
Abstract
Gene targeting is a critical tool for construction of disease models. However, the application of traditional homologous recombination-mediated gene knockout technology is limited by the absence of rapid frequency-guaranteed targeting methods. Although conventional small hairpin RNA (shRNA)-mediated gene silencing offers an alternative for gene targeting, its application is frequently compromised by lower expression efficiency via RNA interference compared to gene knockout. Here we provide an efficient gene targeting strategy involving drug-inducible synergistic silencing with multiple shRNA molecules. On induction, the levels of the target proteins decreased to undetectable levels in all the tested stable transgenic mammalian cell lines, including HEK293 and embryonic stem cell-derived progenies carrying shRNA silencing cassettes. In a transgenic mouse model carrying a silencing cassette targeting the rhodopsin gene, short-time inducer treatment was sufficient to ablate the rhodopsin protein in the retina, resulting in similar retinal phenotypic changes as those observed in rhodopsin mutant mice. Therefore, on a broad basis, this inducible shRNA gene targeting strategy offers a true gene knockout alternative comparable to conventional RNA interference approaches.
Similar content being viewed by others


Introduction
Gene targeting is a key tool for construction of disease models, and it plays a critical role in diverse fields of biological and medical studies, including gene function analysis, gene modification, and drug development. Of note, traditional homologous recombination-mediated gene knockout technology is limited by the absence of rapid frequency-guaranteed targeting methods (Santiago et al. 2008). Recently, zinc-finger nuclease-mediated gene editing or transcription activator-like effector nucleases (TALEN) and Cas9 RNA-guide endonuclease technology appears to be receiving increasing attention for gene manipulation; however, targeted disruption of lethal genes remains an unsolved problem (Cho et al. 2013; Gaj et al. 2012, 2013; Sung et al. 2013; Urnov et al. 2005). The field of developmental biology has greatly benefited from the use of an evolving set of genetic tools. Utilizing conditional, recombination-based strategies, genes can be deleted in a specific cell type and in some instances with temporal control. Although conditional gene inactivation theoretically appears to be an excellent strategy for gene deletion in specific cell types, the maintenance of animal models involves much more labor than that of straight gene knockout models. Although the utilization of interference (RNAi) for targeted gene silencing in mammalian cells has become a benchmark technology, related strategies remain to be developed for higher gene silencing efficiency (Lambeth and Smith 2013). To modify gene inactivation methods, we propose an efficient gene strategy that involves drug-inducible synergistic silencing with multiple small hairpin RNA (shRNA) molecules.
Materials and methods
Constructs
ShRNA silencing vectors were generated using standard cloning procedures. Plasmid DNA was prepared using a QIAprep Spin Miniprep Kit (Qiagen). We constructed the backbone vector pPolyshRNA by replacing the Tet-U6-MCS region of the pSingle-MCS-shRNA vector (Clontech Laboratories, Inc.) with an shRNA silencing cassette (PDRE1–XhoI–shRNA1–AgeI–PDRE2–SalI–shRNA2–MluI–PDRE3–PstI–shRNA3–HindIII), allowing three shRNA subcassettes to be synergistically transcribed under tight control of doxycycline-responsive promoters (PDRE1, PDRE2, and PDRE3). Oligonucleotides matching the sequences of the designed shRNAs were synthesized, annealed, and sequentially subcloned into the pPolyshRNA vector. The sequences of PDRE1, PDRE2, and PDRE3 and the oligonucleotides matching the designed shRNA sequences targeting the genes of interest are listed in supplementary materials (Supplementary Tables S1–6).
