
Abstract
Chinese equine infectious anemia virus (EIAV) attenuated vaccine is the first lentiviral vaccine with a successful application. In order to understand the correlation of viral genomic mutations with viral attenuation and with induced immunoprotective properties, we analyzed the proviral genome sequences of the EIAV-attenuated vaccine strain EIAVFDDV13 (EIAV fetal donkey dermal cell-adapted vaccine) and its highly virulent parental strain EIAVLN40. The sequences of these strains were compared with those of the major foreign EIAV strains. The results indicated a large genetic distance between the Chinese EIAV strain and the major EIAV strains in America and Japan. The Chinese strains belong to an independent phylogenetic branch. The divergence between the entire genome of the Chinese strains and that of other major EIAV strains is ~23%. The divergence rate in LTR is over 14%, whereas that in each open reading frame is over 20%. The gp90 exhibited a divergence of 35% in its nucleotide sequence and 40% in its amino acid sequence. The present study found that after long-term passage in vitro, EIAVFDDV13 has accumulated many stable substitution mutations in each gene. These mutations at multiple sites in multiple genes of the vaccine strain, especially the conserved mutations, provide important references for further understanding the attenuation mechanism of Chinese EIAV-attenuated vaccine and the immunoprotection mechanism of lentiviral vaccines.
Similar content being viewed by others
Introduction
Equine infectious anemia virus (EIAV) belongs to the Lentivirus genus in the Retroviridae family. High frequency of genomic mutations and antigenic drift are common characteristics of all lentiviruses, including human immunodeficiency virus (HIV). These characteristics are the main reasons for persistent infection caused by lentivirus and are major obstacles in lentiviral vaccine development [1].
Although rapid genome replication and high antigenic variation exist in EIAV, most EIAV infections in equine animals enter a latent state under the control of the immune system following the acute and chronic stages of the disease. Infected animals can, thus, become life-long carriers of the virus [2]. Upon immune suppression or stress response, EIAV in the latently infected animals exhibits an increased level of replication, followed by relapse of the disease [2, 3]. Through blood transmission, horses latently infected with EIAV can pass the virus to healthy horses and cause disease [3, 4]. These phenomena indicate that EIAV viral replication can be controlled by the host immune system. However, this control is weak and incomplete.
Consequently, EIAV is considered a unique model for the study of lentiviral replication and host immune defense mechanisms. In the 1970s, Chinese scientists developed the first lentiviral vaccine, attenuated EIAV vaccine, through in vitro passage. The massive immunizations performed using this vaccine contributed tremendously to the prevention and control of equine infectious anemia in China (cited from “Equine infectious anemia and its controlling in China,” a document issued by the Chinese Ministry of Agriculture in 1997; this document is in Chinese and is available by request to the corresponding author; also, see reference [5]). The successful development of this vaccine broke new ground in dealing with existing immunological restrictions on lentivirus and provided hope for the prevention of lentiviral diseases by immunization as well as theoretical and practical foundations for the further investigation of lentiviral vaccines.
HIV has been known for more than 20 years, but few substantive breakthroughs have yet been made in HIV vaccine research. Currently available attenuated lentiviral vaccines are mostly gene-defective strains. These vaccines provide effective immunoprotection only against homologous viral strains and are deficient in immunoprotection against heterologous viral strains [2, 6]. An ideal lentiviral vaccine should not only replicate at low levels in the host, lack clinical manifestations and carry low risk of transmission but, more importantly, should also induce a high level of defense against infection in the host [7].
The Chinese attenuated EIAV vaccine yields an immunoprotection rate of over 80% against the homologous Liao-Ning strain (EIAVLN40) and against the heterologous Chinese Helongjiang strain (EIAVH) and Xinjiang strain (EIAVX), as well as against major American strains including EIAVWyoming and the Cuba strain (cited from “Equine infectious anemia and its controlling in China” as mentioned above; also see [5]). The spectrum of protection offered by this vaccine brought hope to lentiviral vaccine development, especially to HIV-1 vaccine research.
EIAVFDDV13 is the fetal donkey dermal (FDD) cell-adapted strain of the Chinese attenuated EIAV vaccine. In immunized horses, this vaccine strain undergoes a long-term low level of viral replication. Even in the presence of immune suppression, it can still maintain its low level of replication without causing clinical symptoms of equine infectious anemia (EIA) [8], proving its safety and stability. Apparently, the mechanism of low-level replication of the attenuated vaccine is different from that in horses latently infected with EIAV. The low viral load associated with the attenuated vaccine is not caused by pressure from the host immune system, indicating that fundamental changes in genetic characteristics have occurred in the Chinese attenuated EIAV vaccine.
Our previous study showed that the EIAV vaccine strain was highly attenuated and safe. This investigation and some others found that this attenuation of virulence and the enhancement of immunogenicity were closely linked to multiple mutations in multiple viral genes. Using reverse mutations of selected sites of the vaccine strain to the corresponding sequence of the virulent strain, results from our laboratory and others determined that some mutations, especially consensus mutations that consistently existed in most of the successors once raised, in LTR [9, 10], gp90 [11, 12], gp45 [13], and S2 (manuscript in preparation) were responsible for the virulence of EIAV.
