level. These data were consistent with the results of Giemsa staining, indicating that the single-point mutations affect the degree of cell fusion in different levels.
The entire phase of membrane fusion is usually divided into three processes: hemifusion, pore formation, and pore expansion [20]. Hemifusion occurs at the initial stage of membrane fusion. To further verify whether HN mutants have an effect on pore formation or pore enlargement or the hemifusion stage, we conducted a dye transfer experiment to text whether or not these mutants can transfer the lipophilic R18 from the membrane of R18-labeled RBCs to the membrane of cells that were transfected with wt or mutant HN. The result of hemifusion is shown in Fig. 3a, b which almost mirrored the Giemsa staining of wt or mutated HN and F protein. Mutants (like I112A, R122A, I148A, and L151A) that almost lost the ability to form syncytia can just be detected only with very little fluorescence dequenching. In contrast, mutants (like R141A, I142A, and G147A) that enhance the fusion ability had strong fluorescence. These data demonstrated that the effect of mutants on membrane fusion has occurred at the beginning stage of membrane fusion.

Dye transfer of the BHK-21 cells co-expressed with wt or mutated HN and hPIV3 F proteins. The R18 probe is lipophilic. The fluorescence-labeled R18 can transfer from RBC membrane to the cell membrane of the transfected monolayers, then through the range of dye diffusion we detected the extents of mixing of membrane lipids (arrows). We conducted the dye transfer by BHK-21 cells that was co-expressed with wt or mutated HN and hPIV3 F proteins. a Semi-fusion of HN stalk region mutants; b Semi-fusion of HN head region mutants
Cell surface expression efficiency of mutant HN proteins
To explore whether or not the influences of fusion activity of these mutated HN proteins were associated with their levels of protein expression efficiency on the cell surface, fluorescence-activated cell sorting (FACS) was used to quantify the levels of cell surface expression of the wt or mutated HN proteins. Mean fluorescence intensity (MFI) was used to evaluate the cell surface protein expression efficiency of each mutant. The results of FACS suggested that expression efficiency of most mutants were quite comparable to that of wt HN from 86.31 to 106.54%, except for L151A. The MFI of L151A decreased to 38.09% as much as that of wt HN, as shown in Fig. 4c. The data were average result of three independent repeated tests.

Expression efficiency of mutant proteins on cell surface. Cell surface expression (CSE) of the wt or mutant HN proteins was measured by flow cytometry. a Fluorescent histograms for mutants within the HN stalk domain are shown. The x axis shows the fluorescence intensity value that is displayed on a logarithmic scale and the y axis indicates cell counts. The numbers below the graph represent the percentage of cells in M2 gate compared to the total number of cells. b Representative fluorescent histograms for each mutant within the HN head domain are shown. c Quantitation of the cell surface expression level. Values of the mean fluorescent intensity (MFI) are expressed as a percentage. The error bars represent the standard deviation of three independent replicates. Student’s t test was used to calculate the P value
Hemadsorption (HAD) ability and NA activity of HN mutants
HN protein is a multifunctional protein that is important for receptor binding, neuraminidase activity, and F activation. To evaluate whether or not mutations at the stalk/head interface region influence HN to bind to sialic acid receptors, we used an RBC hemadsorption assay to assess its ability. BHK-21 cells expressing with wt or mutant HN were covered with 2% human RBCs and were incubated at 4 °C to be appropriate for binding without fusion. The amounts of bound RBCs are plotted in Fig. 5a, b. To quantify this ability, absorbance was measured at 540 nm after the bound RBCs were lysed. The result is shown in Fig. 5c. Five mutants (E127A, R141A, I142A, D145A, G147A) retained a similar receptor binding ability as the wt hPIV3 HN (from 88.33 to 92.78%). I48A and L151A significantly affected the HAD ability (32.49 and 24.61%, respectively). The other 8 mutants directed approximately half the levels of receptor binding ability of that of the wt HN.

Detection of HAD ability of HN mutation proteins. The HAD activity was used to test the ability of mutant proteins to bind to cell surface sialic acid receptors. Monolayer cells that express the wt or mutant HN protein were separately incubated with 2% human RBCs at 4 °C for 30 min. Then redundant RBCs were washed away, and photomicrographs taken. a The attached human RBCs on monolayer cells that were transfected with mutants within the stalk domain are indicated by arrows. b Monolayer expressing head mutants, arrows refer to the adsorption of human RBCs. c To more accurately quantify the receptor binding capacity of mutant proteins, ammonium chloride was used to lyse the adsorbed RBCs, and the absorbance was measured by centrifugation at 540 nm
The neuraminidase activity of each mutant is shown in Fig. 6. The exception was R141A, which enhanced NA activity. Neuraminidase activity of other mutant proteins that were compared to that of wt HN protein decreased to some extent.

