
Abstract
The interactions between hydrogen bond donors (dimethylamine (DMA) and methanol (MeOH)) and acceptors (formaldehyde dimethylhydrazone, acetaldehyde N,N-dimethylhydrazone and N-nitrosodimethylamine) were theoretically investigated by DFT. The hydrogen bonding interactions were found on several bonding sites of the acceptors. The important properties of structure, binding energy, enthalpy of formation, Gibbs free energy of formation and equilibrium constant were investigated. Compared to the monomer, the DMA complexes show a small red shift of the NH-stretching vibrational transition but a significantly intensity enhancement. On the other hand, the MeOH complexes have a large red shift but a relatively small intensity enhancement of the OH-stretching transition. Atoms-in-molecules analysis revealed that several types of hydrogen bond interaction were present in the complexes. Since natural bond orbital analysis overestimated the effect of charge transfer, the more reliable localized molecular orbital energy decomposition analysis was performed and it shows that the major contribution to the total interaction energy is the electrostatic interaction. All these parameters suggest that the hydrogen bond donor strength of MeOH is substantially greater than DMA.
Similar content being viewed by others

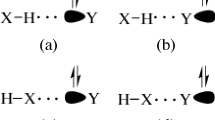
Introduction
Atmospheric aerosol plays an important role in Earth’s climate change and atmospheric chemistry [1, 2]. Nucleation, a fundamental step in atmospheric new particle formation (NPF), is a crucial source of atmospheric aerosols [1]. Numerous investigations have been conducted to study the gas-to-particle nucleation processes. However, little is known about the detailed molecular scale processes behind the observed NPF, due to lack of instruments to measure and characterize the molecular clusters at atmospheric conditions. Sulfuric acid has been accepted as a key molecule in atmospheric nucleation, and coupled with a stabilizing component such as ammonia, amines or organic compounds [3]. It is estimated that 104 − 105 different organic compounds have been identified in the atmosphere, but little is known about the direct involvement of organic compounds in NPF [4]. Water-soluble organic nitrogen compounds have been found to contribute about 18 % to the total mass of fine aerosol particles [5]. The contribution of N-containing compounds to nucleation needs to be studied.
Unsymmetrical 1,1-dimethylhydrazine (UDMH) is often used as fuels in bipropellant combinations in satellites and rockets. The oxidations of UDMH by molecular oxygen at or near room temperature in solutions of diethyl ether or cyclohexane could mainly give rise to formaldehyde N,N-dimethylhydrazone (FDMH, Fig. 1a). Water, N-nitrosodimethylamine (NDMA, Fig. 1b), nitrogen, methane, ammonia were also identified as products [6]. The degradation of UDMH was investigated using atmospheric oxygen and hydrogen peroxide in the presence and absence of copper catalyst and at varying pH. The production of NDMA occurred mostly at neutral and alkaline pH [7]. FDMH, formaldehyde hydrazone (FH), and tetramethyltetrazene (TMT) were also formed. The structure of acetaldehyde N,N-dimethylhydrazone (ADMH) is similar with FDMH. It is also one of the oxidation products during the UDMH storage process, and it was identified by the analysis of impurities and oxidization products with gas chromatography-mass spectrometer (GC–MS) [8]. Both FDMH and ADMH are hydrazones, containing two connected nitrogen atoms of different nature and a C=N double bond that is conjugated with a lone electron pair of the terminal N atom. They have a basic structure of R1R2C=NNR3R4 and formally derived from aldehydes or ketones by replacing =O by =NNH2 (or substituted analogues) [9]. Hydrazones and their derivatives have been characterized by the triatomic group C=N–N, and they are interesting substances because they play an important role in the treatments for biological activities such as antioxidant, anti-inflammatory, anticancer and anti-inflammatory [10–14]. NDMA has been received great attention, because it is a member of a family of extremely potent carcinogens. It has been found in several common food and environmental sources, such as drinking water wells near a rocket engine testing facility [15].

Molecular structures of hydrazone a R1 = H, formaldehyde N,N-dimethylhydrazone (FDMH); R1 = CH3, acetaldehyde N,N-dimethylhydrazone (ADMH) and b N-nitrosodimethylamine (NDMA)
Since hydrogen bonds are involved in the formation of aerosol particles, a number of donor and acceptor atoms have been studied experimentally and theoretically [16]. With the rapid development of computer technology, quantum chemical calculations have already become an essential tool to gain new insights into the very first steps of NPF. Hereinto, OH, NH and SH are some of the most common hydrogen bond donors, while O, N, S, π-electrons are some of the most typical acceptors [16]. Amines, such as dimethylamine (DMA), are good examples of strong hydrogen bond acceptors [17, 18]. Fourier transform infrared (FTIR) and DFT studies have been carried out for the DMA complexes, such as DMA–DMA and DMA–trimethylamine (TMA). The geometries of both DMA–DMA and DMA–TMA have a nonlinear N–H···N hydrogen bond. The IR spectrum of both DMA–DMA and DMA–TMA shows a characteristic small red shift of the NH-stretching vibrational band and a pronounced intensity increase (600–700 times) [19, 20]. Meanwhile, many investigations have been performed using methanol (MeOH) as a hydrogen bond donor [21–23]. A combination of vapor-phase IR spectroscopy and ab initio calculations has been performed for the MeOH–dimethyl sulfide (DMS) and MeOH–dimethyl ether (DME) complexes. The experimental red shifts of the OH-stretching fundamental transition were 103 and 113 cm−1 for MeOH–DME and MeOH–DMS, respectively. The calculated binding energies were −17.6 and −18.8 kJ mol−1 for MeOH–DMS and MeOH–DME, respectively. It was reported that sulfur is weaker than, but nearly equivalent to, oxygen as a hydrogen bond acceptor [24]. Hansen et al. [25] investigated the effect of fluorine substitution in alcohol–DMA complexes. The observed red shifts were 301, 296 and 460 cm−1 for MeOH–DMA, ethanol (EtOH)–DMA and 2,2,2-trifluoroethanol (TFE)–DMA, respectively. The replacement of an H atom by a methyl group has limited effect on the hydrogen bond interaction, but the replacement by a CF3 group has a significant effect.