Although the vaccine was successfully developed more than 20 years ago and a considerable amount of research on its biological characteristics have been performed, detailed whole-genome analysis of this vaccine strain has not been reported. In this study, we report a detailed analysis of the genomic sequences of the FDD cell-adapted vaccine strain EIAVFDDV13 and the parental virulent EIAVLN40 strain and of the comparison of their sequences with those of the major international strains. The clones analyzed were 10 and 5 for EIAVFDDV13 and EIAVLN40, respectively. This study provides a foundation for the further understanding of EIAV gene functions and the molecular mechanisms of virulence attenuation and immunoprotection by the Chinese attenuated EIAV vaccine.
Materials and methods
Viruses
The EIAV high-virulence strain EIAVLN40 and the FDD cell-adapted attenuated vaccine strain EIAVFDDV13 were maintained by Harbin Veterinary Research Institute, Chinese Academy of Agricultural Sciences. EIAVLN40 proviral DNA was isolated from leukocytes of infected horses during viremia. The FDD cells were cultured as described previously [11]. After EIAVFDDV13 inoculation, the cells were cultured at 37°C in medium containing 10% bovine serum. The infected cells were harvested when cytopathic effects were visible.
Sequencing strategy and amplification of proviral genome
Based on the published nucleotide sequences of the EIAV vaccine strain (GenBank# AF327878), two primer pairs (P1: TGTGGGATTAATATAAGATTCTTATAAGTG; P2: CATACTTTCCCTCCCTTTGT; P3: TTGGCAGATGAGGCTACTAA; P4: CCGCTCGAGGGTGTTAGATCTTGAAAACAA) were designed using computer software Oligo 6.0. The 25-μl PCR reaction contained 2.5 μl of 10× LA buffer, 2 μl of dNTPs (2.5 mM), 1 μl each of the upstream and downstream primers (25 pM), 0.5 μl of LA Taq DNA polymerase (1.25 U), 5 μl of template DNA (200 ng), and H2O to make up the volume of 25 μl. PCR program was as follows: 94°C for 4 min, then 30 cycles of 94°C × 20 s, 56°C × 30 s, and 72°C × 4 min 40 s, followed by a 10-min extension at 72°C. All amplifications were performed in triplicate to obtain independent amplification replicates. The recovered PCR products were ligated into the pMD18-T vector. The ligated DNA was transformed into E. coli DH5α competent cells.
Genome analysis
The consensus sequences of EIAVFDDV13 and EIAVLN40 were generated from sequencing data of nine (GU385353-GU385362) and four (GU385362-GU385365) full-length genomes, respectively. Nucleotide sequences were edited and assembled using the SeqManII tool of the Lasergene DNAStar program 7.0 (DNAStar). The Megalign tool (DNAStar) was used for pairwise and multiple alignments of DNA and deduced amino acid sequences. Phylogenetic and molecular evolutionary analyses were conducted using the Molecular Evolutionary Genetics Analysis (MEGA) program 4, using the methods of neighbor joining.
Nucleotide sequences generated in this study were submitted to GenBank as above. Further full-length sequences published in GenBank, including the complete nucleotide sequence of EIAVLN40 (AF327878), EIAVUK (AF016316, the parental EIAVPV infection from which we generated the clone), V70 (AB008196, field strain, virulent), and V26 (AB008197, prepared by 50 passages of the Japanese virulent strain V70 in primary horse macrophage culture) were selected as reference sequences.
When analyzing the viral long terminal repeat (LTR), the LTR sequence of EIAVWyoming (M87580) and Malmquist (M87581, derived by adaptation of the Wyoming strain on equine dermal fibroblasts) [14] were selected as further comparison sequences. The complete gp90 sequences of EIAVWyoming and EIAVPV (AF005014, AF005109, AF005112,AF005114, AF005115, AF005117, AF005118, AF005125, AF005150, and AF033820 pony virulent) strain, an in vitro variant of the EIAVWyoming strain that is infectious but non-pathogenic in experimentally infected ponies, were also selected for comparison with gp90.
Results
Sequence analysis of the whole genome of EIAV
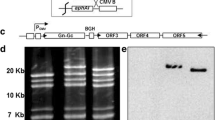
Using the long-PCR method, the full-length genomes of the provirus of the EIAVLN40 and EIAVFDDV13 strains were amplified in two fragments (Fig. 1) with sizes of 4.8 and 4.3 kb, respectively. In the present study, nine and four clones, respectively, were randomly picked up from EIAVFDDV13 and EIAVLN40 for sequencing. Using the results of previous sequencing of the whole genomes of the two strains (EIAVLN40, AF 327878; EIAVFDDV13, unpublished) as reference, we analyzed 15 complete EIAV proviral genomes and compared their sequences with those of EIAVUK, V26, and V70.

Design of primers used for amplification of the EIAV proviral DNA full genome. Long RT-PCR was performed for the amplification of full-length EIAV proviral genome in two segments; a 4.8-kb fragment (p1 and p2) and a 4.4-kb fragment (p3 and p4) were amplified
The results showed that EIAVFDDV13 is 8253–8258 bp in length, with one clone containing an 81-nucleotide deletion in the env gene. EIAVLN40 is 8235–8237 bp in length. The genome of the Chinese EIAV bears 22.7–23.8% difference from the international reference strains (the American strain EIAVUK and the Japanese strains V26 and V70) (Fig. 2). The divergence rates were >20% in all open reading frames. In contrast, the genomic variation between the vaccine strain EIAVFDDV13 and its parental strain EIAVLN40 was 3.2%, with a divergence of 2.2–5.4% in nucleotide sequences and 2.9–5.5% in amino acid sequences for each gene (Tables 1, 2).

