
Abstract
The single crystal structures of two 2-acylaminopyrimidines, where alkyl groups in acyl moiety are iso-propyl (1) and dichloromethyl (2), were solved by X-ray diffraction method. The strength of intermolecular hydrogen bonding interactions depends on the C–H bond polarization increased by exchanging two methyl groups by chlorine atoms in the adjacent substituent. The computational methods provide an additional insight into the intermolecular interactions and are utilized in explaining the differences in the observed crystal structures. The experimental and computational data together explain the differences in the formed aggregates and revealed that these simple substitutions cause crucial changes in the intermolecular interactions.
Graphical Abstract
Introduction of nitrogen atom instead of CH moiety completely changes the aggregation.

Similar content being viewed by others
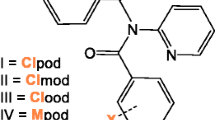

Introduction
The non-covalent interactions are nowadays explored and used in design and preparation of supramolecular assemblies [1–3]. Among the classic examples of strongly bound dimers are amides, carboxylic acids, and a plethora of heteroaromatics such as 2-aminopyridine and 2-[1H]-pyridone [4]. In general the hydrogen bonding depends on properties of hydrogen bond donors (D) and acceptors (A) such as the acidity and basicity of interacting groups. It has been shown how hydrogen bonding can be used in designing the shape of aggregate [5–7] in solution and in solid state [8]. Generally, non-covalent intermolecular interactions are influenced by steric effects [9–12], electronic repulsion [11, 13], position of the heteroatoms in the ring [14–16], and cooperative effects [17]. The atoms able to compete for hydrogen bonds (basic or acidic centers) are responsible for the conformational flexibility of NH–CO bond in amides [18–20]. This conformational freedom is restricted in 2,6-bis(acylamino)pyridines preventing their dimerization via quadruple hydrogen bonding while the conformational flexibility is maintained in related pyrimidines [10, 18] possessing an additional nitrogen in the ring when compared with pyridine. The same is realized in comparison of the solid-state structure of 2-acetylaminopyridine [9] versus 2-acetylaminopyrimidine [16] (Fig. 1).

The solid-state structures of 2-acetylaminopyridine and 2-acetylaminopyrimidine
Furthermore, intramolecular hydrogen bonding stabilizes some of these molecules [19, 20]. An increased flexibility of the 2-acylamino substituent in pyrimidine is argued to be due to electron lone pair repulsions between N-3 and carbonyl oxygen [16]. This was confirmed by comparison of the heteroassociation of 2,2′-dipyridylamine versus 2-acetylaminopyrimidine which both contain ADA hydrogen bond motifs with DAD counterparts capable for triple hydrogen bonding [12]. The weak (usually below 16 kJ/mol [21, 22]) CH···O [23–25], CH···N [26, 27], or CH···π [28] interactions may also influence the geometry [29] of self-assembled molecules. The nonconventional CH···O hydrogen bonds have been found in pyridine–carboxylic acid co-crystals [30–33], in crystals of N,N′-di(2-pyridyl)oxamide [34], and nitromethane entrapped in cyclodextrin [35]. It is commonly known that CH is a weaker hydrogen bond donor than NH/OH due to lower electronegativity of carbon than that of nitrogen and oxygen. During our studies on 2-acylaminopyrimidines an interesting question arises whether a change in C–H bond polarization would result in formation of intramolecular hydrogen bonding as in some heterocyclic urea derivatives [15, 36–38]. The main goals in the present study are: (a) to check if the intramolecular hydrogen bonding by CH donor is able to limit the conformational flexibility in these molecules, (b) to study how polarization of CH bond by chlorines influences the molecular structure, and (c) to model and interpret the molecular properties via computational methods. It is worth mentioning that in Cambridge Structural Database (CSD) only three similar structures were found revealing that the bifurcation of hydrogen bond in pyrimidine derivatives is not common. These contain aromatic CH···N/O [39, 40] or OH···N/O [41] contacts.