Elm et al. has performed an assessment of binding energies of 107 atmospherically relevant clusters containing H2SO4, H2O, NH3 and DMA using DFT. Several different DFT functionals (B3LYP, CAM-B3LYP, M06-2X, PW91, LC-PW91, PBE0 and ωB97X-D) were evaluated against the benchmark high level explicitly correlated coupled cluster methods. They have observed that different methods yielded a large difference in the binding energies [26]. Recently, Bork et al. performed a computational benchmarking study of the Gibbs free binding energies in molecular complexes and clusters based on gas-phase FTIR spectroscopy. Among the tested MP2 and DFT methods, B3LYP-D3 was found to give the most accurate thermal correction terms [27]. We present a computational study of MeOH/DMA–hydrazone/NDMA for comparing the hydrogen bond strength of X–H···Y (X = O, N; Y = O, N). We have used DFT to determine the binding energy (BE), enthalpy of formation (ΔH θ298K ), Gibbs free energy of formation (ΔG θ298K ) and equilibrium constant (\(K_{\text{eq}}^{\text{calc}}\)) at 298 K. Natural bond orbital (NBO) analysis was used to demonstrate the existence of charge transfer (electron delocalization) between lone pair of the acceptor and antibonding orbital of the donor [28, 29]. Localized molecular orbital energy decomposition analysis (LMO-EDA) was performed to study the contribution of different components to the total interaction energy [30]. The atoms-in-molecules (AIM) analysis was applied to investigate the electronic densities and the intermolecular hydrogen bond interactions in the complexes [31, 32].
Computational methods
DFT geometry optimizations for both monomers and complexes were performed with Gaussian 09, using aug-cc-pVTZ basis set [33]. Four functionals were used as follows: B3LYP, M06-2X, ωB97X-D and B3LYP-D3. M06-2X is a hybrid meta exchange–correlation functional, which is parameterized only for nonmetals [34]. Both ωB97X-D and B3LYP-D3 are hybrid functionals, containing Grimme’s D2 and D3 empirical dispersion correction, respectively. The empirical dispersion correction has been shown to give an important contribution for general thermochemistry, kinetics and noncovalent interaction [35]. Several stable conformers for each complex were obtained, and frequency analysis was followed after all the calculations to ensure that the calculated structures were indeed structural minima. Both zero-point vibrational energy (ZPVE) and basis set superposition error (BSSE) were corrected for binding energies (BEs). Counterpoise (CP) method was used for the correction of BSSE [36], which has been shown to improve interaction energies of weakly bound complexes [37–40].
Atoms-in-molecules (AIM) theory as a powerful method is applied to provide an understanding of both covalent and noncovalent molecular interactions, including hydrogen bonding [31, 32]. It is based on topological analysis of the electron charge density (ρ). Electron density properties at bond critical point (BCP) are used for description of bond formation [41]. Two most important parameters in AIM theory are bond critical point charge density ρ(r) and the Laplacian of charge density ∇2 ρ(r), which is used for description of the strength and type of bond. Topological analysis of the charge density was carried out by utilizing the AIM2000 program package.
Natural bond orbital (NBO) analysis was used to understand and describe the donor–acceptor electron delocalization between occupied Lewis type and unoccupied non-Lewis orbitals correspond to a stabilizing donor–acceptor interaction [28, 29]. The energy of these interactions can be estimated by the second-order perturbation theory. The second-order perturbation energy E (2) i→j* is caused by electron density transfer from the lone pair orbital i to the antibonding orbital j*,
where φ (0) i and φ (0) j* are the zeroth-order wave functions of the lone pair orbital and the antibonding orbital, respectively. ɛ (0) i and ɛ (0) j* are the zeroth-order energies of the lone pair orbital and antibonding orbital, respectively. n (0) i is the occupancy in the lone pair orbital i, and \(\hat{F}_{\text{KS}}\) is the Kohn–Sham form of one-electron effective Hamiltonian. NBO analysis implemented in Gaussian 09 was used to compare the different hydrogen bond interactions in the complexes.
Besides, the total interaction energy of the complexes was decomposed into individual energy components at the B3LYP/aug-cc-pVTZ level of theory using the localized molecular orbital energy decomposition analysis (LMO-EDA), which is implemented in the GAMESS program [42]. The total interaction energy (INT) is divided into electrostatic energy (ES), exchange energy (EX), repulsion energy (REP), polarization energy (POL) and dispersion energy (DISP) [30]. The interaction energy in terms of component energy terms is expressed as:
The ES term represents the total Columbic interaction between the free monomer charge distributions. The EX term is the interaction caused by the exchange of electrons between the monomers satisfying Pauli’s principle, and this contribution accounts for the short-range repulsion due to overlap of the electron distribution of one monomer with that of another. The POL term denotes the polarization interaction. The DISP interaction is an attractive interaction between molecules and atoms. The REP interaction is the opposing attractive force between two molecules.
Results and discussion
Geometries and interaction energies
The ADMH monomer has two structures, Z- and E-isomers (supplementary material, Fig. S1). For the Z-isomer, the –Na(CH3)2 and –R1 groups are on the same side of the Nb=C double bond. For the E-isomer, the two groups are on the opposite side of the double bond. The relative energy difference (B3LYP-D3: 17.7 kJ mol−1) shows that the E-isomer is much more stable than the Z-isomer, which is in consistent with previous studies that the N,N-disubstituted hydrazones (e.g. dimethyl-) exist exclusively in the E-isomer in both solid state and liquid solution [43–45]. The Z/E isomerization was proposed through an in-plane inversion of the imine nitrogen atom rather than by using a rotational process round the C=N double bond [46, 47]. In the present study, we only focused on the E-isomer-related complexes.