Cell culture
HEK293 cells and mouse embryonic stem (ES) cells (129/Sv line) were used to generate stable transgenic cell lines. HEK293 cells were cultured in Dulbecco’sModified Eagle Medium (DMEM) supplemented with 10 % fetal bovine serum (FBS). To generate stable transgenic cell lines targeting the SIRT1 gene, HEK293 cells were transfected with shRNA vectors targeting the SIRT1 gene. Stable transgenic cell lines were selected by supplementing with 400 µg/ml G418. ES cells were cultivated on irradiated mouse embryonic feeder (MEFs) in DMEM containing 15 % FBS, leukemia-inhibiting factor (LIF), penicillin/streptomycin, l-glutamine, and non-essential amino acids. Trypsinization (0.25 % trypsin, 1 mM EDTA, 37 °C, 3–5 min) was used to dissociate cell clumps into single cells; these were then transduced with a 40-µg DNA vector by electroporation (250 V, 500 µF). After 48 h, G418 (400 µg/ml) was added to select stable transgenic cells, and culture was continued for 7–10 days. qRT-PCR, immunoblotting, and immunofluorescence staining were performed after at least 7–10 days of doxycycline treatment. Stepwise-differentiation of mouse ES cells into photoreceptor cells was performed as previously described (Osakada et al. 2008). Gene expression was analyzed by qRT-PCR, immunoblotting, and immunofluorescence staining after 7-day doxycycline treatment.
Animals
Wild-type (WT) mice carrying a mixed genetic background of C57BL/6 and 129/Sv strains were used as positive controls. ES cell clones integrated with shRNA silencing alleles were used to microinject C57BL/6 blastocysts. The resultant chimeras were crossed with CB57BL/6 to generate heterozygotes. Genotyping was performed using genomic PCR. Primers (N5: 5′-gctgctctgatgccgccgtgttc; N3: 5′-gatgtttcgcttggtggtcgaatg) matching a partial sequence of the neomycin-resistance gene were used to amplify transgenic alleles. Newborn mice were intraperitoneally administered with or without 2 µg doxycycline per gram body weight every day until they were sacrificed (about 2 months). qRT-PCR, immunoblotting, and immunofluorescence staining were used to evaluate silencing efficiency after doxycycline treatment. Mouse eyes were enucleated and eye cups were subjected to morphological analysis. Retinas were subjected to protein analysis. All experiments involving animals were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Biological Research Ethics Committee, Chinese Academy of Sciences.
Quantitative real-time PCR
Total RNA was isolated and reverse transcription was performed as described, and quantitative real-time PCR (qRT-PCR) was performed to evaluate the mRNA levels of genes of interest (Chen et al. 2013). qRT-PCR was performed using Platinum SYBR Green qPCR SuperMixUDG with Rox (Invitrogen). mRNA levels of actin were determined as internal controls. Corresponding oligonucleotides for determining the mRNA levels of SIRT1, rhodopsin and actin are provided in supplementary materials (Supplementary Table S7).
Immunofluorescence analysis and immunoblotting
Immunoblotting and immunofluorescence staining were performed to determine the levels and distributions of target proteins. Frozen sections were cut after fixation with 4 % formaldehyde at room temperature for 10 min. Paraffin sections were prepared for hematoxylin and eosin (HE) staining to evaluate the morphological changes in transgenic mouse retinas. The antibodies and working dilutions used for immunofluorescence analysis and immunoblotting were as follows: rabbit anti-SIRT1 (1:500–2,000; #1104-1, Epitomics), anti-rhodopsin (1:500–2,000; #sc-57433, Santa Cruz), rabbit anti-GNAT1 (1:200–1,000; # sc-389, Santa Cruz), goat anti-CNGA1 (1:500–1,000; #sc-13694, Santa Cruz), goat anti-PDC (1:500–1,000; #sc-18413, Santa Cruz), anti-PDE6b (1:200–1,000; #sc-30717, Santa Cruz), anti-actin (1:2,000–10,000; #A5228, Sigma-Aldrich). Corresponding IgG antibodies conjugated with Alexa Fluor® dye (594 or 488; Invitrogen) were used as secondary antibodies for the immunofluorescence analysis. In immunoblotting, anti-mouse IgG peroxidase conjugate (1:50,000; #a2304, Sigma-Aldrich), goat anti-rabbit IgG peroxidase conjugate (1:50,000; #a9169, Sigma-Aldrich), and rabbit anti-goat IgG peroxidase conjugate (1:80,000; #a5420, Sigma-Aldrich) were used as probes for the proteins of interest.
