
Abstract
Growing tumors develop additional new blood vessels to meet the demand for adequate nutrients and oxygen, a process called angiogenesis. Cancer is a highly complex disease promoted by excess angiogenesis; interfering with this process poses for an attractive approach for controlling tumor growth. This hypothesis led to the identification of endogenous angiogenesis inhibitors generated from type IV collagen, a major component of vascular basement membrane (VBM). Type IV collagen and the angiogenesis inhibitors derived from it are involved in complex roles, than just the molecular construction of basement membranes. Protease degradation of collagens in VBM occurs in various physiological and pathological conditions and produces several peptides. Some of these peptides are occupied in the regulation of functions conflicting from those of their original integral molecules. Tumstatin (α3(IV)NC1), a proteolytic C-terminal non-collagenous (NC1) domain from type IV collagen α3 chain has been highlighted recently because of its potential role in anti-angiogenesis, however its biological actions are not limited to these processes. α3(IV)NC1 inhibits proliferation by promoting endothelial cell apoptosis and suppresses diverse tumor angiogenesis, thus making it a potential candidate for future cancer therapy. The present review surveys the physiological functions of type IV collagen and discovery of α3(IV)NC1 as an antiangiogenic protein with a comprehensive overview of the knowledge gained by us towards understanding its signaling mechanisms.
Similar content being viewed by others
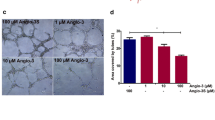
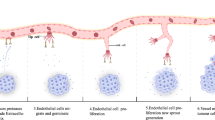

INTRODUCTION
The development of extracellular matrix (ECM), a complex structure comprising different proteins, is a crucial event in evolution of multicellular organisms. In addition to providing mechanical support for cells, ECM also influences cell behavior (1). ECM remodeling during physiological or pathological processes generates new signals, particularly between endothelial cells and basement membranes (BMs) (2). BMs are thin layers of specialized ECM associated closely with different cells (3). Different collagens together with laminins, nidogens, heparan sulfate proteoglycans (HSPG), fibulins, dystroglycan and other glycoproteins, are major constituents of BM (4). Some of the non-collagenous (NC1) C-terminal domains from different collagens were identified to be antiangiogenic and anti-tumorigenic in addition to their biological role in molecular architecture of the BM (5,6).
During angiogenesis, new capillaries sprout from pre-existing blood vessels by an invasive process “neovascularization” that occurs in physiological (embryonic development, reproduction and wound healing) and pathological (tumor growth, metastasis, arthritis, age related macular degeneration etc.) conditions (7). Early reports hypothesized that tumor growth is strictly dependent on neovascularization, and inhibition of vascular supply to growing tumors could suppress tumor growth (7,8). Solid tumors cannot grow beyond 2 to 3 mm in diameter without recruitment of their own blood supply and nutrients, thus tumor growth depends on balance between circulating endogenous pro-angiogenic factors (VEGF, FGF, PDGF etc.) and endogenous angiogenesis inhibitors (antiangiogenic peptides generated from ECM by proteases) (7,9). The ECM derived endogenous angiogenesis inhibitors control three fundamental processes (tumor cell proliferation, apoptosis, and tumor angiogenesis) that are critical for growth of primary tumors and metastases (10). Since these endogenously released circulating peptides play a crucial role in anti-angiogenesis, much effort has been focused on development and identification of endogenous anti-angiogenic molecules from BM in the past decade aiming to produce potential medical treatments. A search for endogenous angiogenesis inhibitors led to the discovery of endostatin, a C-terminal NC1 domain from type XVIII collagen α1 chain (5). To date several endogenous angiogenesis inhibitors were identified, some of them were cryptic fragments released by proteolysis of ECM and the others are of non-ECM origin (10). The angiogenesis inhibitors from ECM includes a large multifunctional glycoproteins such as thrombospondin, anastellin, a fibronectin fragment, fibulins (COOH terminal fragments corresponding to fibulin 1D and the domain 111 of fibulin 5), endorepellin, a COOH terminal end of perlecan, (or perlecan domain V) (11–14). Some of the type IV collagen derived NC1 domains posses characteristic antiangiogenic activities that were first reported by Dr. Brook’s group (6).
This review will illustrate a comprehensive overview of type IV collagen, its physiological functions with specific emphasis on the generation of α3 chain type IV collagen NC1 domain α3(IV)NC1 (tumstatin) an endogenous inhibitor of angiogenesis. This review also highlights important features of α3(IV)NC1 addressing signaling mechanisms in regulation of tumor angiogenesis which would explain how this endogenous angiogenesis inhibitor regulates angiogenic balance in the tumor bed.
PHYSIOLOGICAL FUNCTIONS OF TYPE IV COLLAGEN
Type IV collagen is ubiquitously present in vascular basement membrane (VBM) and is highly conserved among vertebrates and invertebrates, regulating cell adhesion and migration (15–17). Type IV collagen is one of the most abundant constituents of basement membrane (BM) architecture that forms a network like structure in ECM (18,19). Type IV collagen consists of six distinct gene products (α1 to α6 chains) and their genomic localization shows a pair-wise head-to-head arrangement with a bi-directional promoter, that were mapped onto three different chromosomes (20–22). Each α-chain in type IV collagen is composed of three distinct domains, a cysteine-rich N-terminal 7S domain, a central long triple helical domain and a globular C-terminal NC1 domain (Fig. 1). The NC1 domains of type IV collagen are involved in the assembly of α-chains to form heterotrimers and the 7S domain is involved in the covalent assembly of heterotrimers in a complex mesh like network that serves as a scaffold for BM (18,19,23,24). Type IV collagen is found normally in the BM, during certain pathological conditions associated with tumor fibrosis, it accumulates in tumor interstitium (4). α1(IV) and α2(IV) collagen chains are most abundant forms of type IV collagen and are widely expressed and co-localized in numerous tissue types, whereas α3–α6 chains show temporal and spatial distribution, and expression in physiological as well as pathological processes (25–29).

A hypothetical structure of α3 chain type IV collagen. Linear structure of human α3 chain type IV collagen composed of three distinct domains: a cysteine rich N-terminal 7S domain, a central triple helical domain with multiple small interruptions and a globular C-terminal non-collagenous (NC1) domain.
Several human genetic diseases provided insights into the physiological role of type IV collagen. Mutations or deletions in Col4α5, Col4α3 and Col4α4 chains are involved in Alport syndrome (defective glomerular BM) and diffused leiomyomatosis (a benign smooth muscle tumor) (30,31). The C-terminal region of α3 chain of type IV collagen has been identified as an autoantigen involved in Goodpasture syndrome (an immune disease characterized by glomerulonephritis and pulmonary hemorrhage) (29). Mouse models for autosomal Alport syndrome have been developed and a phenocopy of human disease was also reported (32).
