
Abstract
Contact glow discharge electrolysis (CGDE)/plasma electrolysis (PE) which is associated with the formation of a light emitting plasma around an electrode in a high conductivity electrolyte solution at moderate voltages up to ~1 kV, has in recent years attracted considerable interest as a tool for generating a large quantity of heat and a high yield of solvent-split radicals. These potentialities of CGDE/PE have, in fact, been exploited by a large number of investigators for applications ranging over areas as varied as synthetic chemistry, waste water treatment, degradation of polymers, electrosurgical tools, surface engineering, nanoparticle fabrication, machining and micro-machining, hydrogen production with very encouraging results. The article reviews comprehensively these results.
Similar content being viewed by others

Introduction
Electrolysis of a relatively high conductivity electrolyte solution when driven by DC, pulsed DC-, AC- or RF sources at several hundred volts to a maximum of ~1 kV, gives rise to a light emitting gas-plasma around one of the electrodes immersed well inside a liquid electrolyte. This electrolytic phenomenon with plasma formation at an electrode dipped well inside the liquid is referred as contact glow discharge electrolysis (CGDE) or plasma electrolysis (PE) where chemical reactions are brought about by processes additional to a normal electron transfer one between an ion and the electrode leading to strong non-faradaic chemical effects. The non-faradaic effects originate from energy transfer processes between the energized particles emanated from the plasma and the species in the electrolyte near the plasma-liquid interface, and also from the reactions within the plasma around the electrode. These processes give rise to a high yield of solvent-split radicals such as OH· and H· near the electrode plasma-aqueous electrolyte interfacial area. The phenomenon is also associated with generation of considerable heat in both the plasma and near the plasma-liquid interface. These two effects have been explored under the name electrolytic plasma technology (EPT) or electrolytic plasma processing (EPP) for a variety of potential applications ranging over surface treatments (e.g. heating, cleaning, texturing, micro-to-nanostructured coating and alloying metal surfaces), micro-precision machining of non-conducting and conducting materials, fabrication of nanoparticles, synthesis of compounds ranging from simple organics and bio-organics to polymers and superabsorbent composites, treatment of waste waters containing various organic and inorganic pollutants, degradation of polymers, electrosurgical devices, and hydrogen production. Results of studies on the origin, plasma diagnostics and non-faradaic chemical effects of CGDE were reviewed recently [1]. The progress made in several of the applications of CGDE/PE such as surface engineering [2–6], micromachining [7], nanoparticle fabrication [8–12], waste water treatment [13] and hydrogen generation [14] was reviewed over 1999–2015. However, there has not been published any comprehensive review covering the whole range of current applications of CGDE in aqueous and non-aqueous media besides the above ones for a specific application. Further, several more publications in the areas on these published reviews have appeared. Certainly, there is a need for an updated review for the whole range of application areas of CGDE. It may be mentioned that there has not been yet published any review on applications of CGDE for synthetic chemistry, degradation of polymers and bio-polymers. The article is aimed towards bridging these gaps and reviews applications of CGDE in all the areas explored till date. It may be noted here that the use of high-voltage (tens of kilovolts) electrolytic plasmas generated in low-conductivity (<50 μS/cm) water or organic media such as gliding arc plasma discharges (glidarc) [15, 16], electrohydraulic discharges (EHD) [15, 17–19] etc. for purposes like decontamination and sterilization of water, is not covered in this article.
Background Information on Contact Glow Discharge Electrolysis (CGDE)
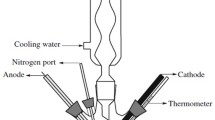
Conventional electrolysis of an electrolyte solution when carried out at several hundred DC, pulsed DC, AC or RF voltages breaks down accompanied by a steep fall in the current after rising to some maximum with simultaneous formation of some vapor envelop anchoring on the smaller electrode, the anode or the cathode immersed well underneath the liquid electrolyte. The threshold voltage for the breakdown of normal electrolysis (NE) is referred as the breakdown voltage (VB). With further increases in the applied voltage, the vapor envelop gets stabilized and at some critical voltage called the mid-point voltage (VD), a light emitting glow-discharge plasma fills the vapor envelop over the electrode and the current records a minimum. At or beyond this voltage, a non-conventional electrolysis generally referred as contact glow discharge electrolysis (CGDE) proceeds. The magnitude of VD (duly corrected for the Ohmic drop in the electrolysis cell) of CGDE at an electrode though insensitive to electrolyte conductivity, temperature and surface tension, was found much higher for anodic CGDE (~420 V) than for cathodic CGDE (~280 V) [1, 20, 21] (Fig. 1).

Current–voltage characteristics of electrolysis [transition from normal electrolysis (NE)] to contact glow discharge electrolysis (AB normal electrolysis, B breakdown voltage, BC transition region, CD partial contact glow discharge electrolysis, D mid-point voltage, DE contact glow discharge electrolysis)
This large difference may be ascribed to the huge difference (~102 to 103 times) in Townsend secondary electron emission coefficient from the cathode element between cathodic CGDE plasma where the cathode is a metal, and the anodic CGDE plasma where the cathode is the liquid near the plasma-anolyte solution interface. Consequently, the cathode voltage drop where the majority of the voltage drop occurs in the plasma, is much higher in anodic CGDE than in cathodic CGDE [1, 21]. The operation of a CGDE plasma, and in particular the cathodic CGDE, one is attended with liberation of intense heat. This strong thermal effect was, in fact, explored successfully for applications in surface engineering, nanoparticle fabrication and machining and micromachining of materials. The effect may be coupled with diffusion of chemical species both inward to the metal surface and outward from the surface. Cathodic CGDE of aqueous electrolyte solutions was explored also as a viable tool for generating steam with an efficiency of the order of 80% with the possibility of its use to various liquid wastes [1, 22].
A detailed mechanism for the breakdown of NE at high enough voltages and for the growth of a stable light-emitting plasma at the anode as the smaller electrode with transition to CGDE was elucidated. The mechanism was mainly based on Joule heating induced local solvent vaporization near the anode surface and on the onset of Helmholtz-Taylor’s hydrodynamic instabilities in the local vaporization there [1, 23] Though small some degree of secondary electron emission from the anolyte solution cathode of the anodic plasma may also contribute. Whereas for the transition of NE-to-cathodic CGDE, in addition to local Joule heating of the electrolyte in the vicinity of the cathode surface and the onset of hydrodynamic stabilities in local vaporization, large emission of secondary electrons from the metal cathode of the plasma with sufficient kinetic energies appears a major contributor to the origin of cathodic CGDE during electrolysis [1, 21].
Studies of diagnostics of CGDE plasmas generated in aqueous solutions by optical emission spectroscopy (OES) showed that the plasmas were highly non-equilibrated (T electron ≫ T gas ), consisted of excited atoms, ions and molecules derived from the breakup of H2O molecules, the electrode material and cationic and anionic constituents of the electrolyte. The plasma characteristics: the electron number density (n e) for the anodic and cathodic plasmas as determined were of the order of 1021–1023 and 1020–1022 m−3 respectively, and the electron temperature (T electron ) was of the order of 104 K for both the plasmas [1, 24–27].
A very remarkable characteristic of CGDE is its highly non-faradaic chemical yield. The products of CGDE at the anode in K2SO4 solution at a cell voltage of 440 V found were: 2.00 mol/mol electron of H2 and H2O2 plus O E2 each, O E2 (O2 in excess of the faradaic yield) being in terms of equivalent H2O2 over and above the faradaic yield of 0.25 mol/mol electron of O2 [1, 28].
On the other hand, the products of CGDE at the cathode in KHSO4 solution at a cell voltage of 300 V were: 0.68 mol/mol electron each of O2 (in terms of equivalent H2O2) plus H2O2, and H E2 (H2 in excess of the faradaic yield) over and above the faradaic yield of 0.50 mol/mol electron of H2 [21]. A model based on two reaction zones viz. one within the CGDE plasma around the electrode (plasma phase zone), and another near the plasma-electrolyte solution interface (liquid phase zone) was proposed to elucidate satisfactorily the mechanism of non-faradaic chemical effects of CGDE in aqueous media. In the plasma phase zone, water (the electrolyte solvent) vapor molecules would break up by energetic plasma particles into H· and OH· radicals and subsequently into H2 and O2 following the mechanism of dissociation of H2O molecules in electrical discharges through water vapor. The voltage drop near the metal cathode in cathodic CGDE plasma being substantially higher than that near the metal anode in anodic CGDE plasma, the plasma phase non-faradaic yields should be more in the former than in the latter [1, 21, 28]. The data calculated from the non-faradaic results described above showed indeed these plasma phase yields in terms of H2 were 0.52 mol/mol electron in cathodic CGDE as compared to 0.40 mol/mol electron in anodic CGDE. On the other hand, in the liquid phase reaction zone liquid water molecules on being bombarded by the charged plasma particles (e −plasma and H2O +gas in cathodic CGDE and anodic CGDE respectively) would break up into H· and OH· radicals. The radicals mutually would interact to form H2 and H2O2 plus O2. The electric field in the plasma-catholyte solution interfacial layer (anode fall) in cathodic CGDE being ~1/3 rd of the field in the plasma-anolyte solution interfacial layer (cathode fall) in anodic CGDE, the energy of the bombarding plasma particles and consequently the liquid phase non-faradaic yields should be much less in the cathodic than in the anodic CGDE. The liquid phase yield is most conveniently expressed as the initial differential yield of H2O2, G0 (H2O2): G0 (H2O2) mol/mol electron each of H2 and H2O2. The data thus obtained in reference to the above results showed indeed these liquid phase yields in terms of G0 (H2O2) were 0.16 mol/mol electron in cathodic CGDE as compared to 1.60 mol/mol electron in anodic CGDE. The distribution of plasma phase and liquid phase non-faradaic yields found in cathodic CGDE: 76% and 24% respectively was, in fact, remarkably different from that for anodic CGDE: 20% and 80% respectively [1, 21, 28].
As seen from the above, the liquid phase yields of anodic CGDE, H2 and H2O2 were found quite high indicating to generation of considerably large yields of H· and OH· radicals in its liquid phase reaction zone keeping in view also the high probability of mutual interaction among these radicals back into H2O molecules. Several estimates made for the primary yield of these radicals through the action of selected H· as well as OH· scavengers, showed that 11 ± 1 mol/mol electron each of H· and OH· radicals were generated in the liquid-phase reaction zone of anodic CGDE [1, 29].
As one would expect, the potentiality of generating high local concentrations of H· and OH· radicals and H2O2 in the liquid phase reaction zone near the anode plasma-anolyte solution interface made anodic CGDE well exploited for applications in synthetic chemistry, waste water treatment, polymer degradation, electrosurgical devices.
It may be noted too that both anodic and cathodic CGDE could give rise to considerably large yields of H2, the former particularly from the liquid phase reaction zone and the latter from the plasma phase zone. The H2 generation by CGDE could be further increased through the use of potential H· scavengers, one H2 molecule being formed from one H· radical by interaction with its scavenger. Among the potential H· scavengers are acetone, methanol, ethanol, iso-propanol, n-propanol, n-butanol, etc. [29–31].
In fact, CGDE particularly the cathodic one has significant potentiality in the application to H2 production [14].
An operation of CGDE gives rise to two prominent effects: (i) localized liberation of a large amount of heat within the plasma and in the liquid electrolyte near the plasma and (ii) production of solvent-split radicals (e.g. OH· and H· radicals from aqueous electrolytes) in high local concentrations near the plasma-electrolyte interface and also to a significant extent within the plasma around the electrode. Whereas the heating effect is considerably more intense in cathodic CGDE, the liquid-phase radical yield is much higher in anodic CGDE. The great potential of both of these chemical and thermal effects of CGDE was exploited efficiently in a number of applications over diverse fields as specified under Introduction (vide Table 1).
In fact, CGDE can offer an economical and green solution to water pollution abatement, surface modification of metals and alloys, and several other technologies. The reactor designs usually employed for exploring applications of CGDE are schematically described in Fig. 2.

Reactor cell designs for studying applications of CGDE: a–c for general purpose and d for machining and micromachining (Reproduced from [7] with permission from Elsevier Publishing Co.)
An overview of the salient aspects of all such applications of CGDE with illustrative examples is presented here.
Synthetic Chemistry
Anodic CGDE is a potential tool for generating solvent split radicals (e.g. H· and OH· from aqueous, H· and \({\text{NH}}_{2}^{ \cdot }\) from ammoniacal electrolytes) in high local concentrations near the plasma-anolyte interface. This has opened up an avenue for synthesizing a variety of compounds through radical medited routes. The first example of the synthesis by anodic CGDE was reported by Hickling et al. who while studying CGDE of liquid NH3 solutions of NH4NO3 at a thin Pt wire anode obtained hydrazine in yields 2 mol/mol electron at 500 V. They explained the finding in terms of mutual interactions between \({\text{NH}}_{2}^{ \cdot }\) radicals generated by the bombardment of gaseous NH3 + ions coming out of the plasma on liquid NH3 molecules in the anolyte [32].
Synthesis of Bio-organics
In an attempt to explore CGDE as a simulation of lightning striking on the hydrosphere under non-reducing atmosphere (mainly H2O, N2 and CO2) on the primitive Earth, Harada et al. synthesized bio-organic compounds. In this endeavor they used a glow discharge anode just touching the liquid surface as against an usually immersed one underneath the liquid electrolyte. They carried out successfully this way simulated pre-biotic synthesis of several amino-acids by applying a current of 50–60 mA at 500–1200 V keeping temperature at 10–20 °C: (i) from saturated aliphatic carboxylic acids (e.g. glycine, alanine, aspartic acid, glutamic acid, α-aminobutyric acid from acetic, propionic, glutaric, succinic acids) in aqueous ammonia with an yield of amino-acids up to 13% for an energy expenditure of ~108 kJ [33]; (ii) from aliphatic amines (or amino acids) by direct carboxylation using formic acid (or formamide) in aqueous solutions at an energy consumption of 4–12 × 105 kJ for each mole of amino-acids [34]; (iii) from ammonium bicarbonate or ammonium formate in ammoniacal aqueous solutions with 0.001% and 0.03% conversion to amino-acids respectively for an energy consumption of ~14 to 15 × 103 kJ for each mole of the ammonium salts [35]; (iv) from elemental carbon in the presence of aqueous ammonia with a conversion of 0.2% of the a carbon rod anode into urea, glycine and other amino-acids for an energy consumption of ~24 × 104 kJ per g atom of carbon [36]; (v) from β-unsaturated carboxylic acids and aqueous ammonia [37]; (vi) from 2-pyrrolidone in aqueous formic acid [38]; (vii) from aliphatic nitriles by amination in aqueous ammonia followed by hydrolysis and from aliphatic amines by cyanization in aqueous NaCN followed by hydrolysis with an energy consumption of ~(9–17) × 105 kJ for each mole of glycine either from amination of acetonitrile or cyanization of methylamine [39]; (viii) from several hydroxy amino acids, β- and γ- amino acids and aliphatic amines by oxidative degradation in aqueous solutions with ~1 to 15% conversions to glycine at ~120 kJ [40–43]. Obviously, the OH· radical produced during anodic CGDE was the key oxidizing agent. CGDE could thus be utilized as a clean and powerful oxidation technique. They demonstrated further that not only the OH· but also the H· radical could participate during the synthesis by anodic CGDE with an appropriate substrates e.g. aqueous CD3COOH gave rise to both OHCD2COOH and HCD2COOH [44]. The authors showed also that maleic acid and acrylic acid solutions could undergo simultaneous hydroxylation, hydration and hydrogenation during CGDE giving rise to succinic acid, malic acid and tartaric acid from maleic acid; and propionic acid, β-hydroxy propionic acid, lactic acid and glyceric acid from acrylic acid in significant quantities with conversions of ~36 and ~20% for maleic acid and acrylic acid respectively at an energy expenditure of 18 kJ for both the instances [45]. They attempted anodic CGDE for abiotic formation of asparagine from alanine in formamide solutions [46]. They could utilize the technique also for the conversion of the alanine residue of poly (alanine) in formic acid and acetic acid solutions to aspartic acid, threonine, serine, glutamic acid and glycine residues to the extent of 20% of the alanine [47]. In their further studies, NH3 and HNO3 were shown to form during anodic CGDE of a very dilute H2SO4 solution under N2 atmosphere. Further, when aliphatic carboxylic acids were added to the anolyte, aliphatic amines and amino acids were found to form. The reaction was considered an experimental model for lightning striking on the hydrosphere under a non reducing atmosphere (H2O, N2 and CO2) on the primitive earth [48]. Harada et al. did not restrict their studies on synthesis by CGDE to amino acids only but to other bio-organic compounds too. The authors could synthesize uracil, thyamine and orotic acid from their respective dihydro forms by CGDE in aqueous solutions and discussed the relation of these reactions to prebiotic formation of pyrimidines [49].
Sen Gupta et al. showed that anodic CGDE at a well immersed thin Pt wire anode in ammonical aqueous solutions of acetates and propionates while applying 420–500 V at 35–45 mA keeping temperature at around 20 °C gave rise to glycine and α-alanine respectively in yields highly exceeding the Faraday law value and that the energy efficiency for the synthesis of glycine increased by one to two order of magnitude by changing the positioning of the anode from just touching the liquid to immersing well inside the liquid. It was found that the energy efficiency of glycine production by this method was 1–5 × 10−4 mol/kJ. This technique could, in fact, produce glycine at an energy efficiency 50–100 times that of spark discharge or X-ray for glycine production from the constituents of a reducing atmosphere [50].
The scavenging action of formic acid for H· and OH· radicals generated in the anolyte ~during anodic CGDE was utilized by Sen Gupta et al. for synthesizing oxalic acid from formic acid in aqueous solutions at 450 V with an ambient temperature of ~85 °C. They showed that oxalic acid was produced in quantities significantly exceeding 1.5 mol/mol electron under this conditions [51].
Synthesis in Aprotic Media
The potentiality of anodic CGDE for synthetic applications in aprotic media was demonstrated by Tezuka et al. The authors carried out cyanation of benzene/monosubstituted benzenes from their solutions in acetonitrile containing lithium perchlorate or tetraethylammonium bromide as the supporting electrolyte. It was noted that 500 V anodic CGDE at 20–30 mA of acetonitrile itself containing either of the supporting electrolytes keeping the cell in an ice-water bath gave rise to propionitrile and succinonitrile each in yields exceeding 100% in moles of product formed for each mol electron of electricity passed. They showed also that N, N-dimethylaminoacetonitrile was formed when N, N-dimethylformamide was used as the solvent. These chemical effects of CGDE appeared to originate from bond cleavage of the solvent molecules by the bombardment of energized gaseous positive ions from glow discharges [52].
Synthesis of Polymers and Superabsorbent Composites
Sen Gupta et al. made an attempt to exploit the radical generating potentiality of anodic CGDE for polymerization of acrylamide in aqueous media at 660 V and 40–50 mA using an ambient temperature of ~45 °C and could synthesize polyacrylamide successfully in considerably high yields. The authors measured percent monomer conversion, polymerization kinetics, productivity/charge efficiency and viscometric molar mass \(\overline{{(M_{V} )}}\) of the polymers produced as functions of the quantities of electricity passed. They showed that the charge efficiency of polymerization of acrylamide by 660 V anodic CGDE was 166 in terms of the number of electrons consumed to generate a molecule of polyacrylamide of \(\overline{{M_{V} }}\) ∼ 11 × 105 as compared to the charge efficiency of 2200 by ordinary electrochemically initiated polymerization, showing efficiency of anodic CGDE an order of magnitude higher than that by ordinary electrolysis for polymerization [53].
Gao et al. made use of the polymerization potentiality of anodic CGDE for successful synthesis of superabsorbent composites through co-polymerization of acrylic acid and acrylamide in the presence of montmorillonite using a voltage 700 V and current of 50 mA at room temperature. The water absorbency (1024 g/g for distilled water and 56 g/g for 0.9% NaCl solution), water retention and thermal stability of the superabsorbent prepared this way were higher than those of the superabsorbent prepared by a conventional method using chemical initiators [54]. They extended further their studies on the utilization of anodic CGDE as a tool of co-polymerization for the synthesis of a number of superabsorbent hydrogels. Examples of these superabsorbent hydrogels include polyvinylpyrrolidone/acrylic acid hydrogels for adsorption of Pb (II) and other heavy metal ions with conformity to the Langmuir adsorption model [55]; poly(methylmethacrylate-butyl acrylate) for oil absorbency of 28.5 g/g for benzene, 25.4 g/g for toluene, 28.0 g/g for xylene, 38.1 g/g for chloroform and 37.0 g/g for carbon tetrachloride [56]; acrylic acid-poly(ethylene glycol) hydrogel for the adsorption of heavy metal ions [57]; a superabsorbing composite[HEC-g-P(AANa-co-AMPS)] made by graft copolymerization of acrylic acid and 2-acrylamido-2-Me propane sulfonic acid onto hydroxyethyl cellulose (HEC) for efficient removal of heavy metal ions [58]; poly(2-acrylamido-2-methy-1-propanesulfonic acid- co-acrylic acid), a potentially smart water superabsorbent with maximum equilibrium swelling (1.685 g H2O/g dry hydrogel) for use in agriculture, horticulture, hygiene products, pharmaceutics and biosensors [59]; poly(acrylic acid) grafted on to carboxymethyl cellulose (CMC), a highly pH-sensitive superabsorbent smart hydrogel as a promising candidate for drug delivery systems, hygiene products, agriculture and horticulture [60]; poly(acrylamide-co-acrylic acid) hydrogel which adsorbs very efficiently cationic dyes e.g. Crystal Violet, Methylene Blue from 5 to 10 pH aqueous solutions through a multi-step diffusion process with chemisorption as the rate-limiting step [61]; and poly (butyl methacrylate-co-butyl acrylate), a highly efficient oil absorptive resin utilizing emulsion polymerization technique [62].