Result
Targeting scheme
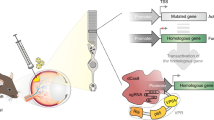
Tetracycline-responsible elements (TREs) or their modified doxycycline-responsive elements (DREs) have been widely applied in inducible expression systems (Bockamp et al. 2007; Freundlieb et al. 1999; Henriksen et al. 2007; Lai et al. 2004; Lamartina et al. 2003; Zhu et al. 2002). Tetracycline-controlled transcriptional silencer (tTS), a fusion protein composed of the tet repressor and the KRAB-AB domain of the kid-1 transcriptional repressor, is known to tightly bind with the RNA polymerase-binding site in the absence of doxycycline, resulting in very low basal expression of directed silencing subcassettes (Zhu et al. 2001). To develop an efficient inducible silencing system, we utilized the tTS silencer in this study, whose expression was constitutively driven by the CMV promoter. We also aligned multiple silencing subcassettes in a single vector with each subcassette controlled by its own independent PDRE. To minimize the potential reciprocal interference of the RNA polymerase-binding domains, we designed the promoters (PDRE1, PDRE2, and PDRE3) by hybridizing a doxycycline-responsive element and U6 promoter with a spacer (100–200 nucleotides) inserted between independent silencing subcassettes, resulting in a backbone vector (pPolyshRNA) (Fig. 1). Thus, this vector allowed multiple shRNA molecules to be synergistically transcribed on induction by doxycycline.

Gene silencing design: Element arrangement for the silencing vector pPolyshRNA. The three shRNA silencing subcassettes are boxed as indicated. Given that the vector carries a neomycin-resistance allele, stable cell lines that integrated the three silencing cassettes could be selected by G418. The tTS regulator is constitutively expressed under the control of the CMV promoter. tTS tetracycline-controlled transcriptional silencer, CMV cytomegalovirus
Gene silencing in mammalian cell lines
To test the silencing efficiency of this strategy, we targeted the human SIRT1 gene (GenBank accession number, NM_012238.4) with three different shRNA subcassettes (SIRT1–shRNA1, SIRT1–shRNA2, and SIRT1–shRNA3) assembled in the pPolyshRNA vector (Fig. 2a) (Supplementary Table S2). To examine the silencing efficiency of each shRNA, we also constructed vectors for each individual shRNA in the absence of the other two shRNA molecules (Fig. 2b–d). qRT-PCR revealed that the gene silencing efficiency ranged from 88 to 93 % for each individual shRNA subcassette in stable transgenic HEK293 cell lines after doxycycline treatment. More strikingly, the silencing efficiency was up to 98 % with all the three shRNA subcassettes placed in a single vector, indicative of the obvious additive effect of these shRNA subcassettes (Fig. 2e). Immunobotting showed that 10–30 % SIRT1 was expressed in the presence of any one of these shRNA subcassettes alone, whereas it was below the detectable level in the presence of all three shRNA subcassettes in a single vector (Fig. 2f), indicating that the residual traceable amount of SIRT1 mRNA might not serve as an effective template for SIRT1 protein production. These observations suggested that the shRNA molecules probably not only targeted SIRT1 mRNA to degradative pathways but also blocked the translation process by binding to the residual SIRT1 mRNA molecules.

Silencing of SIRT1 expression in HEK293 cells. a Targeting schemes for SIRT1 gene silencing with three shRNAs (SIRT1–shRNA1, SIRT1–shRNA2, and SIRT1–shRNA3). b-d Targeting schemes for the vectors harboring individual shRNA subcassettes (SIRT1–shRNA1, SIRT1–shRNA2, or SIRT1–shRNA3). e SIRT1 mRNA levels. f SIRT1 protein levels. HEK–shRNA123, HEK–shRNA1, HEK–shRNA2, and HEK–shRNA3, were integrated with the DNA vectors harboring the silencing subcassettes as indicated in a–d, respectively. Cells were analyzed after 1-week doxycycline treatment. Normal HEK293 cells were included as control. Values are mean ± SEM (n = 3, P < 0.01)
Furthermore, we examined the silencing efficiency of each individual shRNA subcassette by increasing its copy number in a single vector. We constructed three vectors by placing each identical shRNA subcassette under the control of all the silencing subcassettes in the pPolyshRNA vector (Fig. S1a-c). qRT-PCR and immunoblotting showed that silencing efficiency was about 90 % (Fig. S1d,e).