GENERATION OF α3(IV)NC1 (TUMSTATIN) FROM TYPE IV COLLAGEN
The proteolytic pathways involved in generation of NC1 fragments from type IV collagen are not well understood. Several NC1 fragments were detected in the serum suggesting that these fragments exist due to physiological cleavage of ECM by proteases. Presumably, several distinct proteolytic pathways are involved in generation of these NC1 domains; however their generation is not extensively tested like as endostatin, which is generated from type XVIII collagen α1 chain NC1 domain (33). To isolate NC1 domain from type IV collagen, researchers isolated vascular basement membrane (VBM) and extracted type IV collagen that was subjected to different proteases. The different protease degradation fragments in the supernatant were separated by gel filtration, anion exchange chromatograph, HPLC etc. Several small peptides were identified by amino acid sequence analysis and immunoblotting using antibodies specific to type IV collagen NC1 domains (34–37) (Fig. 2). Earlier we reported α3(IV)NC1 as a 28 kDa proteolytic NC1 peptide generated from α3 chain of type IV collagen by MMP-9 and MMP-2 (38,39). Although we gained preliminary clues for in vitro processing of type IV collagen NC1 domains, the exact mechanism of generation of these NC1 domains still needs to be extensively investigated.

Generation of antiangiogenic cryptic domains from VBM. Vascular basement membrane was extracted to isolate type IV collagen. The assembled types IV collagen (monomer or trimer) has no antiangiogenic activity. Interestingly when type IV collagen was digested with matrix metalloproteinases (MMP-2/-9), antiangiogenic fragments were identified. Four out of six non-collagenous (NC1) domains of type IV collagen were discovered as antiangiogenic (α1, α2, α3 and α6(IV)NC1) whereas α4 and α5(IV)NC1 were not reported to be showing similar functions. The lengths of NC1 domain are approximately 231 amino acids shown in the structure. α3(IV)NC1 (tumstatin) a 28 kDa protein was cloned in baculovirus expression system and identified that it binds to cell surface integrins to mediate its antiangiogenic activity (66,100,101).
BIOLOGICAL FUNCTIONS OF α3(IV)NC1 IN ANTI-ANGIOGENESIS
The biological functions of type IV collagen NC1 domains seem to be conserved throughout evolution. In primitive invertebrate Hydra vulgaris, addition of NC1 type (IV) collagen alters morphogenesis, blocking cell aggregation and development (40). In vitro, NC1 type (IV) collagen promotes axonal but not dendritic growth in rat embryos sympathetic neurons, hexameric NC1 supports attachment and migration of chicken neural crest cells, but not intact dimers (41,42). These results confirm that the biological functions of NC1 domains are conformation dependent. Proteolytic degradation of type IV collagen may induce exposure of cryptic sites which involved in binding of integrins, and sends new signals between cells and basement membrane (43). Cells bind to type IV collagen, and this binding was inhibited by type IV collagen derived peptides was demonstrated in several cell types that was first reported in 1986 (44,45). A synthetic peptide encompassing residues 183–205 of α3 chain type IV collagen NC1 domain was shown to specifically inhibit activation of polymorphonuclear leukocytes (46). This peptide binds to an integrin complex, promotes adhesion, chemotaxis and inhibits proliferation of various human cancer cell lines (47,48). In addition a peptide derived from α3(IV)NC1 was shown to prevent glomerular hypertrophy in the early stages of diabetic nephropathy (49).
Direct interaction between α3(IV)NC1 domain and β3 integrin signaling inhibited focal adhesion kinase (FAK) and phosphorylation of phosphatidylinositol 3-kinase (PI-3K) (50). Furthermore, inhibition of cell migration was reported in melanoma and fibrosarcoma cells using native type IV collagen or 185–205 peptide, with a decrease in expression of membrane-bound metalloproteinase (MT1-MMP) and β3 integrin subunit with a decrease in the levels of activated membrane-bound matrix metalloproteinase-2 (MMP-2) (51). MMP-2 is involved in tumor progression and metastasis and its activation is dependent on MT1-MMP/TIMP-2 (tissue inhibitor of metaloproteinase-2) complexes (52,53). α3(IV)NC1 inhibited expression of MT1-MMP in bronchial tumor cell line and 185–205 peptide inhibited their invasion on [α1(IV)]2 α2(IV) collagen (54). Altogether, this data indicates the ability of this peptide to inhibit proliferation and regulates cellular adhesion and motility. The α3 type IV collagen chain has specific interaction with invasive cancer cells. In the context of tumor progression and metastasis, the presence of α3(IV)NC1 may negatively regulate the invasion process. Interestingly, in lungs, where α1, α2, α3, α4 and α5(IV) collagen chains are expressed in normal alveolar BM, development of bronchoalveolar carcinoma correlates with loss of α3, α4 and α5 chain expression and an increase in α1 and 2 chain expression (55). The heterotrimer [α1(IV)]2 α2 is permissive for the invasion of different cancer cell lines and mediates pro-MMP-2 activation (56).
Synthesis of type IV collagen by VBM is a prerequisite for angiogenesis (57,58). Several groups have focused their attention on potential anti-angiogenic properties of NC1 domains. Dr. Brook’s group first generated all six type IV collagen NC1 domains and described the antiangiogenic effects of α2, α3 and α6(IV)NC1 by chorioallantoic membrane (CAM) assays (6). Later several researchers including myself re-conformed anti-angiogenic activities of these domains and nomenclatured as α1(IV)NC1 (arresten), α2(IV)NC1 (canstatin) and α3(IV)NC1 (tumstatin) (50,59–65). These type IV collagen derived NC1 domains inhibit endothelial cell proliferation and migration (38,50,59–62). α3(IV)NC1 seems to be studied more extensively compared to other domains of type IV collagen. Interestingly, none of the whole NC1 domains inhibited proliferation of cancer cell lines, as observed with the 185–205 (α3(IV)NC1) peptide, indicating that this effect is dependent on partial degradation of the NC1 domain.
INHIBITION OF ANGIOGENESIS BY α3(IV)NC1
α3(IV)NC1 inhibits formation of tubular structures in mouse aortic endothelial cells embedded in Matrigel and block the recruitment of capillaries in Matrigel plugs and inhibits growth of different tumors in mouse models (6, 38, 62, 63, 66). But what are the cell surface integrin receptor(s) involved in these antiangiogenic actions of α3(IV)NC1? It is clear that different integrins are key targets of α3(IV)NC1. α3(IV)NC1 binds to αVβ3 integrin in an RGD-dependent and independent manner (67). Integrin αVβ3 interacts with α3(IV)NC1 through two distinct regions, comprising residues 54–132 and 185–203 amino acids. The first site is involved in the anti-angiogenic activity, whereas the second site is involved in anti-proliferative activity on cancer cell lines (68). Adhesion of endothelial cells to α3(IV)NC1 also seem to occur through α6β1 and αVβ5 integrins, but the significance of these integrins interaction is not yet clear (63, 67). It is also known that type IV collagen NC1 domain, interacts with cells via α1β1 and α2β1 integrins (69). During angiogenesis cryptic integrin-binding sites in type IV collagen are exposed that induce a switch in integrin recognition, with a loss of α1β1 binding site and a gain of αVβ3 binding site that might be due to denaturation and concomitant degradation of type IV collagen by MMPs (70,71). These results indicate that α3(IV)NC1-integrin interactions are involved in the regulation of angiogenesis.