The two nitrogen atoms (Fig. 1a: Na and Nb) of hydrazone are nucleophilic, so both of them can be treated as hydrogen bond acceptors. For NDMA, both Nd and O atoms are good hydrogen bond acceptors. In total, there are four bonding sites could be acted as the hydrogen bond acceptors (Fig. 1), i.e., Na, Nb, Nd, O. The Nc bonding site in NDMA is surrounded by two methyl groups as compared with the Nd and O bonding sites, which makes the hydrogen bond donors difficult to bond with it. Several other initial geometries with O–H···π and N–H···π interactions were also attempted. However, these structures converged to the lowest O–H···Nb/Nd and N–H···Nb/Nd structures.
We have optimized the geometries of the complexes and the monomers with four DFT methods. For each complex, several geometries were attempted with the four methods, only the lowest energy structures were discussed in this study, and the other conformational information was presented in the supplementary material (Fig. S2-S3). The optimized structures of the MeOH complexes and DMA complexes are shown in Figs. 2 and 3. The geometric parameters of the complexes optimized at the B3LYP-D3/aug-cc-pVTZ level are given in Table 1. The geometric parameters obtained with the other three methods were presented in the supplementary material (Table S1-S3). The changes in the OH bond length upon complexation vary from 0.008 to 0.016 Å; on the other hand, the changes in the NH bond length upon complexation are only 0.001–0.004 Å. The intermolecular hydrogen bond angles of MeOH complexes deviate within 20° from the ideal linear orientation. However, the angles deviate about 40° for the DMA complexes, which suggests that O–H···N/O hydrogen bond is much stronger than N–H···N/O. In previous study, the OH bond length of MeOH was calculated to lengthen substantially by approximately 0.013 Å upon complexation with TMA, which was identified as a strong hydrogen bond. The O–H···N angle was calculated to be nearly linear at approximately 179°, which is an optimal orientation for an intermolecular hydrogen bond [21]. This is close to the O–H···Nb hydrogen bonds in the present study.

Optimized structures of the MeOH complexes at the B3LYP-D3/aug-cc-pVTZ level of theory. a MeOH–FDMH b MeOH–ADMH c MeOH–NDMA d MeOH–FDMH e MeOH–ADMH f MeOH–NDMA

Optimized structures of the DMA complexes at the B3LYP-D3/aug-cc-pVTZ level of theory. a DMA–FDMH b DMA–ADMH c DMA–NDMA d DMA–FDMH e DMA–ADMH f DMA–NDMA
Based on the geometric parameters, the hydrogen bond strengths of MeOH–ADMH are slightly stronger than MeOH–FDMH on both Na and Nb sites. This indicates that the replacement of an H atom by a methyl group has limited effect on the hydrogen bond interaction. Meanwhile, the effect of =O replacement on the hydrogen bond strength in MeOH–hydrazone molecular complexes was investigated. When the –N=CH(CH3) and –N=CH2 are replaced by =O, the changes in the OH bond length upon complexation with Nd and the intermolecular hydrogen bond angles at Nd become much smaller. This implies that the –N=CH(CH3) and –N=CH2 groups are good electron withdrawing groups with high electronegativity, which makes the hydrogen bond much stronger than those in the NDMA complexes. The similar trend was found for the DMA complexes. The substitution effects were in accordance with several previous investigations [25, 48, 49]. Hansen et al. [25] investigated the effect of fluorine substitution in alcohol–DMA complexes. The obtained red shifts for MeOH–DMA, EtOH–DMA and TFE–DMA were 301, 296 and 460 cm−1, respectively. TFE molecule has a good electron withdrawing CF3 group with high electronegativity, which makes the hydrogen bond much stronger than those in the MeOH and EtOH complexes. Similar substitution effect was observed in alcohol–trimethylphosphine (TMP) complexes with O–H···P hydrogen bonds [48]. On the other hand, Schrems et al. have stepwisely introduced a fluorine substituent in the β-position of the alcohols each time, which causes a stepwise increase of the frequency shift of the OH-stretching vibrations in the O–H···O bonded complexes [49].
In Table 2, the calculated binding energy (BE), enthalpy of formation (ΔH θ298K ), Gibbs free energy of formation (ΔG θ298K ) and equilibrium constant (\(K_{\text{eq}}^{\text{calc}}\)) at 298 K at the B3LYP-D3/aug-cc-pVTZ level of theory are presented. In the study of hydrogen bonding in the carboxylic acid–aldehyde complexes, the B3LYP-D3 functional has been found to give reasonable interaction energies [50]. The results obtained with the other three methods were presented in the supplementary material (Table S4-S6). The enthalpy ΔH θ298K of hydrogen bond formation is directly related to the stabilization energy of a complex [51]. The enthalpies of hydrogen bond formation in the weaker DMA complexes have been determined to be −14.3 to −17.8 kJ mol−1, while the values are in the range from −21.3 to −29.9 kJ mol−1 for the MeOH complexes, which shows that the MeOH complexes are much more stable than the DMA complexes. The BEs were corrected with BSSE and ZPVE. In general, the BSSE and ZPVE values do not vary so much for all the studied complexes. For the MeOH complexes, the BEs are in the range of −22.0 ~ −30.3 kJ mol−1. In previous studies, the BE of O–H···N hydrogen bond was calculated to be −35 kJ mol−1 (QCISD/6-311++G(d,p)) for the MeOH–DMA complex [21]. The calculated BE of the O–H···O hydrogen bond was −15.5 kJ mol−1 (B3LYP/6-31+G*) for MeOH–MeOH [52]. For the MeOH–DME complex, the BE was calculated to be −14.0 kJ mol−1 (B3LYP/aug-cc-pVTZ) [24]. In the present study, the BE of O–H···O bonded complex was slightly larger (−23.3 kJ mol−1). For the N–H···N/O hydrogen bond, the BEs are greater than −20 kJ mol−1. In previous DFT (B3LYP/aug-cc-pVTZ) study, the N–H···N hydrogen bond has smaller binding energies: −9.8 and −10.6 kJ mol−1 for DMA–DMA and DMA–TMA, respectively [19].