In recent years, anodic CGDE at a Pt wire in aqueous solutions was exploited by Zhang et al. with a graphite rod as the cathode in the voltage range of 500–650 V DC to prepare pH and temperature dual sensitivity intelligent hydrogels based on reed hemicellulose as the backbone on which acrylic acid (AA) and N-isopropyl acrylamide(NIPAAm) monomers were copolymerized through N,N-methylene-bis (acrylamide) (MBA) as the crosslinking agent. Among the hydrogels prepared over 500–650 V span, the one obtained at 600 V exhibited the highest sensitivity to temperature and pH, and the maximum de-swelling ratio as well. They observed also that de-swelling behavior of all reed hemicellulose-based hydrogels prepared followed first order kinetics [63]. The authors applied this method further for graft copolymerization of acrylic acid (AA) monomer on to the cellulose network through N,N-methylene-bis (acrylamide) (MBA) as the crosslinker in aqueous NaOH/urea solutions at 500–630 V DC to synthesize novel ionic hydrogels possessing multi-stimulus responses to pH and salts. The swelling behavior and the network structure of the hydrogels thus obtained could be controlled by changing the discharge voltage/time and the highest swelling ratio in distilled water was observed for the hyrdrogel prepared at 570 V and 90 s. It was found that hydrogels prepared under different discharge voltages/times exhibited different response behavior to change of pH, ionic species, and concentrations. Shrinkage of the network hydrogels and lowering in swelling ratio could occur at higher or lower pH. The hydrogels were found more sensitive to Zn2+ and Fe3+ buffers as compared to a Na+ buffer, and showed network shrinkage and lower swelling ratio. Furthermore, these hydrogels showed a reversible on–off switching behavior in acidic–neutral solutions. These smart hydrogels have prospective applications in areas like agriculture, food and drug delivery [64].
A new direction for polymerization potential of CGDE was given by Friedrich et al. The authors could exploit CGDE using right-angled wave AC at 1–3 kV at ~55 °C between a jacketed stainless steel rod electrode with a sintered disc at the bottom and a Pt wire electrode in distilled water containing poly (ethylene glycol) (PEG), N,N′-methylenebis (acryl amide) (MBA), acrylic acid monomer (AA) and pieces of macroporous ultra-high molecular weight poly (ethylene) (UHMWPE) membranes for coating and filling of the membrane pores successfully. OH· radicals generated in the process functionalized the inner (pore) surfaces of the membranes by –OH groups, gave rise to radicals derived from PEG which in turn initiated homopolymerization of AA to PEG-PAA copolymer chains. The latter were crosslinked by MBA and the PAA-PEG-MBA hydrogels so formed could penetrate into the pores of UHMWPE membranes even at the trickiest sites by plasma-induced shock waves and vigorous stirring, and adhere very well to OH-functionalized groups there as demonstrated by SEM of their cross-sections. The polymer hydrogels thus prepared were found held intact and well adhered to the OH functionalized inner PE surfaces through interactions among OH, COOH and other minor functionalities. They further demonstrated the biocompatibilities of these hydrogels by cell adhesion tests on the copolymer coated PE using IEC-6 cells [65].
The energy efficiency data of CGDE (mol/kJ) for various synthetic applications are presented in Table 2.
Waste Water Treatment
The very strong oxidizing power of OH· radicals produced in high local concentrations in the anolyte during anodic CGDE gave rise to an avenue for its utilization as an environmentally benign technology for the treatment of industrial, agricultural and domestic waste waters containing harmful pollutants. H2O2 formed in the liquid phase reaction zone also play an important role in the treatment of waste waters. Even H· radicals may also exhibit their reducing potentiality for an appropriate waste in the water treatment. Interestingly, the technique requires neither any special power source nor an evacuating system for destroying hazardous substances. The method has the potential of degradation of a variety of organics, reduction of Cr(VI), inactivation of algae in waste waters.
Degradation of Aromatics
Effluents from industries manufacturing antioxidants, disinfectants, pesticides, herbicides, synthetic dyes, wood preservatives, detergents, medicines etc. contain considerable quantities of toxic aromatic compounds many of which have carcinogenic character and been notified as priority pollutants. These compounds comprise a substantial majority of the pollutants in waste waters. Anodic CGDE is a potential tool for abatement of such aromatic pollutants through exhaustive oxidation mainly by OH· radicals. A systematic investigation for degradation of selected aromatic compounds as model pollutants in aqueous media by 500–750 V anodic CGDE at 100–400 mA depending on the cell configuration and elctrolyte composition was initiated by Tezuka et al. with the aim of treatment of wastewaters. They studied kinetic and mechanistic aspects of exhaustive oxidation of benzoic acid [13, 66, 67], aniline [13, 68], phenol [13, 69, 70], cresols [13, 71], mono- [13, 72, 73], di-and tri- [74], tetra- [13, 75] and pentachlorophenols [13, 76], benzene- [13, 77], alkylbenzene- [13, 78] and alkane sulphonates [79] in neutral phosphate buffers to inorganic carbon by anodic CGDE. They further observed mineralization of mono- and poly chlorophenols to chloride ion besides inorganic carbon. Obviously the attack of OH· radicals on the benzene ring during anodic CGDE was the key step in these degradations. It was found that by anodic CGDE at 35 W pentachlorophenolate would be removed at an efficiency of 100% in 2 h which was higher than that of other AOPs like photocatalytic UV/TiO2 125 W high pressure Hg lamp, sonolytic ozonization or Fentons reagent [76]. Further, by anodic CGDE at 45 W benzene- and p-toluene- sulfonate could be removed at an efficiency of 100% in 3 h as compared to 100% in 2 h by UV/TiO2 500 W for benzenesulfonate and also to 100% in 3 h by ozone at 24 mg/min, pH = 3.0 for p-toluenesulfonate [77, 78].
The study on phenol degradation by anodic CGDE was also undertaken by Gao et al., Liu et al., and Wang both in the absence and presence of Fe2+ as well as Fe3+ catalyst. They also employed 500–900 V at ~100 mA. Their results on degradation of phenol [13, 80–83], nitrophenols [13, 84–86]. Chlorophenols [13, 87–90] and chloroanilines [91] showed that the rate of the degradation was considerably enhanced by Fe2+ and more so by Fe3+ catalyst to as high as 93.3%. The reaction intermediate found was different in the presence of the catalyst though the final products were carboxylic acids in all cases. They found advantages of acidic or neutral media over a basic one. The energy efficiency of phenol degradation by anodic CGDE was found 0.6 × 10−6 mol/kJ. It rose to 7.4 × 10−6 mol/kJ in the presence of Fe2+ and to 9.5 × 10−6 mol/kJ in the presence of Fe3+ each at an energy input of 50 W. The results could be compared to the efficiency of 2.28 × 10−6 mol/kJ by 2 MW spark discharge [83]. They studied also the efficiency of other electron acceptor catalysts such as Cr(VI), H2O2 and Cu2+ but found Fe3+ the most effective.
A study on degradation of chloro- and nitrobenzenes by anodic CGDE in aqueous media was carried out by Liu et al. They observed p-chloro phenol [13, 92] and o-nitro phenol [13, 93] respectively as the predominant products. In their further studies on degradation of p chloronitrobenzene (PCNB) by 450–550 V anodic CGDE, it was found that PCNB degraded to p-nitro phenol, 2-chloro-5-nitro phenol, oxalic and formic acids, chloride and nitrate ions but no chlorophenol indicating that dechlorination was easier than denitration from the aromatic ring of PCNB [94].
A study on exhaustive breakdown of 4-phenolsulfonic acid (4-PSA) by 500 V anodic CGDE at ~70 mA in a neutral phosphate buffer was undertaken by Yang et al. They observed that most of its total organic carbon (TOC) and sulphonate group was converted to inorganic carbon (IC) and the corresponding sulphate ion respectively. They proposed a reaction pathway involving successive attacks of OH· and H· radicals on the basis of the products and the kinetics observed. It was found also that that the rate of degradation of 4-PSA was enhanced by Fe2+ and Fe3+ ions and influenced considerably by pH of the reaction media [95].
Anodic CGDE at 600 V of water contaminants containing with two aromatic rings degrades them to complex products. Gao et al., Gai et al. observed that 2-naphylamine gave rise to o-phthalic acid, salicylic acid and catechol [13, 96]; diphenylamine to eventually inorganic carbon (IC) [97] and azobenzene to aniline [13, 98]. Interestingly, whereas the primary step in the degradation of an aromatic by anodic CGDE was the attack of OH· at the aromatic ring, formation of aniline from azobenzene pointed clearly to a role of H· generated during CGDE. These findings showed that while proposing the mechanism of degradation of compounds, the combined effect of OH· and H· along with H2O2 should be taken into account.
While investigating pulsed DC CGDE induced degradation of p-benzoquinone, a model organic pollutant in water, Bratescu et al. introduced the coherent anti-Stokes Raman Spectroscopy (CARS). The authors carried out pulsed DC CGDE (with peak values of voltage and current of 4 kV and 4 A respectively) of 10 kHz and 250 ns pulse width between two Pt rod electrodes in a 20 mM aqueous solution of p-benzoquinone with 1 mM KCl and observed initially an increase in the intensity of the CARS signal at 1233 cm−1 corresponding to the ring vibration of p-disubstituted benzene, and at 1660 cm−1 due to the C=C symmetrical stretching vibrations. This indicated to polarization of p-benzoquinone molecules by the plasma electric field and an orientation of the molecules with the electrical dipole arranged along the electric field. This enhancement in the signal intensity, however, decayed after switching off the HV pulse of the plasma with a time constant of about 20 μs. This decrease in the signal intensity was ascribed to degradation of p-benzoquinone by CGDE generated OH· radicals into maleic acid and succinic acid, and by H· radicals into hydroquinone, the latter being attacked further by OH· radicals into oxalic acid. The formation of the final degradation products was confirmed by liquid chromatography analysis. The authors showed further that p-benzoquinone after the CGDE treatment of 30 min might degrade to more than 50% of its initial value [99].
Degradation of Trichloroacetic Acid
During water treatment by chlorine, especially in the chlorination of drinking water, a part of organic compounds gets converted into chloroacetic acids which are toxic and carcinogenic. These acids can get into human body through ingestion, inhalation and dermal contact. Though mono- and di-chloroacetic acids can be decomposed by AOPs, trichloroacetic acid (TCAA) is very stable, un-reactive toward the oxidizing reagents including the most potent hydroxyl radicals (OH·). Wang et al. made an attempt to dechlorinate and decompose TCAA by CGDE at a Pt wire anode at 600 V and 75–85 mA in a Na2SO4 solution at ~27 °C. The authors observed that TCAA underwent dechlorination and decomposition efficiently into inorganic carbon (IC) and \({\text{Cl}}^{ - }\) through the intermediacy of the products: di-and mono-chloroacetic acids besides acetic acid and formic acid by the action of CGDE. It was seen that an energy consumption of ~1200 kJ could dechlorinate completely a 150 ml aliquot of 1 mM TCAA solution into \({\text{Cl}}^{ - }\) ion. Due to the absence of any α-H in trichloroacetic acid, the initial involvement of \({\text{e}}_{\text{aq}}^{ - }\) which would form by H·–H2O reaction followed by the attack of H· and OH· on the intermediates was suggested to explain the total degradation of trichloroacetic acid [100].
Degradation of Bromoform
Bromoform (BF), commonly used in laboratory and industry and generated during the chlorination of drinking water, ozonation of sea water and purification of swimming pools is a widespread stable toxic contaminant in water. BF is a proven carcinogenic and has been classified as a group B2 carcinogen. Wang et al. employed CGDE at a Pt wire anode at 500 V and ~100 mA in a Na2SO4 solution at ~27 °C to debrominate and decompose BF in wastewater efficiently into inorganic carbon (IC) and \({\text{Br}}^{ - }\) through the intermediacy of the products: formic acid and oxalic acid, dibromomethane and bromate ion. It was shown that alkaline conditions and the presence of organic additives promoted both the removal and the debromination of bromoform. An energy consumption of ~300 kJ could debrominate completely a 150 ml aliquot of 0.4 mM bromoform solution into \({\text{Br}}^{ - }\) ion. It was suggested that at alkaline pH, \({\text{e}}_{\text{aq}}^{ - }\), the conjugate base form of H·, was the predominant primary species for the initial attack on bromoform forming \({\text{Br}}^{ - }\) and dibromomethyl radical, and OH· radicals were the ones for oxidation of the intermediate byproducts. In the presence of an OH· radical scavenger e.g. iso-propanol, methanol, tert-butanol etc. as a model organic additive, the effective concentration of \({\text{e}}_{\text{aq}}^{ - }\) was increased and more bromoform was removed as bromomethyl radicals and more \({\text{Br}}^{ - }\) was produced. Hydrolyses and oxidations of the resulting bromo-methyl radicals contributed to the formation of formic acid and oxalic acid for the debromination of the organic bromine [101].
Degradation of Dichloromethane
Dichloromethane (DCM), a widely used solvent for manufacturing various products is present in significant quantities in water bodies due its high water solubility. DCM containing wastewater can harm central nervous system, cause renal and liver dysfunction, liver cancer and lung cancer in humans. DCM has been categorized as a B2 group carcinogen and a priority pollutant in water a B2 group carcinogen and a priority pollutant in water. Wang et al. in their further studies on wastewater treatment by CGDE, subjected aqueous dichloromethane, to CGDE at a pointed Pt wire anode at 560 V and 75–85 mA in a Na2SO4 solution at ~27 °C, for its dechlorination and decomposition and investigated the mechanism of degradation of dichloromethane. It was found that dichloromethane underwent effective dechlorination and decomposition by CGDE. Further, both removal and dechlorination of DCM increased with increasing pH and with addition of iso-propanol, a typical OH· radical scavenger and decreased with the presence of \({\text{NO}}_{3}^{ - }\), a typical quencher of \({\text{e}}_{\text{aq}}^{ - }\). Formic acid and formaldehyde were the major intermediate byproducts. Final products were carbon dioxide and chloride ion. Hydrated electrons were the most important among the primary active species (\({\text{e}}_{\text{aq}}^{ - }\), H· and OH·) for initiation of the reaction, and OH· radicals were the ones for oxidation of the intermediate byproducts. Hydrolysis of the resulting chloromethyl and dichloromethyl radicals played an important role in complete mineralization of the organic chlorine. The authors proposed a reaction mechanism based on the dechlorination kinetics and the distribution of intermediate byproducts. It might be noted that the energy efficiency of dichloromethane degradation by CGDE (1.31 × 10−6 mol/kJ at pH 7.0 and 2.05 × 10−6 mol/kJ at pH 11.0) was 4–5 times that by photocatalysis or ultrasonic degradation, especially in basic media [102].
Treatment of Cr(VI) Solutions
Cr(VI) in Cr-containing effluents from metal finishing, leather tanning, dyeing, electroplating and textile factories impose severe environmental threat and causes health problems due its toxicity, mobility, mutagenecity, and carcinogenicity. Reduction of Cr(VI) to relatively harmless Cr(III) and its removal as Cr(OH)3 is a way to treat Cr(VI) containing effluents. Possibility of wastewater treatment for Cr(VI) solutions by anodic CGDE was investigated by Wang et al. and Liu et al. They applied 500 V with a current of 100 mA and studied the treatment both in absence and presence of OH· scavengers such as phenol, ethanol etc and observed significant reduction of Cr(VI) to Cr(III) in either case at ~27 °C. The rate of reduction was interestingly found enhanced in the presence of phenol etc. They ascribed the observed reduction to the role of H· radicals produced during anodic CGDE and its enhanced rate in the presence of phenol to the increased availability of H· since phenol could scavenge OH· and prevent formation of H2O by the reaction between H· and OH· [13, 103, 104]. The energy efficiency for treatment of Cr(VI) in potassium dichromate solutions by 50 W anodic CGDE was found 0.5 mg/kJ as compared to 0.01 mg/kJ in the treatment by 125 W UV/TiO2 at pH 1.0 [103].
Degradation of Dyes
Organic dye containing effluents from textile dyeing and printing industries besides causing eutrophication of receiving water bodies, incorporates the bio-refractory components as well adding to the difficulty of purifying the wastewater. Generally, dyes are one of the major pollutants in water. Dyes are toxic, some are carcinogenic and some are harmful to aquatic life. A number of researchers Gao et al., Jin et al., Wang, Gong et al. and Ramjaun et al. attempted utilizing 500–600 V anodic CGDE at 60–80 mA for the treatment of dye-containing wastewaters using several dyes as model dyes at ~27 °C. The mechanism of degradation of a sample textile dye Brilliant Green (4,4′ bisdiethylaminodiphenyl triphenyl cation) used in textile industries by anodic CGDE was elucidated by Gao et al in detail by monitoring its degradation by GC-MS technique: OH· radicals, the very powerful oxidant product of anodic CGDE, in very high yields cleaved the dye molecule into benzoic acid and 1,2,3,4,5,6-cyclohexanehexaol at first This is followed by opening the rings of these intermediates further producing lots of smaller molecule of organic acids such as 2-hydroxypropanoic acid, hydroxyacetic acid, 4-oxopentanoic acid, oxalic acid and succinic acid besides (1E,2E)-ethanedial dioxime and glycerol. The finding showed also that the dye Brilliant Green rapidly underwent degradation and eventually mineralized into CO2 and H2O [105].
The other authors studied kinetics of degradation of several other dyes and mechanism in certain cases dyes such as Cationic Blue [13, 106], Methyl Orange [13, 107, 108], Acridine Orange [13, 109], Acid Orange 7 [13, 110, 111], Weak Brilliant Red B and Weak acid Flavine G [13, 112], Polar Brilliant B [13, 113], Crystal Violet [13, 114], Methyl Violet [115], Alizarin Red S [13, 116], Reactive Yellow 176, Reactive Red 239 and Reactive Black 5 [13, 117], Cationic Red [118] and Eosin [13] as target pollutants by anodic CGDE both in the absence and the presence of Fe2+ catalyst (Fenton’s reagent). It was inferred that Fenton’s reagent improved significantly the efficiency of dye degradation. Such enhancing effects on dye degradation were also shown by Fe3+, Cr(VI). The degradation rate was found stable and varies over ~90 to ~100%. However, inhibitory action was shown by OH· scavengers like ethanol and Cl–. As an extension of these studies, a multi anode system was tried for degradation of Acid Orange 7 and an increase in the efficiency of H2O2 formation as well as dye discoloration was observed [119].
Degradation of Ionic Liquids
Ionic liquids (ILs) regarded as a potential “green” alternative to volatile organic compounds have strong toxic effects on aquatic organisms and impose a potential threat to the environment.
For degradation of imidazolium-based ILs in waste waters, Yan et al. attempted CGDE of ILs at a temperature below 70 °C applying 400–750 V at 0.6–1 A with a wolfram stick cathode and a steel plate anode, and found ILs rapidly degrading into \({\text{NO}}_{2}^{ - }\), \({\text{NO}}_{3}^{ - }\), formic acid and acetic acid at an energy efficiency of ∼1 mg/kJ. The degradation followed pseudo-first order kinetics and occurred through the attack of O, OH· and H2O2 generated during CGDE. The degradation efficiency for ionic liquids decreased for different anions in the order Cl− > Br− > CH3COO− ≈ \({\text{BF}}_{4}^{ - }\). Among the 1-alkyl-3-methylimidazolium cations, the highest degradability was shown by the 1-butyl cation and the least by 1-ethyl one [120].
Degradation of Antibiotics
The continuous input of antibiotics into the aquatic environment through anthropogenic sources results in an increasing potential risk for aquatic and terrestrial organisms because antibiotics may cause resistance in bacterial populations, making them ineffective for treatment of several diseases in the near future. Jin et al. chose amoxicillin as the model broad-spectrum antibiotic pollutant and studied kinetics of its degradation in Na2SO4 solutions at ~30 °C by CGDE using a Pt wire anode at 510 V. They showed that the degradation was more than 67% which got enhanced to more than 96% through the use of Fe2+, Fe3+ or stainless steel wire as a single or multi-anode. It was found that OH· radicals only were responsible for the degradation and the energy efficiency could reach 6 g kW/h [121].