A peptide composed of residues 45–132 of α3(IV)NC1 fragment is sufficient to inhibit in vitro and in vivo angiogenesis by increasing apoptosis in endothelial cells (72). These results confirm specific regulatory sub-domains in α3(IV)NC1 controlling adhesion, proliferation or apoptosis in various cell types. The functional specificity of these sub domains from α3(IV)NC1 in endothelial or cancer cells is very interesting. Indeed, the recently published 3D crystal structure of type IV collagen NC1 domain reveals N and C homologous sub domains. The major difference between these sub-domains for each chain is in the region from residues 86–95 in the N sub-domain and 196–209 in the C sub-domain. These regions overlap two sequences that were previously identified to be having anti-angiogenic and anti-proliferative effects in cancer cells (73). α3(IV)NC1 or its peptides interaction with integrins seems to be involved in the disruption of contacts between endothelial or tumor cells and the BM, leading to apoptosis in these cells.
αVβ3/α3β1 INTEGRIN MEDIATED SIGNALING REGULATED BY α3(IV)NCI
The signaling mechanism involved in inhibition of endothelial cell-specific protein synthesis by α3(IV)NC1 by binding to αVβ3 integrin was reported by us (65). In addition soluble α3(IV)NC1 induces endothelial cell apoptosis by interacting with αVβ3 integrins and inhibits adhesion to VEGF in the matrix, and this effect was potentiated by anti-αVβ3 blocking antibody. Immobilized VEGF almost abolished endothelial cell apoptosis through interactions with these integrins. The inhibition of αVβ3 engagement with immobilized VEGF by α3(IV)NC1, inhibited most of its survival activity (74) (Fig. 3). These mechanisms have since been implicated in inhibition of tumor growth from several tumor cell lines such as renal cell carcinoma (786-O), CT26 (colon adenocarcinoma), prostate carcinoma (PC3), Lewis lung carcinoma (LLC), human lung cancer (H1299), human prostate cancer (DU145), human fibrosarcoma (HT1080) and teratocarcinoma (SCC-PSA1) by inhibiting tumor angiogenesis (6,63,66,67,75). The antiangiogenic activity of α3(IV)NC1 was conferred by its interaction with integrin αVβ3 and inhibiting activation of focal adhesion kinase (FAK), PI-3K, Akt/protein kinase B, mammalian target of rapamycin (mTOR) and prevents dissociation of eukaryotic translation initiation factor 4E (eIF4E) from 4E binding protein (4E-BP1) leading to the inhibition of Cap-dependent translation specifically in proliferating endothelial cells (65,76). Furthermore, these findings indicate a specific role for integrins in mediating cell specific inhibition of protein translation suggesting a potential specific mechanism of α3(IV)NC1 on endothelial cells.

Schematic illustration of apoptotic effects of α3(IV)NC1 in endothelial cell. a Proliferating endothelial cells binds to extra cellular matrix (ECM), bound VEGF or to immobilized VEGF by integrins leading to cell survival, whereas floating cells will die. b When integrin αVβ3 is engaged with ECM bound VEGF, α3(IV)NC1 can induce endothelial cell apoptosis. c Whereas the interaction between ECM-bound VEGF and integrin αVβ3 when blocked by anti-αVβ3 integrin antibody, α3(IV)NC1 induced apoptosis is significantly enhanced.
The antiangiogenic activity of α3(IV)NC1 is localized on two distinct integrin binding regions on the molecule that is separate from the region responsible for the anti-tumor cell activity (62,63). αVβ3 integrin binds to the NH2-terminal end comprising amino acid residues 54–132 region of the α3(IV)NC1 that is presumably associated with Cap-dependent translation inhibition and antiangiogenic activity (65). Whereas α3β1 integrin binds to C-terminal region 185–203 residues associated with antitumor activity (66,77). These data correlate with earlier observations that α3(IV)NC1 binds to α3β1 integrin and transdominantly inhibits expression of αVβ3 integrin (73). Several previous studies have investigated the role of α3(IV)NC1 and its peptides in tumor growth suppression due is to direct pro-apoptotic effects on endothelial cells (72,78,79). Interestingly recent studies clearly show that in addition to tumor suppressive action of α3(IV)NC1 or its peptide (T3; C-terminal end comprising amino acid residues 133–244 region of the α3(IV)NC1) directly inhibits growth of glioma cells (80). In addition a cyclopeptide derived from α3(IV)NC1 (YSNSG) was also shown to inhibit human melanoma cell proliferation about 45% (81). These results indicates that α3(IV)NC1 affects endothelial and non-endothelial cells, and does not appears to be specific for endothelial cells.
Surprisingly, abnormal tumorigenesis is observed in mice lacking α3(IV)NC1, the secret behind this abnormal tumor growth needs to be investigated. No significant tumor angiogenesis effect was observed in mice lacking collagen XVIII indicating that α3(IV)NC1 is playing a role in pathological angiogenesis to decrease tumor progression (38,82–85). However, in a controlled angiogenic process such as wound healing, α3(IV)NC1 does not affect the overall neovascularization (38). Although α3(IV)NC1 is efficient in reducing tumor neovascularization, its exact role needs to be deciphered.
Recently we identified that α3(IV)NC1 inhibits hypoxia induced cyclo-oxygenase-2 (COX-2) expression in endothelial cells via FAK/Akt/NFκB (nuclear transcription factor-kappa B) pathways, leading to decreased tumor angiogenesis and tumor growth in an α3β1 integrin dependent manner (66). Hypoxic COX-2 expression was inhibited in β3 integrin null endothelial cells upon treatment with α3(IV)NC1, indicating that COX-2 mediated signaling is not regulated through αVβ3 integrin (66). Interestingly COX-2 expression was also not affected when hypoxic α3 integrin null endothelial cells were treated with α3(IV)NC1 protein, confirming that COX-2 expression was regulated by α3β1 integrin (66). In addition to COX-2 inhibition, the down stream VEGF and bFGF protein expression was also inhibited upon α3(IV)NC1 treatment to endothelial cells (66).
COX-2 is induced by a variety of factors, including cytokines, growth factors, and tumor promoters (86). Hypoxia induced COX-2 expression regulated by NFκB (87). There is ample evidence that COX-2 over expression contributes to carcinogenesis and its disruption can both prevent and treat a variety of solid tumors (88,89). COX-2 was also reported to play a key role in tumor angiogenesis (90). Moreover, several investigators have demonstrated that blockade of COX-2 mediated pathway serves as a therapeutic benefit in different cancer models and potential target for tumor angiogenesis (91,92). These findings indicate that there may be several targets for the inhibitory effects of α3(IV)NC1 in tumor-angiogenesis, including or in addition to COX-2, VEGF and bFGF (66). The above studies supports the antiangiogenic and anti-tumorigenic activity of α3(IV)NC1 mediated through αVβ3 and α3β1 integrins (Fig. 4). Integrin α3β1 mediates signaling events that influence downstream effects of COX-2 expression that is central to the mechanism of α3(IV)NC1 regulating tumor-angiogenesis (66).