Gene silencing in photoreceptor cells differentiated from embryonic stem cells
To further test the general applicability of the present strategy, we also performed similar silencing experiments for cell- or tissue-specific genes in the progenies of ES cells. We targeted the murine rhodopsin gene (GenBank accession number, NM_145383) using a similar shRNA silencing cassette (Fig. 3a). Rhodopsin is a major rod photoreceptor-specific component critical for both phototransduction and outer segment (OS) disk morphogenesis in the retina (Lem et al. 1999; Palczewski 2006). We randomly selected three G418-resistant stable ES cell lines (ES1, ES2 and ES3) for further differentiation to corresponding photoreceptor cells (RES1, RES2 and RES3). qRT-PCR showed that rhodopsin mRNA levels were reduced by more than 94 % in all the photoreceptor cells differentiated from the transgenic ES cell lines after doxycycline treatment (Fig. 3b). Western-blotting assay showed that the differentiated photoreceptor cells expressed multiple retina-specific markers including RHO, GNAT1, PDC, PDE6b, CNGA1, GRK1, and RD12, while these markers were not detectable in the mES cell lines (Fig. 3c). Immunofluorescence analysis further demonstrated that the rhodopsin protein was not detectable in any of the tested cells (Fig. 3d).

Silencing of rhodopsin expression in photoreceptor cells derived from embryonic stem cells. a Targeting scheme. b Rhodopsin mRNA levels in three clusters of cells derived from ES cells. c Interfered expression of the rhodopsin gene in the transgenic retina. Three G418-resistent stable ES cell lines (ES1, ES2 and ES3) carrying the rhodopsin shRNA silencing alleles and their corresponding progeny cultures (RES1, RES2 and RES3) were used in this study. WT mouse ES cells (ES) and their progenies (RES) were included as controls. Values are mean ± SEM (n = 3, P < 0.01); scale bar 10 µm. d Rhodopsin protein levels detected by immunofluorescence. RHO, rhodopsin; ES, embryonic stem cells; ES1–ES3, the randomly selected three G418-resistant stable ES cell lines which could express shRNA targeting Rhodopsin; RES, photoreceptor cells derived from ES; RES1–RES3, photoreceptor cells derived from ES1–ES3, respectively
Gene silencing in transgenic animals
To investigate whether this silencing strategy was also effective in transgenic animals, we generated a rhodopsin silencing mouse model (RhoKD) by using ES1, one of the above transgenic mES cell lines. We found that the transgenic retina (RhoKD) was normally developed with a normal rhodopsin expression level and normal retinal morphology in the absence of doxycycline. On immunoblotting, the rhodopsin protein was not detectable, whereas the expression levels of multiple retina-specific markers (GNAT1, PDC, PDE6b, CNGA1, GRK1, and RD12) were minimally affected prior to obvious loss of photoreceptor cells, suggesting that the silencing effect was highly specific to the rhodopsin gene (Fig. 4a). Using immunofluorescence staining, we found that the rhodopsin protein was highly concentrated in the OS disks of the retinal rod photoreceptor cells of the transgenic mice in the absence of doxycycline. Thus, there was no substantial difference between WT and transgenic retinas in terms of rhodopsin abundance and distribution under normal conditions. However, the rhodopsin protein was not detectable after 2-week doxycycline treatment (Fig. 4a, b). As doxycycline treatment proceeded, photoreceptor degeneration became increasingly severe. Morphological analysis revealed that doxycycline did not cause abnormalities in either photoreceptor number or structure (Fig. 4c, first and second panels). The photoreceptor numbers in the transgenic retina remained comparable to those in the WT control in the absence of doxycycline (Fig. 4c, the third panel) or after the 2-week doxycycline treatment (Fig. 4c, the fourth panel), whereas the OS length was slightly shortened after the 2-week doxycycline treatment (Fig. 4c, the fourth panel). However, after 1-month doxycycline treatment, the OS length was significantly shortened even though a remarkable proportion of photoreceptor cells (>¾) remained (Fig. 4c, fifth panel). After a 2-month doxycycline treatment, less than two rows of photoreceptor cells remained in the transgenic retinas whereas the OS length was negligible (Fig. 4c, sixth panel). These morphological changes were very similar to those reported for rhodopsin knockout and dominant-negative mutant mice (Chadderton et al. 2009; Frederick et al. 2001; Li et al. 1998; Naash et al. 1993; Olsson et al. 1992).