Degradation of Pesticides
The presence of pesticides in food is a concern for human exposure to pesticides, and the removal of pesticide residues from foods has become an important issue with respect to food safety. While considering CGDE as a tool for degradation of pesticides in foods, water should be chosen as the medium. This is most suited for fruits and vegetables. Hong et al. selected two highly water soluble pesticide samples fenothiocarb (an acaricide) and imidacloprid (an insecticide) for studying their degradation by CGDE in water with the particular focus on their kinetics and pattern of degradation. The authors showed that the degradation of fenothiocarb and imidacloprid in water using 220 V and 60 Hz. CGDE achieved figures of 57.8% and 43.2% respectively which in the presence of HCl were enhanced to 100% and 93.02% respectively. Acidic conditions favored either production of OH· radicals or enhancement of the degradation of organic compounds by OH·. However, both the degradation rates and amounts for the pesticides were found reduced in the presence of methanol apparently due to the competition between pesticides and methanol for OH·. CGDE induced degradation of both the pesticides obeyed first-order kinetics. Obviously, CGDE is a potential tool for removal of pesticide residues from fruits and vegetables using water as a reaction medium [122].
Degradation of NH3
Ammonia from various wastes resulting from human and animal activities contaminate water sources and is a threat to aquatic life as it can kill fish and cause algae bloom. Kartohardjono et al. made use of OH· generating potentiality of anodic CGDE to remove NH3 from wastewaters. The optimal condition for NH3 removal was achieved using 0.02 M KOH at 500–700 V and 60 °C when NH3 degradation efficiency and energy efficiency were 89.2% and 1.25 × 10−5 mol/kJ respectively [123].
Degradation of Methyl Tert-Butyl Ether (MTBE)
Methyl tert-butyl ether (MTBE) is a serious ground water pollutant from accidental fuel spills. Wen et al. studied kinetically its degradation by anodic CGDE in Na2SO4 solutions at 530 V into acetic and formic acids. The efficiency of MTBE degradation by this method was found comparable to that of electrocatalytic oxidation [13, 124]. Further, the energy efficiency of MTBE degradation by 53 W anodic CGDE was found 3.0 × 10−6 mol/kJ as compared to 2.65 × 10−6 mol/kJ by 200 W high-density plasma [124].
Degradation of Algae
Algae in waters often pose serious threat to the drinking safety. In an interesting exploratory study, Jin et al. could work out optimum conditions for employing anodic CGDE as a tool for inactivation of microcystis aeruginosa (MA), a notorious dominant species in the fresh and marine water. They achieved an inactivation rate for the algae more than 90% within 5 days of incubation after inoculation under optimal conditions of 530 V, 30 mA and 20 min of treatment time. The results gave strong evidence about the potentiality of CGDE on the inactivation of MA in aqueous solution [13, 125].
The energy efficiency data of CGDE (mol/kJ) for degradation of water pollutants are presented in Table 3.
Degradation of Polymers
The potentiality of anodic CGDE in generating high yields of radicals was exploited by Harada et al. for degradation of polymers also. They studied the effect of applying 750 V anodic CGDE at ~80 mA to poly (ethylene glycol) [126] and also of applying ~700 V anodic CGDE at 60–120 mA to poly (acrylamide) [127] in aqueous solutions. Poly (ethylene glycol) was found to undergo cleavage preferentially in a localized zone of the reaction system. Poly (acrylamide) was found degraded stepwise caused by the cleavages of the main and also of the side chains of the polymer. Further, Sandhir employed 470 V anodic CGDE at 45 mA for degradation of poly (vinyl alcohol) in aqueous solutions [128]. Poly (vinyl alcohol) was found to undergo simultaneous crosslinking and degradation through the fracture of the main chain. Studies on polymer degradation were extended to biopolymers also. Bae et al. studied depolymerization of fucoidan by anodic CGDE in NaCl solutions and found its molecular weight decreased about 40 times [129]. Munegumi et al. investigated degradation of several dipeptides by anodic CGDE and found that C-terminal amino acid residue decomposed faster than the N-terminal amino acid residue [130]. In recent years, as illustrated below, two large biopolymers were chosen for degradation studies by anodic CGDE in aqueous media.
Chitosan, the deacetylated product of chitin, is a renewable biopolymer and has owing to its large number of active hydroxyl (–OH) and amine (–NH2) groups, attracted considerable attention for the adsorptive removal of dyes. In an effort to improve its adsorption capacity, Wen et al. pre-treated chitosan by applying 530 V anodic CGDE to a suspension of chitosan powder in Na2SO4 solution. The authors found that the CGDE treatment changed the morphology and crystallinity of chitosan particles, and increased the number of –CH2 and –CH3 groups in the chitosan samples. It was observed that the CGDE modified chitosan had a higher maximum adsorption capacity for both acid and reactive dyes than the untreated chitosan. The dye uptake of chitosan could increase twice by the CGDE treatment. Thus CGDE is an attractive pretreatment method for environmental adsorption materials. [13, 131].
Water hyacinth (WH), a lignocellulosic biomass through appropriate pre-treatment and subsequent enzymatic hydrolysis may provide sugar for bioconversion to fuel ethanol and biogas. The OH· and H2O2 generating potentiality of CGDE was exploited by Gao et al. as a pre-treatment method for WH. The pre-treatment by CGDE of the biomass in a FeCl3 solution at 450 V between a wolfram stick cathode and a stainless steel plate anode resulted in the substantial reduction of the lignin content and cellulose crystallinity index of WH. Further, the treatment enhanced the surface porosity by highly disrupting the compact framework of the untreated WH. The authors showed that on pre-treatment by CGDE, the sugar yield from WH by enzymatic hydrolysis improved 16.6% to as much as 37.6%. CGDE thus provides an efficient route to obtain high sugar yield from water hyacinth [132].
Electrosurgical Devices
CGDE owing to its OH· and H· radical generating potentiality was exploited as a plasma scalpel in new electrosurgical devices for tissue treatments. Stalder et al. using optical spectroscopy and electrical diagnostics studied microplasmas formed at the active electrode of a probe immersed in a 0.16 M NaCl isotonic solution by 100 kHz RF square wave voltage pulses of 200–300 V rms at discharge currents in the range 10–150 mA rms. The probe consisted of a suitably organized set of small independent Ti elements as the active multi-electrode and a stainless steel cap as the common counter electrode. The optical emission spectra (OES) of the dense microplasmas grown at the active electrode exhibited strong emissions at 306.4 nm characteristic of \({\text{OH}} \, \{ {\text{A}}^{2} \Sigma^{ + } \left( {{\text{v}}^{{\prime }} = \, 0} \right) \, \to {\text{X}}^{2} \Pi \left( {{\text{v}}^{{\prime \prime }} = 0} \right)\}\) and at 653.6 nm characteristic of Hα (n = 3 → n = 2) besides Na D—line at 589 nm. The authors found parameters of these electrosurgical plasmas such as electron temperature (T electron ) as ~4 to 6 eV and of electron density (n e ) as ~1012 cm−3. Obviously these OH· and H· radicals were generated in the process by plasma electron-impact dissociation of water vapor molecules. When a biological tissue was positioned in proximity to the energetic microplasmas localized near the active electrode, the OH· and H· radicals from the plasma might encounter surfaces such as collagen or other tissue structures and react quite strongly with the surfaces through numerous reactions with proteins, collagen molecules of the tissue. Several of these reactions led to abstraction of H atoms from a highly hydrogenated protein surface, and subsequent reactions of the dehydrogenated protein could took place, leading to their fragmentation. Further, energetic plasma electrons could penetrate many monolayers of liquid water before losing significant energy, and induce direct fragmentation of underlying collagen proteins. These plasma surgical devices as compared to traditional electrosurgical tools operated at a much lower temperature (40–70 °C) and thus minimized the damage to the surrounding healthy tissue. These avoided further contact of the electrode with the tissue reducing greatly the charring deposit on the electrode, and prevented current through the body which might lead to the pad site burn and temperature increase in the body. These devices are, in fact, finding increased use in surgical procedures requiring fine control of the tissue excision, cauterization or debulking process [133–136].
Surface Engineering
As early as 1893 CGDE was exploited as a surface engineering tool for electrolytic heat treatment of steel cathodes to improve their surface hardness and wear resistance by Lagrange and Hoho, and later by other investigators. [137–142]. Since then CGDE cathodic as well as anodic has undergone numerous stages of improvisation over the years generally under the name PE (plasma electrolysis), EPT (electrolytic plasma technology) or EPP (electrolytic plasma processing) and emerged an environment friendly high performance surface engineering tool not simply for hardening and welding metal work pieces but mostly for high potential cleaning and coating of metal surfaces with immense promise of applications to biomedical, aerospace, automotive, oil, gas, electric and other tool industries. During these processes, not only heat evolution, diffusion and plasma chemical reactions besides electrochemical ones are occurring, but also cataphoresis of macroparticles away and towards the electrode surface may take place.
Plasma Electrolytic Heat Treatment of Metals
The technique of electrolytic heat treatment of metal cathodes was improvised later by Hoho, Yasanogorodskii and others for high speed and plain carbon steel twist drills [ 137–149 ]. The process is generally carried out at ~500 V and ~25 A DC or AC or DC pulses. In this process the surface temperature of a steel cathode rises with rapid heating rates to temperatures exceeding the austenitization temperatures. When the current is switched off the plasma film breaks and the hardening specimen comes in contact with the cold electrolyte in its vicinity and gets quenched. By the end of the electrolytic heat treatment, the steel surface hardens due mainly to formation of martensite while maintaining the core at its original toughness. The formation of martensite is confirmed by the results of light microscopy. It was found further that higher is the voltage applied, the lower the content of the ferrite-pearlite component in the structure, and the latter is primarily represented by martensite.
Recently Hikino et al. developed anodic PE for applications to surface heat treatment of AZ series magnesium alloys with varying aluminium content in phosphate electrolyte solutions using a voltage range of 250–400 V. The effect of the treatment on microstructure as well as mechanical properties of the alloys was examined in detail. They observed significant changes in tensile strength of only AZ61 and AZ91D alloys having relatively high aluminium content (>6%). Further corrosion resistance on the corrosive magnesium alloy is improved by this treatment. Possibly the metal structure near the surface is made to change by the plasma electrolytic treatment under optimum conditions, and then to improve the mechanical property [150].
Plasma Electrolytic Cleaning of Metal Surfaces
Cathodic PE has proved a highly useful environment friendly single step technique for cleaning surfaces of ferrous and non-ferrous metals and alloys from oxides, dirt, lubricants etc. which is essential to many applications such as coating, drawing etc. Surface cleaning results in surface morphology desirable for improved adhesion and lubrication. Nie et al. developed cathodic PE at 170–200 V in the normal glow region and current density of 0.23–0.45 A/cm2 for effectively removing oxides, mill scales, dirt, lubricants etc. from low carbon steel (AISI 1010) surfaces in NaHCO3 at 75 °C. The cleaned surface had highly desirable anchor profiles and was nano-crystalline. Corrosion tests on the samples when cleaned by PE showed a significant improvement in the ennoblement of the sample surface over the surface cleaned by mechanical grinding. This was attributed to the increased rate of cathodic processes caused by surface homogenization and removal of the oxide layer [3, 151]. In an attempt of further development in PE cleaning, Mathews et al. employed pulsed 100–350 V DC cathodic CGDE for thermally hardened AISI 4340 steel samples in NaHCO3 solutions at 70 °C. The new method as compared to simple DC treatments resulted in reduced surface roughness and in the compressive residual stress at the surface [4, 152].
Plasma Electrolytic Coating of Metal Surfaces: Plasma Electrolytic Oxidation (PEO)
PE or EPT/EPP offers a number of promising surface coating methods such as plasma electrolytic deposition (PED) including plasma electrolytic oxidation (PEO) and plasma electrolytic saturation (PES) covering plasma electrolytic nitriding/carburising/boriding (PEN/PEC/PEB) or their hybrids [2, 3]. It may be noted in this context that the difference in chemical composition between the heated surface of the metal electrode and its vapor envelope drives diffusion processes in PED methods.
Plasma electrolytic oxidation (PEO) of metal anode surfaces is a potential technique for fabricating hard oxide ceramic coatings with reduced friction coefficient under lubricated as well as dry conditions, on valve materials such as Al, Mg, Ti, Zr etc. metals and alloys. The method improves significantly the resistance to abrasive wear even in corrosive environments and is frequently exploited commercially e.g. in textile machines, aerospace components, gas/oil extraction and refining machinery etc. The thickness of PEO coatings can range from tens to hundreds of microns, depending on the power supply, substrate and electrolyte used. PEO is, in fact, an emerging technique to develop firmly adherent, crystalline, porous, relatively rough and thick oxide coatings on valve materials in some environmental friendly electrolyte. The following is an account of varied examples of applications of PEO.
PEO of Aluminium and Its Alloys
Matthews et al. showed that AC-pulse PEO of an Al alloy (e.g. H30T) anode in a suitable passivating electrolyte like Na2SiO3 solutions led to as thick as 500 μm and as hard as 23 GPa well-adhered alumina-based layers on the metal surface with excellent prospects for many tribological applications [2]. They studied also DC PEO of 6082 Al alloy anode in KOH solutions in respect of oxide film growth, anodic dissolution and oxygen liberation at voltages 500–900 V and current densities in the range 467–1407 A/m2. During this process the rate of oxygen liberation at the anode exceeded the Faraday law value. They found that the overall current efficiency of the oxide film formation was estimated to be in the range of 10–30%. These authors demonstrated further that use of pulsed bipolar current in 1–3 kHz range in place of 50 Hz AC led to a qualitative improvement in PEO coating on 2024 Al alloy in an alkaline Na2SiO3 + Na2P2O7 electrolyte [153].
The authors investigated also the influence of interfacial shear strength as an indication of the bond strength between a PEO layer and its aluminium substrate for PEO coated AA2024-T3 Al alloy towards understanding the coating’s failure mechanism to optimize the coating/substrate performance for exploiting fully the capacity of PEO coatings in structural applications. To evaluate the interfacial shear strength in PEO coated AA2024-T3 alloy, they developed a non-direct optical technique using synchronous digital image correlation and digital microscopy which had the benefit of using real time measurements without the requirement for electron microscopy, a single specimen could be used to provide stress strain and interfacial shear data [154].
Zinc–aluminum (ZA) alloys which feature clean, low-temperature, energy saving melting and low production cost are considered alternative materials for aluminum alloy, iron, and brass in areas like pressure tight housings, electronic instrument chassis etc. Towards achieving high-efficient and high-quality surface protective coating on ZA alloys, Wang et al. selected ZA-27 alloy (containing mainly Zn, Al and Cu, a minor constituent) which has the highest strength and the lowest density, as well as excellent bearing and wear resistance properties, for investigating PEO. They carried out PEO of a ZA27 alloy anode in solutions of Na2SiO3, NaAlO2 or a mixture of NaAlO2 and NaBO2 made alkaline to pH 13.2 with KOH, in a stainless steel cell as the cathode with a unipolar pulsed DC in a constant current mode at a current density of 0.1 A/cm2 at 1000 Hz. The method was found successful to produce continuous and dense ceramic coatings on ZA27 alloy. The coatings grown from each of the three electrolytes contained mainly ZnAl2O4, Al2O3, and CuAlO2 phases. Further the coating when produced in silicate solutions contained ZnSiO4. During the process of coating, Al was apparently oxidized prior to Zn and more Al diffused to the outer interface to react with the electrolyte to form the ceramic coatings. The addition of borate was found beneficial to produce a denser coating probably by accelerating the growth rate of the coating. Moreover, coatings grown in silicate or aluminate/borate electrolyte had a structure with porous outer layer, dense layer, and inner barrier layer, whereas the coating produced in aluminate electrolyte showed a dense monolayer structure only. The coating produced in an aluminate/borate composite electrolyte exhibited the best wear performance, the lowest friction coefficient, and the highest wear resistance among the three coatings under both low and high normal loads [155].
Stojadinovic et al. investigated PEO of aluminum in 12-tungstosilicic acid solution at 25 mA/cm2 to develop an innovative procedure for fabrication of silicate tungsten bronzes on aluminium. Their results on detailed surface structure of PEO coatings showed that the outer layer of the oxide coatings (mainly composed of γ-Al2O3 and WO3) was silicate tungsten bronze [156]. The authors investigated also DC PEO of 99.9% Al sheet anode in aqueous boric acid and borax solutions to which ZnO nanoparticles (in the form of commercial ZnO powder of mean size 28 nm) was added at the concentration of 2 gm/l ZnO at a current density of 150 mA/cm2. The resulting mixed Al2O3/ZnO coatings formed on Al were explored for their potential application using photoluminescence and photobleaching of Methyl Orange as a tool. Photoluminescence measurements showed well-pronounced bands native to Al2O3 and ZnO oxides in the formed coatings identified oxygen vacancies as centers of luminescence. Results on photocatalytic decompositionon of Methyl orange under simulated sunlight showed that longer PEO processing times were beneficial for enhanced photocatalytic activity [157].
PEO of Magesium and Its Alloys
Arrabal et al. studied AC PEO of magnesium/aluminium, magnesium/zinc and magnesium/rare earth metal alloys in silicate phosphate electrolytes at 650 mA/cm2 (rms) at 50 Hz with a square waveform and found coatings consisting of MgO and Mg2SiO4. The corrosion rate of the alloys in NaCl solutions was found reduced by 2–4 orders of magnitude by the treatment [4, 158]. To optimize the conditions for PEO, Hussein et al. investigated the mechanism of the growth of PEO coating on AJ62 Mg-alloy discs in alkaline aluminate solutions using optical emission spectroscopy (OES). They concluded that for the growth of coating, three simultaneous processes viz. electrochemical reactions, plasma chemical reactions and thermal diffusion should occur. Further, O2 diffusion into the Mg-alloy substrate played a leading role in the growth of coating [159].
Following Arrabal et al.’s [4, 158] work on AC–PEO coatings on Mg alloys, Yerokhin et al. undertook a thorough study of current regimes for development of protective PEO coatings on Mg–Mn alloys, an important group of medium-strength wrought Mg with good weldability widely used in the form of sheets, plates and forgings at temperatures up to 200 °C. Moreover, the Mg–Mn system shows great promises for development of rare-earth modified Mg alloys with superior creep resistance. The authors carried out deposition of PEO coatings on a rectangular coupon of wrought MA8 magnesium alloy (containing Mg, Mn as a minor constituent) in a Na2SiO3 solution containing NaF additive under both unipolar (at 200 V and 300 Hz) and bipolar (phase A: at 30 → 300 V; phase B: at = −30 V, frequency 300 Hz) current modes. They found the current efficiency of both processes though similar, the protective surface coatings produced in the bipolar mode had higher microhardness (4.8 GPa) and better corrosion resistance (3.3 × 105 Ω cm2). It was shown that the application of the bipolar PEO mode would enable one to synthesize on the alloy’s surface a high-temperature phase of magnesium silicate, forsterite (Mg2SiO4) having good anticorrosion and mechanical properties [160].
Hong et al. showed that cerium conversion coating (CeC) coatings on initially formed PEO coating of Mg2SiO4 on AZ31 Mg alloy caused sealing of its pores and defects by cerium oxides (likely to be amorphous Ce2O3 along with CeO2) and that the composite coating/substrate interface acted as a good barrier to aggressive electrolytes [161].
While investigating the feasibility of Mg alloys having excellent mechanical properties close to natural bone for their use as light weight metallic bio-implants, Liu et al. in an attempt to improve/enhance their anti-corrosion property/corrosion resistance and biocompatibility/cellular/osteoblast response by fabricating ZrO2 ceramic coatings onto the substrate alloy, explored PE at a WE43 Mg alloy disc cathode in a solution of Zr(NO3)4 and ethanol with a graphite anode applying a 400 V pulsed DC of 100 Hz frequency. They found that ZrO2 coatings on native Mg alloy strongly protected the alloy from corrosion when immersed in simulated body fluid (SBF). The authors’ results on cytocompatibility including osteoblasts adhesion and viability indicated that cathodic CGDE induced ZrO2 coatings on Mg alloys were beneficial for cell proliferation and differentiation. The method has significant potentiality for surface modification of Mg-based implants to improve anticorrosion and bone osseointegration [162].
Yerokhin et al. studied anodic PE for PEO of a commercially pure Mg disc in an aqueous solution containing Ca(OH)2 and Na3PO4 with a stainless steel coil cathode by applying a 100–5000 Hz pulsed unipolar current at an average current density of 30 mA/cm2 and found that porous coatings mainly composed of MgO with minor amounts of Na4Ca(PO3)6 formed on the Mg metal substrate. The authors showed that these PEO coatings enhanced the corrosion resistance of Mg in simulated body fluid (SBF) by a factor >10 with the best corrosion protection provided by the coating obtained at the pulse frequency of 3000 Hz which has allowed simultaneous increases in coating thickness, porosity refinement and internal stress relaxation without significant cracking [163].
In an attempt to improve wear and corrosion resistances of PEO coatings of AZ31 Mg alloy, Cheng et al. studied the effect of the incorporation of SiC nanoparticles into the PEO coatings by carrying out PEO on a AZ31 Mg alloy plate in aqueous solutions of Na2SiO3 and (NaPO3)6 containing 2 gm/l SiC nanoparticles (~50 nm) under pulsed bipolar constant current regimes, using a frequency of 1000 Hz. A higher average positive and negative current densities of ~0.22 and 0.09 A/cm2 and a lower average positive and negative current densities of ~0.13 and 0.03 A/cm2 respectively were employed to develop coatings in this work. The SiC nanoparticles were found incorporated into the coatings composed mainly of amorphous materials, possibly silica and phosphorous containing species, and crystalline phases of Mg2SiO4 and MgO, and the incorporated particles distributed relatively uniformly along the coating depth. The incorporation of SiC nanoparticles resulted in an excellent wear performance of the coatings. Further, corrosion resistances of these coatings were greatly improved [164].