Schematic illustration of distinct molecular signaling pathways mediated by α3(IV)NC1. α3(IV)NC1 binds to different cell surface integrins, when it binds to αVβ3 integrin in endothelial cells it inhibits phosphorylation of FAK. Inhibition of FAK activation leads to inhibition of FAK/PI-3K/Akt/mTOR/eIF4E/4E-BP1 cap dependent translation resulting in activation of apoptosis and cell death. When α3(IV)NC1 binds to α3β1 integrin transdominantly inhibits αVβ3 expression in cells and inhibits NFκB mediated signaling in hypoxic conditions leading to inhibition of COX-2/VEGF/bFGF expression, resulting in inhibition of hypoxic tumor angiogenesis.
Three possible conclusions can be drawn from the signaling mechanisms of α3(IV)NC1 in regulating hypoxic tumor-angiogenesis in addition to Cap-dependent translation in endothelial cells. α3(IV)NC1 binds to cell surface integrins and inhibits hypoxia induced tumor angiogenesis by (1) inhibiting NFκB activation, leading to inhibition of COX-2 expression, which in turn results in (2) down regulation of hypoxia induced VEGF/bFGF expression in addition to inhibition of cap dependent translation (Table I). (3) In addition, α3(IV)NC1 possibly by binding to its several receptors, crosstalk with other cell surface receptors such as VEGF and bFGF, and activate specific caspase mediated signaling and regulate cell functions, as similarly shown by another type IV collagen NC1 domain, α2(IV)NC1 (93). The decrease in COX-2 expression under hypoxia results in decreased VEGF/bFGF expression representing one of the primary molecular mechanisms by which α3(IV)NC1 inhibits pathological angiogenesis that is essential for the growth of tumors (66).
Besides αVβ3 and α3β1 integrins, another common target for α3(IV)NC1 seems to be inhibition of MMP-2 activation. A direct interaction with the catalytic domain has been shown in case of endostatin, such an interaction has not been demonstrated for α3(IV)NC1, and needs further investigation in this direction. An increase in the basal level of pro-MMP-2 activity was also observed in α3(IV)NC1 null mice (94). Similar possible interaction with other MMPs might exist in the generation of α3(IV)NC1 and warrants further investigation.
CONCLUSIONS AND PERSPECTIVES
In the last decade several different endogenous angiogenesis inhibitors have been discovered from ECM. Researchers identified that these endogenous angiogenesis inhibitors, through pharmacological studies, showed promising anti-tumor activity, but their mechanism of action and physiological role is not yet understood. Type IV collagen derived endogenous angiogenesis inhibitor, α3(IV)NC1, binds to different cell surface integrins and exerts its effects through multiple mechanisms including induction of endothelial cell apoptosis, inhibition of cell proliferation, tube formation in endothelial cells, and inhibit or alter the functions of pro-angiogenic growth factors. In this regard α3(IV)NC1 demonstrates a genetic evidence for its physiological function in negative regulation of tumor growth and progression in mice (66,73). However, the detailed physiological and biological functions of α3(IV)NC1 are not yet completely identified. The anti-angiogenic and anti-tumorigenic activities of α3(IV)NC1 is dependent on αVβ3 and α3β1 integrins (66,73). In mice deletion of β3 integrin or deletion of α3(IV)NC1 enhanced tumor angiogenesis, suggesting a role for this integrin and this NC1 domain in limiting angiogenesis in vivo (38,95) The anti-angiogenic activities of α3(IV)NC1 is partly dependent on binding to αVβ3 integrin which also supports this hypothesis. Whereas anti-tumorigenic activities of α3(IV)NC1 are dependent on its binding to α3β1 integrin which transdominantly inhibits αVβ3 integrin (73). Another feature of α3(IV)NC1 is inhibition of various signaling molecules which are involved in cell survival. However anti-angiogenic and anti-tumorigenic activities of α3(IV)NC1 also seems to be because of the inhibition of different locally released growth factors (66).
In addition several angiogenic inhibitors including αV integrin antagonist EMD 121974, 2-methoxyestradiol (panzam) and MMP-2, -9 inhibitor COL-3 etc. are currently in one of two phases in human clinical trails (96). A recent study suggests that up-regulation of specific pro-angiogenic factors is a common mechanism for colorectal and renal carcinoma cells to evade inhibition by several of extracellular derived endogenous angiogenesis inhibitors (97). Questions regarding resistance to these angiogenesis inhibitors do remain unanswered; however a combination of radiation therapy with other anti-angiogenic therapies may also prove to be clinically useful and effective (97). Earlier lessons from preclinical trials of angiostatin, endostatin, thrombospondin-1 (ABT-510) and 2-ME suggest that more basic research is required for better understanding of the mechanisms of action associated with each of these endogenous angiogenesis inhibitor molecules. Presently, some of the anti-angiogenic agents such as Bevacizumab and several other VEGFR tyrosine kinase inhibitors; vatalanib (PTK787/ZK 222584), semaxanib (SU5416), sunitinib (SU11248), sorafenib (BAY 43-9006) etc., are in clinical trials (98,99). Further extensive evaluation of α3(IV)NC1 through pharmacokinetic studies is needed to address this molecule as an inhibitor of angiogenesis and to be considered for the clinical trials in the context of tumor angiogenesis and cancer.
Change history
05 June 2020
Editor’s Note: Readers are alerted that this article has been subject to an investigation by The Office of Research Integrity (ORI). The outcome of this investigation is being considered by the Editor-in-Chief. We will update readers once we have further information and all parties have been given an opportunity to respond in full.
15 January 2021
A Correction to this paper has been published: https://doi.org/10.1007/s11095-020-02974-x
References
M. Paulsson. Basement membrane proteins: structure, assembly, and cellular interactions. Crit. Rev. Biochem. Mol. Biol. 27:93–127 (1992).
J. C. Schittny, and P. D. Yurchenco. Basement membranes: molecular organization and function in development and disease. Curr. Opin. Cell Biol. 1:983–988 (1989).
N. A. Kefalides. Isolation of a collagen from basement membranes containing three identical-chains. Biochem. Biophys. Res. Commun. 45:226–234 (1971).
R. Timpl, H. Wiedemann, V. van Delden, H. Furthmayr, and K. Kuhn. A network model for the organization of type IV collagen molecules in basement membranes. Eur. J. Biochem. 120:203–211 (1981).
M. S. O’Reilly, T. Boehm, Y. Shing, N. Fukai, G. Vasios, W. S. Lane, E. Flynn, J. R. Birkhead, B. R. Olsen, and J. Folkman. Endostatin: an endogenous inhibitor of angiogenesis and tumor growth. Cell. 88:277–285 (1997).
E. Petitclerc, A. Boutaud, A. Prestayko, J. Xu, Y. Sado, Y. Ninomiya, M. P. Sarras Jr., B. G. Hudson, and P. C. Brooks. New functions for non-collagenous domains of human collagen type IV. Novel integrin ligands inhibiting angiogenesis and tumor growth in vivo. J. Biol. Chem. 275:8051–8061 (2000).