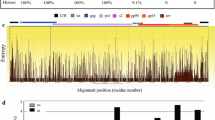
Similarity plots of the full-genome sequences of EIAVLN40, EIAVFDDV13, V26, and V27 and reference strains EIAVUK. The two-parameter (Kimura) distance model, with a sliding window of 200 bp and a step size of 20, was used. The vertical and horizontal axes of the graph indicate nucleotide distance (%) and nucleotide position (bp), respectively
Phylogenetic analyses were performed using LTR, gag, pol, env, gp90, gp45, tat, S2, and rev with the neighbor-joining method using software Mega4.1. All phylogenetic trees showed a similar shape. All the genes from the Chinese strains obviously occupy a different branch than the foreign strains, whereas the genes from the American and the Japanese strains are on the same branch. Genes from EIAVFDDV13 and EIAVLN40 occupy two small branches of a large branch (Fig. 3).

EIAV phylogenetic analysis of LTR and various ORFs, performed using the program MEGA4. Open circle indicates EIAVFDDV13 sequence determined in this study, filled circle indicates foregone EIAVFDDV13, open square indicates EIAVLN40 sequence determined in this study, and filled square indicates foregone EIAVLN40 (AF327878)
Sequence analysis of LTR variations
Long terminal repeat is a common noncoding region in the genome of retroviruses and plays an important regulatory role in viral replication and gene expression. Figure 4 shows the results of sequence analysis of the LTR region conducted in this study. The LTR of EIAVLN40 is 316 bp, whereas that of EIAVFDDV13 is 328 bp; 16 stable variations exist between the two strains. EIAVFDDV13 contains an 11-bp repeat sequence, GATGACTAGCT, inserted in its negative regulatory region, with an insertion of G or A between nucleotides 241 and 242. In EIAVFDDV13, the substitution of 91C → T generates a methylation-dependent binding protein (MDBP) site, the mutation of 101A → G produces an E_box binding site, and the mutation of 108G → A deletes the GATA binding motif. The 11-bp repeat sequence contains an AP-1 binding motif, TGAC. The first three nucleotides of the EIAVLN40 transcription initiation site are GGA, whereas in EIAVFDDV13 they are GTC.

Comparison of the LTR nucleotide sequences of various EIAV strains. Dashes indicate a gap. Putative transcription factor binding sites are boxed or underlined
The LTR sequences of EIAVLN40 and EIAVFDDV13 were compared with those of other reported EIAV strains, including the wild-type EIAVWyoming strain, the Malmquist strain, EIAVUK, V26, and V70. As shown in Fig. 4, the Chinese EIAV lacks PEA-2 and CAAT binding motifs, and EIAVFDDV13 has two AP-1 binding sites in its negative regulatory region. The fibroblast-tropic strains Malmquist and EIAVUK each have one more AP-1 binding site than does the wild-type EIAVWyoming strain.
Analysis of Gag protein variations
The assembly of Gag precursor protein in the plasma membrane is a necessary step in the budding and release of viral particles from the host cell membrane. Following digestion by the viral protease, the precursor protein gives rise to matrix protein (MA, p15), capsid protein (CA, p26), nucleocapsid protein (NC, p11), and core protein (p9) [15, 16].
The gag gene of EIAV is relatively conserved. Gag genes from different strains all have 1461 nucleotides encoding 486 amino acids. Compared with the reference strains, the Chinese EIAV strains exhibit a divergence rate of 23.2–23.7% at the nucleotide level and of 16.2–17.5% at the amino acid level. The gag genes and the resulting proteins of EIAVFDDV13 and EIAVLN40 display differences of 2.5 and 3.1% at the nucleotide and amino acid levels, respectively (Table 1). There are 11 stable amino acid substitutions between these two strains (Table 2), four of them (27N4/T1 → S9/R1, 100A → S, 103T4/Q1 → S, and 328S4/N1 → N) located within the identified CTL epitope region. As for each functional protein, four stable amino acid substitutions were found in the MA of EIAVFDDV13 and EIAVLN40 (27N4/T1 → S9/R1, 100A → S, 103T4/Q1 → S, and 124M4/S1 → E), one stable substitution was identified within the CA of EIAVFDDV13 and EIAVLN40, and five stable substitutions that may contribute to viral replication were discovered within the p9 of EIAVFDDV13 and EIAVLN40 strains.
Analysis of Pol protein variations
The proteolysis products of Pol precursor protein provide the enzymes required by EIAV for replication. They are protease (PR), reverse transcriptase (RT)/RNase H, dUTPase, and integrase (IN) [17–19]. Compared with the reference strains, Chinese EIAV strains display a divergence rate of 20.1–20.7% for Pol at the nucleotide level and of 14.9–15.5% at the amino acid level (Table 1). The pol genes of EIAVFDDV13 and EIAVLN40 are 3405 bp in length and encode 1134 amino acids. They exhibit a 2.8% difference at both the nucleotide and amino acid levels, with 15 stable amino acid substitutions (Table 2). We found no stable substitutions in PR, the function of which is to proteolyze the viral precursor protein. In contrast, there were six stable substitutions discovered in RT, two stable substitutions in the dUTPase and one stable substitution in IN.