For the O–H···N hydrogen bond system, the \(K_{\text{eq}}^{\text{calc}}\) values range from 1.7 × 10−1 to 4.3 × 10−2 whereas for the N–H···N hydrogen bond system, the \(K_{\text{eq}}^{\text{calc}}\) values are much smaller with values of 1.6 × 10−3 to 5.4 × 10−4. In summary, all these parameters suggest that the hydrogen bond donor strength of MeOH is substantially greater than DMA. For MeOH complexes, the observed geometric parameters and interaction energies indicate the following trend for the hydrogen bond acceptor strength: Nb ≫ Na > O > Nd. For DMA complexes, the differences for the geometric parameters and interaction energies are not significant, which is due to the weakness of the DMA donor.
Calculated OH- and NH-stretching vibrational frequencies
The red shift \(\left( {\varDelta \tilde{\nu }} \right)\) is the wavenumber difference between the free and hydrogen bonded XH-stretching (X = O, N) vibrational transitions \(\left( {\varDelta \tilde{\nu } = \tilde{\nu }_{\text{monomer}} - \tilde{\nu }_{\text{dimer}} } \right)\). It has been treated as a measurement of hydrogen bond strength. The calculated OH- and NH-stretching fundamental transition wavenumbers and the red shifts of the MeOH and DMA complexes are summarized in Table 3. In general, the red shifts of MeOH complexes (169–326 cm−1) are much larger than those of DMA complexes (2–69 cm−1), because the hydrogen bond strength of O–H···N/O is much stronger than that of N–H···N/O.
The red shifts of MeOH–ADMH are slightly larger than MeOH–FDMH on both Na and Nb sites. The same observation was found in their DMA complexes. This indicates that the replacement of an H atom by a methyl group has limited effect on the hydrogen bond interaction. This is perhaps expected as the additional CH3 group donates additional electron density to the N atom, which is reflected in the slightly larger red shifts of the MeOH/DMA complexes. When the –N=CH(CH3) and –N=CH2 are replaced by =O, the red shifts become much smaller.
The IR intensity may change significantly upon complexation, which has been considered to be the criterion for hydrogen bonding [53]. It is worth to calculate the increase of the XH-stretching transition intensity upon complex formation (I D/I M). The intensity ratio of O–H···N was calculated to be ~20–30 times stronger than that of the monomer. In comparison, the observed red shift of O–H···N in the much stronger complex formed between MeOH and TMA is 323 cm−1. The \(\tilde{\nu }_{\text{OH}}\) intensity of the complex was calculated to be nearly 70 times stronger than that for free MeOH [21]. In contrast, the increase of the OH-stretching transition intensity of the O–H···O hydrogen bond upon complex formation (I D/I M) was much lower, only about 16. For MeOH–DME, the calculated O–H···O hydrogen bond frequency is 3659 cm−1 (B3LYP/aug-cc-pVTZ) with a red shift of 170 cm−1 and an I D/I M ratio of 18 [24]. For MeOH–MeOH, the calculated O–H···O hydrogen bond frequency is 3613 cm−1 (B3LYP/6-31+G*) with a red shift of 150 cm−1 and an I D/I M ratio of 21 [52]. Moreover, the hydrogen bond acceptor ability of N, P, O and S was carried out for the PhOH–Et3Y and PhOH–Et2Y (Et = ethyl; Y = N, P, O and S) in n-heptane. The observed red shifts of the fundamental OH-stretching frequency were as follows: O–H···N (556 cm−1) > O–H···P (333 cm−1) > O–H···O (274 cm−1) > O–H···S (251 cm−1) [54]. A similar study was carried out for the PhOH–Bu3Y and PhOH–Bu2Y (Bu = butyl) complexes. The experimental red shifts were O–H···N (800 cm−1) > O–H···O (280 cm−1) > O–H···S (254 cm−1) > O–H···P (240 cm−1) [55]. Our results are in consistent with these studies: The O–H···N hydrogen bond strength is significantly stronger than that of the O–H···O hydrogen bonds.
On the other hand, the intensity of N–H···Na was calculated to be ~ 400 stronger than that of the monomer. In the DMA–DMA dimer, the NH-stretching transition intensity was calculated to be 363 times (B3LYP/aug-cc-pVTZ) stronger than that of the monomer, with a red shift of 82 cm−1 [20]. The intensity of the fundamental NH-stretching transition for the DMA–TMA complex was calculated to be ~700 times stronger than that of the monomer, with a red shift of 94 cm−1 (CCSD(T)-F12a/VDZ-F12). For the DMA–DMA complex, it is about 600 times stronger, with a red shift of 90 cm−1 [19]. The large intensity enhancement is due to the very small dipole moment derivative of DMA, which makes the fundamental NH-stretching transition very weak [56]. By comparison, the intensity of N–H···Nb/d was calculated to be ~80–170 times stronger than that of the monomer and only about 34 times for N–H···O. However, the intensity of the NH3–NH3 complex was only a factor of 9 larger than the corresponding transition of single ammonia molecule [57].
NBO, LMO-EDA and AIM analyses
The NBO analysis was performed using the B3LYP-D3/aug-cc-pVTZ wavefunctions, and the interacting donor–acceptor NBOs of the complexes are shown in Figs. 4, 5. The calculations of the second-order interaction energies E (2) i→j* between Lewis and non-Lewis orbitals, occupation numbers in the lone pair orbital (δ) and the antibonding orbital (δ(σ *)), the zeroth-order energy difference of the lone pair orbital and antibonding orbital (ɛ (0) j* − ɛ (0) i ), and the Kohn–Sham matrix element between the orbitals (\(\langle \varphi_{i}^{(0)} |\hat{F}_{\text{KS}} |\varphi_{j*}^{(0)} \rangle\)) are listed in Table 4.