J. Folkman. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1:27–31 (1995).
J. Folkman. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285:1182–1186 (1971).
M. W. Kieran, J. Folkman, and J. Heymach. Angiogenesis inhibitors and hypoxia. Nat. Med. 9:1104 author reply 1104–1105 (2003).
J. Folkman. Tumor suppression by p53 is mediated in part by the antiangiogenic activity of endostatin and tumstatin. Sci. STKE. 2006:pe35 (2006).
D. J. Good, P. J. Polverini, F. Rastinejad, M. M. Le Beau, R. S. Lemons, W. A. Frazier, and N. P. Bouck. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc. Natl. Acad. Sci. U. S. A. 87:6624–6628 (1990).
M. Yi, and E. Ruoslahti. A fibronectin fragment inhibits tumor growth, angiogenesis, and metastasis. Proc. Natl. Acad. Sci. U. S. A. 98:620–624 (2001).
A. R. Albig, and W. P. Schiemann. Fibulin-5 antagonizes vascular endothelial growth factor (VEGF) signaling and angiogenic sprouting by endothelial cells. DNA Cell Biol. 23:367–379 (2004).
P. D. Yurchenco, and J. J. O’Rear. Basal lamina assembly. Curr. Opin. Cell. Biol. 6:674–681 (1994).
B. Blumberg, A. J. MacKrell, P. F. Olson, M. Kurkinen, J. M. Monson, J. E. Natzle, and J. H. Fessler. Basement membrane procollagen IV and its specialized carboxyl domain are conserved in Drosophila, mouse, and human. J. Biol. Chem. 262:5947–5950 (1987).
K. O. Netzer, K. Suzuki, Y. Itoh, B. G. Hudson, and R. G. Khalifah. Comparative analysis of the noncollagenous NC1 domain of type IV collagen: identification of structural features important for assembly, function, and pathogenesis. Protein Sci. 7:1340–1351 (1998).
M. P. Sarras Jr., X. Zhang, J. K. Huff, M. A. Accavitti, P. L. St John, and D. R. Abrahamson. Extracellular matrix (mesoglea) of Hydra vulgaris III. Formation and function during morphogenesis of hydra cell aggregates. Dev. Biol. 157:383–398 (1993).
B. G. Hudson, S. T. Reeders, and K. Tryggvason. Type IV collagen: structure, gene organization, and role in human diseases. Molecular basis of Goodpasture and Alport syndromes and diffuse leiomyomatosis. J. Biol. Chem. 268:26033–26036 (1993).
R. Kalluri. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer. 3:422–433 (2003).
J. D. Filie, P. D. Burbelo, and C. A. Kozak. Genetic mapping of the alpha 1 and alpha 2 (IV) collagen genes to mouse chromosome 8. Mamm. Genome. 6:487 (1995).
R. Momota, M. Sugimoto, T. Oohashi, K. Kigasawa, H. Yoshioka, and Y. Ninomiya. Two genes, COL4A3 and COL4A4 coding for the human alpha3(IV) and alpha4(IV) collagen chains are arranged head-to-head on chromosome 2q36. FEBS Lett. 424:11–16 (1998).
R. Soininen, M. Huotari, S. L. Hostikka, D. J. Prockop, and K. Tryggvason. The structural genes for alpha 1 and alpha 2 chains of human type IV collagen are divergently encoded on opposite DNA strands and have an overlapping promoter region. J. Biol. Chem. 263:17217–17220 (1988).
A. Boutaud, D. B. Borza, O. Bondar, S. Gunwar, K. O. Netzer, N. Singh, Y. Ninomiya, Y. Sado, M. E. Noelken, and B. G. Hudson. Type IV collagen of the glomerular basement membrane. Evidence that the chain specificity of network assembly is encoded by the noncollagenous NC1 domains. J. Biol. Chem. 275:30716–30724 (2000).
P. D. Yurchenco, and G. C. Ruben. Basement membrane structure in situ: evidence for lateral associations in the type IV collagen network. J. Cell Biol. 105:2559–2568 (1987).
P. Dehan, D. Waltregny, A. Beschin, A. Noel, V. Castronovo, K. Tryggvason, J. De Leval, and J.M. Foidart. Loss of type IV collagen alpha 5 and alpha 6 chains in human invasive prostate carcinomas. Am. J. Pathol. 151:1097–1104 (1997).
R. Fleischmajer, K. Kuhn, Y. Sato, E. D. MacDonald 2nd, J. S. Perlish, T. C. Pan, M. L. Chu, Y. Kishiro, T. Oohashi, S. M. Bernier, Y. Yamada, and Y. Ninomiya. There is temporal and spatial expression of alpha1 (IV), alpha2 (IV), alpha5 (IV), alpha6 (IV) collagen chains and beta1 integrins during the development of the basal lamina in an “in vitro” skin model. J. Invest. Dermatol. 109:527–533 (1997).
J. H. Miner, and J. R. Sanes. Collagen IV alpha 3, alpha 4, and alpha 5 chains in rodent basal laminae: sequence, distribution, association with laminins, and developmental switches. J. Cell Biol. 127:879–891 (1994).
K. Tanaka, K. Iyama, M. Kitaoka, Y. Ninomiya, T. Oohashi, Y. Sado, and T. Ono. Differential expression of alpha 1(IV), alpha 2(IV), alpha 5(IV) and alpha 6(IV) collagen chains in the basement membrane of basal cell carcinoma. Histochem. J. 29:563–570 (1997).
B. G. Hudson, K. Tryggvason, M. Sundaramoorthy, and E. G. Neilson. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N. Engl. J. Med. 348:2543–2556 (2003).
L. Heidet, L. Cohen-Solal, E. Boye, P. Thorner, M. J. Kemper, A. David, L. Larget Piet, J. Zhou, F. Flinter, X. Zhang, M. C. Gubler, and C. Antignac. Novel COL4A5/COL4A6 deletions and further characterization of the diffuse leiomyomatosis-Alport syndrome (DL-AS) locus define the DL critical region. Cytogenet. Cell Genet. 78:240–246 (1997).
J. Zhou, T. Mochizuki, H. Smeets, C. Antignac, P. Laurila, A. de Paepe, K. Tryggvason, and S. T. Reeders. Deletion of the paired alpha 5(IV) and alpha 6(IV) collagen genes in inherited smooth muscle tumors. Science. 261:1167–1169 (1993).
D. Cosgrove, D. T. Meehan, J. A. Grunkemeyer, J. M. Kornak, R. Sayers, W. J. Hunter, and G. C. Samuelson. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 10:2981–2992 (1996).
T. H. Lee, T. Rhim, and S. S. Kim. Prothrombin kringle-2 domain has a growth inhibitory activity against basic fibroblast growth factor-stimulated capillary endothelial cells. J. Biol. Chem. 273:28805–28812 (1998).
G. K. Reddy, S. Gunwar, R. Kalluri, B. G. Hudson, and M. E. Noelken. Structure and composition of type IV collagen of bovine aorta. Biochim. Biophys. Acta. 1157:241–251 (1993).