Experimental
The synthesis of 1 and 2 (Fig. 2) was performed as described before [16]. Their dimerization constants were determined by 1H NMR dilution studies fitted to Benesi-Hildebrand equation [42]. X-ray diffraction data of 1 and 2 were collected at 123(2) K on a Bruker-Nonius KappaCCD diffractometer with ApexII detector using graphite monochromated Mo-Kα radiation. The details of X-ray crystallography are given in Electronic Supplementary Material (ESM). The solved geometries of 1 and 2 were used as inputs for quantum chemical calculations to validate the intermolecular interactions including the strength of hydrogen bonding. The M05/6-311G(d,p) level of theory, recommended in studying non-covalent interactions [43–46], was applied to monomers and dimers of 1 and 2. The frequency calculations were run to be sure that the geometry is in energy minima (no negative frequencies were obtained). The transition state for reaction shown in Fig. 3 was optimized with the use of Synchronous Transit-Guided Quasi-Newton algorithm [47, 48]. All optimizations have been run using Gaussian software [49]. The H-BCP properties were calculated with the use of AIM2000 software [50].

The ORTEP [70] plots of 1 and 2 (most hydrogens are omitted for clarity), atom numbering and hydrogen bonding pattern

Open and closed forms of 1 and 2 and the intramolecular hydrogen bonding in the closed form and in a urea derivative
Results and discussion
2-Acylaminopyrimidines either with two methyls (1) or chlorines (2) in acyl moiety (Fig. 1) gave crystals for X-ray structure determination after slow evaporation of CDCl3 in NMR tube. Compounds were characterized also by 1H, 13C, and 15N NMR spectroscopy and by elemental analysis (see ESM).
It has been shown [16] that 2-acetylaminopyrimidine exists in E conformation in crystals as dimers stabilized by two NH···N and two CH···O interactions (Fig. 1). The less limited rotation about N7–C8 in pyrimidine is due to an additional nitrogen (N3 differing from pyridine derivatives [9]). In 1 and 2 such a free rotation is not observed and the amide conformation is Z in crystal structure as also in 1-adamantyl derivative [16]. The substitution of i-Pr group by CHCl2 results in a different crystal structure although the methyl groups and chlorine atoms are comparable in size. In structure of 2 molecules are oriented around four-fold axis (Fig. 2; ESM). This is suspected to result from the electronic repulsion of the chlorine atoms. In structure of 1 two molecules are present in asymmetric unit and they show slightly deviating geometry in acylamino group (see ESM). The hydrogen bonding geometries of 1 and 2 are collected in Table 1.
The geometry data show that the substitution of methyls by chlorine atoms causes only clear shortening in N7···O9′, C10···N3′ intermolecular hydrogen bonding distances (and partly in C10···O9′, Table 1). However, out of four intermolecular distances presented in Fig. 2 the hydrogen bond contact N7···N3′ is longer in 2 than in 1. The chains of molecules along the c-axis formed by these four hydrogen bonds are connected with adjacent chains by weak C–H···N/O type interactions (also one C–H···Cl in 2). The distance between ring centroids of adjacent molecules are ca. 5.4 Å in crystals of 1 and 6.1 Å in crystals of 2 (Table S3), which is perfectly understood since the angle between ring planes are 61° and 90°, respectively. While the intermolecular distances clearly show closer placement of molecules in crystal of 2 than that of 1, an alternative way of showing these relations is the area of triangles (T1–T4, Table S3) delimited by crucial atoms. All triangles are smaller in 2 than in 1. The D–H···A angles are between ca. 125 and 160°. The deviation from linearity (180°) is due to the bifurcated character of hydrogen bond bridges. It is also worth to stress that both hydrogen bond donors and acceptors are bifurcated. Moreover, the solved structures suggest that at times the interactions usually considered as weaker can support stronger ones and together with the ability of additional interactions by extra basic centers (N3) new, hardly predictable structures are formed [51]. To better understand the intermolecular interactions in these molecules computations were performed.
The QTAIM [52] is useful in describing the properties of hydrogen bond bridges, i.e., the electron density (ρ) and Laplacian of electron density (∇2 ρ) at Hydrogen Bond Critical Point (H-BCP). According to this method for the covalent bond the Laplacian of electron density at BCP is negative and for interactions of hydrogen bonding nature (H-BCP), it is positive [52, 53]. QTAIM data for currently studied interactions are collected in Table 2 (the geometry taken from XRD measurements).