Silencing effects of the rhodopsin gene in mouse transgenic retinas. a Determination of levels of rhodopsin (RHO), GNAT1, PDC, PDE6b, CNGA1, GRK1, RD12, and mouse actin (m-actin) in the transgenic retina (RhoKD) by immunoblotting. Retinal proteins were extracted for immunoblotting after a 2-week doxycycline treatment. b Rhodopsin (green color in the OS layers) levels in the transgenic retinas (RhoKD) revealed by immunofluorescence. c Retinal morphology shown by HE staining. Mice were administered with or without doxycycline for periods indicated at the bottom of the corresponding panels (2 weeks, 1 and 2 months). Retinas were taken from mice at 2 months of age. Age-matched WT retinas were included as controls. Scale bar 20 µm. OS, outer segment; IS, inner segment; ONL, outer nuclear layer; INL, inner nuclear layer; GC, ganglion cell layer; RHO, rhodopsin; GNAT1, alpha-transducing 1; PDC, phosducin; PED6b, phosphodiesterase 6b; CNGA1, cyclic nucleotide-gated channel alpha-1; GRK1, G protein-coupled receptor kinase 1; RD12, retinal pigment epithelium 65. (Color figure online)
Discussion
Gene targeting is a powerful tool for gene function analysis and mechanistic understanding of disease conditions. However, the less than 1,000-fold lower frequency of the targeted homologous recombination event relative to random integration can necessitate screening of thousands of clones to identify a correct recombinant. To date, strategies including marker selection, promoter-trap, and viral delivery have been widely applied to boost efficiencies; however, these approaches are not always successful in achieving high-efficiency targeting.
Recently, homologous recombination-mediated disruption of the occludin gene locus in mouse spermatogonial stem cells has been shown to reach high efficiency (Kanatsu-Shinohara et al. 2006). Targeting approaches using engineered zinc-finger nucleases or TALEN technologies have also been reported to achieve biallelic disruption of mammalian genes at higher frequencies in cell lines or animals (Boch et al. 2009; Miller et al. 2011; Moscou and Bogdanove 2009; Urnov et al. 2010; Wood et al. 2011). Recently, the type II bacterial CRISPR/Cas system has been shown to be an efficient gene-targeting technology with the potential for multiplexed genome editing (Cong et al. 2013; Garneau et al. 2010; Wang et al. 2013). However, it remains to be elucidated whether these approaches are widely applicable to many other loci or species or drug-controlled targeting methods.
While the emergence of RNAi technology has led to a new era in gene targeting, most successful cases of conventional RNAi approaches are still limited to transfected cells transiently targeting the genes of interest. Our data indicated that, the silencing efficiency of an increment in both shRNA diversity, and its copy number was more effective than each individual shRNA subcassette, and the copy number increased alone shRNA construct. This observation supports the notion that different shRNA molecules might target different pathways to silence gene expression, whereas the silencing effect of each individual shRNA subcassette could be saturated. Therefore, our results suggest that an effective silencing strategy should combine a copy number increment with diverse shRNA molecules. The modified strategy proposed here presents a remarkable silencing efficiency (close to 100 % at the protein level) through synergistic interference of multiple shRNAs whose expressions are tightly controlled by engineered PDREs. The non-detectable levels of target proteins in all tested mammalian systems, including HEK293 cells, ES cell-derived photoreceptor cells, and the transgenic mouse model, suggest the wide applicability of our modified strategy.