A. Ries, J. Engel, A. Lustig, and K. Kuhn. The function of the NC1 domains in type IV collagen. J. Biol. Chem. 270:23790–23794 (1995).
M. Chen, M. P. Marinkovich, A. Veis, X. Cai, C. N. Rao, E. A. O’Toole, and D. T. Woodley. Interactions of the amino-terminal noncollagenous (NC1) domain of type VII collagen with extracellular matrix components. A potential role in epidermal-dermal adherence in human skin. J. Biol. Chem. 272:14516–14522 (1997).
D. B. Borza, O. Bondar, Y. Ninomiya, Y. Sado, I. Naito, P. Todd, and B. G. Hudson. The NC1 domain of collagen IV encodes a novel network composed of the alpha 1, alpha 2, alpha 5, and alpha 6 chains in smooth muscle basement membranes. J. Biol. Chem. 276:28532–28540 (2001).
Y. Hamano, M. Zeisberg, H. Sugimoto, J. C. Lively, Y. Maeshima, C. Yang, R.O. Hynes, Z. Werb, A. Sudhakar, and R. Kalluri. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cells. 3:589–601 (2003).
W. H. Pearce, and V. P. Shively. Abdominal aortic aneurysm as a complex multifactorial disease: interactions of polymorphisms of inflammatory genes, features of autoimmunity, and current status of MMPs. Ann. N. Y. Acad. Sci. 1085:117–132 (2006).
X. Zhang, B. G. Hudson, and M. P. Sarras Jr. Hydra cell aggregate development is blocked by selective fragments of fibronectin and type IV collagen. Dev. Biol. 164:10–23 (1994).
P. J. Lein, D. Higgins, D. C. Turner, L. A. Flier, and V. P. Terranova. The NC1 domain of type IV collagen promotes axonal growth in sympathetic neurons through interaction with the alpha 1 beta 1 integrin. J. Cell Biol. 113:417–428 (1991).
R. Perris, J. Syfrig, M. Paulsson, and M. Bronner-Fraser. Molecular mechanisms of neural crest cell attachment and migration on types I and IV collagen. J. Cell Sci. 106(Pt 4):1357–1368 (1993).
N. Ortega, and Z. Werb. New functional roles for non-collagenous domains of basement membrane collagens. J. Cell Sci. 115:4201–4214 (2002).
M. Aumailley, and R. Timpl. Attachment of cells to basement membrane collagen type IV. J. Cell Biol. 103:1569–1575 (1986).
M. K. Chelberg, J. B. McCarthy, A. P. Skubitz, L. T. Furcht, and E. C. Tsilibary. Characterization of a synthetic peptide from type IV collagen that promotes melanoma cell adhesion, spreading, and motility. J. Cell Biol. 111:261–270 (1990).
J. C. Monboisse, R. Garnotel, G. Bellon, N. Ohno, C. Perreau, J. P. Borel, and N. A. Kefalides. The alpha 3 chain of type IV collagen prevents activation of human polymorphonuclear leukocytes. J. Biol. Chem. 269:25475–25482 (1994).
J. Han, N. Ohno, S. Pasco, J. C. Monboisse, J. P. Borel, and N. A. Kefalides. A cell binding domain from the alpha3 chain of type IV collagen inhibits proliferation of melanoma cells. J. Biol. Chem. 272:20395–20401 (1997).
T. A. Shahan, A. Fawzi, G. Bellon, J. C. Monboisse, and N. A. Kefalides. Regulation of tumor cell chemotaxis by type IV collagen is mediated by a Ca(2)-dependent mechanism requiring CD47 and the integrin alpha(V)beta(3). J. Biol. Chem. 275:4796–4802 (2000).
Y. Yamamoto, Y. Maeshima, H. Kitayama, S. Kitamura, Y. Takazawa, H. Sugiyama, Y. Yamasaki, and H. Makino. Tumstatin peptide, an inhibitor of angiogenesis, prevents glomerular hypertrophy in the early stage of diabetic nephropathy. Diabetes. 53:1831–1840 (2004).
S. Pasco, J. C. Monboisse, and N. Kieffer. The alpha 3(IV)185–206 peptide from noncollagenous domain 1 of type IV collagen interacts with a novel binding site on the beta 3 subunit of integrin alpha Vbeta 3 and stimulates focal adhesion kinase and phosphatidylinositol 3-kinase phosphorylation. J. Biol. Chem. 275:32999–33007 (2000).
S. Pasco, J. Han, P. Gillery, G. Bellon, F. X. Maquart, J. P. Borel, N. A. Kefalides, and J. C. Monboisse. A specific sequence of the noncollagenous domain of the alpha3(IV) chain of type IV collagen inhibits expression and activation of matrix metalloproteinases by tumor cells. Cancer Res. 60:467–473 (2000).
Y. Itoh, A. Ito, K. Iwata, K. Tanzawa, Y. Mori, and H. Nagase. Plasma membrane-bound tissue inhibitor of metalloproteinases (TIMP)-2 specifically inhibits matrix metalloproteinase 2 (gelatinase A) activated on the cell surface. J. Biol. Chem. 273:24360–24367 (1998).
T. Kinoshita, H. Sato, A. Okada, E. Ohuchi, K. Imai, Y. Okada, and M. Seiki. TIMP-2 promotes activation of progelatinase A by membrane-type 1 matrix metalloproteinase immobilized on agarose beads. J. Biol. Chem. 273:16098–16103 (1998).
C. Martinella-Catusse, M. Polette, A. Noel, C. Gilles, P. Dehan, C. Munaut, A. Colige, L. Volders, J. C. Monboisse, J. M. Foidart, and P. Birembaut. Down-regulation of MT1-MMP expression by the alpha3 chain of type IV collagen inhibits bronchial tumor cell line invasion. Lab. Invest. 81:167–175 (2001).
K. Y. Nakano, K. I. Iyama, T. Mori, M. Yoshioka, T. Hiraoka, Y. Sado, and Y. Ninomiya. Loss of alveolar basement membrane type IV collagen alpha3, alpha4, and alpha5 chains in bronchioloalveolar carcinoma of the lung. J. Pathol. 194:420–427 (2001).
E. Maquoi, F. Frankenne, A. Noel, H. W. Krell, F. Grams, and J. M. Foidart. Type IV collagen induces matrix metalloproteinase 2 activation in HT1080 fibrosarcoma cells. Exp. Cell Res. 261:348–359 (2000).
M. E. Maragoudakis, E. Missirlis, G. D. Karakiulakis, M. Sarmonica, M. Bastakis, and N. Tsopanoglou. Basement membrane biosynthesis as a target for developing inhibitors of angiogenesis with anti-tumor properties. Kidney Int. 43:147–150 (1993).
G. C. Haralabopoulos, D. S. Grant, H. K. Kleinman, P. I. Lelkes, S. P. Papaioannou, and M. E. Maragoudakis. Inhibitors of basement membrane collagen synthesis prevent endothelial cell alignment in matrigel in vitro and angiogenesis in vivo. Lab. Invest. 71:575–582 (1994).