PEO of Titanium and Its Alloys
Titanium alloys, due to their combination of low density, high specific strength and excellent biocompatibility, are widely used in aerospace, automotive, chemical and biomedical industries (e.g. for the replacement of hard human tissues). However, owing to their poor tribological behavior and vulnerability to galvanic corrosion or corrosion embrittlement causing the release of poisonous metal ions, PEO of alloys of titanium (and also of zirconium) emerged as a potential technique for protecting and functionalizing their surfaces. It was found that the properties of PEO coatings depended mainly on the electrolyte composition and the electrical regime such as DC, AC or pulsed DC employed to grow them. Matthews et al. investigated processing and property aspects of the films produced by AC PEO of a Ti-6AI-4V alloy disc in aqueous solutions containing aluminate, phosphate, silicate, and sulphate anions and some of their combinations in a stainless steel bath which served as the counter electrode at an anodic power densities at 4.3, 6.2 and 10.5 kVA/dm2. The authors found that the films formed from aluminate-phosphate electrolytes were dense and uniform, composed mainly of Al2TiO5 and rutile TiO2 phases. These films had the beneficial combination of 50–60 μm thickness, 575 kg/mm2 Knoop microhardness and high adhesion along with a low wear rate (3.4 × 10−8 mm3/Nm) and relatively high friction coefficient (μ) 0.6–0.7 against steel. Further, the films produced from a phosphate electrolyte exhibited a minimum friction coefficient (μ) 0.18 during the testing of thin 2.5–7 μm relatively soft rutile-anatase films. Both of these types of films showed good corrosion resistance in NaCl and physiological solutions, where the corrosion current was approximately 1.5 orders of magnitude lower than that of the uncoated substrate. 60–90 μm thick SiO2/TiO2-based films with high bulk porosity produced from silicate and silicate-aluminate electrolytes demonstrated better corrosion behavior in H2SO4 solutions, due to the greater chemical stability of the film phase components in this environment [165].
Cheng et al. demonstrated how constant voltage PEO treatment of a 2 cm2 Ti-6Al-4V alloy plate in aqueous solutions of Na2SiO3 and (NaPO3)6 with a stainless steel plate counter electrode with a pulsed bipolar waveform of 600 V positive voltage and −70 V negative voltage, at a frequency of 1000 Hz, led to rapid formation of thick, adherent coatings on the surface of the alloy. They found that the coatings were composed of rutile, anatase and amorphous material, and contained relatively large amounts of silicon and phosphorus species. The rutile content increased with extended times of treatment. They used real-time imaging of titanium probably for the first time under pulsed DC or AC regimes to reveal the spatial distribution, size, population density and lifetime of the microdischarges. It was observed that a greater corrosion resistance for coatings was formed with relatively short times of treatment. However, tribological tests showed that coatings formed with long times of treatment had better wear resistance, possibly due to changes in the composition and morphology of the coating during PEO [166].
Shin et al. investigated AC PEO of pure titanium in potassium triphosphate and pyrophosphate solutions. They found that as compared to the oxide film formed in triphosphate solutions the one formed in pyrophosphate solutions had higher surface roughness and more abundant anatase phase. They further demonstrated that the oxide film formed in pyrophosphate solutions exhibited easier growth of denser biomimetic apatite in a simulated body fluid (SBF) [167]. They showed also that PE of KOH solutions without any potassium phosphate but containing hydroxyapatite particles led to pure hydroxyapatite layers with superior bioactive performance on Ti anode [168]. Moreover, the authors studied the effect of current densities in 100–250 mA/cm2 range on surface characteristics and the biological response of the titanium oxide layers formed during AC PEO of a commercially pure Ti plate in alkaline phosphate solutions. They found that with decreasing current densities, whereas the pore size decreased, the mean surface roughness as well as the amount of anatase phase increased, a trend favoring the formation of biomimetic apatite and the proliferation of osteoblast cells. The results of tests in SBF showed that the formation of biomimetic apatite and the proliferation of osteoblasts on the titanium oxide layers produced at 100 mA/cm2 when uniformly distributed ∼ 2 μm crater like pores most suitable for cell anchoring form, was the maximum among the samples evaluated [169]. Keeping in view the broad-spectrum anti-microbial activity of Ag with low human toxicity, the authors probed what effect Ag nanoparticles (in the form of Ag metal powder with a mean size of 50 nm) in 0–0.5 gm/l concentration range if added to an alkaline pyrophosphate electrolyte, would have on the surface structure and in vitro biological properties of the titanium oxide layer prepared by AC PEO of a pure Ti plate at 100 mA/cm2 current density at a frequency of 60 Hz using a stainless steel counter electrode. They observed successful incorporation of Ag nanoparticles into the oxide layers and topographical deformation of the porous surface when Ag nanoparticle concentration exceeded 0.1 gm/l. Based on the evaluation of bone forming-ability in SBF, as well as MC3T3-E1 cell proliferation test and anti-bacterial test against E. Coli bacteria of the oxide layer samples produced, it was found that the one from the electrolyte with 0.1 gm/l Ag nanoparticles exhibited both superior bioactivity and anti-bacterial activity leading to gain beneficial biological performances of titanium implants [170].
Rudnev et al. studied formation of oxide-phosphate coatings on titanium during PEO of titanium alloy VT1-0 in electrolytes containing polyphosphate complexes of Ca (II) and Sr (II) at 5 A/dm2 in unipolar mode using a stainless steel bath as the counter electrode. They inferred that the coatings containing calcium phosphate with small inclusions of strontium phosphate were promising for the application to titanium implants [171].
Chung et al. synthesized specific surface coatings of anatase-TiO2, rutile-TiO2, hydroxyapatite, strontium-containing hydroxyl apatite and dual-phase hydroxyapatite-TiO2 on Grade II pure Ti (CP Ti) implants by PEO in NaH2PO4 plus Ca (CH3COO)2 electrolyte in various concentrations with or without Sr (CH3COO)2 by adjusting Ca/P ratio, pH (4.81–12.00) and discharge voltage (430–480 V) for studying their relative osseointegration performance. They found that the dual-phase hydroxyapatite-TiO2 coating formed at 480 V and pH 4.94 outperformed all the other coatings in effectively promoting osseointegration [172].
Clyne et al. utilized PEO of grade 2 commercially pure titanium in sodium phosphate solutions at 6000 A/m2 to produce coatings of highly porous surface with relatively high (>90% by volume) anatase content and examined their photocatalytic efficiency for degradation of Methylene Blue dye under UV irradiation. They found an enhancement by a factor of as large as 10 for photocatalytic rate constant could be obtained through PEO treatment of a bare Ti substrate (thin native oxide layer) and correlated enhanced photoactivity of PEO coatings to their relatively high anatase content and high levels of surface-connected porosity as well [173].
Ito et al. employed PEO of a Ti plate using an electrolyte mixture of H2O2, H3PO4 and H2SO4 containing 30 nm anatase TiO2 and 300 nm of Fe2O3 particles and a Ti sheet cathode as a one pot low temperature synthesis of micrometer thick Fe3+ doped anatase/anatase/rutile/TiO2 film on Ti metal using a current density of 3 A/dm2. This Fe-doped TiO2 film as compared to a pure TiO2 film exhibited enhanced photocatalytic activity under UV illumination for acetaldehyde decomposition [174].
To avoid the toxic effects of V in Ti based bio-implants e.g. Ti-6Al-4V, the use of vanadium free biomaterials was stressed. In an attempt to enhance the potential of a vanadium free Ti alloy, gamma titanium aluminide intermetallic alloy, (γ TiAl: Ti-48Al-2Cr-2Nb at.%) originally developed for aircraft engine applications was explored recently for biomedical applications. Sundaram et al. modified successfully its surface characteristics carrying DC PEO of anode rods of vanadium free Ti alloy (γ TiAl: Ti-48Al-2Cr-2Nb) in a calcium and phosphorous rich aqueous solution containing high purity Na2-EDTA, Ca(CH3COO)2 and Ca(H2PO4)2 using a stainless steel beaker cell as the cathode at current density over 133–171 mA/cm2. The results observed showed the formation of a submicron scale porous structure and a concomitant increase in the surface roughness. The resulting oxide formed on the surface of γ TiAl was found mainly composed of rutile and anatase phases. It was demonstrated that the technique had the feasibility of incorporating Ca and P into the oxide matrix in a gradient pattern with the purpose of increasing osseointegration. Nanoindentation data confirmed the formation of a fairly compact oxide layer on γ TiAl alloy during the process. The authors made a parallel study on a vanadium containing Ti alloy, Ti-6Al-4V and a comparison of the findings with those for γ TiAl showed that the oxide layer formed on the latter alloy was less porous, less rough and more stable. The oxide layers for both the alloys were, however, composed of Ti and O. The lack of Al and other elements in the oxide for both γ TiAl and Ti6Al4V would be very beneficial and would represent a minimal risk biomaterial in causing neurological disorders associated with the presence of aluminum [175].
A duplex α + β phase Ti-based alloy, Ti-6Al-4V which has significantly higher elastic modulus (100–110 GPa) than cortical bone (10–40 GPa), can cause stress shielding and is commonly used as an non-cement hip replacement. A number of reports about aluminum and vanadium ions as toxic, soluble in blood and causing long-term health problems led to studies on novel titanium alloys composed of non-allergic elements such as Ti–13Nb–13Zr. This β-phase titanium alloy exhibits mechanical properties close to the natural bone and is considered as one of the future, promising materials for bone implants. To enhance the bioactivity of the Ti–13Nb–13Zr alloy (TNZ), Simka et al. investigated modification of its surface by carrying out plasma electrolytic oxidation of a TNZ sample as the anode in a bath composed of calcium hypophosphite and osteoinductive additives like tricalcium phosphate, wollastonite (CaSiO3) or silica powder with a titanium mesh cathode, at 350, 400 and 450 V DC. The authors observed the growth of porous oxide layers on TNZ surface in each of the solutions employed containing tricalciumphosphate, wollastonite or silica. PEO of TNZ in calcium hypophosphite solution with wollastonite at 450 V gave the highest atomic ratio of bioactive compounds, Ca/P. The roughness of the alloy surface increased after the surface treatment. The surface modification improved the alloy’s corrosion resistance in Ringer’s solution and would improve the long-term outcomes for implants in biological environments [176].
Yerokhin et al. investigated very recently the size effect of hydroxylapatite, Ca10(PO4)6(OH)2 mineral particles in electrolyte suspension on the characteristics and biological properties of thin titania –based coatings on Ti-6Al-4V alloy discs produced by PEO in Na2HPO4 solution. The PEO treatment was performed using the two-step control method, wherein a potentiostatic anodic polarisation, 250 V was applied to the surface for 15–30 s to allow surface passivation to occur, then galvanostatic control at a mean average anodic current density of 300 mA/cm2 was employed using pulsed unipolar or pulsed bipolar mode. The hydroxylapatite particles used were in form of micro- or nano-powder. The micro-particles deposited on the surface altered the porous morphology of the TiO2 matrix, reducing the pore size and surface roughness, which led to a highly irregular morphology formed by interpenetrating networks of the voids and the coating material. The nanoparticles migrated more readily towards the surface and penetrated into the coating, partly filling the pores but not altering significantly the porous matrix morphology. Regardless of the particle size, hydroxylapatite increased the coatings’ scratch resistance, however, all coatings suffered cohesive failure in scratch tests, but no adhesive failure could be observed. The biological assessment of hydroxyapatite doped coatings included in vitro evaluation of the coating bioactivity in simulated body fluid (SBF) as well as studies of spreading, proliferation and osteoblastic differentiation of MC3T3-E1 cells. Both the hydroxylapatite micro- and nano-particles doped PEO coatings’ surfaces revealed enhanced bioactivity in vitro and induced formation of apatite precipitates during exposure in SBF. Furthermore, the cell proliferation/morphometric tests on all the coatings showed their good biocompatibility. Fluorescence microscopy revealed a well-organized actin cytoskeleton and focal adhesions in MC3T3-E1 osteoblastic cells cultivated on these substrates. The cell alkaline phosphatase activity in the presence of ascorbic acid and β-glycerophosphate was significantly higher on hydrooxylapatite nanoparticle-doped TiO2 coating as compared to both the microparticle-doped TiO2 and undoped TiO2. This could be to be due to the combination of more abundant presence of hydrooxylapatite nanoparticles on the surface, as evidenced by corresponding FTIR spectrum rich of specific phosphate bands, and characteristic porous morphology, which provided a synergic effect on the osteoblastic differentiation [177].
PEO of Zirconium and Its Alloys
Rameshbabu et al. explored PEO as a potential surface treatment for fabricating oxide films on Zr with a good combination of corrosion resistance, bioactivity and cell adhesion for improving their biocompatibility and functionality towards the use of these films on Zr as implant materials in orthopedic and dental restoration fields. The authors carried out pulsed DC PEO of a Zr coupon work piece in methodically varied concentrations of Na3PO4, Na2SiO3 and KOH based electrolytes at 150 mA/cm2 current density and 50 Hz frequency. The oxide films thus fabricated on Zr were uniform with 6–11 μm thickness and predominantly in monoclinic zirconia phase. They found further the PEO film formed in the phosphate (5 gm/l) + silicate (5 gm/l) + KOH (3 gm/l) mixed electrolyte among the different electrolyte systems employed, the optimum one with higher surface energy and roughness, good wettability and bioactivity, resistance toward pitting corrosion and superior osteoblast cell attachment and spreading [178].
Zirconium alloys owing to their low neutron absorption cross-section, satisfactory mechanical properties and excellent corrosion resistance, are one of the best structural materials for water-cooled nuclear power reactors. However, during the operation of nuclear reactors, the severe environmental conditions make the alloys vulnerable to nodular corrosion and erosion in flowing conditions, waterside corrosion and hydrogen uptake. To improve upon the surface properties of Zr alloys and to restrict hydrogen ingress, Cheng et al. investigated PEO of a 1 cm2 Zircaloy-2 alloy (Fe: 0.19, C: 0.02, Sn: 1.31, Hf: <0.01, Cr: 0.089, Ni: <0.002, and Zr: the balance in wt%) foil in an alkaline Na2SiO3 solution with a stainless steel plate as the cathode using a 5 kW pulsed DC source under pulsed bipolar current regime with the initial positive and negative current densities set at 0.4 and 0.3 A/cm2 respectively at a frequency of 1000 Hz. The authors found that the formation of the solidification structures in the PEO coatings formed on the surface of Zircaloy-2 alloy was related to the long lasting discharges during the PEO process and the very low thermal conductivity of zirconium oxide. PEO coatings were found to be zirconia phases consisting of monoclinic-ZrO2 and tetragonal-ZrO2, with the monoclinic-ZrO2 dominating the coatings in dilute solutions of silicate electrolyte. With increased silicate concentration in the solution, the tetragonal-ZrO2 content in the coating increased. Besides the content of silicate in the electrolyte, thermal effects in the coating would affect phase compositions of the coatings. During plasma discharges at the coating surface, its upper layer as compared to the lower one had a higher temperature, resulting in an increased tetragonal–ZrO2 presence in the upper coating layer. Thick coatings appeared to have a higher level of tetragonal-ZrO2 because more heat was retained due to its low thermal conductivity. Further, the PEO processing improved remarkably the corrosion resistance of the substrate, Zircaloy-2 in 0.5 M NaCl solution. Among these PEO coatings, the long duration (∼30 min) treatment specimen though had an increased thickness, owing to increased porosity its corrosion resistance was lower than that provided by the moderate duration (∼10 min) treatment specimen [179].
Cheng et al. extended their studies on PEO of another Zr alloy, Zircaloy-4 (composition: 1.50 Sn, 0.20 Fe, 0.10 Cr, Ni <0.007, Zr balance in wt%) of 1 cm2 working area under 50 and 100 Hz AC current regimes (Hz) using a square waveform (positive to negative current density ratio = 3:1), in alkaline Na2SiO3 solution at a current density of 300 mA/cm2 (rms). They observed that coatings developed an outer compact region and an inner more porous region, with a thin barrier region next to the substrate. For treatment times >1 min, the surfaces of the coatings revealed pancake-like zirconia-rich features and surrounding silica-rich material. The coatings formed with a relatively long treatment time (∼60 min with a thickness of 50–100 μm) were more porous. Further, results of corrosion studies in 6 M HNO3 solution at 100 °C, an environment relevant to reprocessing of nuclear fuel, demonstrated the efficacy of PEO treatment in improving remarkably the corrosion resistance of the alloy, and indicated that corrosion protection was mainly provided by the thin barrier layer (∼0.2 to 0.5 μm) next to the substrate [180]. For a better understanding of the underlying mechanisms of plasma generation and coating formation the authors made also a comparative investigation on growth kinetics, formation efficiencies and microstructures of the PEO coatings on the plates of two alloys, Ti-6Al-4V and Zircaloy-2 formed by applying a pulsed bipolar, constant current regime with average positive and negative current densities of 0.20 and 0.15 A/cm2 respectively at a frequency of 1000 Hz in an aqueous Na2SiO3–(NaPO3)6 solution. Their results suggested that the high electronic resistance of the ZrO2 coating on Zircaloy-2 promoted the formation of pancake structures which formed in type B (metal-oxide interface) discharges as per the model by Hussein et al. when discharge channels led to the substrate and afforded the main current. Whereas, the semiconducting nature of the TiO2 coating on Ti-6Al-4V alloy promoted O2 evolution, reduced the coating growth efficiency and led to “coral reef” coating features attributed to type A (oxide-electrolyte interface discharges at the upper coating or gas attached to the coating surface) or type C (discharges within micropores below the surface) discharges of the model by Hussein et al. The authors reported anomalous gas evolution in quantities far exceeding the estimated faradaic yield of oxygen under the charge passed, though observed on both the alloys after the appearance of strong plasma discharges, was more abundant with Zircaloy-2. However, more current was consumed by O2 evolution during PEO of the Ti-6Al-4V alloy in comparison with Zircaloy-2 [181].
Plasma Electrolytic Coating of Metal Surfaces: Plasma Electrolytic Saturation (PES)
Cathodic PE can be utilized as a plasma electrolytic saturation (PES) technique: plasma electrolytic nitriding (PEN), carburizing (PEC), boriding (PEB) or their hybrids where the elements C, N, B undergo interstitial diffusion into the surface of the cathode metal, the work piece specimen for achieving surface coats of high hardness, reduced friction coefficient, improved fatigue strength and excellent wear and corrosion resistances. PES coatings find potential uses in textile machine, cutting and processing tool components.
Employing cathodic PE at less than 415 V a current densities 0.5–1.0 A/cm2, Mathews et al. could obtain a 10–20 μm thick surface compound layer on steel substrates in NaNO2, or glycerol in Na2CO3 solutions with very good mechanical and corrosion resistant properties in only 3–5 min by the use of plasma electrolytic nitriding (PEN)/plasma electrolytic carburising (PEC) saturation techniques. They also found that plasma electrolytic boriding (PEB) of a medium C steel work piece in a NaOH solution containing Na2B4O7 led to a 100 to 150 μm thick coating of B4C with 4–5 GPa [2].
Xue et al. employed 150 Hz 340 V pulsed DC cathodic PE as a plasma electrolytic carburising (PEC) technique at a pure Al plate in an aqueous solution containing 80% glycerol and a little KCl in a stainless steel container as the anode, and obtained in 5 min a 2 μm thick carburized layer on Al. The electron temperature (T electron ) of the cathodic plasma was found fluctuating around 7000 K. Under the plasma discharge condition over the Al cathode, glycerol molecules underwent extensive decomposition and fragmentation into active carbon radicals and ionic components which under the strong electric field near the cathode was accelerated towards Al cathode generating an enhanced interstitial diffusion into the crystal lattice of Al. As a result, the carbon containing solid solution and Al4C3 precipitation phase were formed subsequently followed by the growth of a compact carburized layer containing 38.5% carbon. PEC treatment was found to improve the corrosion resistance of pure Al, increased its hardness by nearly two and reduced its frictional coefficient by ∼40%. PEC treatment, thus, has the potential of fabricating carburized layer with good wear and corrosion resistances on low melting point metals [182].
Tavakoli et al. applied ∼200 V DC CGDE at a St 14 steel cathode work piece surrounded by a sixty times larger stainless steel 316 cylinder as the anode within a cell containing a 25% aqueous borax solution and obtained a boride layer of Fe2B phase on the steel surface with considerably improved hardness and corrosion resistance [183].
Mathews et al. investigated the feasibility of cathodic PE as a DC plasma electrolytic nitrocarburising (PEN/C) technique for AISI 316 stainless steel using an aqueous solution of urea at 250 V. The corrosion behavior of the layer formed was found closely related to their microstructure and composition, and could be significantly improved, particularly if a thin but dense magnetite-based iron-chromium oxide layer be produced at the surface [3, 4, 184].