The hydrogen bond donor NBO (left), acceptor NBO (middle) and interacting donor–acceptor NBOs (right) of the MeOH complexes. For MeOH–NDMA, both n p → σ *O–H (top) and n sp2 → σ *O–H (bottom) are shown. The isovalue of the NBO plots is ±0.02 a.u. a MeOH–FDMH, b MeOH–ADMH, c MeOH–NDMA, d MeOH–FDMH, e MeOH–ADMH, f MeOH–NDMA

The hydrogen bond donor NBO (left), acceptor NBO (middle) and interacting donor–acceptor NBOs (right) of the DMA complexes. For DMA–NDMA, both n p → σ *N–H (top) and n sp2 → σ *N–H (bottom) are shown. The isovalue of the NBO plots is ±0.02 a.u. a DMA–FDMH, b DMA–ADMH, c DMA–NDMA, d DMA–FDMH, e DMA–ADMH, f DMA–NDMA
The p-type lone pair (LP) orbital of the acceptor N atoms has a major overlap with OH/NH antibonding orbital. However, O atom has two LP orbitals; thus, the overlaps of the LP orbitals of O atom with the OH/NH antibonding orbital are divided into two types, the p-type and sp2-type LP orbitals, which is similar with the previous NBO analysis for the p-cresol–tetrahydrofuran (O–H···O hydrogen bond) and p-cresol–tetrahydrothiophene (O–H···S hydrogen bond) complexes [58]. Surprisingly, the N–H···O hydrogen bond in the DMA–NDMA complex is very weak and the interaction of n sp2 → σ * N–H is very small (Fig. 4f).
The increased natural charges on the H atom and the decreased natural charges on the O or N atoms could be seen clearly (Table 4), which indicates the existence of charge transfer (electron delocalization). The delocalization of electron density takes place between lone pair of the acceptor Y (O or N) and proximal antibonding σ * X–H orbital of the donor, which corresponds to stabilizing interactions on donor–acceptor and is estimated by E (2) i→j* [28, 59]. E (2) i→j* as a consequence of the n Y → σ * X–H interaction reflects the attractive interaction in the H···Y bonding and thus can be used to characterize the strength of the H···Y bond. The n N → σ *O–H interaction between the nitrogen LP orbital at Nb and the OH antibonding orbital is seen to give the strongest stabilization of ~50–60 kJ mol−1. The n N → σ *O–H interactions at Na/Nb and the n O → σ *O–H interaction were smaller. The n N → σ *N–H interactions were even smaller (less than 30 kJ mol−1) than their OH analogues. When the monomer size increases from FDMH to ADMH, the second-order interaction energy increases by 4–8 kJ mol−1 for both the MeOH and DMA complexes. The extent of overlap between the nitrogen/oxygen LP and σ *O–H is greater than that between the nitrogen/oxygen LP and σ *N–H overlap, which suggests that the nitrogen/oxygen LP and σ *N–H overlap attributed to relatively poor overlap upon complexation. This implies that the interaction of O–H···O/N is stronger than N–H···O/N.
In the NBO analysis, the charge transfer effect is greatly overestimated. Therefore, to get more insight into the nature of molecular interactions as well as for quantification of the interactions, the more reliable localized molecular orbital energy decomposition analysis (LMO-EDA) has been carried out for the MeOH complexes and DMA complexes. The decomposition of the total interaction energy of the complexes is presented in Table 5. The LMO-EDA shows that the electrostatic energy (ΔE ES) is the main driving force for the formation of the complexes while the repulsion (ΔE REP) term is not favorable for the formation of complexes. Meanwhile, the ES term was found to play an important role in determining the relative stabilities in van der Waals complexes of benzene–methane and benzene–benzene [60]. Exchange energy (ΔE EX), polarization energy (ΔE POL) and dispersion energy (ΔE DISP) terms also give large contribution to the formation of complexes. The interaction energy (sum of all the components, ΔE INT) obtained from LMO-EDA is found to be comparable to the corrected interaction energies calculated at the B3LYP/aug-cc-pVTZ level of theory.
AIM analysis was performed using the wavefunctions calculated at the B3LYP-D3/aug-cc-pVTZ level of theory for the monomers and complexes. The molecular graphs of MeOH and DMA complexes with bond critical points (BCPs), ring critical points (RCPs) and electron density paths are shown in Figs. 6, 7. The molecular graph shows the BCPs along the lines joining the OH (or NH) and Y (O, N) atoms for the MeOH and DMA complexes, which clearly prove the presence of a hydrogen bond between the MeOH (or DMA) and hydrazones (or NDMA). The topological parameters, including electron density ρ(r) and Laplacian ∇2 ρ(r) at the BCPs, change in atomic charge Δq(H) and atomic energy ΔE(H) at the H atom with the B3LYP-D3 method are listed in Table 6. Hydrogen bond exists if the electron density at the BCPs is in the range of 0.002–0.040 a.u. and the Laplacian of electron density is in the range of 0.024–0.139 a.u. [61, 62]. The electron densities at the BCPs are in the ranges of 0.0077–0.0364 and 0.0099–0.0215 a.u. for the MeOH and DMA complexes, respectively. The Laplacian of the electron density is in the range of 0.0297–0.0717 and 0.0374–0.0605 a.u. for the MeOH and DMA complexes, respectively.

The molecular graphs of the MeOH complexes obtained at the B3LYP-D3/aug-cc-pVTZ level. The bond critical points and ring critical points are presented by the red and yellow balls, respectively. a MeOH–FDMH b MeOH–ADMH c MeOH–NDMA d MeOH–FDMH e MeOH–ADMH f MeOH–NDMA

The molecular graphs of the DMA complexes obtained at the B3LYP-D3/aug-cc-pVTZ level. The bond critical points and ring critical points are presented by the red and yellow balls, respectively. a DMA–FDMH b DMA–ADMH c DMA–NDMA d DMA–FDMH e DMA–ADMH f DMA–NDMA
Upon formation of a hydrogen bond, it is usually accompanied by charge transfer from the hydrogen bond acceptor to the donor, which causes the decrease in the charge on the hydrogen atom [63]. The atomic charge at the H atoms (Δq(H)) for the MeOH complexes is slightly changed, but significant increase of 0.025–0.053 a.u. is observed for the DMA complexes. Meanwhile, the increase in the atomic energy at the H atom (ΔE(H)) is similar for the MeOH and DMA complexes. The atomic energy increases in the following order: N/O–H···Nb > N/O–H···O ≈ N/O–H···Nd > N/O–H···Na.