Analysis of Env protein variations
Lentiviral envelope protein (Env) comprises the main region for the binding of the virus to receptors on target cells. The env gene of EIAV encodes a surface unit (SU) glycoprotein, gp90, and a transmembrane (TM) glycoprotein, gp45. The genetic distance of env between the Chinese strains and the reference strains ranges from 30.1 to 30.9%. EIAVFDDV13 and EIAVLN40 display a difference of 3.4% for env at the nucleotide level. The env gene of EIAVLN40 is 2598 bp in length and encodes 865 amino acids, whereas that of EIAVFDDV13 is 2592 bp in length but encodes only 709 amino acids due to an emerged stop codon at the gp45 gene fragment. There are two frequently identified deletions in the EIAVFDDV13 env, which are located at 205–207 nt and 705–708 nt of the gp90 gene fragment.
The variations of Env occur mainly in gp90. This protein is divided into eight highly variable regions (V1–V8) [20]. Considering the diversity of EIAV genes, we obtained, from Genbank, the previous EIAVFDDV13 and EIAVLN40 sequencing results and the complete gp90 gene sequences of other foreign strains (EIAVWyoming, Malmquist, EIAVPV, EIAVUK, V26, and V70) and performed a comparative analysis of these sequences. The results show that the genetic distance between the Chinese strains and the foreign strains is 35.0–35.9% at the nucleotide level and 35.9–40.6% at the amino acid level. The average divergence rate between EIAVFDDV13 and EIAVLN40 is 3.5% at the nucleotide level and 5.2% at the amino acid level. Despite the large divergence of the genes in the Chinese EIAV strains and the foreign strains, the genetic distance between EIAVLN40 and the foreign strains is smaller than that between EIAVFDDV13 and those strains (Table 1).
Although gp90 proteins from different strains show significant differences in primary structure, the cysteine residues and the glycosylation sites, which are important for higher order protein structure, were highly conserved, reflecting the consistency of protein conformational structure on the EIAV surface. In the gp90 protein, the American and Japanese strains have 19 conserved cysteine residues and the Chinese strains have 20 conserved cysteine residues. Among these strains, 18 cysteine residues are conserved in their sequence positions, indicating that these 18 residues are indispensable in maintaining the protein conformation of gp90. Significant differences between the Chinese strains and the foreign strains were identified in the three known neutralizing epitopes, especially in the neutralizing epitope ENT. The reference strains possess several amino acid deletions and point mutations in the principle neutralizing domain (PND), whereas the Chinese strains have mainly point mutations.
Twenty-seven stable amino acid substitutions occur between EIAVFDDV13 and EIAVLN40, four in the V3 and V4 regions. The 193S → N substitution in the V3 region and the 239N → K substitution in the V4 region cause EIAVFDDV13 to lose glycosylation sites NSSN and NNTW, respectively. The 256N → K mutation leads to loss of the glycosylation site NETW in most EIAVFDDV13 sequences. The V3 region is the major target of neutralizing antibodies. Glycosylation sites in this region play a role in shielding PND neutralizing epitopes and Th epitopes. However, the conformation of the V3 region is very susceptible to changes in the V4 region [21].
Glycosylation is an important feature of the EIAV envelope protein. The number of glycosylation sites on gp90 of the American and Japanese strains range from 13 to 18, whereas 16–21 sites are present on gp90 of the Chinese strains. However, the positions of most glycosylation sites in the amino acid sequences are consistent or similar among these strains (Fig. 5). The 40N → D mutation in the variable region V1 of the Japanese strain causes a loss of the glycosylation site in this region. The foreign strains and EIAVLN40 generally have 1–2 glycosylation sites in the PND region, whereas most EIAVFDDV13 sequences contain three glycosylation sites in this region. Although significantly different in primary sequences, EIAVLN40 and the foreign strains have glycosylation sites at corresponding positions. In contrast to the Chinese attenuated vaccine strain, the Japanese strains V26 and V70 exhibit a difference of only 1.0% in their surface proteins. The only difference in glycosylation sites occurs at the 360 residue and results in the presence or absence of NESI.



Multiple comparisons of the gp90 amino acid sequences of EIAVLN40 (LN) and EIAVFDDV13 (FDDV) with those of EIAVMalmquist (Malm), EIAVWyoming (Wyoming), EIAVUK (UK), EIAVPV (PV), V26, and V70. Dots indicate identical residues with the sequence of EIAVLN40 (AF327878); dashes indicate deletions. Eight gp90 variable regions (V1–V8) are boxed. Potential N-linked glycosylation sites are defined by shaded boxes. The different sequences compared are shown on the left, indicated by the name of the sequence. The principal neutralizing domain (PND) is marked by a gray line. The major neutralizing epitopes (ENT, DNT, and CNT) are indicated by a thick gray underline. Conserved cysteine residues are indicated by arrows
Gp45 is the major form of EIAV transmembrane glycoprotein (TM) in infected cells [22]. A further cleavage event occurs within the N-terminus of the cytoplasmic tail (CT) of gp45 and yields the N-terminal 32–35 kDa glycosylated peptide gp35 and the C-terminal 20 kDa unglycosylated peptide p20 [23]. The gp35 and p20 peptides cannot be detected in infected cells. Therefore, hydrolysis is thought to take place in viral particles. The two cysteine residues in the gp35 region are highly conserved and are crucial for maintaining gp45 conformation. The p20 peptide displays, if any, very weak and uncertain immunogenicity. EIAV cell-adapted strains do not express p20 at all during their replication in certain cells and generate a truncated form of gp45 [24]. For gp45, the Chinese strains and the reference strains showed a difference of 24.2–25.1% at the nucleotide level, whereas EIAVLN40 and the reference strains displayed a difference of 33.4% at the amino acid level. An average difference of 2.8% in gp45 at the nucleotide level was found between EIAVFDDV13 and EIAVLN40. The env gene in EIAVFDDV13 has a G → A mutation at nucleotide 2130, generating a TGA stop codon in the gp45 gene (Fig. 6). Therefore, the TM of EIAVFDDV13 has only 260 amino acid residues. Western blotting analysis of the lysis products of EIAVFDDV13 confirmed the existence of truncated TM (unpublished). EIAVFDDV13 and EIAVLN40 in total have four stable amino acid substitutions.