Bao et al. made a systematic investigation on the growth of PEN/C layer on a Q235 steel sample in aqueous urea using a pulsed DC (150–250 V at 0.8 A/cm2), and showed that the PEN/C sample had a thiOck diffusion layer consisting mainly of Fe4N, Fe3C, and Fe5C2 phases. The technique was found to improve greatly wear resistance of the low carbon steels and proved to be efficient, energy effective and environment friendly [185].
Xue et al. could grow a carbonitrided (PEC/N) layer on pure aluminium for the first time by applying 150 Hz 260 V pulsed DC PE plasma discharge on a pure Al plate cathode in a solution of glycerol and urea contained in a stain steel vessel as the anode. The electron concentration (n e ) and electron temperature (T electron ) of the cathodic plasma were calculated as 6 × 1021 m−3 and 4000 K respectively. The carbonitrided layer was found to contain Al4C3, AlN, Al7C3N3 phases. PEC/N coating on pure Al improved greatly its corrosion resistance. This was found related to the formation of nitride phases. This study has opened the application of EPT on surface modification to low melting point metals [186].
Ivanov et al. developed cathodic PE at 150–160 V for improving hardness and wear resistance of cutting tools from high speed steel R6M5 by a plasma borosulfocarbonitriding. They employed aqueous solutions of sodium thiosulfate, carbamide, and ammonium sulfate for complex saturation of the surface and added additionally carbide and borax powders as suspensions to the electrolyte bath, and the steel tool as the cathode. They observed formation of a composite borosulfocarbonitride layer on the cutting tool cathode. The endurance of the tools after such a treatment was found 1.5–2 times higher than after simple plasma carbonitriding, nitriding, cyaniding or sulphidizing [3, 4, 187]. Very recently, Belkin et al. made a comprehensive review examining the progress made in plasma electrolytic carburising, nitriding, and nitrocarburising of steels of various composition, including treatment modes, electrolyte compositions, structures, and properties of hardened materials. Analysis of the results obtained up to date indicates that pulse plasma electrolytic saturation treatments leading formation of surface nano-structures appear to be the most promising to advance further this type of electrolytic plasma technology [188].
Plasma Electrolytic Coating by Metals and Alloys
In an attempt to explore PE as a tool for depositing a metal or an alloy on the clean metal cathode surface, Nie et al. could develop coatings of Zn and Zn + Al on PE cleaned surface of AISI 1010 low carbon steel cathodes by PE in ZnSO4 and ZnSO4 + Al2(SO4)3 solutions respectively at 180–220 V and 0.3–1.2 A/cm2. The treatment led to a large improvement in the adhesion strength (>70 MPa) [3, 4, 151]. Zhang et al. successfully employed PE to deposit Ni-P coatings on a carbon steel cathode for achieving significant improvement in both wear and corrosion resistance of the steel [189].
Towards exploring a fast treatment for improving the corrosion resistance of low carbon steels, Meletis et al. applied PE to carry out very short depositions of Ni coating on a low carbon steel (AISI 1018) cathode in 20% NiSO4 solution at 75 °C with pH 3.8 at 200 V and 2.6 A/cm2. The authors observed a positive shift in the corrosion potential by 200 mV and more than an order of magnitude reduction in the corrosion rate for a deposition time of 30 s only. The electrochemical impedance spectroscopy studies revealed that a similar corrosion mechanism prevailed between pure Ni and PE-coated Ni. The results showed that PE deposition could be used to form a dense, nano-grained surface coating of Ni and an effectively full coverage at a very short deposition time [190].
Nano-structured Coating by Plasma Electrolytic Process
In an attempt to explore PE towards developing nano-structured films on metal surfaces, Bell et al. observed the growth of a very adherent nano-crystalline titanium dioxide film on a Cu rod cathode during cathodic PE of a solution containing titanium tetraisopropoxide, absolute ethanol and hydrochloric acid at 650–1200 V and 30 mA/cm2. They demonstrated that through the adjustment of the composition of solutions by a post-processing calcination, these films could be made very promising for some useful industrial benefits. They were also successful in developing a method for the growth of titanium dioxide coatings composed of nano-rods and nano-particles on a Cu cathode in the above bath. By controlling the applied voltage (at 400–700 V), the treatment time and initial titanium concentrations they could produce a large range of different crystallinities and morphologies promising in applications such as nanoelctronics, photonics, dye sensitized solar cells, gas sensors and photocatalysts for solar cell technologies [4, 191, 192]. In an extension of the study they deposited nano-crystalline graphite films on a 3 mm dia. titanium rod as the cathode from an ethanolic solution of a neutral phosphate buffer at 900–1200 V using a much larger cylindrical graphite counter electrode (40 mm inner dia.) and studied thoroughly the deposition mechanism. These coatings were found very attractive for production of inexpensive cold and high field emission cathodes in solid state electronic devices [4, 193]. They tried to correlate the structure and composition of different films of TiO2 and of graphite with the composition and physical characteristics of the glow discharge plasma [4, 194].
Cai et al. made use of AC PEO technique to nucleate a seed layer of TiO2 film on a pure Ti sheet anode with porous structure, remarkable thickness and well adhesion to the substrate in Na3PO4 solution at the positive voltage, negative voltage, and frequency of 400, −50 V and 700 Hz respectively with a stainless steel plate cathode. The authors utilized the PEO prepared seed layer of TiO2 film for the synthesis of TiO2 nanotube films with well adhesion and large-area through a hydrothermal method on it. Calcination resulted in crystallization of the nanotubes from amorphous state to single-crystalline anatase phase. Furthermore, the increase of calcination temperature was found beneficial to the improvement of crystallization degree. However, when the calcination temperature increased to 700 °C, the nanotubes collapsed completely to form short nanorods. The TiO2 nanotube film calcined at 600 °C exhibited the narrowest band gap energy, the strongest UV light absorption, the weakest band–band PL intensity and the best photocatalytic activity due to the combined effects of appropriate crystallinity and surface states. The method thus introduced is a potential one for synthesizing TiO2 nanotubes for the abatement of environmental pollution [195].
Diamond Like Carbon (DLC) Coating by Plasma Electrolytic Process
Plasma electrolysis (PE) was exploited also for depositing diamond like carbon (DLC) on metal cathodes. Kong et al. applied cathodic PE at 130 V and 1–2 A in the name micro-arc discharges for deposition of uniform 200 nm thick DLC and silicon doped DLC films on pure nickel plate cathodes from aqueous ethanolic solutions of NaCl and NaCl containing Na2SiO3 respectively. They found that such DLC films improved markedly the corrosion resistance of the Ni substrate [196]. Chen et al. extended the technique by using 1-propanol solutions at 700–900 V to obtain sub-micron diamonds [197].
Ban et al. showed that DLC films deposited by PE in 50% methanol solutions and KNO3 aqueous solution on a AZ31 Mg alloy plate cathode at 130 V with a Pt plate as the counter electrode improved markedly the corrosion resistance of the alloy [198]. Xue et al. demonstrated the formation of a composite film of DLC with iron oxides (mainly FeO) on a T8 high-carbon steel cathode by PE of aqueous glycerol solutions at 400 V. It was found that carbon particles in the film were uniformly embedded into the oxides and the sp3 C content of DLC was nearly 35%. The technique provides an approach to fabricate highly wear resistant DLC composite films on steel materials [199].
Xue et al. produced a DLC-containing carburized layer containing martensite, retained austenite and Fe3C phases on T8 high carbon steel plate cathode by DC-pulse PE of an aqueous 80% glycerol solution at 380 V and 150 Hz. The technique reduced significantly the activation energy of carbon diffusion and increased the diffusion coefficient of carbon. The sp3 C content for DLC-components was found up to 67%. The treatment improved both the wear resistance and the corrosion resistance of the steel [200].
These PE coating techniques would be potential precursor treatments in combination with other coating techniques to develop hybrid or duplex processes, which may extend the capabilities of both PE and other coating techniques.
Plasma Electrolytic Texturing and Alloying of Metal Surfaces
EPT has proved an effective tool to texture metal surfaces by creating unique surface morphology and microroughness for many applications such as coating adhesion, tribological performance, etc. Gupta et al. employed cathodic EPT to texture brass coated steel cord (high carbon steel, 350 μm dia.) used in automobile tyres leading to its improved adhesion with tyre rubber without any removal of brass coating necessary for protection against corrosion [3, 201]. Fan et al. investigated PE at a mirror-polished n-type (100) silicon wafer cathode (650 μm thickness) in NaHCO3 solutions containing glycerol at 400 V and showed that the process led to anisotropic chemical etching which created highly ordered textured silicon surface to realize light trapping which could significantly improve solar cell efficiency [202].
Surface Alloying
EPT is found also a potential tool for alloying metal surfaces for improving their mechanical and tribological performances. Gupta et al. could alloy Mo on AISI 4330 V steel and Inconel 718 by using cathodic EPT in Na2MoO4 solution in which Mo powder was added at a power density of 139.5 W/cm2. Mo alloyed steel thus obtained showed up to a two-fold increase in surface hardness (with variation in concentration of Mo) and a threefold increase in wear resistance. The increase in hardness was attributed to the presence of Fe0.54Mo0.73 phase in the alloyed sample [3, 203].
Recently, Pogrebnyak et al. reviewed the chemical and physical processes involved in various plating protective coatings and in modifying surfaces on metals by EPT [5].
Nanoparticle Fabrication
During cathodic CGDE, the thermal energy released in the plasma discharge may cause local melting and evaporation at relatively rough and electrothermally unstable surface spots of the cathode. The molten and evaporated metal clusters sputter down from the cathode and resolidify by rapid quenching in the catholyte solution to form ultrafine spheres. Parallel to this process, metal ions derived either from the electrolyte or from the dissolution of the anode may undergo reduction at the plasma-catholyte interface by \({\text{e}}_{\text{aq}}^{-}\), H·, H2O2 to zero-valent metal atoms which progressively agglomerate to ultrafine particles. Depending upon the electrode material and catholyte pH, aggregates of metal oxides, hydroxides may also form. The size of such ultrafine spheres ranges over micro-to-nanometers. The size distribution of the particles depends on the applied voltage, time of CGDE treatment and the composition of the electrolyte bath. This method for fabrication of nanoparticles offers certain advantages such as simple equipment, no need of gas supply in general, easy mass production, applicability to any metal/alloy and ease of controlling the size of the product. In fact, CGDE is a new generation green technology for the synthesis of nanomaterials.
A successful formation of metal nanoparticles by cathodic CGDE was shown by Toriyaba et al. The authors produced controlled formation of relatively monodispersed nanospheres (‘nanoballs’) of Ni, Ti, Ag and Au of diameters between 100 and 400 nm in a gas/liquid mixed dual phase system by cathodic CGDE of K2CO3 solutions between thin wires of respective metal cathodes and an anode of a Pt mesh of much larger surface area in a glass cell at a cell voltage <160 V with a certain size controllability. The trend in their data of particle size vs cell voltage suggested that particles of sub-100 nm mean diameter could be produced at voltages exceeding 200 V. The electro and thermal conductivity, chemical potential, and surface energy of the electrode material played an important role in determining the particle formation processes and diameter distribution. TEM studies indicated that the electrode surface was locally heated and induced by electrothermal instability momentarily exceeded the melting point, and the molten metal yielded to a spherical shape due to surface tension. The metal immediately cooled down to form fine spherical particles ‘nanoballs’ without contamination from the electrolyte. This glow discharge plasma electrolytic method in liquid was achieved extremely easily, quickly and inexpensively, and had the prospect to meet the demand for nanoballs to meet industrial needs. Further, it had a number of features that enabled it to control such nanoball properties and one could obtain complex composition or multilayered balls by utilizing the salient feature of the gas/liquid dual phase [9, 10, 204].
In further investigations Lal et al. fabricated nanoparticles of Cu, Au, Pt metals and Pt + Au alloy by cathodic CGDE between a Pt wire cathode and a large Pt ring anode using CuSO4 + H2SO4, NaAuCl4 + HClO4, H2PtCl6 + HClO4 and H2PtCl6 + NaAuCl4 + HClO4 electrolytes respectively using a high concentration of supporting electrolyte (~1 M) and a low concentration of metal salt (≤10 mM) generally at a large current density (~1 A/mm2 at plasma discharge voltage ~20 V). They introduced a ‘rotating cathode’ (rotations up to 5000 rpm) method for applying Coriolis forces to the process and controlled successfully the size of the particles. Whereas the range of the particle size for Pt was 30–200 nm without electrode rotation, it narrowed down to 80–140 nm by 2000 rpm [8, 10].
In this endeavor, Wuthrich et al. carried out cathodic CGDE in H2SO4 and KOH media containing ethanol as an OH· scavenger and polyvinyl-pyrrolidone (PVP 40) as the nanoparticle stabilizing agent between a large Ni plate anode and a Ni wire or a Pt wire cathode. They obtained nanoparticles of NiO (70 nm) in 2 M H2SO4 media with a Ni cathode and of Ni, NiO (<100 nm) and Pt (<10 nm) in the same media with a Pt cathode at the discharge voltage of 36 V. However, they obtained nanoparticles of β-Ni (OH)2, NiO, Ni (<100 nm) from 5 M KOH media with a Ni cathode but only of Pt (<10 nm) from the same media with a Pt cathode at 50 V. The results of Wuthrich et al. was a clear testimony to the simultaneous operation of two mechanisms, melting-quenching of the cathode metal and reduction of metal ions at the plasma-catholyte interface [205–207]. The authors probed further the size-dependence of specific capacitance of the NiO nanoparticles synthesized by cathodic CGDE. They carried out CGDE at a Ni wire cathode in 2 M H2SO4 + 0.5 M ethanol + 2.5 mg/ml of PVP 40 with large sheet of Ni anode at three voltages: 30, 36 and 42 V and obtained nanoparticles of 70, 91 and 107 nm average diameter respectively which were tested for electrochemical energy storage. It was found that a maximum specific capacitance of 218 F/g was observed with the 70 nm NiO nanoparticles at 2.7 A/g. Their corresponding energy and power densities were 98.1 Wh/kg and 0.7 kW/kg, respectively showing the performance characteristics of these NiO nanomaterials were clearly that of a supercapacitor. Larger nanoparticles of 91 and 107 nm diameter exhibited specific capacitances of 106 and 63 F/g, respectively, suggesting a size dependent capacitive performance enhanced with decreasing particles size [208].
The study was extended by Akiyama et al. who applied cathodic CGDE on a 1 mm Cu wire in K2CO3 solution or citrate buffer (pH 4.8) with a counter electrode of 0.5 mm Pt wire bent into a half-round mesh of 1 m length at ~140 V for synthesizing copper nanoparticles. They studied the relationship between the experimental conditions and the formation of copper/copper oxide nanoparticles and found that flower-like CuO <100 nm dia. nanorods with [010] orientation in the growth direction form in K2CO3 electrolyte. This was ascribed to the formation of [Cu (OH)4]2− ions and to oriented crystal growth. However, when the citrate buffer was used as the electrolyte copper nanoparticles were obtained: spherical particles at 105 V and porous spherical nanoparticles at 130 V. The technique offers an easy and quick method for production of copper/copper oxide nanoparticles [209].
The authors attempted synthesis of Ni nanoparticles by applying cathodic CGDE with full plasma at 173 ± 2 V in NaOH solutions between wires of a Ni cathode and a Pt anode and could, however, obtain a mixed product of Ni and NiO particles of 890 nm mean diameter. Towards preventing the oxidation of Ni, they introduced a technique of edge-shielding the cathode by shielding of the tip of Ni wire with a glass tube which allowed the growth of CGDE to a partial plasma only on the unshielded part even at 180 V. The technique led interestingly to the formation of exclusively metallic Ni particles free from any oxide particle. The edge-shielding of the cathode led also to significant reduction in the average size of Ni particles from 890 to 220 nm [210].
Towards optimization of the experimental conditions and studying the formation mechanism of metallic nanoparticle formation by cathodic CGDE, Akiyama et al. extended their studies to the surface morphology of a Ni wire cathode during nanoparticle fabrication by CGDE in a NaOH solution with a long Pt wire (bent into a semicircular mesh) counter electrode by examining the influence of electrolysis time, solution temperature, voltage, electrolyte concentration, and surface area on the size and the amount of nanoparticles formed. The results demonstrated that the amount of nanoparticles produced increased proportionally with the electrolysis time and current. At voltages below 140 V, surfaces with nanoparticles attached, called “Particles” type surfaces, formed on the cathode. These surfaces changed to display ripples, turning into “Ripple” type surfaces, and the nanoparticle sizes increased with increasing amount of nanoparticles produced. In contrast, at voltages over 160 V the surfaces of the electrodes were found “Random” or “Hole” type and the particle sizes remained constant with different amounts of nanoparticles produced [211]. They demonstrated further that cathodic CGDE was applicable not only for nanoparticle synthesis of pure metals but also that of various alloys. They described a facile CGDE synthesis of bimetallic nanoparticles, including solid solution alloys (Ni–Cu and Ni–Cr system), eutectic alloys of Sn–Pb, and intermetallic alloys (SnSb and Ni3Sn), at a metallic alloy wire cathode with a semicircular Pt wire mesh as the anode using 0.1 M NaOH as the electrolyte for Ni based substrates at 160 V or 0.1 M KCl for Sn based ones at 150 V [212].
With an objective to control the size of nanoparticles and their formation during cathodic CGDE, Saito et al. estimated the excitation temperature of the plasma from its emission spectrum, the plasma being generated around 1 mm dia. cathode wires of Ti, Fe, Ni, Cu, Zn, Zr, Nb, Mo, Pd, Ag, W, Pt, Au metals, and various alloys of stainless steel and Cu–Ni alloys in NaOH solutions with a Pt wire mesh anode, by applying the Boltzmann plot method with the assumption of local thermodynamic equilibrium(LTE). They studied also the effects of edge shielding, applied voltage, and electrode material on the plasma. Their results showed that with a Ni wire cathode edge-shielded by a quartz glass tube, a uniform plasma was formed and Ni nanoparticles were generated. OES of the plasma showed OH, Hα, Hβ, Na, O, and Ni lines. Without electrode edge-shielding, oxidized coarse particles formed due to high enough temperature of the electrode surface radiating continuous IR emission from the edge. It was observed that the excitation temperature increased as the voltage at the edge-shielded electrode was increased. The size of the Ni nanoparticles was found decreased at high excitation temperatures. Although the formation of nanoparticles via melting and solidification of the electrode surface was generally considered, vaporization of the electrode surface would occur at a high excitation temperature to produce small particles. Moreover, the results of the effects of different cathode materials: Ti, Fe, Ni, Zn, Nb, Mo, Ag, Pt, Au, and various alloys of stainless steel and Cu–Ni alloys on nanoparticle synthesis in 0.1 M NaOH solution at 140 V showed that with the exception of Ti (T excitation , 8151 K), the excitation temperatures ranged from 3500 to 5500 K and the particle size depended on both the excitation temperature and electrode-material [213].
With the aim of synthesizing nanoparticles containing photocatalytically active oxides, Watanabe et al. applied cathodic CGDE at 1 mm dia JIS SUS316 stainless steel wire in K2CO3 solution with a large area Pt wire mesh anode at 130 V. They could this way synthesize uniformly spherical stainless steel ‘nanoballs’ having <200 nm diameter containing photocatalytically active oxide species: Fe2O3 and Cr2O3 distributed uniformly over the surfaces of nanoballs. It appeared that nanosize molten materials ejected during repeated occurrence of localized melting spots on the cathode surface in CGDE were solidified into spherical nanoballs because of the surface tension and quenching effect of the electrolyte. They investigated the efficacy of stainless steel as photocatalytic materials towards the decomposition of 0.1% Methylene Blue dye solution under UV irradiation with the wavelength of 354 nm and found the dye concentration reduced to the baseline level in 72 h. They found further that in the presence of stainless steel nanoballs, the decomposition of Methylene Blue followed two different pathways: fast and slow decompositions. The dye was observed to oxidize into sulfoxide first, before being decomposed into lighter products. This was different compared to its decomposition pathway occurring in the presence of TiO2 and ZnO nanoballs where the dye was decomposed directly into products with lower mass number without being oxidized into sulfoxide first [214].
For synthesizing TiO2 nanoparticles with wide spectral absorption, Guo et al. utilized CGDE in NH4NO3 solutions using a Ti wire cathode at 100 V very successfully for the synthesis of high quality TiO2 nano/microspheres that absorbed in the full range of optical wavelengths over 240–2600 nm. They showed both from experimental and theoretical studies that oxygen vacancies in the bulk TiO2 spheres were primarily responsible for this excellent absorption. In an attempt to improve the diametric distribution of the TiO2 particles they employed electropolishing of the Ti wire cathode and a lower voltage CGDE at 70 V, and obtained TiO2 nanospheres (20–200 nm) with more anatase phase but with full retention of wide optical absorption [215, 216].