The AIM analysis indicates the existence of RCPs in some of the structures, with the formation of a C–H···O/N hydrogen bond. The C–H···O hydrogen bond lengths are quite large in the two MeOH–NDMA conformers (2.407–2.444 Å (B3LYP-D3/aug-cc-pVTZ)), while the O–H···O/N hydrogen bond lengths are much shorter by at least 0.4 Å. In general, the C–H···O/N hydrogen bond makes positive contribution to stabilize the complex. Hence, the formation of hydrogen bonded ring structure stabilizes the complexes.
Conclusions
We have presented the results of hydrogen bond interactions of DMA and MeOH with hydrazone and its derivatives by DFT. There were four sites that were treated as hydrogen bond acceptors. Comparable red shifts (169–326 cm−1) of the OH-stretching transition were found for the MeOH complexes. On the other hand, the red shifts of the NH-stretching transition were much smaller for the DMA complexes (below 70 cm−1). The NH-stretching vibrational bands have pronounced intensity increase. In some cases, the intensity increases over 400 times as compared with that of monomer. Both AIM and NBO analyses show the existence of hydrogen bond in the complexes, and LMO-EDA demonstrates that the electrostatic interaction (ES) plays a major role in the total interaction energy. The calculated enthalpy, Gibbs free energy and all the parameters indicate the order of the hydrogen bond strength: O–H···Nb > O–H···Na > O–H···O > O–H···Nd > N–H···Nb ≈ N–H···Na ≈ N–H···O ≈ N–H···Nd.
References
Zhang R, Khalizov A, Wang L, Hu M, Xu W (2012) Nucleation and growth of nanoparticles in the atmosphere. Chem Rev 112(3):1957–2011. doi:10.1021/cr2001756
Rosenfeld D, Sherwood S, Wood R, Donner L (2014) Climate effects of aerosol-cloud interactions. Science 343(6169):379–380. doi:10.1126/science.1247490
Kulmala M, Kontkanen J, Junninen H, Lehtipalo K, Manninen HE, Nieminen T, Petaja T, Sipila M, Schobesberger S, Rantala P, Franchin A, Jokinen T, Jarvinen E, Aijala M, Kangasluoma J, Hakala J, Aalto PP, Paasonen P, Mikkila J, Vanhanen J, Aalto J, Hakola H, Makkonen U, Ruuskanen T, Mauldin RL III, Duplissy J, Vehkamaki H, Back J, Kortelainen A, Riipinen I, Kurten T, Johnston MV, Smith JN, Ehn M, Mentel TF, Lehtinen KEJ, Laaksonen A, Kerminen V-M, Worsnop DR (2013) Direct observations of atmospheric aerosol nucleation. Science 339(6122):943–946. doi:10.1126/science.1227385
Goldstein AH, Galbally IE (2007) Known and unexplored organic constituents in the earth’s atmosphere. Environ Sci Technol 41(5):1514–1521. doi:10.1021/es072476p
Zhang Q, Anastasio C, Jimemez-Cruz M (2002) Water-soluble organic nitrogen in atmospheric fine particles (PM2.5) from northern california. J Geophys Res. doi:10.1029/2001jd000870
Mathur MA, Sisler HH (1981) Oxidation of 1,1-dimethylhydrazine by oxygen. Inorg Chem 20(2):426–429. doi:10.1021/ic50216a021
Lunn G, Sansone EB (1994) Oxidation of 1,1-dimethylhydrazine (UDMH) in aqueous solution with air and hydrogen peroxide. Chemosphere 29(7):1577–1590. doi:10.1016/0045-6535(94)90287-9
Cao Y, Wang L, Zhang G-Y (2006) Analysis of impurities and oxidization products in hydrazine fuels by GC-MS. Chin J Anal Lab 25(12):62–64. doi:10.13595/j.cnki.issn1000-0720.2006.0362
Lazny R, Nodzewska A (2010) N, N-dialkylhydrazones in organic synthesis. From simple N, N-dimethylhydrazones to supported chiral auxiliaries. Chem Rev 110(3):1386–1434. doi:10.1021/cr900067y
Belkheiri N, Bouguerne B, Bedos-Belval F, Duran H, Bernis C, Salvayre R, Nègre-Salvayre A, Baltas M (2010) Synthesis and antioxidant activity evaluation of a syringic hydrazones family. Eur J Med Chem 45(7):3019–3026. doi:10.1016/j.ejmech.2010.03.031
Radwan MAA, Ragab EA, Sabry NM, El-Shenawy SM (2007) Synthesis and biological evaluation of new 3-substituted indole derivatives as potential anti-inflammatory and analgesic agents. Bioorgan Med Chem 15(11):3832–3841. doi:10.1016/j.bmc.2007.03.024
Kumar D, Maruthi Kumar N, Ghosh S, Shah K (2012) Novel bis(indolyl)hydrazide–hydrazones as potent cytotoxic agents. Bioorgan Med Chem Lett 22(1):212–215. doi:10.1016/j.bmcl.2011.11.031
Effenberger K, Breyer S, Schobert R (2010) Modulation of doxorubicin activity in cancer cells by conjugation with fatty acyl and terpenyl hydrazones. Eur J Med Chem 45(5):1947–1954. doi:10.1016/j.ejmech.2010.01.037
Kajal A, Bala S, Sharma N, Kamboj S, Saini V (2014) Therapeutic potential of hydrazones as anti-inflammatory agents. Int J Med Chem 2014:11. doi:10.1155/2014/761030
Mitch WA, Sharp JO, Trussell RR, Valentine RL, Alvarez-Cohen L, Sedlak DL (2003) N-nitrosodimethylamine (NDMA) as a drinking water contaminant: a review. Environ Eng Sci 20(5):389–404. doi:10.1089/109287503768335896
Arunan E, Desiraju GR, Klein RA, Sadlej J, Scheiner S, Alkorta I, Clary DC, Crabtree RH, Dannenberg JJ, Hobza P, Kjaergaard HG, Legon AC, Mennucci B, Nesbitt DJ (2011) Defining the hydrogen bond: an account (IUPAC technical report). Pure Appl Chem 83(8):1619–1636. doi:10.1351/pac-rep-10-01-01