The mutation of 2130G → A of EIAVFDDV13 env created a premature stop TGA (indicated by the shaded box) in gp45. As a result, EIAVFDDV13 TM had only 260 residues, leading to truncated gp45. LN sequences from molecular clones of EIAVLN40, FDDV sequences from molecular clones of EIAVFDDV13
Analysis of accessory protein variations
EIAV encodes three accessory regulatory proteins: the Tat protein encoded by the tat (S1) gene, the S2 protein encoded by S2, and the Rev protein encoded by the rev (S3) gene [25]. The tat gene in Chinese EIAV strains is 240 nucleotides in length and encodes 79 amino acids. The average divergence rates in the nucleotide and amino acid sequences of EIAVFDDV13 and EIAVLN40 are 2.7 and 5.2%, respectively. There are three major amino acid substitutions between these two strains. None of these three stable mutations occurs in identified functional regions.
The genetic distance for S2 between the Chinese strains and the reference strains is 50.0–58.5% at the nucleotide level and 50.6–56.0% at the amino acid level. The S2 gene in the Chinese strains contains 207 bp and encodes 68 amino acids. EIAVFDDV13 and EIAVLN40 show divergence rates of 2.1 and 5.2% at the nucleotide and amino acid levels, respectively, with four major amino acid substitutions (Fig. 7). Additionally, Rev in the Chinese and reference strains exhibits an average difference of 20.6–23.0% at the nucleotide level and 29.0–34.2% at the amino acid level. The rev gene in EIAVLN40 is 498 bp in length, whereas that in EIAVFDDV13 is 495 bp in length, with a deletion at positions 64–66. The differences in the rev gene in these two strains are 2.2 and 5.5% at the nucleotide and amino acid levels, respectively, with eight major amino acid substitutions.

Comparison of the deduced S2 amino acid sequences of various EIAV strains. Points indicate identity with EIAVLN40 at that position and dashes indicate a gap. LN EIAVLN40, FDDV EIAVFDDV13, UK EIAVUK
Discussion
The observed genetic divergence between the Chinese EIAV vaccine and virulent strains and the American and Japanese strains is much larger than that among HIV-1 clades, which can be as high as 15%. Differences in amino acid sequence between genes from the high-virulence Chinese strains and those from the attenuated Chinese vaccine strains can also be >5%. Study of the differences in the nucleotide sequences, the protein primary sequences, and the three-dimensional conformations of specific proteins of these EIAV strains and determination of the relationship of these differences to the biology of these viruses will facilitate understanding of the molecular bases of viral virulence and immunoinductivity of the virus. For example, the Shao Laboratory has modified the Env protein of HIV-1 according to the predicted 2D structural characteristics of gp90 from the Chinese attenuated EIAV vaccine. The neutralizing titer and spectrum of antibodies induced in mice by this modified protein are significantly higher than those of the native unmodified protein (Yiming Shao, personal communication). Therefore, stable mutations in genes and their encoded proteins relevant to the virulence and immunogenicity of the Chinese attenuated EIAV strain have provided a useful model for study of the virulence and the induction of immunoprotection for EIAV and lentiviruses.
The natural target cells of EIAV are macrophages. The LTR regions affect both cellular tropism and virulence of these viruses. LTR regions are conserved during the persistent infection stage. However, in cultured cells, the enhancer region of LTR undergoes a high frequency of nucleotide substitutions, insertions, and deletions [9, 26–28]. These mutations cause changes in the transcription factor binding sites in LTRs and, subsequently, lead to alterations in the cell tropism and virulence of the virus [10]. The analysis of LTR in our study shows that the fibroblast-tropic strains have more AP-1 binding sites in the U3 region than their parental strains. AP-1 controls a number of cellular processes, including differentiation, proliferation, and apoptosis. Similar to findings from other studies, the in vitro transcription activity of the in-vitro-adapted strain EIAVFDDV13 is significantly higher than that of the high-virulence strain EIAVLN40 (data not shown).
Gag protein is an important target of the immune response. During the acute phase of EIAV infection, the initial cytotoxic T lymphocyte (CTL) response contributes to the clearance of the primary viremia. Epitopes most frequently recognized by CTLs are mainly located on the MA and CA. MA is a major retroviral structural protein. Lentiviral MA may help carry the viral genome into the nucleus after infection [15, 16] It may function as a membrane attachment regulator to enhance viral attachment to the cell membrane, which, in turn, regulates viral replication efficiency. CA is the major core protein of EIAV and is also one of the most important for immunogenicity. It is the major antigen component used in commercial diagnosis. It can stabilize unintegrated proviral DNA during the early stage of infection and uncoating and also plays an important role in viral assembly and budding at late stages of infection. The p9 protein is now known to facilitate the release of budding viral particles from the host cell plasma membrane through a YPDL motif that functions as a late assembly domain (L domain). As CTL is considered to play a key role in sterile protection to lentiviral infection, and also because the gag is relatively conserved between the Chinese EIAV vaccine and virulent strains, the correlation of mutation sites with the difference in cell-mediated immune response is presumed, providing useful information about the enhanced immunogenicity of the vaccine strain.