Detection of NA ability of HN mutation proteins. NA activity can cleave the receptor that is conducive to the release of progeny virus particles. The percentages were used to quantify the neuraminidase activity of each mutant protein. Results were expressed as mean ± SD of 3 independent experiments
Discussion
Similar to the NDV and the PIV5 HN proteins, the arrangement of hPIV3 HN is in a tetrameric, dimer-of-dimers conformation on the virion. The HN stalk provides an important driving force for tetramerization, but the TM domains are also considered to play a role [11, 23]. The helical stalk region of hPIV3 forms 4-helix bundles. Based on the crystal structure of the hPIV3 HN protein, some researchers have already recognized the locations of the receptor binding residue and neuraminidase active site residues on the globular head [12, 24]. It is crucial to clarify the relationship between the head and stalk domains of multifunctional HN protein because the specific residues on the stalk domain are considered to trigger F protein and after binding to the receptor the head of HN protein can transmit the activation signal to F [10, 14, 24,25,26].
It has been shown that the global head of HN in the down position are responsible for masking the activating residues on the stalk region [26, 27]. According to the result of electron microscopy data, the heads of NDV, PIV5, and hPIV3 HN proteins can adopt different conformations relative to the stalk of each other, that is to say HN heads can present different arrangements on the viral envelope, and these structural rearrangements may be related to fusion activation [11, 14, 28, 29]. The evidence suggests that hPIV3 HN protein in a tetrameric, dimer-of-dimers can adopt two conformation, including a “heads-down,” in which two out of the four HN head domains have interactions with the HN stalk while the other two near to the target membrane (Fig. 1), and a “heads-up” form. F protein is observed irrelevant to HN protein in a heads-down position. In addition, “heads-up” HN may have interaction with F protein prior to receptor engagement but this interaction is not sufficient to activate F [11]. To explore whether or not mutations at the stalk/head interface of hPIV3 HN protein would affect fusion, we made single-point mutations of residues located within both the stalk and head domains.
The expression efficiency of 15 mutants on the cell surface was determined by FACS. It was noted that while L151A was significantly reduced in surface expression compared with wt HN, all the other mutants were expressed at the cell surface in nearly similar amounts to wt HN (Fig. 4). In other words, except L151A, the other 14 mutants have the same expression efficiency as wt HN protein. They can be translated accurately, folded correctly in the cytoplasm, and successfully transported to the cell membrane surface. The results suggest that these 14 mutated proteins’ structure can remain intact and the impact on other functions could not be accounted for the efficiency change of surface proteins expression.
Within the stalk region, only I112A and R122A were significantly impaired for cell–cell fusion, while mutations D120A and E127A were fusion competent. Other four mutants (Q116A, M118A, K123A, S126A) in this domain retained this activity around half of that of wt HN level (Fig. 2), suggesting that I112 and R122 residues play an important role in maintaining the fusion function of HN protein. In addition, compared with dye transfer assay, it was found that the reduction of cell fusion activity at these amino acid sites has occurred in the early stage of membrane fusion. The decrease or loss of fusion ability may be caused by various factors at the structural or functional level.
The mutations demonstrated that except for E127A, seven mutations (I112A, Q116A, M118A, D120A, R122A, K123A, and S126A) affected both the receptor binding ability and NA activity (Figs. 5, 6). When the E127 residue was mutated to alanine, there was no difference in fusion activity, receptor binding activity, and neuraminidase activity between E127 and wt HN. We have proved that the decrease of receptor binding ability for the mutants I112A (67.1%), D120A (56.7%), and R122A (59.3%) correlated with the loss of NA activity (70.4, 65.6, and 61.7%, respectively) (Figs. 5c, 6). The extent of fusion activity of I112, D120A, and R122A proteins co-expressing with the F protein was 23.4, 82.1, and 28.7%, respectively (Figs. 5c, 6). As for these three mutant proteins, there was an imbalance in the degree of reduction of the three functions such as fusion promotion, HAD, and NA activity. A variety of biochemical and virological data for paramyxoviruses verify an important role for HN stalk in activating F protein. Specific residues on the hPIV3 stalk domain have been implicated in triggering F protein [28, 30]. In addition, some mutations of stalk may affect the 4HB stalk structure, and would disrupt the underlying oligomer scaffold that forms the putative F interaction surface, and in the end result in reducing F protein interactions and activation [14, 31]. We guess that I112A, D120A, and R122A mutants of the stalk domain disrupt the head–stalk interaction, and I112A and R122A may be defective in fusion activation. In contrast, when D120 was replaced by alanine, after binding to the receptor, it may enhance the interaction with the F protein. The mutational data further approve the significance of this stalk region in F activation and fusion, given the functional effects of the I112A, D120A, and R122A mutations. It is less clear how HN stalk mutations could affect NA and HAD activities. It is possible that full NA and HAD activity is dependent on activating interactions with the stalk [32].