Du L, Mackeprang K, Kjaergaard HG (2013) Fundamental and overtone vibrational spectroscopy, enthalpy of hydrogen bond formation and equilibrium constant determination of the methanol-dimethylamine complex. Phys Chem Chem Phys 15(25):10194–10206. doi:10.1039/c3cp50243k
Hunter CA (2004) Quantifying intermolecular interactions: guidelines for the molecular recognition toolbox. Angew Chem Int Ed 43(40):5310–5324. doi:10.1002/anie.200301739
Du L, Lane JR, Kjaergaard HG (2012) Identification of the dimethylamine-trimethylamine complex in the gas phase. J Chem Phys 136(18):184305. doi:10.1063/1.4707707
Du L, Kjaergaard HG (2011) Fourier transform infrared spectroscopy and theoretical study of dimethylamine dimer in the gas phase. J Phys Chem A 115(44):12097–12104. doi:10.1021/jp206762j
Howard DL, Kjaergaard HG (2006) Vapor phase near infrared spectroscopy of the hydrogen bonded methanol-trimethylamine complex. J Phys Chem A 110(31):9597–9601. doi:10.1021/jp061547o
Jovan Jose KV, Gadre SR, Sundararajan K, Viswanathan KS (2007) Effect of matrix on IR frequencies of acetylene and acetylene-methanol complex: infrared matrix isolation and ab initio study. J Chem Phys 127(10):104501. doi:10.1063/1.2752159
Nedic M, Wassermann TN, Larsen RW, Suhm MA (2011) A combined raman- and infrared jet study of mixed methanol-water and ethanol-water clusters. Phys Chem Chem Phys 13(31):14050–14063. doi:10.1039/c1cp20182d
Howard DL, Kjaergaard HG (2008) Hydrogen bonding to divalent sulfur. Phys Chem Chem Phys 10(28):4113–4118. doi:10.1039/b806165c
Hansen AS, Du L, Kjaergaard HG (2014) The effect of fluorine substitution in alcohol-amine complexes. Phys Chem Chem Phys 16(41):22882–22891. doi:10.1039/c4cp02500h
Elm J, Bilde M, Mikkelsen KV (2013) Assessment of binding energies of atmospherically relevant clusters. Phys Chem Chem Phys 15(39):16442–16445. doi:10.1039/c3cp52616j
Bork N, Du L, Reiman H, Kurten T, Kjaergaard HG (2014) Benchmarking ab initio binding energies of hydrogen-bonded molecular clusters based on FTIR spectroscopy. J Phys Chem A 118(28):5316–5322. doi:10.1021/jp5037537
Reed AE, Curtiss LA, Weinhold F (1988) Intermolecular interactions from a natural bond orbital, donor–acceptor viewpoint. Chem Rev 88(6):899–926. doi:10.1021/cr00088a005
Reed AE, Weinhold F, Curtiss LA, Pochatko DJ (1986) Natural bond orbital analysis of molecular interactions: theoretical studies of binary complexes of HF, H2O, NH3, N2, O2, F2, CO, and CO2 with HF, H2O, and NH3. J Chem Phys 84(10):5687–5705. doi:10.1063/1.449928
Su P, Li H (2009) Energy decomposition analysis of covalent bonds and intermolecular interactions. J Chem Phys 131(1):014102. doi:10.1063/1.3159673
Lane JR, Contreras-Garcia J, Piquemal J-P, Miller BJ, Kjaergaard HG (2013) Are bond critical points really critical for hydrogen bonding? J Chem Theory Comput 9(8):3263–3266. doi:10.1021/ct400420r
Parthasarathi R, Subramanian V, Sathyamurthy N (2006) Hydrogen bonding without borders: an atoms-in-molecules perspective. J Phys Chem A 110(10):3349–3351. doi:10.1021/jp060571z
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd J, Brothers EN, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam NJ, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian 09, revision d.01. Gaussian, Inc., Wallingford, CT
Zhao Y, Truhlar DG (2008) The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor Chem Acc 120(1–3):215–241. doi:10.1007/s00214-007-0310-x
Goerigk L, Grimme S (2011) A thorough benchmark of density functional methods for general main group thermochemistry, kinetics, and noncovalent interactions. Phys Chem Chem Phys 13(14):6670–6688. doi:10.1039/c0cp02984j
Boys SF, Bernardi F (1970) The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol Phys 19(4):553–566. doi:10.1080/00268977000101561
Hippler M (2005) Quantum-chemical study of CHCl3-SO2 association. J Chem Phys 123(20):204311. doi:10.1063/1.2121609
Chung S, Hippler M (2006) Infrared spectroscopy of hydrogen-bonded CHCl3–SO2 in the gas phase. J Chem Phys 124(21):214316. doi:10.1063/1.2207617
Simon S, Duran M, Dannenberg JJ (1999) Effect of basis set superposition error on the water dimer surface calculated at hartree–fock, møller–plesset, and density functional theory levels. J Phys Chem A 103(11):1640–1643. doi:10.1021/jp9842188
Hippler M, Hesse S, Suhm MA (2010) Quantum-chemical study and FTIR jet spectroscopy of CHCl3–NH3 association in the gas phase. Phys Chem Chem Phys 12(41):13555–13565. doi:10.1039/c0cp00530d
Bader RF (1991) A quantum theory of molecular structure and its applications. Chem Rev 91(5):893–928. doi:10.1021/cr00005a013
Schmidt MW, Baldridge KK, Boatz JA, Elbert ST, Gordon MS, Jensen JH, Koseki S, Matsunaga N, Nguyen KA, Su S, Windus TL, Dupuis M, Montgomery JA (1993) General atomic and molecular electronic structure system. J Comput Chem 14(11):1347–1363. doi:10.1002/jcc.540141112