Pol provides enzymes that are essential for viral replication. PR plays a key role during the early stage of the viral life cycle. RT has RNA-dependent DNA polymerase activity, DNA-dependent DNA polymerase activity, and RNase H activity. It can reverse transcribe viral RNA into viral cDNA. Its RNase H activity degrades the RNA strand in the RNA–DNA duplex during reverse transcription. The dUTPase can promote viral replication in non-dividing cells and is required for viral replication in macrophages. IN can catalyze the integration of the linear double-stranded DNA produced by reverse transcription from the viral RNA genome into the host chromosomal DNA. Pol is the most conserved protein found in the virus. This fact implicates the importance of the non-structure EIAV protein. Among these viral encoded enzymes, the dUTPases of non-primate lentiviruses such as EIAV, visna virus, caprine arthritis–encephalitis virus (CAEV), and feline immunodeficiency virus (FIV) play key roles in viral virulence and mutation frequency [29]. Mutational analysis of EIAV dUTPase clones has shown that it determines viral virulence [17].
The envelope glycoprotein is the major component responsible for the induction of immunoprotection in the host [2, 30]. Current studies have found that mutations in Env play an important role in persistent viral infection, especially in escaping from the established neutralizing antibody response. Slight changes in the Env amino acid sequence can cause variations in host antibody and cellular immune responses. In persistent infections of simian immunodeficiency virus (SIV), there is a temporal correlation between Env sequence changes and increased tolerance to a neutralizing antibody [31, 32]. Similar results have also been obtained from SIV/HIV chimeric virus (SHIV) studies [33, 34].
Env is a major factor affecting the immunization efficacy of lentiviral vaccine. Effective lentiviral vaccines must be able to induce the body to produce humoral and cellular immune responses against the conserved region of Env. Studies on the immunoprotective rate of S2 gene-deleted EIAV-attenuated vaccine (EIAVD9) by the Montelaro Laboratory have shown a negative correlation between envelope variations and the immunoprotective efficacy of vaccine [2]. Change in glycosylation sites caused by env gene mutation is a common feature of lentivirus. Glycosylation sites can shield immune epitopes, especially neutralizing epitopes. The surface glycoproteins are highly glycosylated, and the position and number of N-linked glycosylation sites are related to the biological characteristics of the virus. Studies on SIV showed that virulence was increased with the increase in number of N-linked glycosylation sites. The loss of glycosylation sites can expose a larger immunorecognition region that contains neutralizing epitopes, thereby stimulating production of neutralizing antibodies and producing a stronger and more sensitive immune response. Previous studies have shown that the neutralizing region outside PND is important for the ability of the virus to escape monitoring by the host immune system. Laryssa et al. has reported that the V3 and V4 regions have similar functions in allowing escape from neutralizing antibodies. In fact, mutations in the V4 region have more impact on tolerance to neutralization than those in the V3 region. It is easier for glycosylation sites in the V4 region to affect neutralizing epitopes in the V3 region [21]. Different variable regions not only cooperate but also affect each other functionally; thus, coordination and compatibility between the V3 and V4 regions are necessary for resistance to immunorecognition.
The TM of lentivirus plays important roles in viral replication, virus fusion, cytopathic effects, surface envelope glycoprotein internalization, immunogenesis, and pathogenesis. Studies on both EIAV and HIV-1 have reported TM truncation during in vitro passage [24, 35]. Studies on CT mutants have shown that the ability to replicate with a truncated CT is cell type and truncation length dependent. EIAV with truncated TM is more suited to growth in canine cell lines, such as Cf2TH cells [24]. Luciw et al. [36] found that SIV with truncations in the cytoplasmic domain of TM replicated in low copy number in infected monkeys, whereas animals infected with the virus carrying revertant mutations showed a relatively high level of viral load and evident SIV clinical symptoms. Studies by Jia et al. [37] demonstrated that the in vivo replication ability of EIAV with truncated TM was 10–1000-fold lower than that of the parental strain. These findings confirmed that an intact TM is an important factor for in vivo viral replication. Thus, we hypothesize that the TM truncation in EIAVFDDV13 is possibly an important reason for its low level of replication [13].
Lentivirus employs complex mechanisms to regulate viral gene expression at transcriptional and post-transcriptional levels. Mutations in viral regulatory elements can modify the replication level of viruses. Limited expression of viral structural genes is a common strategy employed by a virus to sustain persistent infections [38]. In this strategy, viral gene expression is down-regulated and replication is decreased to escape an effective immune response [39]. Many studies have proven that genetic variations in regulatory regions can alter viral gene expression level and viral virulence.
The Tat protein is an essential factor for viral replication. It binds to the functional TAR region in the LTR, increases the efficiency of viral gene transcription, and consequently plays an important role in the regulation of viral gene expression [40–42]. There are three predicted functional regions in the S2 protein, the nucleoporin motif, the SH3 domain binding motif, and the nuclear localization sequence. However, the detailed functions of the S2 protein have not yet been clarified. Previous studies have shown that S2 is localized in the cytoplasm of infected cells, where it probably facilitates the aggregation of Gag protein during the process of viral assembly. Studies have also indicated that S2 is not necessary for viral replication in vitro [43]. However, in vivo assays have found that the S2 gene is highly conserved in horses with persistent infection. The in vivo replication levels of the attenuated strain V26, which lost the S2 gene start codon during in vitro passage in macrophages, and the attenuated vaccine strain EIAVD9, which lacks the S2 gene, are significantly decreased. These results suggest that the S2 gene plays an important role in viral replication in vivo and in viral virulence. These two attenuated strains generate effective immunity against infection by their respective virulent parental strains, but cannot produce immunoprotection against heterologous viral strains [2, 44–46].