Of the seven mutants within the head domain, three mutants (R141A, I142A, and G147A) have shown increasing fusion activity (178.0, 106.9, 124.6% respectively), in contrast to significant reduction in I148A, L151A fusion levels(less than 30%). D145A and N152A retained this ability by 67.9% and 44.7%, respectively, of that of wt HN (Fig. 2c). Previous reports suggested that receptor binding activity is closely related to neuraminidase activity. In the same mutant, the impact on HAD ability, NA activity, and cell fusion is different [14]. Our experimental results show that for mutants I148A, L151A, and N152A, the degree of decline between HAD ability and NA activity is related, respectively (Figs. 5c, 6). It was concluded that I148A, L151A, and N152A play almost the same role in the receptor binding and receptor lysis process.
Even though surface expression for the L151A mutant was reduced to 60% compared with wt HN, the formation ability of syncytia was completely inhibited (Fig. 4). NA activity and receptor binding activity are also reduced to a low degree. It is possible that the reason why L151A mutant lacks fusion promotion, HAD, and NA activity is due to defects in processing or protein transport.
Further analysis of the three mutations (R141A, I142A, and G147A) that improved the formation abilities of syncytia and increase the contents mixing abilities, with the initial fusion enhancement suggested that mutants enhanced fusion activity in the initial stage of membrane fusion. Researchers have proposed a model for hPIV3 fusion, in which they assumed that pre-fusion F and HN already interacts with each other before binding to the reception and after the process HN’s globular head transmits activating signal to F protein [11]. For the head domains that directly interact with the stalk 4HB, the first observed residue is R141. The current experimental results show that R141 did not improve protein expression or receptor binding, while in the other hand it obviously enhanced fusion promotion activity (178.04%). Our results show that the increasing neuraminidase activity for R141A (103.51%) can enhance the ability to cleave receptors and do good to the release of virus particles. Moreover, when R141, I142, and G147 residues are replaced by alanine, every single site may be beneficial for the transduction of activation signals from HN head domain to the stalk.
In sum, the data above suggest that the stalk/head interface may be important for hPIV3 HN fusion-promoting activities with four features pointing to potential functional significance of the stalk/head interface. First, the arrangement is symmetrical, and two of the four head domains interact with two opposite sides of the 4HB. Secondly, mutations at this interface play an important role in membrane fusion, receptor binding ability, and neuraminidase ability without affecting F protein triggering. Then, L151A of the HN stalk domain affects protein surface expression. Finally, functional studies suggest that specific residues of the HN stalk domain are related to the activation of F protein.
References
M.V. Villaran, J. Garcia, J. Gomez, A.E. Arango, M. Gonzales, W. Chicaiza, W. Aleman, I. Lorenzana de Rivera, F. Sanchez, N. Aguayo, T.J. Kochel, E.S. Halsey, Influenza Respir. Viruses 8, 217–227 (2014)
M.E. Counihan, D.K. Shay, R.C. Holman, S.A. Lowther, L.J. Erson, Pediatr. Infect. Dis. J. 20, 646–653 (2001)
K.J. Henrickson, Clin. Microbiol. Rev. 16, 242–264 (2003)
S. Bose, C.M. Heath, P.A. Shah, M. Alayyoubi, T.S. Jardetzky, R.A. Lamb, J. Virol. 87, 13520–13531 (2013)
N.G. Xiao, Z.J. Duan, Z.P. Xie, L.L. Zhong, S.Z. Zeng, H. Huang, H.C. Gao, B. Zhang, J. Med. Virol. 88, 2085–2091 (2016)
R. Xu, S.G. Palmer, M. Porotto, L.M. Palermo, S. Niewiesk, I.A. Wilson, A. Moscona, mBio 4, e00803–e00813 (2013)
C. Godoy, P. Peremiquel-Trillas, C. Andrés, L. Gimferrer, S.M. Uriona, M.G. Codina, L. Armadans, M.D.C. Martín, F. Fuentes, J. Esperalba, M. Campins, T. Pumarola, A. Antón, Diagn. Microbiol. Infect. Dis. 86, 153–159 (2016)