One disadvantage of RNAi strategies is the potential effect of off-target silencing, while this problem can be solved by designing ShRNA silencing subcassettes matching the 5′UTR or 3′UTR of target genes in combination with phenotype reversal with their coding regions. Overall, this strategy not only achieves an ideal silencing efficiency compared to conventional RNAi approaches but also saves considerable time and labor compared to the traditional gene knockout strategy. Therefore, this synergistic shRNA silencing system may serve as a truly powerful gene targeting alternative by avoiding laborious screening of thousands of clones required by traditional knockout strategies.
References
Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U (2009) Breaking the code of DNA binding specificity of TAL-type III effectors. Science 326:1509–1512
Bockamp E, Christel C, Hameyer D, Khobta A, Maringer M, Reis M, Heck R, Cabezas-Wallscheid N, Epe B, Oesch-Bartlomowicz B et al (2007) Generation and characterization of tTS-H4: a novel transcriptional repressor that is compatible with the reverse tetracycline-controlled TET-ON system. J Gene Med 9:308–318
Chadderton N, Millington-Ward S, Palfi A, O’Reilly M, Tuohy G, Humphries MM, Li T, Humphries P, Kenna PF, Farrar GJ (2009) Improved retinal function in a mouse model of dominant retinitis pigmentosa following AAV-delivered gene therapy. Mol Ther 17:593–599
Chen GF, Shi XJ, Sun C, Li M, Zhou Q, Zhang C, Huang J, Qiu Y, Wen XY, Zhang Y et al (2013) VEGF-mediated proliferation of human adipose tissue-derived stem cells. PloS One 8:e73673
Cho SW, Kim S, Kim JM, Kim JS (2013) Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31:230–232
Cong L, Ran FA, Cox D, Lin SL, Barretto R, Habib N, Hsu PD, Wu XB, Jiang WY, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339:819–823
Frederick JM, Krasnoperova NV, Hoffmann K, Church-Kopish J, Ruther K, Howes K, Lem J, Baehr W (2001) Mutant rhodopsin transgene expression on a null background. Invest Ophthalmol Vis Sci 42:826–833
Freundlieb S, Schirra-Muller C, Bujard H (1999) A tetracycline controlled activation/repression system with increased potential for gene transfer into mammalian cells. J Gene Med 1:4–12
Gaj T, Guo J, Kato Y, Sirk SJ, Barbas CF 3rd (2012) Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods 9:805–807
Gaj T, Gersbach CA, Barbas CF (2013) ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol 31:397–405
Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, Moineau S (2010) The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468(7320):67–71
Henriksen JR, Lokke C, Hammero M, Geerts D, Versteeg R, Flaegstad T, Einvik C (2007) Comparison of RNAi efficiency mediated by tetracycline-responsive H1 and U6 promoter variants in mammalian cell lines. Nucleic Acids Res 35:e67
Kanatsu-Shinohara M, Ikawa M, Takehashi M, Ogonuki N, Miki H, Inoue K, Kazuki Y, Lee J, Toyokuni S, Oshimura M et al (2006) Production of knockout mice by random or targeted mutagenesis in spermatogonial stem cells. Proc Natl Acad Sci USA 103:8018–8023
Lai JF, Cheng HY, Cheng TL, Lin YY, Chen LC, Lin MT, Jou TS (2004) Doxycycline- and tetracycline-regulated transcriptional silencer enhance the expression level and transactivating performance of rtTA. J Gene Med 6:1403–1413
Lamartina S, Silvi L, Roscilli G, Casimiro D, Simon AJ, Davies ME, Shiver JW, Rinaudo CD, Zampaglione I, Fattori E et al (2003) Construction of an rtTA2(s)-M2/tTS(kid)-based transcription regulatory switch that displays no basal activity, good inducibility, and high responsiveness to doxycycline in mice and non-human primates. Mol Ther 7:271–280
Lambeth LS, Smith CA (2013) Short hairpin RNA-mediated gene silencing. Methods Mol Biol 942:205–232
Lem J, Krasnoperova NV, Calvert PD, Kosaras B, Cameron DA, Nicolo I, Makino CL, Sidman RL (1999) Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci USA 96:736–741