Use of Cathodic GDE in Aqueous Electrolytes
A well-known variation of cathodic CGDE where a DC glow discharge is struck between the cathode positioned in the gas space above the electrolyte surface and the electrolyte itself with the anode immersed in the liquid, is cathodic glow discharge electrolysis (GDE). Sankaran et al. applied a DC power supply to ignite such non-contact GDE in the form of a continuous-flow of a gas (Ar at 25 sccm), atmospheric-pressure microplasma as the cathode positioned 1–2 mm from the surface of a dilute acid solution, operated with a metal foil anode dipped in the electrolyte at a constant discharge current of 5 mA. They produced thus colloidal dispersions of uniform, high-purity metal nano-scale particles directly in the aqueous phase, and showed this emerging method a low cost, scalable and general one for the synthesis of a wide range of nanoparticle materials in the liquid phase. The particles were formed from the metal cations generated from anodic dissolution of a bulk metal (Ag, Au) or metal ions present in the electrolyte (AgNO3, HAuClO4) with a Pt anode. When these metal ions reached the cathode, defined by the microplasma– liquid interface, reduction to the zero oxidation state would occur by the species created at the solution interface (e −aq , H., H2O2) through interactions of the energetic particles from the microplasma with the solvent molecules there. Metal (Ag, Au) nanoparticles freely nucleated, grew to ~10 nm size and dispersed into the solution. Undesirable agglomeration was prevented by the addition of a stabilizer molecule (fructose) that could interact with and bind to the metal nanoparticle surface. The authors showed that the relative growth rate of nanoparticles between those formed from metal foil or metal electrolyte depended upon the metal itself. These noble metal nanoparticles have attracted significant interest for surface enhanced Raman scattering (SERS), catalytic, and medical applications. [11, 217, 218].
Use of Cathodic GDE in Ionic Liquids
Ionic liquids as compared to conventional solvent-based systems, offer several distinct advantages for carrying studies on synthesis of nanoparticles by GDE: (i) the low vapor pressure of ionic liquids allows the process to be operated at low pressure (<1 atm) where nonthermal plasmas are more stable, (ii) ionic liquids are, thus, safer than organic solvents from an environmental perspective, and (iii) the electrochemical window for ionic liquids is much wider than the 1.23 V window for water electrolysis and assuming that the plasma creates a potential significantly higher than this value, the current efficiency for the desired electrochemical reaction should be higher. Endres et al. demonstrated first the synthesis of nanoparticles from an ionic liquid by GDE technique at ~470 V and current density of 2 mA/cm2. They prepared crystalline Ag nanoparticles of 8–30 nm size by subjecting AgNO3 and AgCF3SO3 dissolved in the ionic liquid, 1-butyl-3-methylimidazolium trifluoromethylsulfonate ([BMIm][TfO]), to a low-pressure DC cathodic GDE plasma [11, 219]. The authors extended the study to the synthesis of Cu nanoparticles from CuCl and CuCl2 in the ionic liquid, 1-butyl-2-methylimidazolium dicyanamide ([BMIm]dca) at 400–500 V [11, 220]. Hatakeyama et al synthesized Au nanoparticles from HAuCl4 dissolved in the ionic liquid, 1-butyl-3-methylimidazolium tetrafluoroborate ([C8H15N2]+ [BF4]−). They found further that not only cathodic GDE but also anodic GDE both at 1 mA would give rise to Au nanoparticles that too with a substantially higher growth rate from the same HAuCl4 in ([C8H15N2]+ [BF4]−) system [11, 221–224]. This was not unexpected as the yield of Au3+/AuCl4 − reducing species, e −aq , H·, H2O2 from plasma particle-solvent/ionic liquid interactions are several times higher at the anolyte interface than at the catholyte one [1, 21]. Cathodic GDE in ionic liquids has the potential of being the cleanest method for preparing metal nanoparticles since only electrons are involved [225].
Use of Cathodic GDE in Molten Electrolytes
With a view to see the feasibility of molten electrolyte media for fabricating nanoparticles by cathodic GDE, Ito et al. applied the technique in a eutectic of LiCl-KCl-CsCl melt containing FeCl2 and PtCl2 with a W-rod cathode placed in Ar gas at 1 atmosphere and a sacrificial Fe plate as the anode by applying a voltage of 20 V. This way they could synthesize successfully sub-50 nm magnetic nanospheres of FePt intermetallic compound having high coercivity (245 mA/m). Metal ions from the electrolyte melt and/or from the anodic dissolution were reduced by electrons emitted from the cathode at the Ar-melt interface into ultrafine particles [226]. They employed the technique at 300 V for fabrication of Ag, Ni, Ti nanoparticles also [227–229].
Sankaran et al. published a review covering mainly charge transfer processes in both GDE and CGDE with the focus on nanoparticle synthesis [11].
Machining and Micro-machining of Materials
Owing to its potentiality for generating intense localized heat in a controlled manner cathodic CGDE under the title electrochemical discharges (ECD) has emerged a non-conventional machining process for conducing and non-conducting materials. The resulting technology is referred as electrochemical discharge machining (ECDM) or electrochemical spark machining (ECSM). Among the earliest reports on exploring successfully cathodic CGDE/ECD for machining materials were by Kurafuji et al. for drilling holes in glass work pieces [7, 230] and by Kubota for drilling steel plates [7, 231]. Cook et al. carried out pioneering studies on the influence of electrolytic properties, applied voltage and others parameters on the material removal rate during ECD of glass and some other ceramics [7, 232].
Subsequently, Ghosh et al. showed that the technique could be employed for micro-welding very thin twisted wires of a thermocouple (chromel-alumel and several other pairs as the cathode) in a 25% NaCl solution using a sufficiently thicker stainless steel rod anode, to produce satisfactory joints at 80–100 V. They proposed a model predicting the critical voltage and the critical current density for initiating the glow discharge between the tool cathode and the electrolyte considering that the rate of bubble production at the cathode was given by the sum of the rates of electrolytic gas production and the vapor production by local Joule heating. The critical parameters calculated compared well with experimental values [7, 233, 234]. They further developed a simplified model to predict the characteristics of the material removal rate for varying input parameters with the objective of finding the possibility of enhancing the capability of the process. They analyzed ECDM as a switching process between the tool electrode and the electrolyte, and it was found that an extra control parameter can be obtained by introducing an additional inductance in the circuit. The theoretical and experimental results indicated that a substantial increase in the material removal rate could be achieved due to the additional inductance introduced. [7, 235, 236].
In further developments Ghosh et al., Jain et al, Bhattacharya et al, Fascio et al., Wuthrich et al. demonstrated that cathodic CGDE had the potentiality of machining glass, quartz, various ceramics and other non-conducting materials for holes, wires and even threads by positioning the workpiece just below the glow discharge cathode (usually of Cu) in an alkaline electrolyte (20–30% NaOH solution) at 70–90 V pulsed DC at 50 Hz using Cu anode [7, 235–250]. The technique made use of intense thermal effects of cathode glow enough for melting and even evaporating the work material below the tool cathode in combination with etching action by the alkaline electrolyte and was referred as spark assisted chemical engraving (SACE). By controlling the gas film thickness around the cathode reproducibly to a few μm resolution through the use of a surfactant e.g. a liquid soap additive to the catholyte. It was found that the size of the hole in a glass plate by SACE technique was found reduced from ~172 to ~9 μm on adding a liquid soap to 30% NaOH electrolyte with reduction in critical voltage from 21.2 to 13.7 V and reduction in critical current from 1.30 to 0.53 A. Thus method holds the potential of upgradation as a micro-precision machining for non-conducting materials.
The machining accuracy in drilling quartz and glass plates by SACE in a KOH solution was improved by Hourng et al. using a W rod as the tool through the addition of 6.5% ethyl alcohol to the electrolyte [251].
Not only the cathode but also the anode was employed as the tool electrode in the machining process [232, 252, 253]. Interestingly, as shown by West et al. drilling a glass plate with an anode-tool of stainless needle and a stainless steel rod as the auxiliary electrode in 30% NaOH solution at 40 V led to 367 μm dia. hole as compared to 976 μm dia. hole on reversing the polarity of the electrodes. Interestingly, anodic machining led to a more spherical hole shapes than with cathodic machining, where V-shaped profiles were obtained. They attributed this to the possible formation of a large number of OH· radicals at the gas film-solution interface during anodic ECD when the concentration of OH· radicals as a function of hole depth appeared more or less uniform. The work piece was etched isotropically resulting in a spherical shape profile [253].
In a challenging working on drilling a micro-hole in a 304 stainless steel plate as the workpiece anode, Huang et al. employed ECDM at 10–14V successfully with a high speed rotating (24,000–42,000 rpm) tungsten carbide helix micro-tool cathode (250–400 μm dia.) in pure water [254].
Moreover, a number of hybrid micromachining techniques involving cathodic CGDE has been developed for machining of conducting, non conducting and hard to cut materials [255–257].
Hydrogen Production
Currently, hydrogen is regarded as one of the most suitable ways to store renewable electricity, because of its high energy density and hence high energy capacity. Combined with fuel cell technology, this makes hydrogen storage most appropriate for compensation of long-term fluctuations. Based on its non-faradaic chemical effects, CGDE may provide a feasible technology for hydrogen generation. This method of hydrogen production in recent years has attracted significant attention as a sufficiently rapid and amenable one to incorporation into cost-effective point-of-use systems. The first report on non-faradaic H2 liberation during cathodic CGDE at 200 V in KCl solutions at a Pt wire was made by Palit in 1967–68 [258, 259]. The phenomenon was systematically investigated by Sen Gupta et al. at 220–240 V who showed that substantially excess H2 over the faradaic yield (200%) was obtained from the plasma phase reaction zone of cathodic CGDE of aqueous solutions of many salts, acids and alkalis through the recombination of H· radicals produced there [1, 260, 261]. It was expected that in the presence of a H·/OH· scavenger such as a lower alcohol, non-faradaic yield of H2 should enhance further due to their scavenging action on a H· radical in cathodic as well as anodic CGDE producing a H2 molecule straight plus a decrease in the extent of the recombination of H· with OH·. This was shown for several propyl and butyl alcohols [29]. In fact, Yan et al. demonstrated cathodic CGDE of methanol/ethanol solutions of NaOH, a promising method for generation of hydrogen. Cathodic CGDE was found more than 9–10 times more productive than anodic CGDE in terms of mol/mol electron. The energy consumption for H2 production was found 4.2–5.2 kJ/l for cathodic CGDE of a methanol/ethanol solution at a W rod cathode for an applied voltage of 1000–1150 V as compared to 10–12 times higher value for anodic CGDE. The main bye-product was an aldehyde and H2 percentage in the cathode gas after the separation of aldehyde was as high as 80–88%. Thus, the recovery of the aldehyde would effectively reduce the cost of hydrogen production [30, 31].
Mizuno et al. attempted continuous production of non-faradaic hydrogen by optimizing the electrode surface condition, electrolyte temperature, current density and the input voltage. They achieved an electric power efficiency of 10% at an input voltage of 325 V. They further claimed that by plasma electrolysis using a W wire cathode in K2CO3 solutions at 350 V as much as 80 times more hydrogen than by conventional electrolysis could be generated [262, 263].
Bespalko reviewed recently the foregoing developments in an article and made a relative assessment of CGDE with methanol steam reforming, conventional water electrolysis and water vapor electrolysis for H2 generation. CGDE was found to have the superiority over methanol steam reforming in virtual freedom from CO2 emission, over both normal electrolysis of water and a modern water vapor electrolyzer in lesser net energy consumption. However, its power efficiency was rather low. Thus, CGDE as a H2 generating process needs development to overcome this main drawback [14].
Conclusions and Future Outlook
The article provides a review of the state-of-the-art in the applications of contact glow discharge electrolysis (CGDE) as a reference document for researchers in this and related areas. It illustrates how the appreciation of remarkable potentialities of CGDE in generating strong thermal effects and high local yield of H· and OH· radicals has led to its judicious exploitation for economical and green solutions to certain existing problems across diverse technologies ranging over synthetic chemical, polymer, water pollution remediation, hydrogen production, electroceramic, magnetic, photoactive and catalytic surface materials, surgical and biomedical, aerospace and automotive, oil and gas, electric and tools, nanoparticle, film and coatings, machining and micro-machining technologies. The challenge is now to expand its domain of applications, explore it for advanced applications, optimize and tune best its operational conditions for specific applications and improvise CGDE as a commercially viable tool particularly for polymer, waste water management and metal including surgical and bio-implant industries. For every application, the goal is naturally to minimize energy consumption for generation and sustenance of electrolytic plasmas by working out the electrolysis conditions, and to make this green technology economically competitive with the traditional ones. It has been shown that electrolyte temperature, surface tension, resistivity in the vicinity of the working electrode and electrode geometry, specific resistance and surface properties of electrode have considerable influences on the initiation of a plasma in the liquid around the electrode [1, 20, 23, 264, 265].
It is a complex and multidisciplinary area of research which requires a combined effort of specialists from different scientific fields such as plasma physics, plasma chemistry, electrochemistry, materials science, nanotechnology, bio-medicine, engineering, advanced manufacturing, environmental science, analytical sciences and instrumentation etc. The subject interfaces readers from various backgrounds and diverse interests. No doubt, there is yet a long way to go towards the full exploration of the potential of CGDE.
References
Sen Gupta SK (2015) Contact glow discharge electrolysis: its origin, plasma diagnostics and non-faradaic chemical effects. Plasma Sources Sci Technol 24:063001
Yerokhin AL, Nie X, Leyland A, Matthews A, Dowey SJ (1999) Plasma electrolysis for surface engineering. Surf Coat Technol 122:73–93
Gupta P, Tenhundfeld G, Daigle EO, Ryabkov D (2007) Electrolytic plasma technology: science and engineering—an overview. Surf Coat Technol 201:8746–8760
Aliofkhazraei M, Rouhaghdam AS, Gupta P (2011) Nano-fabrication by cathodic plasma electrolysis. Crit Rev Solid State Mater Sci 36:174–190
Pogrebnyak AD, Sh Kaverina A, Kylyshkanov MK (2014) Electrolytic plasma processing for plating coatings and treating metals and alloys. Prot Met Phys Chem Surf 50:72–87
Parfenov EV, Yerokhin A, Nevyantseva RR, Gorbatkov MV, Liang C-J, Matthews A (2015) Towards smart electrolytic plasma technologies: an overview of methodological approaches to process modeling. Surf Coat Technol 269:2–22
Wuthrich R, Fascio V (2005) Machining of non-conducting materials using electrochemical discharge phenomenon—an overview. Int J Mach Tools Manuf 45:1095–1108
Lal A, Bleuler H, Wuthrich R (2008) Formation of metallic nanoparticles by electrochemical discharges. Electrochem Commun 10:488–491
Graham WG, Stalder KR (2011) Plasmas in liquids and some of their applications in nanoscience. J Phys D Appl Phys 44:174037
Kareem TA, Kaliany AA (2012) Glow discharge plasma electrolysis for nanoparticles synthesis. Ionics 18:315–327
Akolkar R, Sankaran RM (2013) Charge transfer processes at the interface between plasmas and liquids. J Vac Sci Technol A 31:050811
Chen Q, Li J, Li Y (2014) A review of plasma–liquid interactions for nanomaterial synthesis. http://arxiv.org/ftp/arxiv/papers/1404/1404.2515.pdf
Wang X, Zhou M, Jin X (2012) Application of glow discharge plasma for wastewater treatment. Electrochim Acta 83:501–512
Bespalko S (2014) Recent advances in hydrogen generation by contact glow discharge electrolysis: review. J Int Sci Publ Mater Methods Technol 8:355–363
Bruggeman P, Leys C (2009) Non-thermal plasmas in and in contact with liquids. J Phys D Appl Phys 42:053001
Brisset J-L, Moussa D, Doubla A, Hnatiuc E, Hnatiuc B, Youbi GK, Herry J-M, Naitali M, Bellon- Fontain M-N (2008) Chemical reactivity of discharges and temporal post-discharges in plasma treatment of aqueous media: examples of gliding discharge treated solutions. Ind Eng Chem Res 47:5761–5781
Locke BR, Sato M, Sunka P, Hoffmann MR, Chang JS (2006) Electrohydraulic discharge and nonthermal plasma for water treatment. Ind Eng Chem Res 45:882–905
Malik MA, Ghaffar A, Malik SA (2001) Water purification by electrical discharges. Plasma Sources Sci Technol 10:82–91
Sun B, Sato M, Clements JS (1997) Optical study of active species produced by a pulsed streamer corona discharge in water. J Electrostat 39:189–202
Sen Gupta SK, Singh OP (1991) Contact glow discharge electrolysis: a study of its onset and location. J Electroanal Chem 301:189–197
Sen Gupta SK, Singh R (2017) Cathodic contact glow discharge electrolysis: its origin and non-faradaic chemical effects. Plasma Sources Sci Technol 26:015005
Sharma N, Diaz G, Leal-Quiros E (2013) Evaluation of contact glow-discharge electrolysis as a viable method for steam generation. Electrochim Acta 108:330–336
Gangal U, Srivastava M, Sen Gupta SK (2009) Mechanism of the breakdown of normal electrolysis and the transition to contact glow discharge electrolysis. J Electrochem Soc 156:F131–F136
Guilpin Ch, Garbarz-Olivier J (1974) Analyse de la lumiere emise aux electrodes durant les effets d’electrode, dans quelques solutions aqueuses d’electrolyte. J Chim Phys 71:1491–1498
Guilpin Ch, Garbarz-Olivier J (1977) Analyse de la lumiere emise aux electrodes pendant les effets d’electrode, dans des solutions aqueuses. Spectrochim Acta B 32:155–164
Azumi K, Mizuno T, Akimoto T, Ohmori T (1999) Light emission from Pt during high-voltage cathodic polarization. J Electrochem Soc 146:3374–3377
Liu Y, Sun B, Wang L, Wang D (2012) Characteristics of light emission and radicals formed by contact glow discharge electrolysis of an aqueous solution. Plasma Chem Plasma Process 32:359–368
Sen Gupta SK, Singh R, Srivastava AK (1998) A study on the origin of non-faradaic behaviour of anodic contact glow discharge electrolysis: the relationship between power dissipated in glow discharges and non-faradaicyields. J Electrochem Soc 145:2209–2213