Conclusions
The present study compares Chinese EIAV strains with American and Japanese strains and demonstrates that the Chinese EIAV strains reside on an independent evolutionary branch with a whole-genome genetic distance of ~23%. The proviral genomes of the attenuated EIAV vaccine strain EIAVFDDV13 and its parental virulent strain EIAVLN40 both contain multiple stable mutations in the LTR and in various genes. The differences in LTR, gp90, tat, S2, and rev between these two genomes are >5%. These multiple-gene and multiple-site mutations in the vaccine strain greatly decrease the potential risk of reverse mutations in virulence, enhancing the safety of the vaccine.
Analysis of the genetic variations and conserved sequences, as well as of known gene functions, suggests that multiple mutations in multiple genes of the EIAV-attenuated vaccine strain determine the non-pathogenicity of this vaccine. We presume that mutations in LTR, S2, Rev, and the dUPTase regions of pol in EIAVFDDV13 resulted in the stable and low-level replication of this vaccine strain [47]. This reduced in vivo replication is the fundamental feature of the vaccine that enabled the induction of proper host immune responses. In addition, the stable amino acid substitutions in Env, MA, CA, and Pol proteins, especially those in the neutralizing domains of the V3 and V4 region of gp90 glycoprotein, are considered the molecular bases for the vaccine strain to induce broad and solid immune protection [11, 12]. The genomic comparison and analysis of the EIAV vaccine have provided valuable information on implications of specific gene fragments and sites that are responsible for virulence and immunogenicity. New methods, such as fragment substitutions between vaccine and virulent strains and site-directed mutagenesis, will be applied to further investigate the mechanism of the unique lentiviral vaccine on inducing protective immunity.
References
B. Gaschen, J. Taylor, K. Yusim, B. Foley, F. Gao, D. Lang, V. Novitsky, B. Haynes, B.H. Hahn, T. Bhattacharya, B. Korber, Science 296, 2354–2360 (2002)
J.K. Craigo, B. Zhang, S. Barnes, T.L. Tagmyer, S.J. Cook, C.J. Issel, R.C. Montelaro, Proc. Natl. Acad. Sci. USA 104, 15105–15110 (2007)
J.B. Henson, T.C. McGuire, Am. J. Clin. Pathol. 56, 306–313 (1971)
C.J. Issel, L. Coggins, J. Am. Vet. Med. Assoc. 174, 727–733 (1979)
R. Shen, Z. Wang, Development and Use of an Equine Infectious Anemia Donkey Leucocyte Attenuated Vaccine. EIAV: A National Review of Policies, Programs, and Future Objectives (American Quarter Horse Association, Amarillo, TX, 1985), pp. 135–148
W.C. Koff, P.R. Johnson, D.I. Watkins, D.R. Burton, J.D. Lifson, K.J. Hasenkrug, A.B. McDermott, A. Schultz, T.J. Zamb, R. Boyle, R.C. Desrosiers, Nat. Immunol. 7, 19–23 (2006)
F. Li, J.K. Craigo, L. Howe, J.D. Steckbeck, S. Cook, C. Issel, R.C. Montelaro, J. Virol. 77, 7244–7253 (2003)
J. Ma, C. Jiang, Y. Lin, X. Wang, L. Zhao, W. Xiang, Y. Shao, R. Shen, X. Kong, J. Zhou, Arch. Virol. 154, 867–873 (2009)
L. Wei, X. Fan, X. Lu, L. Zhao, W. Xiang, X. Zhang, F. Xue, Y. Shao, R. Shen, X. Wang, Virus Genes 38, 285–288 (2009)
T. Zhou, X.F. Yuan, S.H. Hou, Y.B. Tu, J.M. Peng, J.X. Wen, H.J. Qiu, D.L. Wu, H.C. Chen, X.J. Wang, G.Z. Tong, Virus Res. 128, 58–64 (2007)
T. Shen, H. Liang, X. Tong, X. Fan, X. He, Y. Ma, W. Xiang, R. Shen, X. Zhang, Y. Shao, Vaccine 24, 738–749 (2006)
H. Liang, X. He, R.X. Shen, T. Shen, X. Tong, Y. Ma, W.H. Xiang, X.Y. Zhang, Y.M. Shao, Arch. Virol. 151, 1387–1403 (2006)
S. Jiang, R. Song, S. Popov, S. Mirshahidi, R.M. Ruprecht, Vaccine 24, 6356–6365 (2006)
W.A. Malmquist, D. Barnett, C.S. Becvar, Arch. Gesamte Virusforsch. 42, 361–370 (1973)
M.I. Bukrinsky, S. Haggerty, M.P. Dempsey, N. Sharova, A. Adzhubel, L. Spitz, P. Lewis, D. Goldfarb, M. Emerman, M. Stevenson, Nature 365, 666–669 (1993)