Li TS, Sandberg MA, Pawlyk BS, Rosner B, Hayes KC, Dryja TP, Berson EL (1998) Effect of vitamin A supplementation on rhodopsin mutants threonine-17 → methionine and proline-347 → serine in transgenic mice and in cell cultures. Proc Natl Acad Sci USA 95:11933–11938
Miller JC, Tan SY, Qiao GJ, Barlow KA, Wang JB, Xia DF, Meng XD, Paschon DE, Leung E, Hinkley SJ et al (2011) A TALE nuclease architecture for efficient genome editing. Nat Biotechnol 29:143–149
Moscou MJ, Bogdanove AJ (2009) A simple cipher governs DNA recognition by TAL effectors. Science 326:1501
Naash MI, Hollyfield JG, al-Ubaidi MR, Baehr W (1993) Simulation of human autosomal dominant retinitis pigmentosa in transgenic mice expressing a mutated murine opsin gene. Proc Natl Acad Sci USA 90:5499–5503
Olsson JE, Gordon JW, Pawlyk BS, Roof D, Hayes A, Molday RS, Mukai S, Cowley GS, Berson EL, Dryja TP (1992) Transgenic mice with a rhodopsin mutation (Pro23His): a mouse model of autosomal dominant retinitis pigmentosa. Neuron 9:815–830
Osakada F, Ikeda H, Mandai M, Wataya T, Watanabe K, Yoshimura N, Akaike A, Sasai Y, Takahashi M (2008) Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat Biotechnol 26:215–224
Palczewski K (2006) G protein-coupled receptor rhodopsin. Annu Rev Biochem 75:743–767
Santiago Y, Chan E, Liu PQ, Orlando S, Zhang L, Urnov FD, Holmes MC, Guschin D, Waite A, Miller JC et al (2008) Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc Natl Acad Sci USA 105:5809–5814
Sung YH, Baek IJ, Kim DH, Jeon J, Lee J, Lee K, Jeong D, Kim JS, Lee HW (2013) Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol 31:23–24
Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC (2005) Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435:646–651
Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD (2010) Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11:636–646
Wang HY, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R (2013) One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153:910–918
Wood AJ, Lo TW, Zeitler B, Pickle CS, Ralston EJ, Lee AH, Amora R, Miller JC, Leung E, Meng XD et al (2011) Targeted genome editing across species using ZFNs and TALENs. Science 333:307
Zhu Z, Ma B, Homer RJ, Zheng T, Elias JA (2001) Use of the tetracycline-controlled transcriptional silencer (tTS) to eliminate transgene leak in inducible overexpression transgenic mice. J Biol Chem 276:25222–25229
Zhu Z, Zheng T, Lee CG, Homer RJ, Elias JA (2002) Tetracycline-controlled transcriptional regulation systems: advances and application in transgenic animal modeling. Semin Cell Dev Biol 13:121–128
Acknowledgments
This work was supported by the Ministry of Science and Technology of China (2010CB945600, 2011CB965100), the National Natural Science Foundation of China (81271498, 81202550, 81100673), the Shanghai Science Foundation (#11PJ1407800), the Ministry of Education of China (IRT1168).
Conflict of interest
The authors declare they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ming Ying and Guangfeng Chen have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
11248_2014_9841_MOESM1_ESM.tif
Fig. S1 Silencing of SIRT1 expression in HEK293 cells by three identical shRNAs. a A silencing cassette for the vector harboring three SIRT1-shRNA1 subcassettes. b A silencing cassette for the vector harboring three SIRT1-shRNA2 subcassettes. c A silencing cassette for the vector harboring three SIRT1-shRNA3 subcassettes. d SIRT1 mRNA levels. e SIRT1 protein levels. Values are mean ± SEM (n = 3, P < 0.01) (TIFF 655 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Ying, M., Chen, G., Qiu, Y. et al. Drug-inducible synergistic gene silencing with multiple small hairpin RNA molecules for gene function study in animal model. Transgenic Res 24, 309–317 (2015). https://doi.org/10.1007/s11248-014-9841-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11248-014-9841-9