Gangal U, Srivastava M, Sen Gupta SK (2010) Scavenging effects of aliphatic alcohols and acetone on H· radicals in anodic contact glow discharge electrolysis: determination of the primary yield of H· radicals. Plasma Chem Plasma Process 30:299–309
Yan ZC, Li C, Lin WH (2009) Hydrogen generation by glow discharge plasma electrolysis of methanol solutions. Int J Hydrog Energy 34:48–55
Yan Z, Chen L, Wang H (2008) Hydrogen generation by glow discharge plasma electrolysis of ethanol solutions. J Phys D Appl Phys 41:155205
Hickling A, Newns GR (1961) Glow-discharge electrolysis: Part V. The contact glow-discharge electrolysis of liquid ammonia. J Chem Soc 5186–5091
Harada K, Iwasaki T (1974) Synthesis of amino acids from aliphatic carboxylic acid by glow discharge electrolysis. Nature 250:426–428
Harada K, Iwasaki T (1975) Synthesis of amino acids from aliphatic amines by contact glow discharge electrolysis. Chem Lett 185–188
Harada K, Suzuki S (1977) Formation of amino acids from ammonium bicarbonate or ammonium formate by contact glow discharge electrolysis. Naturwissenschaften 64:484
Harada K, Suzuki S (1977) Formation of amino acids from elemental carbon by contact glow discharge electrolysis. Nature 266:275–276
Harada K, Suzuki S, Ishida H (1978) Synthesis of amino acids from unsaturated aliphatic carboxylic acid by contact glow discharge electrolysis. Experientia 34:17–18
Harada K, Suzuki S, Ishida H, Matsuyama M, Tamura M (1978) Amino acid synthesis by contact glow discharge electrolysis: II A possible route for prebiotic synthesis of amino acids. Origin Life 2:141–151
Harada K, Suzuki S, Ishida H (1978) Formation of amino acids from aliphatic nitriles and aliphatic amines by contact glow discharge electrolysis. Bio Syst 10:247–251
Harada K, Terasawa J, Gunji H (1980) Oxidative degradation of hydroxyl amino acids by contact glow discharge electrolysis. Chem Lett 1545–1548
Harada K, Terasawa (1980) Oxidative degradation of β- and γ-amino acids by contact glow discharge electrolysis. Chem Lett 441–444
Suzuki S, Tamura M, Terasawa J, Harada K (1978) Formation of α-amino acids from β- and γ-amino acids by contact glow discharge electrolysis. Bio-org Chem 7:111–113
Harada K, Nomoto MM, Gunji H (1981) Formation of amino acids from aliphatic amines by contact glow discharge electrolysis. Tetrahedron Lett 22:769–772
Kokufuta E, Sodeyama T, Fujimori K, Harada K, Nakamura I (1984) D–H exchange and hydroxylation of (2H3) acetic acid in aqueous solution during glow discharge electrolysis. J Chem Soc Chem Commun 269–270
Kokufuta E, Sodeyama T, Harada K (1985) Simultaneously occurring hydroxylation, hydration, and hydrogenation of the C=C bond of aliphatic carboxylic acids. Chem Lett 1569–1572
Munegami T, Shimoyama A, Harada K (1997) Abiotic asparagines formation from simple amino acids by contact glow discharge electrolysis. Chem Lett 393–394
Nomoto MM, Sakai F, Harada K (1981) Introduction of functional groups to polyalanine and alanine by contact glow discharge electrolysis. Polym Bull 5:451–456
Harada K, Igari S, Takasaki M, Shimoyama A (1986) Reductive fixation of molecular nitrogen by glow discharge against water. J Chem Soc Chem Commun 1384–1385
Harada K, Terasawa J, Suzuki S (1978) Synthesis of uracil and thymine by contact glow discharge electrolysis. Naturwissenschaften 65:259
Sen Gupta SK, Srivastava M (2006) Chemical effects of anodic contact glow discharge electrolysis: formation of amino acid. J Indian Chem Soc 83:787–797
Sen Gupta SK, Singh R, Srivastava AK (1995) Chemical effects of anodic contact glow discharge electrolysis in aqueous formic acid solutions: formation of oxalic acid. Indian J Chem 34A:459–461
Tezuka M, Yajima T, Tsuchiya A (1987) Proc Int Symp Plasma Chem Tokyo Jpn 8:1816
Sen Gupta SK, Sandhir U, Misra N (2001) A study on acrylamide polymerization by anodic contact glow discharge electrolysis: a novel tool. J Polym Sci Part A Polym Chem 39:1584–1588
Gao J, Wang A, Li Y, Fu Y, Wu J, Wang Y, Wang Y (2008) Synthesis and characterization of superabsorbent composite by using glow discharge electrolysis plasma. React Funct Polym 68:1377–1383
Lu Q, Yu J, Gao J, Yang W, Li Y (2011) Glow-discharge electrolysis plasma induced synthesis of polyvinylpyrrolidone/acrylic acid hydrogel and its adsorption properties for heavy-metal ions. Plasma Process Polym 8:803–814
Gao J, Li X, Lu Q, Li Y, Ma D, Yang W (2012) Synthesis and characterization of poly (methyl methacrylate-butyl acrylate) by using glow-discharge electrolysis plasma. Polym Bull 68:37–51
Lu Q, Yu J, Gao J, Yang W, Li Y (2012) A promising absorbent of acrylic acid/poly (ethylene glycol) hydrogel prepared by glow-discharge electrolysis plasma. Cent Eur J Chem 10:1349–1359
Wang X, Gao J, Yang W (2012) Polymeric superabsorbing composite prepared using a glow-discharge electrolysis plasma for the removal of divalent heavy metal ions from aqueous solutions and its swelling properties. Polym Eng Sci 52:2217–2227
Yu J, Pan Y, Lu Q, Yang W, Gao J, Li Y (2013) Synthesis and swelling behaviors of P(AMPS-co-AAC) superabsorbent hydrogel produced by glow-discharge plasma. Plasma Chem Plasma Process 33:219–235
Yu J, Yang G, Li Y, Wang W, Gao J, Lu Q (2014) Synthesis, characterization, and swelling behaviors of acrylic acid/carboxymethyl cellulose superabsorbent hydrogel by glow-discharge electrolysis. plasma. Polym Eng Sci 54:2310–2320
Yu J, Yang G, Pan Y, Lu Q, Yang W, Gao J (2014) Poly (acrylamide-co-acrylic acid) hydrogel induced by glow-discharge electrolysis plasma and its adsorption properties for cationic dyes. Plasma Sci Technol 16:767–776
Li Y, Yao M, Liao R, Yang W, Gao J, Ren J (2014) Synthesis of poly (butyl methacrylate/butyl acrylate) highly absorptive resin using glow discharge electrolysis. Plasma Sci Technol 16:777–781
Zhang W, Zhu S, Bai Y, Xi N, Wang S, Bian Y, Li X, Zhang Y (2015) Glow discharge electrolysis plasma initiated preparation of temperature/pH dual sensitivity reed hemicellulose-based hydrogels. Carbohydr Polym 122:11–17
Zhang W, Zhu S, Huang Y, Bai Y, Xi N, Zhang Y (2015) Glow discharge electrolysis plasma induced synthesis of cellulose-based ionic hydrogels and their multiple response behaviors. RSC Adv 5:6505–6511
Joshi R, Friedrich J, Subramanian SK (2013) Surface modification of ultra-high molecular weight polyethylene membranes using underwater plasma polymerization. Plasma Chem Plasma Process 33:921–940
Tezuka M, Iwasaki M (1999) Liquid-phase reactions induced by gaseous plasmas. Decomposition of benzoic acids in aqueous solution. Plasma Ions 2:23–26
Ogata A, Miyamae K, Mizuno K, Kushiyama K, Tezuka M (2002) Decomposition of benzene in air in a plasma reactor: effect of reactor type and operating conditions. Plasma Chem Plasma Process 22:537–552
Tezuka M, Iwasaki M (2001) Plasma-induced degradation of aniline in aqueous solution. Thin Solid Films 386:204–207
Tezuka M, Iwasaki M (1997) Oxidative degradation of phenols by contact glow discharge electrolysis. Denki Kagaku Electrochemistry 65:1057–1060
Tomizawa S, Tezuka M (2007) Kinetics and mechanism of the organic degradation in aqueous solution irradiated with gaseous plasma. Plasma Chem Plasma Process 27:486–495
Tomizawa S, Tezuka M (2006) Oxidative degradation of aqueous cresols induced by gaseous plasma with contact glow discharge electrolysis. Plasma Chem Plasma Process 26:43–52
Tezuka M, Iwasaki M (1998) Plasma induced degradation of chlorophenols in an aqueous solution. Thin Solid Films 316:123–127
Yang H, Matsumoto Y, Tezuka M (2009) Exhaustive breakdown of aqueous monochlorophenols by contact glow discharge electrolysis. J Environ Sci 21(Suppl.):S142–S145
Caixia Yang H, Tezuka M (2013) Plasma induced decomposition of dichlorophemols and trichlorophenols in water by means of anodic contact glow discharge electrolysis. Plasma Chem Plasma Process 33:1043–1052
Yang H, Tezuka M (2011) Plasma-induced complete destruction of tetrachlorophenols in an aqueous solution. J Phys D Appl Phys 44:155203
Yang H, Tezuka M (2011) Mineralization of aqueous pentachlorophenolate by anodic contact glow discharge electrolysis. J Environ Sci 23:1044–1049
Amano R, Tomizawa S, Tezuka M (2004) Mineralization of aqueous benzenesulfonates by contact glow discharge electrolysis. Electrochemistry 72:836–838
Amano R, Tezuka M (2006) Mineralization of alkylbenzenesulfonates in water by means of contact glow discharge electrolysis. Water Res 40:1857–1863
Hattori M, Amano R, Tezuka M (2006) Oxidative degradation of aqueous alkanesulfonates by contact glow discharge electrolysis. Electrochemistry 74:632–634
Gao J, Liu Y, Yang W, Pu L, Yu J, Lu Q (2003) Oxidative degradation of phenol in aqueous electrolyte induced by plasma from a direct glow discharge. Plasma Sources Sci Technol 12:533–538
Liu Y, Jiang X (2005) Phenol degradation by a nonpulsed diaphragm glow discharge in an aqueous solution. Environ Sci Technol 39:8512–8517
Wang L, Liu Y (2012) Enhancement of phenol degradation by electron acceptors in anodic contact glow discharge electrolysis. Plasma Chem Plasma Process 32:715–722
Wang L, Jiang X (2009) Unusual catalytic effects of iron salts on phenol degradation by glow discharge plasma in aqueous solution. J Hazard Mater 161:926–932
Liu Y, Wang D, Sun B, Zhu X (2010) Aqueous 4-nitrophenol decomposition and hydrogen peroxide formation induced by contact glow discharge electrolysis. J Hazard Mater 181:1010–1015
Gao J, Pu L, Yang W, Yu J, Li Y (2004) Oxidative degradation of nitrophenols in aqueous solution induced by plasma with submersed glow discharge electrolysis. Plasma Process Polym 1:171–176
Gao J, Wang A, Fu Y, Wu J, Ma D, Guo X, Li Y, Yang W (2008) Analysis of energetic species caused by contact glow discharge electrolysis in aqueous solution. Plasma Sci Technol 10:30–38
Wang L (2009) 4-Chlorophenol degradation and hydrogen peroxide formation induced by DC diaphragm glow discharge in an aqueous solution. Plasma Chem Plasma Process 29:241–250
Gao J, Yang W, Liu Y, Chen P, Na P, Lu Q (2003) Oxidative degradation of o-chlorophenol with contact glow discharges in aqueous solution. Plasma Sci Technol 5:1609–1614
Jin X, Zhao X, Wang X, Wang Z (2013) Degradation and toxicity change of 4-chlorophenolin aqueous solution during CGDE treatment. Water Sci Technol 67:2190–2194
Lu Q, Yu J, Gao J (2006) Degradation of 2, 4-dichlorophenol by using glow discharge electrolysis. J Hazard Mater 136:526–531
Gao J, Hu Z, Lu Q, Na P, Chen P, Liu Y, Yu J (2003) Degradation of chloroanilines in aqueous solution by contact glow discharge electrolysis. Plasma Sci Technol 5:1721–1727
Liu Y, Jiang X (2008) Plasma-induced degradation of chlorobenzene in aqueous solution. Plasma Chem Plasma Process 28:15–24
Wang L, Jiang X, Liu Y (2007) Efficient degradation of nitrobenzene induced by glow discharge plasma in aqueous solution. Plasma Chem Plasma Process 27:504–515
Liu Y (2009) Aqueous p-chloronitrobenzene decomposition induced by contact glow discharge electrolysis. J Hazard Mater 166:1495–1499
Yang H, An B, Wang S, Li L, Jin W, Li L (2013) Destruction of 4-phenosulfonic acid in water by anodic contact glow discharge electrolysis. J Environ Sci 25:1063–1070
Lu Q, Yu J, Gao J, Yang W (2005) Glow discharge induced hydroxyl radical degradation of 2-naphthylamine. Plasma Sci Technol 7:2856–2859
Gai K (2007) Plasma-induced degradation of diphenylamine in aqueous solution. J Hazard Mater 146:249–254
Gai K, Dong Y (2005) Plasma induced degradation of azobenzene in water. J Chin Chem Soc 52:273–276
Bratescu MA, Hieda J, Umemura T, Saito N, Takai O (2011) Analysis of organic pollutant degradation in pulsed plasma by coherent anti-Stokes Raman spectroscopy. J Vac Sci Technol A 29:031302
Wang L, Zeng H, Xin Y (2014) Dechlorination and decomposition of trichloroacetic acid by glow discharge plasma in aqueous solution. Electrochim Acta 115:332–336
Wang L, Liu P, Zhang S (2015) Debromination and decomposition of bromoform by contact glow discharge electrolysis in an aqueous solution. Electrochim Acta 165:390–395
Wang L, Liu P, Chen T (2016) Glow discharge plasma induced dechlorination and decomposition of dichloromethane in an aqueous solution. Plasma Chem Plasma Process 36:615–626
Wang L, Jiang X (2008) Plasma-induced reduction of chromium (VI) in an aqueous solution. Environ Sci Technol 42:8492–8497
Liu Y (2009) Simultaneous oxidation of phenol and reduction of Cr(VI) induced by contact glow discharge electrolysis. J Hazard Mater 168:992–996
Gao J, Yu J, Li Y, He X, Bo L, Pu L, Yang W, Lu Q, Yang Z (2006) Decoloration of aqueous Brilliant Green by using glow discharge electrolysis. J Hazard Mater B137:431–436
Jin X, Wang X, Wang Q, Yue J, Cai Y (2010) Plasma degradation of cationic blue dye with contact glow discharge electrolysis. Water Sci Technol 62:1457–1463
Gong J, Cai W (2007) Degradation of methyl orange in water by contact glow discharge electrolysis. Plasma Sci Technol 9:190–193
Gong J, Wang J, Xie W, Cai W (2008) Enhanced degradation of aqueous methyl orange by contact glow discharge electrolysis. J Appl Electrochem 38:1749–1755
Gao J, Hu Z, Wang X, Hou J, Liu X, Kang J (2001) Oxidative degradation of acridine orange induced by plasma with contact glow discharge electrolysis. Thin Solid Films 390:154–158
Jin X, Bai H, Wang F, Wang X, Wang X, Ren H (2011) Plasma degradation of acid orange 7 with contact glow discharge electrolysis. IEEE Trans Plasma Sci 39:1099–1103
Jin X, Xia Q, Zhang H, Wang X (2011) The role of electrolyte constituents and metal ions on dye discoloration with contact glow discharge electrolysis. IEEE Trans Plasma Sci 39:3218–3221
Gao J, Wang X, Hu Z, Deng H, Ho J, Liu X, Kang J (2003) Plasma degradation of dyes in water with contact glow discharge electrolysis. Water Res 37:267–272
Wang L (2009) Aqueous organic dye discoloration induced by contact glow discharge electrolysis. J Hazard Mater 171:577–581
Gao J, Ma D, Cuo X, Li Y, Yang W (2007) Degradation of cationic dye crystal violet in wastewater using glow discharge plasma. Chinese J Appl Chem 24:534–539
Gao J, Guo X, Ma D, Yang W (2007) The role of Fe(II) in the contact glow discharge electrolysis. Plasma Sci Technol 9:431–435
Gao J, Yu J, Lu Q, He X, Yang W, Li Y, Pu L, Yang Z (2008) Decoloration of alizarin red S in aqueous solution by glow discharge electrolysis. Dyes Pigm 76:47–52
Ramjaun SN, Yuan R, Wang Z, Liu J (2011) Degradation of reactive dyes by contact glow discharge electrolysis in the presence of Cl− ions: kinetics and AOX formation. Electrochim Acta 58:364–371
Gao J, Wang X, Hu Z, Yang W (2004) Kinetic behavior of plasma degradation of Catioic Red in aqueous solution. Curr Top Electrochem 10:197–201
Jin X, Zhang H, Wang X, Zhou M (2012) An improved multi-anode contact glow discharge electrolysis reactor for dye discoloration. Electrochim Acta 59:474–478
Gao J, Chen L, He YY, Yan ZC, Zheng XJ (2014) Degradation of imidazolium-based ionic liquids in aqueous solution using plasma electrolysis. J Hazard Mater 265:261–270
Jin X, Wang X, Wang Yu, Ren H (2013) Oxidative degradation of amoxicillin in aqueous solution with contact glow discharge electrolysis. Ind Eng Chem Res 52:9726–9730
Hong S, Min ZW, Mok C, Kwon H, Kim T, Kim D (2013) Aqueous degradation of imidacloprid and fenothiocarb using contact glow discharge electrolysis: degradation behavior and kinetics. Food Sci Biotechnol 22:1773–1778
Saksona N, Adiwidodo BP, Karamah EF, Kartohardjono S (2013) Contact glow discharge electrolysis for treatment of wastewater containing ammonia. J Environ Sci Technol 6:41–49
Tong S, Ni Y, Shen C, Wen Y, Jiang X (2011) Degradation of methyl tert-butyl ether (MTBE) in water by glow discharge plasma. Water Sci Technol 63:2814–2819
Jin X, Xia Q, Wang X, Yue J, Wei D (2011) 1Inactivation of Microcystis aeruginosa with contact glow discharge electrolysis. Plasma Chem Plasma Process 31:697–705
Kokufuta E, Shibasaki T, Nakamura I, Harada K, Sodeyama T (1985) Degradation of polyethylene glycol in a localized reaction zone during glow discharge electrolysis. J Chem Soc Chem Commun 100–102
Kokufuta E, Fujii S, Ishibashi H, Yokoi H, Harada K, Nakamura I (1980) Degradation of poly(acrylamide) in aqueous solution by glow discharge electrolysis. Polym Bull 3:173–178
Sandhir U (1998) Ph.D. Thesis, A study on origin and chemical effects of contact glow discharge electrolysis in aqueous electrolytes. Banaras Hindu University, Varanasi
Bae JS, Lee JS, Kim YS, Sim WJ, Lee H, Chun JY, Park K (2008) Depolymerization of fucoidan by contact glow discharge electrolysis. Hwahak-konghak (Korean Chem Eng Res) 46:886–891
Munegami T, Shimoyama A (1998) Degradation of dipeptides by contact glow discharge electrolysis. Viva Origino 26:103–108
Wen Y, Shen C, Ni Y, Tong S, Yu F (2012) Glow discharge plasma in water: a green approach to enhancing ability of chitosan for dye removal. J Hazard Mater 201–202:162–169
Gao J, Chen L, Zhang J, Yan Z (2014) Improved enzymatic hydrolysis of lignocellulosic biomass through pretreatment with plasma electrolysis. Bioresource Technol 171:469–471
Stalder KR, Woloszko J, Brown IG, Smith CD (2001) Repetitive plasma discharges in saline solutions. Appl Phys Lett 79:4503–4505
Woloszko J, Stalder KR, Brown IG (2002) Plasma characteristics of repetitively-pulsed electrical discharges in saline solutions used for surgical procedures. IEEE Trans Plasma Sci 30:1376–1383
Stalder KR, Woloszko J (2007) Some physics and chemistry of electrosurgical plasma discharges. Contrib Plasma Phys 47:64–71
Stalder KR, McMillen DF, Woloszko J (2005) Electrosurgical plasmas. J Phys D Appl Phys 38:1728–1738
Lagrange MM, Hoho M (1893) C R Acad Sci (Paris) 116:575
Hoho M (1929) Elec Rev 104:18
Yasanogorodskii IZ (1954) Avto Trakt Prom 6:21
Sato T, Mii H (1956) Rep Gov Ind Res Inst Nagoya 5:313, 415, 586
Sato T, Mii H (1957) Rep Gov Ind Res Inst Nagoya 6:179, 338, 610
Hickling A (1971) Electrochemical processes in glow discharge at the gas-solution interface. In: Bockris JO’M, Conway BE (eds) Modern aspects of electrochemistry. Plenum Press, New York, pp 329–373
Yasnogorodskii IZ (1949) Nagrev Metallov i splavov v elektrolite (Electrolytic heating of metals and alloys). Masghiz, Moscow
Owaku S, Kuroyanagi K (1956) Electrolytic hardening (Immersion method-Report 1). J Jpn Inst Met 20:63–67
Yasanogorodskii IZ (1971) Elektrokhimicheskaya i elektromekhanicheskaya obrabotka metallov (Electrochemical and electromechanical treatment of metals). Mashinostroenie, Moscow
Slovetskii DI, Terent’ev SD, Plekhanov VG (1986) The mechanism of plasma–electrolytic heating of metals. Teplofiz Vys Temp 24:353–363
Tyurina YN, Pogrebnjakb AD (2001) Electric heating using a liquid electrolyte. Surf Coat Technol 142–144:293–299
Belkin PN (2005) Electrochemico-thermal treatment of metals in alloys.[In Russian] Moscow Cross Ref:[149]
Bayati MR, Molaei R, Janghorban K (2011) Surface alloying of carbon steels from electrolytic plasma. Met Sci Heat Treat 53:91–94
Hino M, Murakami K, Saijo A, Hikino S, Kanadani T, Tsujikawa M (2011) Surface heat treatment of magnesium alloys by plasma electrolysis from phosphate electrolyte solution. Mater Trans 52:2168–2173
Meletis EI, Nie X, Wang FL, Jiang JC (2002) Electrolytic plasma processing for cleaning and metal-coating of steel surfaces. Surf Coat Technol 150:246–256
Yerokhin AL, Pilkington AP, Matthews A (2010) Pulse current plasma assisted electrolytic cleaning of AISI 4340 steel. J Mater Process Technol 210:54–63
Snizhko LO, Yerokhin AL, Pilkington AP, Gurevina NL, Misnyankin DO, Leyland A, Matthews A (2004) Anodic processes in plasma electrolytic oxidation of aluminium in alkaline solutions. Electrochim Acta 49:2085–2095
Asquith DT, Tai YH, Wong CX, Yates JR, Matthews A, Yerokhin AL (2008) Measurement of the interfacial shear strength of a PEO coated aluminium alloy. In: 17th European conference on fracture Brno Czech Republic, pp 2105–2111
Bian G, Wang L, Wang J, Zheng J, Sun H, DaCosta H (2015) Effects of electrolytes on the growth behavior, microstructure and tribological properties of plasma electrolytic oxidation coatings on a ZA27 alloy. Surf Coat Technol 277:251–257