M. Stevenson, Trends Cell Biol. 6, 9–15 (1996)
D.L. Lichtenstein, K.E. Rushlow, R.F. Cook, M.L. Raabe, C.J. Swardson, G.J. Kociba, C.J. Issel, R.C. Montelaro, J. Virol. 69, 2881–2888 (1995)
W.K. Steagall, M.D. Robek, S.T. Perry, F.J. Fuller, S.L. Payne, Virology 210, 302–313 (1995)
D.S. Threadgill, W.K. Steagall, M.T. Flaherty, F.J. Fuller, S.T. Perry, K.E. Rushlow, S.F. Le Grice, S.L. Payne, J. Virol. 67, 2592–2600 (1993)
C. Leroux, C.J. Issel, R.C. Montelaro, J. Virol. 71, 9627–9639 (1997)
L. Howe, C. Leroux, C.J. Issel, R.C. Montelaro, J. Virol. 76, 10588–10597 (2002)
N.R. Rice, L.E. Henderson, R.C. Sowder, T.D. Copeland, S. Oroszlan, J.F. Edwards, J. Virol. 64, 3770–3778 (1990)
R.L. Schiltz, D.S. Shih, S. Rasty, R.C. Montelaro, K.E. Rushlow, J. Virol. 66, 3455–3465 (1992)
N.R. Rice, A.S. Lequarre, J.W. Casey, S. Lahn, R.M. Stephens, J. Edwards, J. Virol. 63, 5194–5200 (1989)
C. Leroux, J.L. Cadore, R.C. Montelaro, Vet. Res. 35, 485–512 (2004)
S.L. Payne, K. La Celle, X.F. Pei, X.M. Qi, H. Shao, W.K. Steagall, S. Perry, F. Fuller, J. Gen. Virol. 80(Pt 3), 755–759 (1999)
D.L. Lichtenstein, J.K. Craigo, C. Leroux, K.E. Rushlow, R.F. Cook, S.J. Cook, C.J. Issel, R.C. Montelaro, Virology 263, 408–417 (1999)
W. Maury, S. Perryman, J.L. Oaks, B.K. Seid, T. Crawford, T. McGuire, S. Carpenter, J. Virol. 71, 4929–4937 (1997)
S.L. Payne, J.H. Elder, Curr. Protein Pept. Sci. 2, 381–388 (2001)
T.L. Tagmyer, J.K. Craigo, S.J. Cook, D.L. Even, C.J. Issel, R.C. Montelaro, J. Virol. 82, 4052–4063 (2008)
B. Chackerian, L.M. Rudensey, J. Overbaugh, J. Virol. 71, 7719–7727 (1997)
L.M. Rudensey, J.T. Kimata, E.M. Long, B. Chackerian, J. Overbaugh, J. Virol. 72, 209–217 (1998)
C. Cheng-Mayer, A. Brown, J. Harouse, P.A. Luciw, A.J. Mayer, J. Virol. 73, 5294–5300 (1999)
Z. Si, M. Cayabyab, J. Sodroski, J. Virol. 75, 4208–4218 (2001)
S.S. Chen, S.F. Lee, C.T. Wang, J. Virol. 75, 9925–9938 (2001)
P.A. Luciw, C.P. Mandell, S. Himathongkham, J. Li, T.A. Low, K.A. Schmidt, K.E. Shaw, C. Cheng-Mayer, Virology 263, 112–127 (1999)
C. Jiang, J. Ma, Xu. Gao, Y. Lin, L. Zhao, Y. Hua, D. Liu, J. Zhou, Prog. Biochem. Biophys. 37, 261–268 (2010)
M.B. Oldstone, Cell 56, 517–520 (1989)
M. Belshan, M.E. Harris, A.E. Shoemaker, T.J. Hope, S. Carpenter, J. Virol. 72, 4421–4426 (1998)
M. Carvalho, D. Derse, J. Virol. 65, 3468–3474 (1991)
D.W. Hoffman, S.W. White, Nucl. Acids Res. 23, 4058–4065 (1995)
D.W. Hoffman, R.A. Colvin, M.A. Garcia-Blanco, S.W. White, Biochemistry 32, 1096–1104 (1993)
F. Li, B.A. Puffer, R.C. Montelaro, J. Virol. 72, 8344–8348 (1998)
Y.H. Zheng, H. Sentsui, M. Sugita, T. Nakaya, M. Kishi, K. Hagiwara, Y. Inoshima, C. Ishihara, Y. Kono, J.L. Lu, K. Ikuta, Virology 266, 129–139 (2000)
J. Zielonka, I.G. Bravo, D. Marino, E. Conrad, M. Perkovic, M. Battenberg, K. Cichutek, C. Munk, J. Virol. 83, 7547–7559 (2009)
F. Li, C. Leroux, J.K. Craigo, S.J. Cook, C.J. Issel, R.C. Montelaro, J. Virol. 74, 573–579 (2000)
S.L. Payne, F.J. Fuller, Curr. HIV Res. 8, 66–72 (2010)
Acknowledgments
This study was funded by the National Special Found for Control and Treatment of Major Infectious Diseases (Grant 2008ZX1001-1010 to JZ), the National Natural Science Foundation of China (30771994 to JZ and 30901349 to YL), and the Research Foundations of the Department of Education and the Department of Health (11541119 and 2007-218 to QX), Heilongjiang Province, China.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Xu Qi and Xuefeng Wang have contributed equally to this study.
Rights and permissions
About this article
Cite this article
Qi, X., Wang, X., Wang, S. et al. Genomic analysis of an effective lentiviral vaccine-attenuated equine infectious anemia virus vaccine EIAVFDDV13 . Virus Genes 41, 86–98 (2010). https://doi.org/10.1007/s11262-010-0491-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11262-010-0491-6