P. C. Colorado, A. Torre, G. Kamphaus, Y. Maeshima, H. Hopfer, K. Takahashi, R. Volk, E. D. Zamborsky, S. Herman, P. K. Sarkar, M. B. Ericksen, M. Dhanabal, M. Simons, M. Post, D. W. Kufe, R. R. Weichselbaum, V. P. Sukhatme, and R. Kalluri. Anti-angiogenic cues from vascular basement membrane collagen. Cancer Res. 60:2520–2526 (2000).
A. Sudhakar, P. Nyberg, V. G. Keshamouni, A. P. Mannam, J. Li, H. Sugimoto, D. Cosgrove, and R. Kalluri. Human alpha1 type IV collagen NC1 domain exhibits distinct antiangiogenic activity mediated by alpha1beta1 integrin. J. Clin. Invest. 115:2801–2810 (2005).
G. D. Kamphaus, P. C. Colorado, D. J. Panka, H. Hopfer, R. Ramchandran, A. Torre, Y. Maeshima, J. W. Mier, V. P. Sukhatme, and R. Kalluri. Canstatin, a novel matrix-derived inhibitor of angiogenesis and tumor growth. J. Biol. Chem. 275:1209–1215 (2000).
Y. Maeshima, P. C. Colorado, A. Torre, K. A. Holthaus, J. A. Grunkemeyer, M. B. Ericksen, H. Hopfer, Y. Xiao, I. E. Stillman, and R. Kalluri. Distinct antitumor properties of a type IV collagen domain derived from basement membrane. J. Biol. Chem. 275:21340–21348 (2000).
V. Pedchenko, R. Zent, and B. G. Hudson. Alpha(v)beta3 and alpha(v)beta5 integrins bind both the proximal RGD site and non-RGD motifs within noncollagenous (NC1) domain of the alpha3 chain of type IV collagen: implication for the mechanism of endothelia cell adhesion. J. Biol. Chem. 279:2772–2780 (2004).
A. G. Marneros, and B. R. Olsen. The role of collagen-derived proteolytic fragments in angiogenesis. Matrix Biology. 20:337–345 (2001).
Y. Maeshima, A. Sudhakar, J. C. Lively, K. Ueki, S. Kharbanda, C. R. Kahn, N. Sonenberg, R. O. Hynes, and R. Kalluri. Tumstatin, an endothelial cell-specific inhibitor of protein synthesis. Science. 295:140–143 (2002).
C. S. Boosani, A. P. Mannam, D. Cosgrove, R. Silva, K. M. Hodivala-Dilke, V. G. Keshamouni, and A. Sudhakar. Regulation of COX-2 mediated signaling by {alpha}3 type IV noncollagenous domain in tumor angiogenesis. Blood. 110:1168–1177 (2007).
Y. Maeshima, P. C. Colorado, and R. Kalluri. Two RGD-independent alpha vbeta 3 integrin binding sites on tumstatin regulate distinct anti-tumor properties. J. Biol. Chem. 275:23745–23750 (2000).
T. A. Shahan, Z. Ziaie, S. Pasco, A. Fawzi, G. Bellon, J. C. Monboisse, and N. A. Kefalides. Identification of CD47/integrin-associated protein and alpha(v)beta3 as two receptors for the alpha3(IV) chain of type IV collagen on tumor cells. Cancer Res. 59:4584–4590 (1999).
J. A. Eble, R. Golbik, K. Mann, and K. Kuhn. The alpha 1 beta 1 integrin recognition site of the basement membrane collagen molecule [alpha 1(IV)]2 alpha 2(IV). EMBO J. 12:4795–4802 (1993).
J. Xu, D. Rodriguez, E. Petitclerc, J. J. Kim, M. Hangai, Y. S. Moon, G. E. Davis, and P. C. Brooks. Proteolytic exposure of a cryptic site within collagen type IV is required for angiogenesis and tumor growth in vivo. J. Cell Biol. 154:1069–1079 (2001).
J. A. Eble, A. Ries, A. Lichy, K. Mann, H. Stanton, J. Gavrilovic, G. Murphy, and K. Kuhn. The recognition sites of the integrins alpha1beta1 and alpha2beta1 within collagen IV are protected against gelatinase A attack in the native protein. J. Biol. Chem. 271:30964–30970 (1996).
Y. Maeshima, M. Manfredi, C. Reimer, K. A. Holthaus, H. Hopfer, B. R. Chandamuri, S. Kharbanda, and R. Kalluri. Identification of the anti-angiogenic site within vascular basement membrane-derived tumstatin. J. Biol. Chem. 276:15240–15248 (2001).
C. M. Borza, A. Pozzi, D. B. Borza, V. Pedchenko, T. Hellmark, B. G. Hudson, and R. Zent. Integrin alpha3beta1: a novel receptor for alpha 3(IV) noncollagenous domain and a trans-dominant inhibitor for integrin alphavbeta3. J. Biol. Chem. 281:20932–20939 (2006).
H. Hutchings, N. Ortega, and J. Plouet. Extracellular matrix-bound vascular endothelial growth factor promotes endothelial cell adhesion, migration, and survival through integrin ligation. FASEB J. 17:1520–1522 (2003).
T. Miyoshi, S. Hirohata, H. Ogawa, M. Doi, M. Obika, T. Yonezawa, Y. Sado, S. Kusachi, S. Kyo, S. Kondo, Y. Shiratori, B. G. Hudson, and Y. Ninomiya. Tumor-specific expression of the RGD-alpha3(IV)NC1 domain suppresses endothelial tube formation and tumor growth in mice. FASEB J. 20:1904–1906 (2006).
A. Sudhakar, H. Sugimoto, C. Yang, J. Lively, M. Zeisberg, and R. Kalluri. Human tumstatin and human endostatin exhibit distinct antiangiogenic activities mediated by alpha v beta 3 and alpha 5 beta 1 integrins. Proc. Natl. Acad. Sci. U. S. A. 100:4766–4771 (2003).
N. Floquet, S. Pasco, L. Ramont, P. Derreumaux, J. Y. Laronze, J. M. Nuzillard, F. X. Maquart, A. J. Alix, and J. C. Monboisse. The antitumor properties of the alpha3(IV)-(185–203) peptide from the NC1 domain of type IV collagen (tumstatin) are conformation-dependent. J. Biol. Chem. 279:2091–2100 (2004).
Y. Maeshima, P. C. Colorado, A. Torre, K. A. Holthaus, J. A. Grunkemeyer, M. D. Ericksen, H. Hopfer, Y. Xiao, I. E. Stillman, and R. Kalluri. Distinct anti-tumor properties of a type IV collagen domain derived from basement membrane. J. Biol. Chem. 275:21340–21348 (2000).
Y. Maeshima, U. L. Yerramalla, M. Dhanabal, K. A. Holthaus, S. Barbashov, S. Kharbanda, C. Reimer, M. Manfredi, W. M. Dickerson, and R. Kalluri. Extracellular matrix derived peptide binds to alphavbeta3 integrin and inhibits angiogenesis. J. Biol. Chem. 276:31959–31968 (2001).