A.C. Schmidt, A. Schaap-Nutt, E.J. Bartlett, H. Schomacker, J. Boonyaratanakornkit, R.A. Karron, P.L. Collins, Exp. Rev. Respir. Med. 5, 515–526 (2014)
M. Porotto, S.G. Palmer, L.M. Palermo, A. Moscona, J. Biol. Chem. 287, 778–793 (2012)
T.S. Jardetzky, R.A. Lamb, Curr. Opin. Virol. 5, 24–33 (2014)
L. Gui, E.M. Jurgens, J.L. Ebner, M. Porotto, A. Moscona, K.K. Lee, MBio 6, e02393-02314 (2015)
M.C. Lawrence, N.A. Borg, V.A. Streltsov, P.A. Pilling, V.C. Epa, J.N. Varghese, J.L. McKimm-Breschkin, P.M. Colman, J. Mol. Biol. 335, 1343–1357 (2004)
R.A. Lamb, R.G. Paterson, T.S. Jardetzky, Virology 344, 30–37 (2006)
P. Yuan, K.A. Swanson, G.P. Leser, R.G. Paterson, R.A. Lamb, T.S. Jardetzky, Proc. Natl. Acad. Sci. USA 108, 14920–14925 (2011)
B.D. Welch, P. Yuan, S. Bose, C.A. Kors, R.A. Lamb, T.S. Jardetzky, PLoS Pathog. 9, e1003534 (2013)
T.R. Fuerst, E.G. Niles, F.W. Studier, B. Moss, Proc. Natl. Acad. Sci. USA 83, 8122–8126 (1986)
C. Sun, H. Wen, Y. Chen, F. Chu, B. Lin, G. Ren, Y. Song, Z. Wang, BioSci Trends 9, 56–64 (2015)
G. Ren, Z. Wang, X. Hu, Intervirology 50, 115–122 (2007)
F.L. Chu, H.L. Wen, G.H. Hou, B. Lin, W.Q. Zhang, Y.Y. Song, G.J. Ren, C.X. Sun, Z.M. Li, Z. Wang, Virus Res. 174, 137–147 (2013)
W. Xie, H. Wen, F. Chu, S. Yan, B. Lin, W. Xie, Y. Liu, G. Ren, L. Zhao, Y. Song, C. Sun, Z. Wang, PLoS ONE 10, e0136474 (2015)
E. Adu-Gyamfi, L.S. Kim, T.S. Jardetzky, R.A. Lamb, J. Virol. 90, 7778–7788 (2016)
E. Adu-Gyamfi, L.S. Kim, T.S. Jardetzky, R.A. Lamb, J. Virol. 90, 9172–9181 (2016)
P. Yuan, T.B. Thompson, B.A. Wurzburg, R.G. Paterson, R.A. Lamb, T.S. Jardetzky, Structure 13, 803–815 (2005)
V.A. Streltsov, P. Pilling, S. Barrett, J.L. McKimm-Breschkin, Antiviral Res. 123, 216–223 (2015)
S.A. Connolly, G.P. Leser, T.S. Jardetzky, R.A. Lamb, J. Virol. 83, 10857–10868 (2009)
C.K. Navaratnarajah, N. Oezguen, L. Rupp, L. Kay, V.H. Leonard, W. Braun, R. Cattaneo, Nat. Struct. Mol. Biol. 18, 128–134 (2011)
A. Chang, R.E. Dutch, Viruses 4, 613–636 (2012)
S. Bose, A. Zokarkar, B.D. Welch, G.P. Leser, T.S. Jardetzky, R.A. Lamb, Proc. Natl. Acad. Sci. 109, E2625–E2634 (2012)
T. Hashiguchi, T. Ose, M. Kubota, N. Maita, J. Kamishikiryo, K. Maenaka, Y. Yanagi, Nat. Struct. Mol. Biol. 18, 135–141 (2011)
M.A. Brindley, R. Suter, I. Schestak, G. Kiss, E.R. Wright, R.K. Plemper, J. Virol. 87, 11693–11703 (2013)
V.R. Melanson, R.M. Iorio, J. Virol. 80, 623–633 (2006)
M.M. Tappert, J.Z. Porterfield, P. Mehta-D’Souza, S. Gulati, G.M. Air, J. Virol. 87, 8962–8970 (2013)
Acknowledgements
We gratefully acknowledge Dr. Ronald Iorio for providing the recombinant plasmid vectors and Dr. Bernard Moss for vTF7-3.
Funding
This work was supported by a Grant from National Natural Science Foundation of China (No. 81672011) and the Fundamental Research Funds of Shandong University (2015JC044).
Author information
Authors and Affiliations
Contributions
JJ performed all parts of experiments and wrote the manuscript. MC and YL helped in experimental design, JL and ZC helped in the analysis of the results. HW, LZ, YS, NL, and LC supervised this study. ZW directed the experiments and revised the manuscript.
Corresponding author
Ethics declarations
Conflicts of interest
All authors in this paper declare they have no conflict of interest.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Research involving human participants and/or animals
Present paper does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Edited by Zhen F. Fu.
Rights and permissions
About this article

Cite this article
Jiang, J., Wen, H., Chi, M. et al. Functional analysis of amino acids at stalk/head interface of human parainfluenza virus type 3 hemagglutinin-neuraminidase protein in the membrane fusion process. Virus Genes 54, 333–342 (2018). https://doi.org/10.1007/s11262-018-1546-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-018-1546-3