It is worth to note that except for N7H7···N3′ interaction (hydrogen bond distances, Table 1) the polarization of CH bond by chlorine atoms in 2 makes its hydrogen bonds stronger. Moreover, the hydrogen bond energies [54, 55] (E HB) were calculated according to Espinosa approach [54, 55], while the overall interaction energy (E int.) is the difference between energy of the complex and the sum of energies of monomers. Espinosa’s approach based on QTAIM and properties of hydrogen bond critical point properties was used on a variety of non-covalent interactions including XH···O (X = C, N, O), H···F [54–59] hydrogen bonds. Moreover, the same approach has been used to study the hydrogen bond bridges in which the hydrogen bond acceptor carries three (FH···FF [58], NH···F, CH···F [59]), two (NH···O, or OH···O[54]), and one lone electron pair (FH···NH3 [59]). This method have also been used in explaining the properties of intramolecular hydrogen bonds (NH···N [60]) and intermolecular ones that stabilize dimer, trimers and tune the properties of rotamers in supramolecular assemblies [16, 61]. It is fair to mention that we used this approach to highlight the differences in energy of interactions between NH···N/O and CH···N/O contacts, while the QTAIM was shown to be applicable for many structures where the hydrogen bonding is described as purely non-covalent interaction (even a weak one [62]) or it is considered as partially covalent one with relatively high bond orders [63]. The same methodology may be applied to study hydrogen bonding in π-electron conjugated structures [64] or in investigations on halogen bonding [65]. E int.s are corrected to basis set superposition error (BSSE) with the counterpoise method [47, 66] as implemented in Gaussian [49] with default settings (see later in text).
To study the association of these molecules in solution the 1H NMR dilution experiments (in CDCl3) were performed as before [9]. The dimerization constants (K dim). at 293 K are 1.4 for 1 and 1.7 M−1 for 2, respectively, when both NH and CH protons were used as probe nuclei [12].
It has been argued that generally the intramolecular hydrogen bonding is stronger than intermolecular [67]. In addition, a weak CH···N interaction may have some influence on the conformational freedom in these compounds (Fig. 3).
The intramolecular hydrogen bond is a dominant interaction stabilizing the closed form in heterocyclic urea derivatives (Fig. 3). In 1 and 2 the open form is ca. 16 kJ/mol higher in energy than closed one. The energy of CH···N interaction, according to Espinosa [54, 55], is −12.8 kJ/mol in 1 and −17.4 kJ/mol in 2. On the other hand, the closed–open form equilibrium may take place as the rotation about N7–C8 bond is easy and passes through the energetically low-lying transition state. The calculated barrier to rotation (transition state) about N7-C8 bond is ca. 59 kJ/mol with respect to the closed form.
In order to explain the intermolecular interactions in 1 and 2 and resulting solid-state structure the dimerization was considered as the initial step on way to higher aggregates. Therefore, the open and closed forms (Fig. 3) were optimized as dimers and the interaction energy together with QTAIM parameters were calculated. Figure 4 shows six dimers used in these calculations.

Dimers D1–D6 of 1 and 2
In Table 3 the results of calculations are collected, i.e., the interaction energy between molecules in kJ/mol corrected to BSSE (E int.), electron density at H-BCP (ρ), Laplacian of electron density (∇2 ρ), and energy of hydrogen bonds (E HB). The geometry optimized in vacuum was used as initial geometry for calculations with the use of PCM [68] model of solvation and higher basis set (6–311+G(2d,2p), Table 3 in italics) for the associate that was found by X-ray crystallography (D2, Fig. 4). The same basis set was applied on the X-ray structural geometry as a single point runs (Table 2).
In general the data for H-BCP calculated in vacuum, solvent, and from X-ray structure are in agreement. The CH···N/O interaction is stronger in 2 than in 1 due to the C–H bond polarization caused by the adjacent electronegative chlorines.
The data in Table 3 are in agreement with experimental findings. The dimer observed in 2-isobutyroylaminopyridine [9] and stabilized by NH···N hydrogen bond was not found in respective pyrimidine derivatives. The calculated data explain the stability of the 2-isobutyroylaminopyrimidine and respective dichloro derivative in crystal. The current data also show that the bifurcated character of hydrogen bonds is crucial in association of 1 and 2. This is especially true for 2 where the weak interactions by CH group are strengthened by bond polarization. This conclusion is supported by the deshielding in 1H NMR chemical shifts (ESI). The broadened 1H signals of CH moieties in 1 and 2 suggest that this proton is interacting with nitrogen by weak hydrogen bonding.