T. Kawaguchi, Y. Yamashita, M. Kanamori, R. Endersby, K.S. Bankiewicz, S. J. Baker, G. Bergers, and R. O. Pieper. The PTEN/Akt pathway dictates the direct alphaVbeta3-dependent growth-inhibitory action of an active fragment of tumstatin in glioma cells in vitro and in vivo. Cancer Res. 66:11331–11340 (2006).
J. Thevenard, N. Floquet, L. Ramont, E. Prost, J. M. Nuzillard, M. Dauchez, H. Yezid, A. J. Alix, F. X. Maquart, J. C. Monboisse, and S. Brassart-Pasco. Structural and antitumor properties of the YSNSG cyclopeptide derived from tumstatin. Chem. Biol. 13:1307–1315 (2006).
Y. Hamano, and R. Kalluri. Tumstatin, the NC1 domain of alpha3 chain of type IV collagen, is an endogenous inhibitor of pathological angiogenesis and suppresses tumor growth. Biochem. Biophys. Res. Commun. 333:292–298 (2005).
A. Sudhakar, and C. S. Boosani. Signaling mechanisms of endogenous angiogenesis inhibitors derived from type IV collagen. Gene Regulation and Systems Biology. 1:217–226 (2007).
T. M. Mundel, and R. Kalluri. Type IV collagen-derived angiogenesis inhibitors. Microvasc. Res. 74:85–89 (2007).
N. Fukai, L. Eklund, A. G. Marneros, S. P. Oh, D. R. Keene, L. Tamarkin, M. Niemela, M. Ilves, E. Li, T. Pihlajaniemi, and B. R. Olsen. Lack of collagen XVIII/endostatin results in eye abnormalities. EMBO J. 21:1535–1544 (2002).
R. N. DuBois, M. Tsujii, P. Bishop, J. A. Awad, K. Makita, and A. Lanahan. Cloning and characterization of a growth factor-inducible cyclooxygenase gene from rat intestinal epithelial cells. Am. J. Physiol. 266:G822–827 (1994).
J. F. Schmedtje Jr., Y. S. Ji, W. L. Liu, R. N. DuBois, and M. S. Runge. Hypoxia induces cyclooxygenase-2 via the NF-kappaB p65 transcription factor in human vascular endothelial cells. J. Biol. Chem. 272:601–608 (1997).
A. W. Wu, J. Gu, Z. F. Li, J. F. Ji, and G. W. Xu. COX-2 expression and tumor angiogenesis in colorectal cancer. World J. Gastroenterol. 10:2323–2326 (2004).
K. Subbaramaiah, D. Zakim, B. B. Weksler, and A. J. Dannenberg. Inhibition of cyclooxygenase: a novel approach to cancer prevention. Proc. Soc. Exp. Biol. Med. 216:201–210 (1997).
A. L. Harris. Hypoxia—a key regulatory factor in tumour growth. Nat. Rev. Cancer. 2:38–47 (2002).
D. J. Panka, and J. W. Mier. Canstatin inhibits Akt activation and induces Fas-dependent apoptosis in endothelial cells. J. Biol. Chem. 278:37632–37636 (2003).
M. Kunz, S. Moeller, D. Koczan, P. Lorenz, R. H. Wenger, M. O. Glocker, H. J. Thiesen, G. Gross, and S. M. Ibrahim. Mechanisms of hypoxic gene regulation of angiogenesis factor Cyr61 in melanoma cells. J. Biol. Chem. 278:45651–45660 (2003).
C. Magnon, A. Galaup, B. Mullan, V. Rouffiac, C. Bouquet, J. M. Bidart, F. Griscelli, P. Opolon, and M. Perricaudet. Canstatin acts on endothelial and tumor cells via mitochondrial damage initiated through interaction with alphavbeta3 and alphavbeta5 integrins. Cancer Res. 65:4353–4361 (2005).
M. Zeisberg, M. Khurana, V. H. Rao, D. Cosgrove, J. P. Rougier, M. C. Werner, C. F. Shield, Z. Werb, and R. Kalluri. Stage-specific action of matrix metalloproteinases influences progressive hereditary kidney disease. PLoS Med. 3:e100 (2006).
L. E. Reynolds, L. Wyder, J. C. Lively, D. Taverna, S. D. Robinson, X. Huang, D. Sheppard, R. O. Hynes, and K. M. Hodivala-Dilke. Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat. Med. 8:27–34 (2002).
M. Jansen, P. C. de Witt Hamer, A. N. Witmer, D. Troost, and C. J. van Noorden. Current perspectives on antiangiogenesis strategies in the treatment of malignant gliomas. Brain Res. Brain Res. Rev. 45:143–163 (2004).
N. T. Fernando, M. Koch, C. Rothrock, L. K. Gollogly, P. A. D’Amore, S. Ryeom, and S. S. Yoon. Tumor escape from endogenous, extracellular matrix-associated angiogenesis inhibitors by up-regulation of multiple proangiogenic factors. Clin. Cancer Res. 14:1529–1539 (2008).
H. Hurwitz, L. Fehrenbacher, W. Novotny, T. Cartwright, J. Hainsworth, W. Heim, J. Berlin, A. Baron, S. Griffing, E. Holmgren, N. Ferrara, G. Fyfe, B. Rogers, R. Ross, and F. Kabbinavar. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N. Engl. J. Med. 350:2335–2342 (2004).
A. Morabito, E. De Maio, M. Di Maio, N. Normanno, and F. Perrone. Tyrosine kinase inhibitors of vascular endothelial growth factor receptors in clinical trials: current status and future directions. Oncologist. 11:753–764 (2006).
C. S. Boosani, and A. Sudhakar. Cloning, purification, and characterization of a non-collagenous anti-angiogenic protein domain from human alpha1 type IV collagen expressed in Sf9 cells. Protein Expr. Purif. 49:211–218 (2006).
C. S. Boosani, and A. Sudhakar. Molecular cloning and functional characterization of mouse {alpha}3(IV)NC1 Clinical Medicine. Oncology. 2:73–81 (2008).
Acknowledgements
We would like to apologize to those of our colleagues whose work we were unable to cite in the review due to journal restrictions. Research related to this work in the authors’ laboratory is supported by the Flight Attendant Medical Research Institute Young Clinical Scientist Award Grant (FAMRI No. 062558 to S. A). We also acknowledge the generous financial support from Dobleman Head and Neck Cancer Institute and startup research funds of Cell Signaling and Tumor Angiogenesis Laboratory at Boys Town National Research Hospital to S. A. We thank the support of AACR-AstraZeneca Scholar-in-Training Award (2008) to Dr. Boosani in recognition of promising cancer research and Dr. Cosgrove in editing and proofreading this review article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Akulapalli Sudhakar and Chandra S. Boosani contributed equally to this manuscript.
Rights and permissions
This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article
Cite this article
Sudhakar, A., Boosani, C.S. RETRACTED ARTICLE: Inhibition of Tumor Angiogenesis by Tumstatin: Insights into Signaling Mechanisms and Implications in Cancer Regression. Pharm Res 25, 2731–2739 (2008). https://doi.org/10.1007/s11095-008-9634-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11095-008-9634-z