Karabatsos GJ, Shapiro BL, Vane FM, Fleming JS, Ratka JS (1963) Structural studies by nuclear magnetic resonance. II. Aldehyde 2,4-dinitrophenylhydrazones. J Am Chem Soc 85(18):2784–2788. doi:10.1021/ja00901a025
Karabatsos GJ, Vane FM, Taller RA, Hsi N (1964) Structural studies by nuclear magnetic resonance. VIII. Ring-substituted phenylhydrazones, semicarbazones, and thiosemicarbazones. J Am Chem Soc 86(16):3351–3357. doi:10.1021/ja01070a029
Clarke LF, O’Sullivan F, Hegarty AF (1991) Photoisomerisation of (E)- to (Z)-N, N-dimethylhydrazones and thermal reversion. J Chem Soc Perkin Trans 2(11):1649–1652. doi:10.1039/p29910001649
Ramanathan S, Lemal DM (2007) Conformational and configurational dynamics of a highly fluorinated hydrazone. J Org Chem 72(5):1566–1569. doi:10.1021/jo061945n
Shvo Y, Nahlieli A (1970) Detection of thermal isomerization of hydrazones by NMR spectroscopy. Tetrahedron Lett 11(49):4273–4274. doi:10.1016/S0040-4039(00)89463-0
Hansen AS, Du L, Kjaergaard HG (2014) Positively charged phosphorus as a hydrogen bond acceptor. J Phys Chem Lett 5(23):4225–4231. doi:10.1021/jz502150d
Schrems O, Oberhoffer HM, Luck WA (1984) Hydrogen bonding in low-temperature matrices: 1. Proton donor abilities of fluoroalcohols. Comparative infrared studies of ROH O(CH3)2 complex formation in the gas phase, in CCl4 solution, and in solid argon. J Phys Chem 88(19):4335–4342. doi:10.1021/j150663a029
Zhang Q, Du L (2016) Hydrogen bonding in the carboxylic acid–aldehyde complexes. Comput Theor Chem 1078:123–128. doi:10.1016/j.comptc.2016.01.007
Curtiss LA, Blander M (1988) Thermodynamic properties of gas-phase hydrogen-bonded complexes. Chem Rev 88(6):827–841. doi:10.1021/cr00088a002
Hagemeister FC, Gruenloh CJ, Zwier TS (1998) Density functional theory calculations of the structures, binding energies, and infrared spectra of methanol clusters. J Phys Chem A 102(1):82–94. doi:10.1021/jp963763a
Iogansen AV (1999) Direct proportionality of the hydrogen bonding energy and the intensification of the stretching ν(XH) vibration in infrared spectra. Spectrochim Acta Mol Biomol Spectrosc 55(7–8):1585–1612. doi:10.1016/S1386-1425(98)00348-5
Chojnowski J (1970) Triethylphosphine, triethylarsine, and triethylstibine as hydrogen-acceptors in hydrogen bonds II. The association with phenol and methanol. Bull Acad Polon Sci Ser Sci Chim 18:317–324
Épshtein LM, Ashkinadze LD, Gorelik SO, Gambaryan NP, Bochvar DA, Kazitsyna LA (1974) Peculiarities of the interaction of phosphorus with the aromatic ring and their consideration in connection with the problem of conjugation. Bull Acad Sci USSR Div Chem Sci 23(1):58–63. doi:10.1007/bf00922312
Miller BJ, Du L, Steel TJ, Paul AJ, Soedergren AH, Lane JR, Henry BR, Kjaergaard HG (2012) Absolute intensities of NH-stretching transitions in dimethylamine and pyrrole. J Phys Chem A 116(1):290–296. doi:10.1021/jp209118p
Slipchenko MN, Sartakov BG, Vilesov AF, Xantheas SS (2007) Study of NH stretching vibrations in small ammonia clusters by infrared spectroscopy in he droplets and ab initio calculations. J Phys Chem A 111(31):7460–7471. doi:10.1021/jp071279+
Biswal HS, Wategaonkar S (2011) OH X (X = O, S) hydrogen bonding in tetrahydrofurane and tetrahydrothiophene. J Chem Phys 135(13):134306. doi:10.1063/1.3645107
Weinhold F, Landis CR (2005) Valency and bonding: a natural bond orbital donor–acceptor perspective. Cambridge University Press, Cambridge
Sakaki S, Kato K, Miyazaki T, Musashi Y, Ohkubo K, Ihara H, Hirayama C (1993) Structures and binding energies of benzene–methane and benzene–benzene complexes. An ab initio scf/mp2 study. J Chem Soc Farad Trans 89(4):659–664. doi:10.1039/ft9938900659
Koch U, Popelier P (1995) Characterization of CHO hydrogen bonds on the basis of the charge density. J Phys Chem 99(24):9747–9754. doi:10.1021/j100024a016
Grabowski SJ (2004) Hydrogen bonding strength—measures based on geometric and topological parameters. J Phys Org Chem 17(1):18–31. doi:10.1002/poc.685
Bushmarinov IS, Lyssenko KA, Antipin MY (2009) Atomic energy in the ‘atoms in molecules’ theory and its use for solving chemical problems. Russ Chem Rev 78(4):283–302. doi:10.1070/RC2009v078n04ABEH004017
Acknowledgments
This work was supported by Shandong Provincial Natural Science Foundation, China (ZR2014BQ013), National Natural Science Foundation of China (21407095, 21577080) and the Fundamental Research Funds of Shandong University (2015JC045).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhao, H., Tang, S., Li, S. et al. Theoretical investigation of the hydrogen bond interactions of methanol and dimethylamine with hydrazone and its derivatives. Struct Chem 27, 1241–1253 (2016). https://doi.org/10.1007/s11224-016-0749-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-016-0749-2