Conclusions
The single crystal X-ray structures of two 2-acylaminopyrimidines 1 and 2 reveal interesting NH···O/N and CH···O/N interactions that have bifurcated character. The strength of intermolecular interaction depends on the presence of chlorines in the acyl moiety, which polarize the adjacent C–H bond. The quantum chemical calculations based on X-ray structures explain experimental findings. When compared with 2-acylaminopyridines an extra nitrogen (N3) in the heterocyclic ring increases the number of energetically relevant dimeric structures. However, the crucial point is that the C–H bond polarization by chlorines in 2 results in the most stable structure being the dimer stabilized by bifurcated hydrogen bonds. Based on Jorgensen’s approach the secondary interactions [69] rely mostly on the repulsive or attractive interactions in coplanar complexes. Dimers of 1 and 2 with bifurcated hydrogen bonds are nonplanar and do not favor the secondary interactions, especially strong repulsions. Moreover, the dimers with bifurcated hydrogen bonds are able to further aggregate in chain structures. Also, it is worth to mention that although the closed conformer of the monomer is the most stable form, the formation of intermolecular interactions can favor the open form in crystals due to relatively high rotational barrier between open and closed forms.
References
Prins LJ, Timmerman P, Reinhoudt DN (1998) Pure Appl Chem 70:1459
De Greef TFA, Smulders MMJ, Wolffs M, Schenning APHJ, Sijbesma RP, Meijer EW (2009) Chem Rev 109:5687
Prins LJ, Reinhoudt DN, Timmerman P (2001) Angew Chem Int Ed 40:2382
Szyc Ł, Guo J, Yang M, Dreyer J, Tolstoy PM, Nibbering ETJ, Czarnik-Matusewicz B, Elsaesser T, Limbach H–H (2010) J Phys Chem A 114:7749
Etter MC (1991) J Phys Chem 95:4601
Aakeröy CB, Champness NR, Janiak C (2010) Cryst Eng Commun 12:22
Sherrington DC, Taskinen KA (2001) Chem Soc Rev 30:83
Steiner T (2002) Angew Chem Int Ed 41:48
Ośmiałowski B, Kolehmainen E, Dobosz R, Gawinecki R, Kauppinen R, Valkonen A, Koivukorpi J, Rissanen K (2010) J Phys Chem A 114:10421
Ośmiałowski B, Kolehmainen E, Gawinecki R, Dobosz R, Kauppinen R (2010) J Phys Chem A 114:12881
Ośmiałowski B, Gawinecki R, Kolehmainen E, Kalenius E, Behera B, Kauppinen R, Sievanen E (2011) Struct Chem 22:1143
Ośmiałowski B, Kolehmainen E, Kauppinen R, Kowalska M (2011) Supramol Chem 23:579
Ośmiałowski B, Kolehmainen E, Gawinecki R, Kauppinen R, Koivukorpi J, Valkonen A (2010) Struct Chem 21:1061
Gooch A, McGhee AM, Renton LC, Plante JP, Lindsay CI, Wilson AJ (2009) Supramol Chem 21:12
Corbin PS, Zimmerman SC, Thiessen PA, Hawryluk NA, Murray TJ (2001) J Am Chem Soc 123:10475
Ośmiałowski B, Kolehmainen E, Ikonen S, Valkonen A, Kwiatkowski A, Grela I, Haapaniemi E (2012) J Org Chem 77:9609
Hunter C, Anderson H (2009) Angew Chem Int Ed 48:7488
Manesiotis P, Hall AJ, Sellergren B (2005) J Org Chem 70:2729
Jeoung E, Augier de Cremiers H, Deans R, Cooke G, Heath SL, Vanderstraeten PE, Rotello VM (2001) Tetrahedron Lett 42:7357
Schmuck C, Wienand W (2001) Angew Chem Int Ed 40:4363
Turi L, Dannenberg JJ (1993) J Phys Chem 97:7899
Green RD (1974) Hydrogen bonding by C–H groups. Macmillan, London
Desiraju GR (1991) Acc Chem Res 24:290
Pierce AC, ter Haar E, Binch HM, Kay DP, Patel SR, Li P (2005) J Med Chem 48:1278
Desiraju GR (1996) Acc Chem Res 29:441
Xing L, Wiegert C, Petitjean A (2009) J Org Chem 74:9513
Mambanda A, Jaganyi D, Munro OQ (2007) Acta Crystallogr C63:o676
Nishio M (2004) CrystEngComm 6:130
Desiraju GR (2002) Acc Chem Res 35:565
Dale SH, Elsegood MRJ, Hemmings M, Wilkinson AL (2004) CrystEngComm 6:207