Petkovic M, Stojadinovic S, Vasilic R, Belca I, Nedic Z, Kasalica B, Mioc UB (2011) Preparation of silicate tungsten bronzes on aluminium by plasma electrolytic oxidation process in 12-tngstosilicic acid. Appl Surf Sci 257:9555–9561
Stojadinović S, Tadić N, Radić N, Stojadinović B, Grbić B, Vasilić R (2015) Synthesis and characterization of Al2O3/ZnO coatings formed by plasma electrolytic oxidation. Surf Coat Technol 276:573–579
Arrabal R, Matykina E, Hashimoto Skeldon P, Thompson GE (2009) Characterization of AC PEO coatings on magnesium alloys. Surf Coat Technol 203:2207–2220
Hussein RO, Nie X, Nothwood DO (2013) An investigation of ceramic coating growth mechanism in plasma electrolytic oxidation (PEO) processing. Electrochim Acta 112:111–119
Gnedenkov SV, Khrisanfova OA, Zavidnaya AG, Sinebryukhov SL, Egorkin VS, Nistratova MV, Yerokhin AL, Matthews A (2010) PEO coatings obtained on an Mg–Mn type alloy under unipolar and bipolar modes in silicate-containing electrolytes. Surf Coat Technol 204:2316–2322
Lim T, Ryu HS, Hong S (2013) Plasma electrolytic oxidation/cerium conversion composite coatings for the improved corrosion protection of AZ31Mg alloys. J Electrochem Soc 160:C77–C82
Liu C, Zhao Y, Chen Y, Liu P, Cai K (2014) Surface modification of magnesium alloy via cathodic plasma electrolysis and its influence on corrosion resistance and cytocompatibility. Mater Lett 132:15–18
Gao Y, Yerokhin AL, Parfenov E, Matthews A (2014) Application of voltage pulse transient analysis during plasma electrolytic oxidation for assessment of characteristics and corrosion behaviour of Ca- and P-containing coatings on magnesium. Electrochim Acta 149:218–230
Yu L, Cao J, Cheng Y (2015) An improvement of the wear and corrosion resistances of AZ31 magnesium alloy by plasma electrolytic oxidation in a silicate–hexametaphosphate electrolyte with the suspension of SiC nanoparticles. Surf Coat Technol 276:266–278
Yerokhin AL, Nie X, Leyland A, Matthews A (2000) Characterization of oxide films produced by plasma electrolytic oxidation of a Ti-6Al-4V alloy. Surf Coat Technol 130:195–206
Cheng Y, Wu X, Xue Z, Matykina E, Skeldon P, Thompson GE (2013) Microstructure, corrosion and wear performance of plasma electrolytic oxidation coatings formed on Ti-6Al-4V alloy in silicate-hexametaphosphate electrolyte. Surf Coat Technol 217:129–139
Shin KR, Ko YG, Shin DH (2011) Effect of electrolyte on surface properties of pure titanium coated by plasma electrolytic oxidation. J Alloys Compd 509:S478–S481
Shin KR, Yoon SI, Ko YG, Shin DH (2013) Deposition of hydroxyl-apatite on titanium subjected to electrochemical plasma coating. Electrochim Acta 109:173–180
Shin KR, Kim YS, Yang HW, Ko YG, Shin DH (2014) In vitro biological response to the oxide layer in pure titanium formedat different current densities by plasma electrolytic oxidation. Appl Surf Sci 314:221–227
Shin KRYS, Kim GW, Yang HW, Ko YG, Shin DR (2015) Effects of concentration of Ag nanoparticles on surface structure and in vitro biological responses of oxide layer on pure titanium viaplasma electrolytic oxidation. Appl Surf Sci 347:574–582
Rudnev VS, Medkov MA, Yarovaya TP, Nedozorov PM (2012) Calcium and strontium phosphates coatings on titanium formed by the plasma electrolytic oxidation. Russian J Appl Chem 85:1856–1860
Chung C-J, Su R-T, Chu H-J, Chen H-T, Tsou H-K, He J-L (2013) Plasma electrolytic oxidation of titanium and improvement in osseointegration. J Biomed Mater Res 101B:1023–1030
Mirelman LK, Curran JA, Clyne TW (2012) The production of anatase-rich photoactive coatings by plasma electrolytic oxidation. Surf Coat Technol 207:66–71
Soejima T, Yagyu H, Ito S (2011) One-pot synthesis and photocatalytic activity of Fe-doped TiO2 films with anatase-rutile nanojunction prepared by plasma electrolytic oxidation. J Mater Sci 46:5378–5384
Rodriguez LL, Sundaram PA, Rosim-Fachini E, Padovani AM, Diffoot-Carlo N (2014) Plasma electrolytic oxidation coatings on γTiAl alloy for potential biomedical applications. J Biomed Mater Res B Appl Biomater 102B:988–1001
Kazek-Kęsik A, Dercz G, Suchanek K, Kalemba-Rec I, Piotrowski J, Simka W (2015) Biofunctionalization of Ti–13Nb–13Zr alloy surface by plasma electrolytic oxidation. Part I Surf Coat Technol 276:59–69
Yeung WK, Sukhorukova IV, Shtansky DV, Levashov EA, Zhitnyak IY, Gloushankova NA, Kiryukhantsev-Korneev PV, Petrzhik MI, Matthews A, Yerokhin A (2016) Characteristics and in vitro response of thin hydroxyapatite–titania films produced by plasma electrolytic oxidation of Ti alloys in electrolytes with particle additions. RSC Adv 6:12688–12698
Sandhyarani M, Prasadrao T, Rameshbabu N (2014) Role of electrolyte composition on structural, morphological and in-vitro biological properties of plasma electrolytic oxidation films formed on zirconium. Appl Surf Sci 317:198–209
Cheng Y, Wu F, Matykina E, Skeldon P, Thompson GE (2012) The influences of microdischarge types and silicate on the morphologies and phase compositions of plasma electrolytic oxidation coatings on Zircaloy-2. Corros Sci 59:307–315
Cheng Y, Matykina E, Arrabal R, Skeldon P, Thompson GE (2012) Plasma electrolytic oxidation and corrosion protection of Zircaloy-4. Surf Coat Technol 206:3230–3239
Cheng Y, Peng Z, Wua X, Cao J, Skeldon P, Thompson GE (2015) A comparison of plasma electrolytic oxidation of Ti-6Al-4V and Zircaloy-2 alloys in a silicate-hexametaphosphate electrolyte. Electrochim Acta 165:301–313
Wu J, Liu R, Wang B, Yang C, Qu Y, Xue W (2015) Preparation and characterization of carburized layer on pure aluminum by plasma electrolysis. Surf Coat Technol 269:119–124
Tavakoli H, Mousavi Khoie SM, Rasooli F, Marashi SPH, Momeni F (2015) Electrochemical and physical characteristics of the steel treated by plasma-electrolysis boronizing. Surf Coat Technol 276:529–533
Nie X, Tsotsos C, Wilson A, Yerokhin AL, Leyland A, Matthews A (2001) Characteristics of a plasma electrolytic nitrocarburising treatment for stainless steels. Surf Coat Technol 139:135–142
Jiang Y, Geng T, Bao Y, Zhu Y (2013) Electrolyte-electrode interface and surface characterization of plasma electrolytic nitrocarburizing. Surf Coat Technol 216:232–236
Wu J, Zhang Y, Liu R, Wang B, Hua M, Xue W (2015) Anti-corrosion layer prepared by plasma electrolytic carbonitriding on pure aluminum. Appl Surf Sci 347:673–678
Ivanov SV, Salmanov NS, Salmanov MN (2002) Borosulfocarbonitriding of cutting tools in electrolyte plasma. Met Sci Heat Treat 44:405–406
Belkin PNA, Yerokhin A, Kusmanov SA (2016) Plasma electrolytic saturation of steels with nitrogen and carbon. Surf Coat Technol 307:1194–1218
Zhang P, Wang D, He Y (2013) Preparation and properties of cathodic plasma electrolysis Ni-P coatings. Adv Mater Res 634–638:2984–2988
Smith A, Kelton R, Meletis EI (2015) Deposition of Ni coatings by electrolytic plasma processing. Plasma Chem Plasma Process 35:963–968
Paulmier T, Bell JM, Fredericks PM (2007) Development of a novel cathodic plasma/electrolytic deposition technique part 1: production of titanium dioxide coatings. Surf Coat Technol 201:8761–8770
Paulmier T, Bell JM, Fredericks PM (2008) Plasma electrolytic deposition of titanium dioxide nanorods and nano-particles. J Mater Process Technol 208:117–123
Paulmier T, Bell JM, Fredericks PM (2007) Deposition of nano-crystalline graphite films by cathodic plasma electrolysis. Thin Solid Films 515:2926–2934
Paulmier T, Bell JM, Fredericks PM (2007) Development of a novel cathodic plasma/electrolytic deposition technique part 2: physico-chemical analysis of the plasma discharge. Surf Coat Technol 201:8771–8781
Luo Q, Cai Q, Li X, Chen X (2014) Characterization and photocatalytic activity of large-area single crystalline anatase TiO2 nanotube films hydrothermal synthesized on plasma electrolytic oxidation seed layers. J Alloys Compd 597:101–109
Kong X, Wang S, Zhao H, He Y (2010) Preparation of diamond-like carbon films by cathodic micro-arc discharge in aqueous solutions. Thin Solid Films 518:4211–4214
Yan Z, Deng L, Chen L (2013) Cathodic plasma electrolysis in 1-propanol solutions for preparation of submicron diamond particles. Electrochim Acta 105:612–617
Ban C, Huang B, Wang C, Wang L, Shao X (2013) Electrodeposition of diamond-like carbon (DLC) films on Mg by plasma electrolysis. Electrochemistry (Japen) 81:977–980
Wu J, Xue W, Jin X, Wang B, Du J, Wu Z (2013) Preparation and characterization of diamond-like carbon/oxides composite film on carbon steel by cathodic plasma electrolysis. Appl Phys Lett 103:031905
Wu J, Xue W, Wang B, Jin X, Du J, Li Y (2014) Characterization of carburized layer on T8 steel fabricated by cathodic plasma electrolysis. Surf Coat Technol 245:9–15
Gupta P, Tenhundfeld G, Daigle EO (2005) Wire J Int 38:50 Cross Ref: [3]
Zhang Z, Dubey M, Galipeau D, Fan QH, Hoefelmeyer JD, Al-Qaradawi IY (2013) Creating textured surfaces using plasma electrolysis. MRS Commun 3:255–258
Gupta P, Tenhundfeld G, Daigle EO, Schilling PJ (2005) Synthesis and characterization of hard metal coatings by electro-plasma technology. Surf Coat Technol 200:1587–1594
Toriyabe Y, Watanabe S, Yatsu S, Shibayama T, Mizuno T (2007) Controlled formation of metallic nanoballs during plasma electrolysis. Appl Phys Lett 91:041501
Wuthrich R, Allagui A (2010) Building micro and nanosystems with electrochemical discharges. Electrochim Acta 55:8189–8196
Allagui A, Wuthrich R (2011) The electrochemical discharges for the synthesis of nickel oxide nanoparticles: characterization and mechanism. Electrochim Acta 58:12–18
Allagui A, Baranova EA, Wuthrich R (2013) Synthesis of Ni and Pt nanomaterials by cathodic contact glow discharge electrolysis in acidic and alkaline media. Electrochim Acta 93:137–142
Allagui A, Alami AH, Baranova EA, Wüthrich R (2014) Size-dependent capacitance of NiO nanoparticles synthesized with cathodic contact glow discharge electrolysis. J Power Sources 262:178–182
Saito G, Hosokai S, Tsubota M, Akiyama T (2011) Synthesis of copper/copper oxide nanoparticles by solution plasma. J Appl Phys 110:023302
Saito G, Hosokai S, Tsubota M, Akiyama T (2011) Nickel nanoparticles formation from solution plasma using edge-shielded electrode. Plasma Chem Plasma Process 31:719–728
Saito G, Hosokai S, Tsubota M, Akiyama T (2012) Surface morphology of a glow discharge electrode in a solution. J Appl Phys 112:013306
Saito G, Nakasugi Y, Yamashita T, Akiyama T (2014) Solution plasma synthesis of bimetallic nanoparticles. Nanotechnology 25:135603
Saito G, Nakasugi Y, Akiyama T (2014) Excitation temperature of a solution plasma during nanoparticle synthesis. J Appl Phys 116:083301
Julaihia MRM, Yatsu S, Jeem M, Watanabe S (2015) Synthesis of stainless steel nanoballs via submerged glow-discharge plasma and its photolytic performance in methylene blue decomposition. J Exp Nanosci 10:965–982
Zhang ZK, Bai ML, Guo DZ, Hou SM, Zhang GM (2011) Plasma-electrolysis synthesis of TiO2 nano/microspheres with optical absorption extended into the infra-red region. Chem Commun 47:8439–8441
Wu Z, Zhang ZK, Guo DZ (2013) Titanium dioxide nanospheres with wide spectral absorption prepared by low-voltage plasma electrolysis. J Colloid Interface Sci 392:463–464
Chiang W-H, Richmonds C, Sankaran RM (2010) Continuous-flow, atmospheric-pressure microplasmas: a versatile source for metal nanoparticle synthesis in the gas or liquid phase. Plasma Sources Sci Technol 19:034011
Mariotti D, Sankaran RM (2011) Perspectives on atmospheric-pressure plasmas for nanofabrication. J Phys D Appl Phys 44:174023
Meiss SA, Rohnke M, Kienle L, El Abedin SZ, Endres F, Janek J (2007) Employing plasmas as gaseous electrodes at the free surface of ionic liquids: deposition of nanocrystalline silver particles. Chem Phys Chem 8:50–53
Kulbe N, Hofft O, Ulbrich A, El Abedin SZ, Krischok S, Janek J, Polleth M, Endres F (2011) Plasma electrochemistry in 1-butyl-3-methylimidazolium dicyanamide: copper nanoparticles from CuCl and CuCl2. Plasma Process Polym 8:32–37
Baba K, Kaneko T, Hatakeyama R (2009) Efficient synthesis of gold nanoparticles using ion irradiation in gas–liquid interfacial plasmas. Appl Phys Exp 2:035006
Kaneko T, Baba K, Hatakeyama R (2009) Gas–liquid interfacial plasmas: basic properties and applications to nanomaterial synthesis. Plasma Phys Control Fusion 51:124011
KanekoT Baba K, Harada T, Hatakeyama R (2009) Novel gas-liquid interfacial plasmas for synthesis of metal nanoparticles. Plasma Process Polym 6:713–718
Chen Q, Kaneko T, Hatakeyama R (2012) Reductants in gold nanoparticle synthesis using gas–liquid interfacial discharge plasmas. Appl Phys Exp 5:086201
Dupont AJ, Scholten JD (2010) On the structural and surface properties of transition-metal nanoparticles in ionic liquids. Chem Soc Rev 39:1780–1804
Tokushige M, Nishikiori T, Lafouresse MC, Michioka C, Yoshimura K, Fukunaka Y, Ito Y (2010) Formation of FePt intermatallic compound nanoparticles by plasma-induced cathodic discharge electrolysis. Electrochim Acta 55:8154–8159
Kawamura H, Moritani K, Ito Y (1998) Discharge electrolysis in molten chloride: formation of fine silver particles. Plasmas Ions 1:29–36
Kawamura H, Moritani K, Ito Y (1998) Formation of fine nickel particles by discharge electrolysis in molten chloride system. J Jpn Soc Powder Metall 45:1142–1147
Oishi T, Kawamura H, Ito Y (2002) Formation and size control of titanium particles by cathode discharge electrolysis of molten chloride. J Appl Electrochem 32:819–824
Kurafuji H, Suda K (1968) Electrical discharge drilling of glass. Ann CIRP 16:415–419
Kubota M (1974) Proceedings of the international conference on production engineering Tokyo Japan, p 51
Cook NH, Foote GB, Jordan P, Kalyani BN (1973) Experimental studies in electro-machining. Trans ASME J Eng Ind 945–950
Basak I, Ghosh A (1996) Mechanism of spark generation during electrochemical discharge machining: a theoretical model and experimental verification. J Mater Process Technol 62:46–53
Ghosh A, Muju MK, Parija S, Allesu K (1997) Microwelding using electrochemical discharges. Int J Mach Tools Manuf 37:1303–1312
Allesu K, Ghosh A, Muju MK (1992) Preliminary qualitative approach of a proposed mechanism of material removal in electrical machining of glass. Eur J Mech Eng 36:202–207
Basak I, Ghosh A (1997) Mechanism of material removal in electrochemical discharge machining: a theoretical model and experimental verification. J Mater Process Technol 71:350–359
Jain VK, Rao PS, Chaudhury SK, Rajurkar KP (1991) Experimental investigations into travelling wire electrochemical spark machining (TW-ECSM) of composites. Trans ASME J Eng Ind 113:75–84
Singh YP, Jain VK, Kumar P, Agrawal DC (1996) Machining piezo-electric (PZT) ceramics using an electrochemical spark machining (ECSM) process. J Mater Process Technol 58:24–31
Gautam N, Jain VK (1998) Experimental investigations into ECSD process using various tool kinematics. Int J Mach Tools Manuf 38:15–27
Jain VK, Dixit PM, Pandey PM (1999) On the analysis of the electrochemical spark machining process. Int J Mach Tools Manuf 39:165–186
Jain VK, Chak SK (2000) Electrochemical spark trepanning of alumina and quartz. Mach Sci Technol 4:277–290
Jain VK, Chaudhury SK, Ramesh KM (2002) On the machining of alumina and glass. Int J Mach Tools Manuf 42:1269–1276
Bhattacharyya B, Doloi BN, Sorkhel SK (1999) Experimental investigations into electrochemical discharge machining (ECDM) of non-conductive ceramic materials. J Mater Process Technol 95:145–154
Fascio V, Langen HH, Bleuler H, Comninellis Ch (2003) Investigations of the spark assisted chemical engraving. Electrochem Commun 5:203–207
Fascio V, Wuthrich R, Bleuler H (2004) Spark assisted chemical engraving in the light of electrochemistry. Electrochim Acta 49:3997–4003
Wuthrich R, Hof L, Lal A, Fujisaki A, Bleuler K, Mandin Ph H, Picard G (2005) Physical principles and miniaturization of spark assisted chemical engraving (SACE). J Micromech Microeng 15:S268–S275
Wuthrich R, Hof LA (2006) The gas film in spark assisted chemical engraving (SACE) a key element for micro-maching applications. Int J Mach Tools Manufact 46:828–835
Wuthrich R, Spaelter U, Wu Y, Bleuler H (2006) A systematic characterization method for gravity feed micro hole drilling in glass with spark assisted chemical engraving (SACE). J Micromech Microeng 16:1891–1896
Mailard P, Despont B, Bleuler H, Wuthrich R (2007) Geometrical characterization of micro holes drilled in glass by gravity feed with spark assisted chemical engraving (SACE). J Micromech Microeng 17:1343–1349
Jalali M, Mailard P, Wuthrich R (2009) Towards a better understanding of glass gravity-feed micro-hole drilling with electrochemical discharges. J Micromech Microeng 19:045001
Hourng LW, Lin CI, Lee BG (2014) The improvement of machining accuracy on quartz and glasses by electrochemical discharge machining. Appl Mech Mater 72:682–687
Jain VK, Adhikary S (2008) On the mechanism of material removal in electrochemical spark machining of quartz under different polarity conditions. J Mater Process Technol 200:460–470
West J, Jadhav A (2007) ECDM methods for fluidic interfacing through thin glass substrates and the formation of spherical microcavities. J Micromech Microeng 17:403–409
Huang SF, Liu Y, Li J, Hu HX, Sun LY (2014) Electrochemical discharge machining micro-hole in stainless steel with high-speed rotating tool electrode. Mater Manuf Process 29:634–637
Karnik M, Ghosh A, Shekhar R (2011) Polarity dependence of the electrochemical discharge (ECD). Key Eng Mater 486:131–134
Coteata M, Schulze HP, Slatineanu L (2011) Drilling of difficult-to-cut steel by electrochemical discharge machining. Mater Manuf Process 26:1466–1472
Huang S, Zhu D, Zeng Y, Wang W, Liu Y (2011) Micro-hole machined by electrochemical discharge machining (ECDM) with high speed rotating cathode. Adv Mater Res (Durnten-Zurich) 295–297:1794–1799
Palit SR (1967) Electrode glow during electrolysis and liberation of hydrogen and oxygen together on the electrodes. Indian J Phys 41:860–861
Palit SR (1968) Liberation of hydrogen and oxygen together on the electrodes during electrolysis accompanied by electrode glow. Indian J Phys 42:414–418
Sen Gupta SK, Palit SR (1975) Studies in galvanoluminescence: part II chemical effects of cathodic glow. J Indian Chem Soc 52:91–97
Sen Gupta SK, Singh OP (1994) Contact glow discharge electrolysis: a study of its chemical yields in aqueous inert-type electrolytes. J Electroanal Chem 369:113–120
Mizuno T, Akimoto T, Azumi K, Ohmori T, Aoki Y, Takahashi A (2005) Hydrogen evolution by plasma electrolysis in aqueous solution. Jpn J Appl Phys 44:396–401
Mizuno T, Aoki Y, Chung DY, Sesftel F (2006) Generation of heat and products during plasma electrolysis. In: Biberian JP (ed) Proceedings of the 11th international conference of cold fusion marseilles France 2004 in condensed matter nuclear science. World Scientific Publishing Company, Singapore, pp 161–177
Sen Gupta SK, Srivastava AK, Singh R (1997) Origin of contact glow discharge electrolysis in aqueous solutions: effects of electrolyte temperature and surface tension. Indian J Chem 36A:945–950
Sen Gupta SK, Srivastava AK, Singh R (1997) Contact glow discharge electrolysis: a study on its origin in the light of the theory of hydrodyanamic instabilities in local solvent vaporization by Joule heating during electrolysis. J Electroanal Chem 427:23–27
Acknowledgements
The author thanks Banaras Hindu University, Varanasi for financial and infrastructural support. The author thanks Mr. Vishal Tiwari for his meticulous assistance in computer work.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sen Gupta, S.K. Contact Glow Discharge Electrolysis: A Novel Tool for Manifold Applications. Plasma Chem Plasma Process 37, 897–945 (2017). https://doi.org/10.1007/s11090-017-9804-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11090-017-9804-z