Singh D, Baruah JB (2009) CrystEngComm 11:2688
Vishweshwar P, Nangia A, Lynch VM (2001) J Org Chem 67:556
Bhogala BR, Nangia A (2003) Cryst Growth Des 3:547
Hsu Y-F, Chen J-D (2004) Eur J Inorg Chem 2004:1488
Nakagawa T, Immel S, Lichtenthaler FW, Lindner HJ (2000) Carbohydr Res 324:141
Singha NC, Sathyanarayana DN (1997) J Chem Soc Perkin Trans 2:157
Sudha LV, Sathyanarayana DN (1985) J Mol Struct 131:253
Jiménez Blanco JL, Benito JM, Mellet CO, García Fernández JM (1999) Org Lett 1:1217
Oertli AG, Meyer WR, Suter UW, Joho FB, Gramlich V, Petter W (1992) Helv Chim Acta 75:184
Chantrapromma S, Fun H-K, Jana S, Hazra A, Goswami S (2008) Acta Crystallogr E 64:o267
Wu X, Delgado G, Krishnamurthy R, Eschenmoser A (2002) Org Lett 4:1283
Benesi H, Hildebrand J (1949) J Am Chem Soc 71:2703
Zhao Y, Schultz NE, Truhlar DG (2005) J Chem Phys 123:161103
Zhao Y, Truhlar DG (2006) J Chem Theory Comput 2:1009
Zhao Y, Truhlar DG (2008) J Chem Theory Comput 4:1849
Zhao Y, Truhlar DG (2008) Theor Chem Acc 120:215
Peng C, Schlegel HB (1993) Isr J Chem 33:449
Peng C, Ayala PY, Schlegel HB, Frisch MJ (1996) J Comput Chem 17:49
Gaussian 09, Revision A.02, Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery J, J. A., Peralta JE, Ogliaro F, Bearpark M, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Martin RL, Morokuma K, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas O, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ (2009) Gaussian, Inc., Wallingford
Biegler-König F, Schönbohm J, Bayles D (2001) J Comput Chem 22:545
Sureshan KM, Gonnade RG (2013) CrystEngComm 15:1676
Bader RFW (1990) Atoms in molecules: a quantum theory. Oxford University Press, New York
Popelier LA (2000) Atoms in molecule. An introduction. Pearson Education, London
Espinosa E, Molins E, Lecomte C (1998) Chem Phys Lett 285:170
Espinosa E, Souhassou M, Lachekar H, Lecomte C (1999) Acta Crystallogr B55:563
Espinosa E, Lecomte C, Molins E (1999) Chem Phys Lett 300:745
Espinosa E, Molins E (2000) J Chem Phys 113:5686
Espinosa E, Alkorta I, Rozas I, Elguero J, Molins E (2001) Chem Phys Lett 336:457
Espinosa E, Alkorta I, Elguero J, Molins E (2002) J Chem Phys 117:5529
Ośmiałowski B, Krygowski TM, Dominikowska J, Palusiak M (2011) New J Chem 35:1433
Ośmiałowski B, Kolehmainen E, Kowalska M (2012) J Org Chem 77:1653
Koch U, Popelier PLA (1995) J Phys Chem 99:9747
Grabowski SJ (2011) Chem Rev 111:2597
Sobczyk L, Grabowski SJ, Krygowski TM (2005) Chem Rev 105:3513
Grabowski SJ (2012) J Phys Chem A 116:1838
Boys SF, Bernardi F (1970) Mol Phys 19:553
Etter MC (1990) Acc Chem Res 23:120
Tomasi J, Mennucci B, Cammi R (2005) Chem Rev 105:2999
Jorgensen WL, Pranata J (1990) J Am Chem Soc 112:2008
Farrugia L (1997) J Appl Crystallogr 30:565
Acknowledgments
The financial support from the National Science Centre in Kraków (Grant No. N N204 356840) is gratefully acknowledged. We are very much indebted to the ICM in Warsaw for providing computer time and programs. Also, Academy Professor Kari Rissanen is gratefully acknowledged for funding to A.V. (from Academy of Finland grant nos. 130629, 122350, and 140718) and the University of Jyväskylä (postdoc grant to A.V.) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Ośmiałowski, B., Kolehmainen, E., Valkonen, A. et al. The influence of CH bond polarization on the self-association of 2-acylaminopyrimidines by NH/CH···O/N interactions: XRD, NMR, DFT, and AIM study. Struct Chem 24, 2203–2209 (2013). https://doi.org/10.1007/s11224-013-0283-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11224-013-0283-4